

Abstract
EpsinR is a clathrin-coated vesicle (CCV)-associated protein that binds to vti1b, suggesting that it may be a vti1b-selective adaptor. Depletion of epsinR to undetectable levels in HeLa cells using siRNA causes vti1b to redistribute from the perinuclear region to the cell periphery, but vti1a also redistributes in epsinR-depleted cells, and both vti isoforms redistribute in AP-1–depleted cells. As a more direct assay for epsinR function, we isolated CCVs from control and siRNA-treated cells and then looked for differences in cargo content. In clathrin-depleted cells, both coat and cargo proteins are greatly reduced in this preparation. Knocking down epsinR causes a ∼50% reduction in the amount of AP-1 copurifying with CCVs and vice versa, indicating that the two proteins are dependent on each other for maximum incorporation into the coat. In addition, vti1b, but not vti1a, is reduced by >70% in CCVs from both epsinR- and AP-1–depleted cells. Because AP-1 knockdown reduces the amount of epsinR in CCVs, it is possible that its effect on vti1b may be indirect. These findings provide in vivo evidence that epsinR is an adaptor for vti1b, and they also show that CCV isolation can be used as an assay for adaptor function.
INTRODUCTION
Transport vesicles contain many different types of cargo proteins, but none are more important than the SNAREs, because without SNAREs the vesicles would be unable to fuse with the target membrane. To get packaged into vesicles, cargo proteins must contain specific sorting signals that interact with components of the vesicle coat. For instance, tyrosine- and dileucine-based motifs (YXXϕ and [D/E]XXXL[L/I]) interact with adaptor protein (AP) complexes such as AP-1 and AP-2, which are associated with clathrin-coated vesicles (CCVs) budding from TGN/endosomal membranes and from the plasma membrane, respectively (Bonifacino and Traub, 2003). However, with the exception of VAMP4, which contains a dileucine motif and binds to AP-1 (Peden et al., 2001), most of the SNAREs that are packaged into CCVs lack defined sorting signals.
Although AP-1 and AP-2 were originally assumed to be the only cargo adaptors for clathrin-mediated trafficking, recent studies suggest that, at least at the plasma membrane, there are other, more minor adaptors that select cargo proteins lacking binding sites for AP-2. These cargo-selective adaptors all bind to the appendage or “ear” domains of the large subunits of the AP-2 complex, and they also all interact with clathrin, with PIP2, which helps to target them to the plasma membrane, and with different types of internalization signals. For instance, the epsins have ubiquitin-interacting motifs, which may facilitate the internalization of ubiquitinated cargo proteins such as activated growth factor receptors. Dab2 and ARH are two cargo-selective adaptors that bind to sorting signals found in members of the LDL receptor family (Traub, 2003). We have recently shown that these adaptors can function even in the absence of any detectable AP-2. When we knock down AP-2 in HeLa cells using siRNAs, clathrin-mediated endocytosis of the transferrin receptor goes down to background levels, but clathrin-mediated endocytosis of the EGF receptor and of an LDL receptor chimera (as assayed by binding the ligand to the cell surface at 4°C and then warming the cells to 37°C) still proceed normally, even though there are 12-fold fewer clathrin-coated pits at the plasma membrane (Motley et al., 2003; see also Conner and Schmid, 2003; Hinrichsen et al., 2003; Huang et al., 2004). Thus, it now appears that AP-2 is just one, albeit very abundant, adaptor for clathrin-mediated endocytosis.
Although less is known about clathrin-mediated trafficking from intracellular membranes, we and others have identified a number of binding partners for the appendage domain of the AP-1 γ subunit, which are candidates for alternative adaptors on the AP-1 pathway. One of these proteins, which has been variously named epsinR (Hirst et al., 2003; Mills et al., 2003), enthoprotin (Wasiak et al., 2002), and Clint (Kalthoff et al., 2002), is a particularly promising candidate, because it binds not only to AP-1, but also to clathrin and to phosphoinositides. The phosphoinositide interaction is mediated by the ENTH (epsin N-terminal homology) domain of epsinR, but unlike the ENTH domains of epsins 1–3, which bind PIP2, the ENTH domain of epsinR prefers PI(4)P, a phosphoinositide mainly generated on TGN membranes. AP-1 also binds PI(4)P, and in both cases there is evidence that this interaction is important for recruiting the proteins onto the appropriate membrane (Wang et al., 2003). Another difference between epsinR and the epsins is that epsinR has no ubiquitin interacting motif, suggesting that if it is indeed an adaptor, it probably does not recognize ubiquitinated proteins but some other type of cargo. In a recent yeast two-hybrid library screen for binding partners for the endosomal SNARE protein, vti1b, Chidambaram et al. (2004) isolated epsinR, and they suggested that this interaction might help to sort vti1b into budding vesicles.
In our previous study on AP-2, we were able to quantify the packaging of different types of cargo into CCVs at the plasma membrane by using ligand uptake assays (Motley et al., 2003). However, this method cannot be used to study intracellular trafficking, nor can it be used to monitor the trafficking of SNAREs because most of the SNAREs have little or no extracellular or lumenal domain. Therefore, we have developed a method for isolating CCVs from HeLa cells so that cargo content could be assayed in cells that were depleted of particular coat components by RNAi. In this way, we have been able to test the hypothesis that epsinR is an adaptor for vti1b.
