

Abstract
We recently reported that differential gene expression and DNA methylation profiles in blood leukocytes of apparently healthy smokers predicts with remarkable efficiency diseases and conditions known to be causally associated with smoking, suggesting that blood-based omic profiling of human populations may be useful for linking environmental exposures to potential health effects. Here we report on the sex-specific effects of tobacco smoking on transcriptomic and epigenetic features derived from genome-wide profiling in white blood cells, identifying 26 expression probes and 92 CpG sites, almost all of which are affected only in female smokers. Strikingly, these features relate to numerous genes with a key role in the pathogenesis of cardiovascular disease, especially thrombin signaling, including the thrombin receptors on platelets F2R (coagulation factor II (thrombin) receptor; PAR1) and GP5 (glycoprotein 5), as well as HMOX1 (haem oxygenase 1) and BCL2L1 (BCL2-like 1) which are involved in protection against oxidative stress and apoptosis, respectively. These results are in concordance with epidemiological evidence of higher female susceptibility to tobacco-induced cardiovascular disease and underline the potential of blood-based omic profiling in hazard and risk assessment.
Exposure to tobacco smoke is one of the best studied examples of an exposure with proven causal association with a large number of human diseases1. Although the relevant epidemiological evidence is not completely consistent, many studies have provided evidence of differential sex susceptibility to the health effects of tobacco, especially in relation to smoking-induced cardiovascular disease (CVD; including acute myocardial infarction and coronary heart disease)2,3,4, chronic obstructive pulmonary disease (COPD)1,5,6 and lung cancer7,8,9. Focusing in particular on CVD, a systematic review and meta-analysis of data from 86 prospective studies and nearly 4 million subjects came to the conclusion that female smokers have a 25% higher risk of developing coronary heart disease than males with the same exposure to tobacco smoke and after allowing for other risk factors2, a conclusion supported by the results of a recent meta-analysis of the available data3. Another recent systematic review and meta-analysis covering data from 81 cohorts and nearly 4 million subjects concluded that the risk of stroke in Western populations is 10% higher in female smokers4.
Biomarker-based investigations have contributed significantly to our understanding of the disease risks associated with exposure to environmental hazards. Currently such biomarker studies are benefiting from the expanding use of genome-wide profiling (omics). We have recently reported the results of a study of the impact of tobacco smoke exposure on genome-wide gene expression and DNA methylation in white blood cells (WBCs) of apparently healthy smokers10 and identified large numbers of transcripts and DNA CpG sites whose expression and methylation, respectively, differ significantly between current and never smokers. Furthermore we used disease connectivity analysis to show that the corresponding gene profiles can identify with remarkable efficiency (specificity 94%, positive predictive value 86%) most diseases and conditions independently known to be causally associated with smoking10. In view of this finding, we decided to look for sex-related differences in these profiles which might possibly reflect differential disease susceptibility.
Results
In order to minimise statistical power problems, we focused our search for sex-related differences in the effects of tobacco smoking on the transcriptomic and epigenetic features which we previously found to differ significantly between mixed-sex groups of current and never smokers10. These features consist of 1,273 CpG sites (FDR < 0.05; associated with 725 differentially methylated genes - DMGs) and 350 transcripts (FDR < 0.10; associated with 271 differentially expressed genes - DEGs) which were derived from the comparison of genome-wide transcriptomic and epigenetic profiles of 143 current and 311 never smokers (including 134 males and 320 females) derived from 2 cohorts, the Northern Sweden Health and Disease Study and EPIC Italy (Suppl. Tables S1 and S2).
The tobacco exposure data available to us (Table 1) included the number of cigarettes smoked per day and smoking duration in both cohorts, the smoking intensity measured in pack-years (only in the Italian cohort), as well as plasma cotinine levels for a fraction of the study subjects from both cohorts. Inspection of this data did not reveal statistically significant differences between the two sexes, although it did suggest possibly higher exposure intensity (pack-years) in males (Table 1). While the above parameters provide an approximate picture of smoking exposure, they do not allow an accurate quantitative estimation of the long-term exposure to tobacco smoke of the different subjects suitable for use in statistical adjustment for the purpose of sub-group comparison. For this reason, and also having in mind our previous observation of the highly skewed distribution of the expression or methylation differences between current and never smokers (effect size), we opted to base our sex comparisons not on the effect size, which is expected to be strongly dependent on exposure level and duration, but on the ranking of the various features by statistical significance (i.e. p-values in current vs never smoker comparisons) in sex-stratified analyses. Thus we compared the full set of signals separately for males and females, ranking them according to the statistical significance of their current-versus-never smoker differences in each sex and, finally, compared the rankings in the two sexes of the limited number of signals of interest. The methodology is described in detail in Methods while the workflow of the procedure is shown diagrammatically in Fig. 1. We adopted this approach on the reasonable assumption that any differences between the sexes with regard to the level or duration of exposure would in general affect the effect size for the different features in a similar manner but would be unlikely to alter their ranking within each sex group. This non-parametric approach has the added advantage that it minimizes the impact of non-normal data distribution, outliers and differences in statistical power arising from the different sizes of the male and female populations.