MATERIALS AND METHODS
Plasmid Construction and GST Pull-downs
Standard molecular biology techniques were used throughout this study (Sambrook and Russell, 2001). For pull-down experiments, GST fusion proteins were made with the NH2-terminal domains of epsinR (amino acids 1–166) and vti1b (amino acids 1–206) and pull-downs were carried out essentially as previously described (Hirst et al., 2003), using HeLa extracts in PBS containing 0.1% NP-40 and the protease inhibitor AEBSF, at a protein concentration of 1.5 mg/ml. Bound proteins were eluted with sample buffer and subjected to SDS-PAGE. Gels were transferred onto nitrocellulose for Western blotting.
Antibodies and Blotting
Rabbit antibodies against epsinR, AP-1 γ, clathrin, CI-MPR, and μ1A were raised in house and have already been described (Page et al., 1999; Hirst et al., 2003). Monoclonal antibodies were purchased from BD Transduction Laboratories (Lexington, KY; anti-vti1a, anti-vti1b, and anti-μ2), from Santa Cruz Biotechnology (Santa Cruz, CA; anti-EF-2), from Sigma (St. Louis, MO; antig), and from Qbiogene (Carlsbad, CA; anti-CD8). Western blots were probed with these antibodies, followed by a rabbit anti-mouse linker where appropriate and then by 125I-protein A as previously described (Hirst et al., 2003). Samples included GST pull-downs, whole cell extracts, and CCVs isolated from tissue culture cells. Quantifications were carried out using a Packard Cyclone phosphorimager (Meriden, CT).
Microscopy
For immunofluorescence, HeLa cells were fixed with 3% paraformaldehyde, followed by permeabilization with 0.1% Triton X-100. The CD8-furin–expressing cell line was a gift from Matthew Seaman (CIMR, University of Cambridge, United Kingdom; Seaman, 2004). Primary antibodies are listed above; secondary antibodies were purchased from Molecular Probes (Eugene, OR). Cells were viewed using a Zeiss Axiophot fluorescence microscope (Thornwood, NY) equipped with a CCD camera (Princeton Instruments, Princeton, NJ) and photographs were recorded using IP Labs software (Scanalytics, Fairfax, VA). The GFP-conjugated clathrin light-chain construct was a kind gift of Stephen Royle (MRC Laboratory of Molecular Biology). Transfections with this construct were carried out using Fugene (Roche Diagnostics, Lewes, East Sussex, United Kingdom) according to the manufacturer's instructions, and specimens were viewed without fixation.
For electron microscopy, control and siRNA-treated HeLa cells were prepared as previously described (Motley et al., 2003) and observed in a Philips CM100 transmission electron microscope (Mahwah, NJ). For morphometric analyses, electron micrographs were taken of the Golgi region of control, epsinR-depleted, and μ1-depleted cells. To choose the regions to be photographed in an unbiased way, Golgi stacks were found at low magnification, under conditions where clathrin-coated buds and vesicles could not be resolved (×7,900) and then photographed at higher magnification (×39,000). Two measurements were made. First, individual clathrin-coated budding profiles within 1 μm of a Golgi stack were counted in 20 micrographs for each of the three conditions. Second, the diameters of 25 clathrin-coated budding profiles that had been caught in cross section were measured using analySIS 3.0 image analytical software (Soft Imaging System, Lakewood, CO).
RNA Interference
siRNA duplexes against conserved sequences of the target cDNAs were purchased from Dharmacon (Boulder, CO). An epsinR sequence, AACCAUUGAUCUUGGAGCAGC, which had previously been found to be ineffective for knockdown experiments (Hirst et al., 2003), was used as a control. For epsinR knockdowns, a new siRNA with the sequence AAUACAGAUAUGGUCCAGAAA was found to be more effective than the one used in our previous article (Hirst et al., 2003). siRNAs against clathrin and μ1A have been described (Hirst et al., 2003; Motley et al., 2003). HeLa cells were transfected using Oligofectamine (Invitrogen, Carlsbad, CA) as specified by the manufacturer. For efficient knockdown two transfections were performed 2 d apart, and experiments were carried out 2 d after the second knockdown.
CCV Isolation
CCVs were isolated from HeLa cells growing on six to eight 9-cm2 tissue culture dishes (for small-scale preparations) or on four 500-cm2 tissue culture dishes (for large-scale preparations). The cells were rinsed briefly with ice-cold PBS followed by ice-cold buffer A (0.1 M MES, pH 6.5, 0.2 mM EGTA, 0.5 mM MgCl2, 0.02% NaN3, 0.2 mM PMSF) and then scraped with a sliced rubber stopper into either ∼1 ml buffer A per dish (small scale) or ∼5 ml buffer A per dish (large scale). Homogenization was carried out using 15 strokes of a motorized Potter glass homogenizer. Other methods of homogenization, e.g., using a ball-bearing homogenizer or a sonicator, did not significantly improve the yield or purity of the CCVs. The homogenates were centrifuged in a Beckman S4180 rotor (Fullerton, CA) at 4800 rpm for 32 min, after which the supernatant was treated with 50 mg/ml RNase A (MP Biomedicals, Orangebay, NJ) for 30 min at 4°C. The membranes were pelleted by spinning at 50,000 rpm for 30 min in a Beckman TLA100.4 rotor and then resuspended in 300 μl-1 ml buffer A using a 1-ml hand-held glass homogenizer. The suspension was mixed with an equal volume of 12.5% Ficoll/12.5% sucrose in buffer A and centrifuged in a TLA100.4 rotor at 20,000 rpm for 25 min. The supernatant was diluted with four volumes of buffer A, and the CCVs were pelleted by spinning in a TLA100.4 rotor at 50,000 rpm for 30 min. The pellets were resuspended in 30–100 μl buffer A using a 0.1-ml hand-held glass homogenizer. Yield was determined by quantifying the volume and protein concentration at each step and then probing Western blots of equal protein loadings of the various fractions with anticlathrin heavy chain followed by 125I-protein A and measuring the bound radioactivity using a phosphorimager.
Further purification of the CCVs was achieved by sucrose density gradient centrifugation. A step gradient of 40, 43, 46, 49, 52, 55, and 60% sucrose in buffer A (400 μl each) was prepared, and the resuspended pellet from a large scale preparation was layered on the top. The sample was centrifuged for 2 h at 52,000 rpm in a Beckman SW55 Ti rotor, and 200-μl fractions were collected from the top. The fractions were diluted with five volumes of buffer A, pelleted by spinning in a TLA100.4 rotor at 50,000 rpm for 30 min, and resuspended in 75 μl buffer A using a 0.1-ml hand-held glass homogenizer. The fractions were assayed by SDS-PAGE followed by MALDI-TOF mass spectrometry, by Western blotting, by fluorescence microscopy, and by electron microscopy of negatively stained preparations.
RESULTS
Morphology of the epsinR-depleted Cells
We have previously shown that we can deplete epsinR to ∼15% of normal levels by RNAi in both HeLa and COS cells without impairing the sorting of the lysosomal enzyme cathepsin D, although knocking down AP-1 caused cathepsin D to be secreted (Hirst et al., 2003; see also Supplementary Figure 1). However, if epsinR is a cargo-selective adaptor, then we would expect that knocking it down would cause vti1b to be missorted without necessarily affecting the trafficking of other proteins. To optimize our knockdowns, we have now synthesized a different epsinR siRNA, because the original one was somewhat toxic, and we have also modified our procedure so that the cells are now treated twice over a period of 5 d. In this way we are able to deplete epsinR to levels that are undetectable by both immunofluorescence and Western blotting, with nearly 100% of the cells affected (see Figures 1d, 2, and 7).
Figure 1.

Morphology of control and siRNA-treated cells. (a–c) Electron micrographs of epsinR-depleted (a), AP-1–depleted (b), and control cells (c). In both knockdowns, the overall morphology of the Golgi region looks normal and clathrin-coated budding profiles (arrowheads) can be seen. Morphometric analyses are shown in Table 1. Scale bar, 200 nm. (d and e) A mixture of control and epsinR-depleted cells was double-labeled for epsinR (d) and AP-1 (e). Perinuclear labeling of AP-1 is more pronounced in the epsinR-depleted cells. Scale bar, 10 μm.
Figure 2.

Effects of knockdown on the steady state distribution of vti1b and vti1a. (a–d) Mixtures of control and epsinR-depleted cells were double-labeled for epsinR (a) and vti1b (b) or for epsinR (c) and vti1a (d). Both vti1 isoforms redistribute to the cell periphery in the epsinR-depleted cells. (e–h) Mixtures of control and μ1-depleted cells were double-labeled for the AP-1 γ subunit (e) and vti1b (f) or for the AP-1 γ subunit (g) and vti1a (h). Again, both vti1 isoforms redistribute to the cell periphery in the knockdown cells. Scale bar, 10 μm.
Figure 7.

Effect of AP-1 and epsinR knockdown on the composition of the CCVs. a, Western blots of homogenates of control and siRNA-treated cells show that the knockdowns are specific. (b) Western blots of the CCV preparation from the cells shown in a were probed with the indicated antibody. The bar graph shows phosphorimager data pooled from 4–6 independent experiments, ±SE. Both epsinR and AP-1 knockdowns greatly reduce the amount of vti1b in the preparation, with little or no effect on the amount of vti1a.
By electron microscopy, the epsinR-depleted cells look essentially normal (Figure 1a), unlike clathrin-depleted cells, which have swollen post-Golgi membrane compartments and no detectable clathrin-coated buds or vesicles (Motley et al., 2003). AP-1–depleted cells also have a normal post-Golgi morphology (Figure 1b), and clathrin-coated membranes can be seen in the Golgi region in both epsinR and AP-1 knockdowns (arrowheads). However, we could detect some subtle differences between the siRNA-treated cells and controls (Figure 1c and Table 1). The number of clathrin-coated budding profiles in the vicinity of the Golgi stack is increased by ∼50% in the cells that lack epsinR and is decreased by ∼50% in the cells that lack μ1. In addition, in the cells that lack epsinR, the mean diameter of these profiles is ∼50% larger. This apparent increase in the amount of coating of TGN and post-TGN membrane in the cells that lack epsinR could also be seen by immunofluorescence. Figure 1, d and e, shows that AP-1 labeling in the Golgi region is more pronounced in the epsinR-depleted cells than in controls. Nevertheless, we saw no gross changes in the appearance of the epsinR-depleted cells (see also Figure 3c), suggesting that the enlarged perinuclear compartments seen by light microscopy in cells overexpressing epsinR (Mills et al., 2003; Saint-Pol et al., 2004) may be indirect effects of overexpression rather than direct consequences of interfering with epsinR's normal function.
Table 1.