Table 1. Study population and sex comparison of smoking exposure-related parameters.
| Males | Females | ||
|---|---|---|---|
| N | 236 | 413 | |
| age; mean (range) | 51.0 (30.2–65.0) | 52.8(29.6–74.9) | |
| cohort | NSHDS; N | 171 | 228 |
| EPIC Italy; N | 65 | 185 | |
| BMI; mean (range) | 26.5 (19.0–39.5) | 25.4(18.8–55.28) | |
| smoking status | current smokers; N | 43 (18.2%) | 100 (24.2%) |
| former smokers; N | 102 (43.2%) | 93 (22.5%) | |
| never smokers; N | 91 (38.6%) | 220 (53.3%) | |
| duration of smoking | N | 40 | 90 |
| years, mean (s.d) | 31.8 (8.7) | 30.6 (18.1) | |
| p | 0.49 | ||
| pack-years* | N | 14 | 45 |
| mean (S.D) | 582.9 (429.0) | 356.6 (266.0) | |
| p | 0.06 | ||
| cigarettes/day | total N | 42 | 98 |
| ≤4, N | 7 (16.7%) | 24 (24.5%) | |
| 5–15, N | 17 (40.5%) | 50 (51%) | |
| ≥25, N | 18 (42.9%) | 24 (24.4%) | |
| p# | 0.2 | ||
| plasma cotinine | N | 40 | 35 |
| AUC; mean (s.d.) | 33.7 (8.7) | 28.1 (8.5) | |
| p | 0.85 | ||
*Only EPIC Italy; #chi-squared test.
Figure 1. Workflow of the statistical analysis.

Suppl. Fig. S1 shows the distribution of the p-values of the rank differences in the two sexes. It can be seen that a biomodal distribution is observed, with the anticipated pattern of decreasing numbers of signals as p-values decrease being observed along with an increased number of signals with very low p-values (<0.05) increases. The latter indicates the existence, among both the transcriptomic and the epigenetic signals, of sub-groups of signals whose ranking differences between the sexes are more significant than expected.
Tables 2 and 3 show the lists of expression probes and CpG sites, respectively, whose significance rankings differ significantly (p < 0.05) between the two sexes. There was no significant difference in the corresponding rankings of these signals between male and female never smokers (results not shown), indicating that the observed sex-related differences reflect the differential impact of tobacco smoke exposure.
Table 2. Expression probes and DEGs ranked by significance of sex specificity.
| Expression probe | Gene symbol | Gene name | Rank by p-value in sex-stratified analysis |
Effect size (ratio current/never smokers) in sex-stratified analysis |
|||
|---|---|---|---|---|---|---|---|
| Males | Females | p rank difference | Males | Females | |||
| A_23_P120883 | NMOX1 | heme oxygenase (decycling) 1 | 29,411 | 31 | 2.82E-05 | 1.00 | 0.77 |
| A_23_P87013 | TAGLN | transgelin | 29,013 | 74 | 3.86E-05 | 1.00 | 0.80 |
| A_24_P410453 | SYNE1 | spectrin repeat containing. nuclear envelope 1 | 28,573 | 120 | 5.42E-05 | 1.00 | 0.81 |
| A_23_P23834 | LGR6 | leucine-rich repeat-containing G protein-coupled receptor 6 | 27,005 | 26 | 1.46E-04 | 1.49 | 0.72 |
| A_23_P136347 | EPS8 | epidermal growth factor receptor pathway substrate 8 | 26,954 | 16 | 1.50E-04 | 0.99 | 0.75 |