Morphometric analyses of clathrin-coated budding profiles in control and siRNA-treated cells
| Control | EpsinR knockdown | μ1 knockdown | |
|---|---|---|---|
| Frequency (per Golgi stack) | 1.06 | 1.56 | 0.5 |
| Mean diameter (nm) (±SD) | 63.95 ± 10.47 | 94.61 ± 16.83 | 70.51 ± 10.41 |
Electron micrographs were taken of the Golgi region of control, epsinR-depleted, and μ1-depleted cells. To determine the frequency of clathrin-coated budding profiles, individual profiles within 1 μm of a Golgi stack were counted in 20 micrographs for each of the three conditions. The numbers were as follows: for the control cells, 35 profiles and 33 stacks; for the epsinR-depleted cells, 39 profiles and 25 stacks; and for the μ1-depleted cells, 15 profiles and 30 stacks. To determine the mean diameters of the clathrin-coated budding profiles, 25 profiles that had been caught in cross section were measured for each of the three conditions.
Figure 3.

Effects of knockdown on the steady state distribution of a CD8-furin chimera. In control cells (a), the chimera is localized mainly to the TGN. In μ1-depleted cells (b), it redistributes to more peripheral membranes and to the cell surface. However, in epsinR-depleted cells (c), its localization is unchanged. Scale bar, 10 μm.
Localization of Cargo Proteins in siRNA-treated Cells
To investigate the effect of epsinR depletion on the steady state distribution of vti1b, we double-labeled mixtures of control and siRNA-treated cells with antibodies against epsinR and vti1b. Figure 2, a and b, shows that in untreated cells vti1b (b) has a strongly perinuclear distribution, similar although not identical to that of epsinR (a). In the cells that lack epsinR, the vti1b labeling is much more scattered, although by Western blotting the total amount of vti1b was found to be unchanged (see Figure 7a). We also examined the effect of epsinR depletion on vti1a, which is 33% identical to vti1b, but which is unable to bind to epsinR in vitro or in the yeast two-hybrid system (Chidambaram et al., 2004). Surprisingly, we found that knocking down epsinR had a similar effect on the distribution of vti1a (Figure 2, c and d). In addition, when we knocked down the μ1 subunit of the AP-1 adaptor complex, we also saw a redistribution of both vti1b and vti1a away from the perinuclear region (Figure 2, e–h). These results indicate either that both vti1 isoforms interact with both epsinR and AP-1, or alternatively that at least some of the effects of knockdown on the steady state distribution of vti1b and vti1a may be indirect.
We also looked at the effects of epsinR and AP-1 knockdown on other membrane proteins that are known to cycle between the TGN and endosomes in a clathrin-dependent manner. We saw a striking redistribution of a CD8-furin chimera in AP-1–depleted cells. Normally this construct resides mainly in the TGN (Figure 3a; see also Seaman, 2004), but in the absence of AP-1, much of it is found in peripheral vesicles and at the plasma membrane (Figure 3b). In contrast, epsinR depletion had no apparent effect on the localization of the CD8-furin chimera (Figure 3c). The localization of TGN46 and of the cation-independent mannose 6-phosphate receptor were also found to be unaffected by epsinR knockdown (unpublished results). Thus, so far we have only been able to detect an effect of epsinR depletion on the steady state distribution of vti1b and vti1a.
Because previous binding studies on epsinR, vti1b, and vti1a had been done using entirely recombinant proteins (Chidambaram et al., 2004), we could not rule out the possibility that interactions that occur in the context of the whole cell might have been missed. Therefore, we used a GST-epsinR construct to pull down binding partners from HeLa cell extracts, then probed with antibodies against either vti1b or vti1a. Figure 4 shows that under these conditions, only vti1b comes down with epsinR. We also did pull-downs using a GST-vti1b construct, then probed with antibodies against either epsinR or AP-1, but we were only able to detect epsinR (Figure 4). Thus, there is a discrepancy between the data shown in Figures 2 and 4. Knocking down either epsinR or AP-1 strongly affects the steady state distribution of both vti1b and vti1a, yet we can only detect an interaction in vitro between epsinR and vti1b.
Figure 4.

GST pull-downs from HeLa cell extracts. Cell extracts were incubated with either a GST-epsinR construct or a GST-vti1b construct, and binding partners were pulled down with glutathione-Sepharose. The lefthand lane contains the starting material; the other lanes contain the material that was pulled down. Western blots were probed with anti-vti1b, anti-vti1a, anti-epsinR, and anti-AP-1 (γ subunit). The epsinR band is indicated with an arrowhead; the band indicated with an asterisk is the GST-vti1b construct, which cross-reacts with the antibody because it was raised against GST-epsinR. The only interaction we can detect in the pull-downs is between epsinR and vti1b.
Isolation of CCVs from HeLa Cells
Because the steady state distribution of a membrane protein can be influenced by more than one trafficking pathway and by more than one protein-protein interaction, we needed a more direct assay for determining whether epsinR or AP-1 knockdown was preventing either vti1b or vti1a from getting packaged into CCVs. Therefore, we decided to develop a method for isolating CCVs from HeLa cells, either with or without first treating the cells with siRNAs.
One advantage of using cultured cells is that they can be experimentally manipulated, and we were able to exploit this by using HeLa cells that had been transiently transfected with GFP coupled to clathrin light chain to monitor the release of CCVs after cell breakage. Figure 5a shows that after disrupting the cells (either by homogenization or by sonication), many clathrin-coated spots remained associated with membrane fragments, and by Western blotting (Figure 5b) we found that ∼50% of the total clathrin came down in the first low-speed spin and had to be discarded. Most of the remaining clathrin was soluble and remained in the supernatant when centrifuged at high speed. After resuspending the high-speed pellet and centrifuging at moderate speed in a Ficoll/sucrose solution to remove the remaining larger particles, we were able to isolate a CCV-enriched fraction that contained ∼1% of the total clathrin from the starting material (Figure 5b, lane 4). This low yield presumably reflects the situation in the intact cell, where free CCVs are relatively scarce compared with coated buds still attached to membranes and nonassembled clathrin.
Figure 5.

Isolation of CCVs from HeLa cells. (a) Cells expressing GFP-clathrin light chain were disrupted by homogenization. Many clathrin dots remain associated with membrane fragments. Scale bar, 10 μm. (b) Coomassie blue–stained gel and Western blot of equal protein loadings of fractions from the isolation procedure. Lane 1, total cell homogenate. Lane 2, low-speed supernatant. Lane 3, high-speed pellet. Lane 4, Ficoll/sucrose-enriched preparation. The position of the band containing clathrin heavy chain (which also contains glycogen debranching enzyme) is indicated. (c) Coomassie blue–stained gel of the Ficoll/sucrose-enriched preparation (F/S) and fractions from a sucrose density gradient. The indicated bands were excised and identified by mass spectrometry as clathrin heavy chain (1), β1 (2), glycogen synthase (3), tripeptidyl peptidase (4), EF-2 (associated with ribosomes) (5), and ribosomal proteins (6). (d) Western blot of the samples shown in c, probed with antibodies against clathrin heavy chain (CHC), the AP-1 γ and AP-2 μ2 subunits, epsinR, the cation-independent mannose 6-phosphate receptor (CI-MPR), vti1a, and vti1b. (e) Fluorescence micrograph of fraction 5, containing GFP-labeled clathrin-coated vesicles. Scale bar, 10 μm. (f–h) Electron micrographs of negatively stained samples from the sucrose gradient: fraction 5, containing clathrin-coated vesicles (f); fraction 1, containing smooth membranes (g); and pooled fractions from the bottom of the gradient, containing glycogen particles (h). Scale bar, 500 nm.
The CCVs could be further purified by sucrose density gradient centrifugation (Figure 5, c–f), which effectively separated them from smooth membranes (Figure 5g) and glycogen particles (Figure 5h) and partially separated them from ribosomes (Figure 5c). However, this was at the expense of yield and only ∼20% of the clathrin added to the gradient could be recovered, possibly because many of the vesicles lost their coats during the procedure. Western blots of the gradient fractions (Figure 5d) showed that clathrin, AP-1, AP-2, and epsinR all had essentially identical profiles, whereas vesicle cargo proteins including the cation-independent mannose-6 phosphate receptor, vti1a, and vti1b all fractionated as two peaks, one at the light end of the gradient with the uncoated membranes and one in the middle of the gradient with clathrin and the other coat proteins.
We next examined the effect of knocking down clathrin heavy chain on the composition of the “CCV-enriched” preparation (i.e., after the Ficoll/sucrose step but without the gradient). Because there is little or no detectable clathrin left in these cells, this preparation should still contain contaminants, but it should be devoid of CCVs. Figure 6 shows that although Coomassie blue–stained gels of the preparation do not look strikingly different in the clathrin-depleted cells when compared with controls, by Western blotting there are no detectable coat proteins after clathrin knockdown (see also Figure 7b). With the exception of clathrin, these proteins are still expressed at normal levels (Figure 7a), so this indicates that without clathrin they fractionate differently because they cannot be incorporated into vesicles. Surprisingly, cargo proteins including the cation-independent mannose 6-phosphate receptor, vti1a, and vti1b are also reduced to very low levels in the preparation after clathrin knockdown (Figure 6; see also Figure 7b), even though by sucrose gradient fractionation ∼50% of the cargo proteins are associated with smooth membranes (see Figure 5d). This suggests that the smooth vesicles containing the cargo are mainly derived from CCVs, which became uncoated either during the preparation or inside the cell.
Figure 6.