| A_24_P173754 | C1orf21 | chromosome 1 open reading frame 21 | 24,586 | 24 | 6.54E-04 | 0.98 | 0.78 |
| A_23_P354387 | MYOF | myoferlin | 24,542 | 111 | 7.06E-04 | 0.98 | 0.79 |
| A_23_P66881 | RGS9 | regulator of G-protein signaling 9 | 21,736 | 118 | 3.29E-03 | 0.96 | 0.79 |
| A_24_P76644 | 20,804 | 121 | 5.24E-03 | 0.98 | 0.88 | ||
| A_23_P86682 | MYOF | myoferlin | 19,999 | 56 | 7.46E-03 | 0.96 | 0.78 |
| A_23_P43679 | ZNF618 | zinc finger protein 618 | 19,852 | 22 | 7.86E-03 | 0.96 | 0.84 |
| A_23_P1962 | RARRES3 | retinoic acid receptor responder (tazarotene induced) 3 | 19,775 | 113 | 8.49E-03 | 0.97 | 0.87 |
| A_23_P113161 | C1orf21 | chromosome 1 open reading frame 21 | 19,632 | 47 | 8.80E-03 | 0.95 | 0.75 |
| A_24_P921823 | TCF7L2 | transcription factor 7-like 2 (T-cell specific. HMG-box) | 18,329 | 7 | 1.53E-02 | 0.96 | 0.77 |
| A_23_P45831 | CHD1L | chromodomain helicase DNA binding protein 1-like | 19,187 | 1,428 | 1.94E-02 | 0.98 | 0.93 |
| A_23_P48175 | TMEM106C | transmembrane protein 106C | 18,096 | 410 | 2.00E-02 | 0.96 | 0.88 |
| A_23_P39251 | PLIN5 | perilipin 5 | 229 | 17,725 | 2.16E-02 | 1.34 | 1.05 |
| A_23_P200780 | TGFBR3 | transforming growth factor. beta receptor III | 17,259 | 94 | 2.47E-02 | 0.94 | 0.80 |
| A_23_P110791 | CSF1R | colony stimulating factor 1 receptor | 16,516 | 49 | 3.23E-02 | 0.95 | 0.80 |
| A_32_P140475 | KIAA1377 | KIAA1377 | 72 | 16,014 | 3.93E-02 | 0.82 | 0.97 |
| A_23_P213620 | PPP2R2B | protein phosphatase 2. regulatory subunit B. beta | 16,049 | 256 | 4.14E-02 | 0.94 | 0.86 |
| A_32_P75141 | 15,969 | 240 | 4.24E-02 | 0.94 | 0.85 | ||
| A_23_P385105 | PLCD4 | phospholipase C. delta 4 | 15,930 | 354 | 4.48E-02 | 0.94 | 0.86 |
| A_24_P88763 | LOXL3 | lysyl oxidase-like 3 | 15,979 | 413 | 4.50E-02 | 0.95 | 0.89 |
| A_24_P9883 | DKFZp761E198 | DKFZp761E198 protein | 27 | 15,524 | 4.61E-02 | 1.49 | 1.05 |
| A_23_P109171 | BFSP1 | beaded filament structural protein 1. filensin | 15,892 | 481 | 4.75E-02 | 0.93 | 0.84 |
Table 3. CpG sites and DMGs ranked by significance of sex specificity.
| Probe | Gene symbol | Gene name | Rank by p-value in sex-stratified analysis |
Males |
Females |
absΔΔβ = abs(Δβfem −Δβm) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Males | Females | p rank difference | β* current smokers | β* never smokers | Δβm# | β* current smokers | β* never smokers | Δβfem# | ||||
| cg03378003 | SETD2 | SET domain containing 2 | 410,615 | 375 | 1.73E-08 | 84.23 | 84.24 | 0.00 | 84.92 | 83.59 | 1.33 | 1.33 |
| cg05713794 | GP5 | glycoprotein V (platelet) | 399,753 | 108 | 4.17E-08 | 81.42 | 81.44 | −0.02 | 80.63 | 78.46 | 2.17 | 2.19 |
| cg13411554 | CACNA1D | calcium channel. voltage-dependent. L type. alpha 1D subunit | 398,828 | 206 | 4.53E-08 | 29.34 | 29.25 | 0.08 | 34.69 | 28.22 | 6.47 | 6.39 |