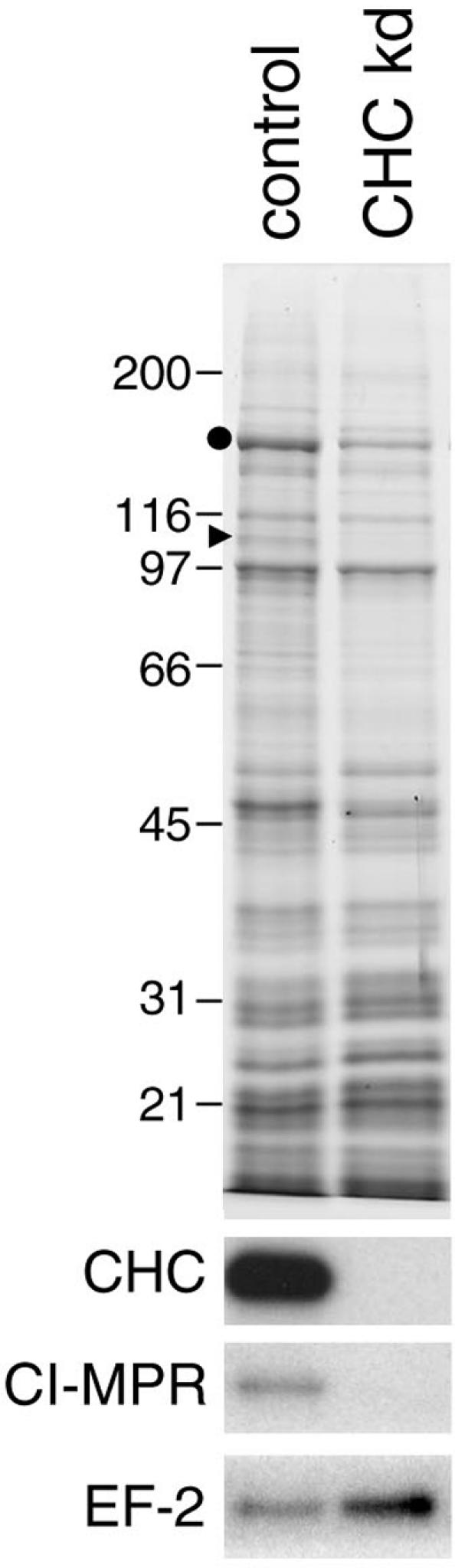
CCVs isolated from control and clathrin-depleted cells. The Coomassie blue–stained gel shows that all of contaminants are still present after clathrin knockdown. The bands containing clathrin heavy chain and β1 (identified by mass spectrometry) are indicated with a circle and a triangle, respectively. On the Western blots, there is little or no signal in the clathrin knockdown lane for either coat proteins (e.g., clathrin heavy chain) or cargo proteins (e.g., the CI-MPR), whereas contaminants (e.g., EF-2) are still present.
CCVs from epsinR-depleted Cells
Having established that clathrin knockdown reduces the amount of both vti1b and vti1a to very low levels in the fraction normally containing CCVs, we repeated the procedure on control cells, cells depleted of clathrin, cells depleted of the μ1 subunit of AP-1, and cells depleted of epsinR. Western blots of equal protein loadings of total cell homogenates and of the CCV-enriched fraction were then probed with antibodies against all of these proteins as well as with antibodies against vti1b and vti1a. Figure 7a shows that in every case, the cell homogenates contained either undetectable levels or <5% of normal levels of the protein that was knocked down, with little or no effect on the other proteins.
Figure 7b shows the CCV-enriched preparation probed with the same antibodies. Knocking down either μ1 or epsinR does not significantly affect the amount of clathrin recovered in this fraction, although (as also shown in Figure 6) the signal from the clathrin heavy-chain band goes down to <5% of control when it is itself knocked down. μ1 and epsinR go down to a similar extent in the CCV-enriched fraction when clathrin is depleted as well as when they are themselves depleted. In addition, knocking down epsinR caused a ∼50% reduction in the amount of μ1 in the CCV-enriched fraction, and knocking down μ1 caused a ∼50% reduction in the amount of epsinR. This suggests that the two proteins are to some extent dependent on each other for incorporation into the coat.
When the blots were probed with anti-vti1b, the signal after epsinR depletion was down to 16.6 ± 3.4% of control levels, thus approaching the background level seen after clathrin depletion (6.4 ± 2.7% of control). The signal for vti1b in the CCV preparation was also strongly reduced after μ1 depletion, to 23.2 ± 3.0% of control. Because AP-1 depletion reduces the amount of epsinR associated with the CCVs and vice versa, it is difficult to dissociate the effects of the two knockdowns. Thus, it is possible that the effect of μ1 depletion on the amount of vti1b in CCVs may be indirect, mediated by epsinR. As a control, we also analyzed CCV preparations from cells depleted of the μ2 subunit of AP-2 and saw no effect on vti1b (Supplementary Figure 2).
In contrast to vti1b, vti1a is only slightly reduced in the CCVs after μ1 depletion (82.9 ± 5.1%, as compared with 20.3 ± 4.8% in the clathrin-depleted cells), and it is not significantly reduced after epsinR depletion (99.1 ± 8.0%). This indicates that vti1a is still getting packaged efficiently into CCVs even in the absence of any detectable AP-1 or epsinR. Thus, neither epsinR nor AP-1 appears to act as an essential cargo adaptor for vti1a.
DISCUSSION
We have used a novel method to investigate the role of three different coat components, clathrin, AP-1, and epsinR, in CCV formation and cargo selection. CCVs were isolated from control and siRNA-treated cells, and the protein composition of the preparations was compared by Western blotting. We found that clathrin knockdown caused every coat protein we tested to be depleted to <5% of control levels. With the exception of clathrin, these proteins are still expressed normally, and they are also still associated with membranes (unpublished observations), so their loss from the preparation indicates that without clathrin they cannot be incorporated into vesicles. Cargo proteins were also depleted from the preparation, although less severely: typically they went down to ∼20% of control levels. Thus, clathrin knockdowns can be used as a baseline when examining the effects of knocking down other coat components on CCV composition.
We found that AP-1 depletion caused a ∼50% reduction in the amount of epsinR associated with CCVs. This is consistent with our EM observations, which showed that there were only about half the number of clathrin-coated budding profiles in the Golgi region in AP-1–depleted cells when compared with controls. The remaining clathrin-coated buds are presumably nucleated by other intracellular clathrin adaptors, including epsinR, which we have previously shown can be recruited onto membranes in an AP-1–independent manner (Hirst et al., 2003). Thus, the AP-1 knockdown phenotype is similar in this regard to the AP-2 knockdown phenotype. AP-2 depletion using siRNA causes a reduction in the number of clathrin-coated pits at the plasma membrane, but it does not completely abolish their formation, and the remaining coated pits are still positive for alternative endocytic adaptors, such as epsin 1 (Motley et al., 2003). Somewhat surprisingly, depleting AP-1 did not cause any apparent decrease in the amount of clathrin we recovered. This suggests that, even though AP-1 is the major adaptor in our preparations, other less abundant adaptors are able to facilitate CCV formation so that the final yield is not affected.
We also found that epsinR depletion caused an ∼50% reduction in the amount of AP-1 associated with CCVs. This is more difficult to explain than the effect of AP-1 depletion on epsinR, especially because electron micrographs indicate that there are actually more clathrin-coated budding profiles in the Golgi region in epsinR-depleted cells than in controls. However, we found that the mean diameter of the buds was larger in these cells, suggesting that there may be a block in the formation of free coated vesicles. Epsin 1 has been proposed to play a role in membrane curvature (Ford et al., 2002), and the same may be true for epsinR. Another possibility is that loss of epsinR, which is the major γ ear binding partner in CCVs (Hirst et al., 2003), may cause an increase in the relative amounts of other γ ear partners at sites of CCV formation, and this might in turn lead to changes in vesicle invagination and/or scission. In any case, the epsinR knockdown phenotype is clearly very different from the phenotype described for epsinR-overexpressing cells, which have an enlarged TGN compartment, mislocalization of TGN46 and of the cation-independent mannose 6-phosphate receptor, and increased secretion of cathepsin D (Mills et al., 2003; Saint-Pol et al., 2004). This may be because overexpression of epsinR can lead to indirect effects that are unrelated to epsinR function. For instance, there may be sequestration of some of epsinR's binding partners, such as AP-1 and clathrin.
When we assayed for vti1b in CCVs isolated from epsinR-depleted cells, we found that it was reduced nearly to background levels. This observation provides in vivo evidence to support the hypothesis that epsinR is a cargo adaptor for vti1b, which was originally proposed on the basis of its ability to bind to vti1b in vitro. Further evidence comes from our observation that epsinR depletion alters the steady state distribution of vti1b, causing it to move away from the Golgi region toward the cell periphery. EpsinR has recently been implicated in the retrograde trafficking of internalized Shiga toxin (Saint-Pol et al., 2004), and the same may be true for vti1b. Although vti1b has been implicated mainly in fusion events involving late endosomes, it must be returned to earlier compartments to be incorporated into outgoing vesicles, and this may be a function of epsinR.
Although the effects of epsinR depletion on vti1b are entirely consistent with its proposed role as a vti1b-selective adaptor, two of our other observations are more difficult to explain. First, AP-1 depletion has an effect very similar to epsinR depletion on vti1b, yet there is no evidence from our pull-down experiments for an interaction between AP-1 and vti1b. One possibility is that such an interaction exists but it has not yet been detected. The interaction would not even have to be direct; normally vti1b is associated with other SNAREs (syntaxin 7, syntaxin 8, and—in the complete v-/t-SNARE complex —VAMP-8), so AP-1 could be an adaptor for one of these other proteins. Alternatively, AP-1 may be interfering with vti1b trafficking through its effects on epsinR. Depleting AP-1 causes an ∼50% reduction in the amount of epsinR associated with isolated CCVs, and although AP-1 depletion has a more profound effect on the amount of vti1b in CCVs, it is possible that relatively small changes in epsinR concentration may lead to larger changes in vti1b concentration. We are now in the process of mapping the epsinR-binding site on vti1b, with the aim of introducing mutated constructs into HeLa cells. If we can show that epsinR-binding mutants are no longer incorporated into CCVs, this should help us to dissociate the requirements for epsinR and AP-1 in vti1b sorting.
The second puzzling observation is our finding that epsinR and AP-1 knockdowns alter the steady state distribution of vti1a as well as vti1b. Unlike vti1b, vti1a has not been demonstrated to interact with either epsinR or AP-1, and moreover it is not lost from CCVs when either epsinR or AP-1 is knocked down. Thus, there must be other cargo adaptors for vti1a, which can still package it into CCVs even when its subcellular distribution is altered. Why then would epsinR and AP-1 knockdown cause vti1a to redistribute? Possibly there may be changes in the localization of the entire vti1a-containing compartment. Knocking down either epsinR or AP-1 prevents vti1b from being packaged into CCVs and changes its steady state distribution, and once a SNARE gets mislocalized there may be a number of knock-on effects. However, not all of the proteins that cycle between the TGN and endosomes are affected by epsinR knockdown. For instance, we have shown that a CD8-furin chimera gets mislocalized in AP-1–depleted cells, but its distribution is normal in epsinR-depleted cells.
It is clear from all of these experiments that knocking down a coat component and then looking for changes in the steady state distribution of potential cargo proteins can sometimes give misleading information and that more direct assays are needed for adaptor function. The CCV isolation procedure is one such assay, and it can potentially be used to investigate the role of any CCV protein in cargo selection. In addition to epsinR, there are a number of other γ ear binding partners whose function is still unknown, including γ-synergin (Page et al., 1999), p200 (Hirst et al., 2003), aftiphilin, and NECAP (Mattera et al., 2004), and these are all candidates for alternative adaptors on the AP-1 pathway. There are also several α ear binding partners whose function is not entirely clear, and these may be alternative adaptors on the AP-2 pathway (Traub, 2003). In some cases, it is possible to make educated guesses about what the cargo proteins might be, and here it will be possible to use Western blotting to quantify these proteins in CCVs after siRNA knockdown, as we have done in the present study. For other candidate adaptors, an unbiased proteomics approach may be the best way forward. We have recently begun a systematic analysis of the protein composition of HeLa cell CCVs, comparing preparations from control and clathrin-depleted cells. This enables us to distinguish bona fide components from contaminants, which is always a problem when trying to establish the proteome of an organelle. By using the same approach of treating cells with an siRNA and then carrying out a comparative proteomics analysis on their CCVs, we hope to be able to establish the functions of even very minor adaptors, because when they are knocked down, their cargo proteins should be depleted from the preparation as well.
Supplementary Material
Acknowledgments
We thank Steve Royle for the GFP clathrin light-chain construct, Matthew Seaman for the CD8-furin–expressing cells; Mike Harbor for mass spectrometry; Nick Bright for advice on EM; Nicola Berg for help with the knockdowns; and Paul Luzio, John Kilmartin, Matthew Seaman, David Owen, and members of the Robinson lab for reading the manuscript and for helpful discussions. This work was supported by grants from the Wellcome Trust and the Medical Research Council.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E04–06–0468. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E04–06–0468.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Bonifacino, J.S., and Traub, L.M. (2003). Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395-447. [DOI] [PubMed] [Google Scholar]
- Chidambaram, S., Mullers, N., Wiederhold, K., Haucke, V., and von Mollard, G.F. (2004). Specific interaction between SNAREs and epsin N-terminal homology (ENTH) domains of epsin-related proteins in trans-Golgi network to endosome transport. J. Biol. Chem. 279, 4175-4179. [DOI] [PubMed] [Google Scholar]
- Conner, S.D., and Schmid, S.L. (2003). Differential requirements for AP-2 in clathrin-mediated endocytosis. J. Cell Biol. 162, 773-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford, M.G., Mills, I.G., Peter, B.J., Vallis, Y., Praefcke, G.J., Evans, P.R., and McMahon, H.T. (2002). Curvature of clathrin-coated pits driven by epsin. Nature 419, 361-366. [DOI] [PubMed] [Google Scholar]
- Hinrichsen, L., Harborth, J., Andrees, L., Weber, K., and Ungewickell, E.J. (2003). Effect of clathrin heavy chain- and alpha-adaptin-specific small inhibitory RNAs on endocytic accessory proteins and receptor trafficking in HeLa cells. J. Biol. Chem. 278, 45160-45170. [DOI] [PubMed] [Google Scholar]
- Hirst, J., Motley, A., Harasaki, K., Peak Chew, S.Y., and Robinson, M.S. (2003). EpsinR: an ENTH domain-containing protein that interacts with AP-1. Mol. Biol. Cell 14, 625-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, F., Khvorova, A., Marshall, W., and Sorkin, A. (2004). Analysis of clathrin-mediated endocytosis of epidermal growth factor receptor by RNA interference. J. Biol. Chem. 279, 16657-16661. [DOI] [PubMed] [Google Scholar]
- Kalthoff, C., Groos, S., Kohl, R., Mahrhold, S., and Ungewickell, E.J. (2002). Clint: a novel clathrin-binding ENTH-domain protein at the Golgi. Mol. Biol. Cell 13, 4060-4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattera, R., Ritter, B., Sidhu, S.S., McPherson, P.S., and Bonifacino, J.S. (2004). Definition of the consensus motif recognized by γ-adaptin ear domains. J. Biol. Chem. 279, 8018-8028. [DOI] [PubMed] [Google Scholar]
- Mills, I.G., Praefcke, G.J., Vallis, Y., Peter, B.J., Olesen, L.E., Gallop, J.L., Butler, P.J., Evans, P.R., and McMahon, H.T. (2003). EpsinR: an AP1/clathrin interacting protein involved in vesicle trafficking. J. Cell Biol. 160, 213-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley, A., Bright, N.A., Seaman, M.N.J., and Robinson, M.S. (2003). Clathrin-mediated endocytosis in AP-2-depleted cells. J. Cell Biol. 162, 909-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, L.J., Sowerby, P.J., Lui, W.W.Y., and Robinson, M.S. (1999). γ-Synergin: an EH domain-containing protein that interacts with γ-adaptin. J. Cell Biol. 146, 993-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden, A.A., Park, G.Y., and Scheller, R.H. (2001). The di-leucine motif of vesicle-associated membrane protein 4 is required for its localization and AP-1 binding. J. Biol. Chem. 276, 49183-49187. [DOI] [PubMed] [Google Scholar]
- Saint-Pol, A. et al. (2004). Clathrin adaptor epsinR is required for retrograde sorting on early endosomal membranes. Dev. Cell 6, 525-538. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., and Russell, D.W. (2001). Molecular Cloning, A Laboratory Manual, New York: Cold Spring Harbor Laboratory Press.
- Seaman, M.N.J. (2004). Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J. Cell Biol. 165, 111-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub, L.M. (2003). Sorting it out: AP-2 and alternate clathrin adaptors in endocytic cargo selection. J. Cell Biol. 163, 203-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y.J. et al. (2003). Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell 114, 299-310. [DOI] [PubMed] [Google Scholar]
- Wasiak, S. et al. (2002). Enthoprotin: a novel clathrin-associated protein identified through subcellular proteomics. J. Cell Biol. 158, 855-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.