| cg19028369 | C3orf19 | chromosome 3 open reading frame 19 | 395,672 | 613 | 6.06E-08 | 4.45 | 4.46 | −0.01 | 4.29 | 4.59 | −0.29 | −0.29 |
| cg12611488 | SKI | SKI proto-oncogene | 394,498 | 63 | 6.38E-08 | 78.07 | 77.99 | 0.08 | 82.74 | 78.36 | 4.39 | 4.31 |
| cg00436663 | LOC400927 | 388,154 | 171 | 1.07E-07 | 4.89 | 4.88 | 0.01 | 4.58 | 5.04 | −0.45 | −0.47 | |
| cg17993335 | DNMBP | dynamin binding protein | 387,580 | 886 | 1.19E-07 | 79.14 | 79.20 | −0.06 | 80.01 | 78.03 | 1.99 | 2.05 |
| cg15743533 | FAM110A | family with sequence similarity 110. member A | 369,751 | 234 | 4.50E-07 | 11.46 | 11.50 | −0.04 | 10.67 | 11.72 | −1.05 | −1.01 |
| cg17098415 | 366,343 | 190 | 5.79E-07 | 1.83 | 1.84 | −0.02 | 1.93 | 2.20 | −0.27 | −0.25 | ||
| cg17532753 | HDAC4 | histone deacetylase 4 | 370,727 | 5.606 | 6.26E-07 | 67.37 | 67.24 | 0.13 | 67.71 | 66.06 | 1.65 | 1.52 |
| cg25115829 | SUZ12P | SUZ12 polycomb repressive complex 2 subunit pseudogene 1 | 179,095 | 698 | 7.57E-07 | 73.04 | 72.23 | 0.81 | 75.55 | 72.97 | 2.58 | 1.77 |
| cg13543915 | 360,064 | 247 | 9.28E-07 | 61.82 | 61.91 | −0.09 | 62.91 | 64.51 | −1.60 | −1.52 | ||
| cg09257526 | IL6R | interleukin 6 receptor | 359,954 | 1.607 | 1.03E-06 | 20.44 | 20.52 | −0.08 | 20.04 | 20.67 | −0.62 | −0.55 |
| cg12689529 | KIRREL3 | kin of IRRE like 3 (Drosophila) | 341,191 | 406 | 3.63E-06 | 6.84 | 6.91 | −0.07 | 6.50 | 7.19 | −0.69 | −0.62 |
| cg13379236 | EGF | epidermal growth factor | 332,044 | 256 | 6.73E-06 | 60.43 | 60.24 | 0.19 | 62.51 | 60.67 | 1.84 | 1.65 |
| cg06459104 | EPB41L3 | erythrocyte membrane protein band 4.1-like 3 | 331,586 | 341 | 6.98E-06 | 34.31 | 34.76 | −0.46 | 33.82 | 38.07 | −4.25 | −3.79 |
| cg10232140 | ERCC6 | excision repair cross-complementation group 6 | 328,904 | 141 | 8.25E-06 | 1.54 | 1.50 | 0.05 | 1.78 | 1.32 | 0.46 | 0.41 |
| cg27430293 | 152,757 | 2.427 | 8.33E-06 | 34.11 | 33.26 | 0.85 | 36.95 | 34.92 | 2.03 | 1.18 | ||
| cg04985185 | MBTPS1 | membrane-bound transcription factor peptidase. site 1 | 321,636 | 229 | 1.34E-05 | 83.83 | 83.59 | 0.24 | 84.90 | 82.76 | 2.14 | 1.89 |
| cg27171474 | CDCA8 | cell division cycle associated 8 | 329,032 | 404 | 2.05E-05 | 84.89 | 84.78 | 0.11 | 86.22 | 85.01 | 1.21 | 1.09 |
| cg08202265 | TAC1 | tachykinin. precursor 1 | 314,885 | 272 | 2.09E-05 | 87.55 | 87.23 | 0.31 | 88.45 | 85.15 | 3.30 | 2.99 |
| cg08528970 | 311,563 | 11.616 | 5.23E-05 | 13.11 | 13.34 | −0.22 | 13.88 | 14.30 | −0.41 | −0.19 | ||
| cg02003183 | CDC42BPB | CDC42 binding protein kinase beta (DMPK-like) | 296,555 | 442 | 6.59E-05 | 8.34 | 8.14 | 0.21 | 6.33 | 5.57 | 0.76 | 0.55 |
| cg10665960 | EPC2 | enhancer of polycomb homolog 2 | 295,295 | 491 | 7.13E-05 | 4.71 | 4.66 | 0.05 | 4.57 | 4.90 | −0.33 | −0.38 |
| cg26724967 | IL32 | interleukin 32 | 184,670 | 577 | 9.65E-05 | 59.76 | 60.34 | −0.58 | 59.95 | 61.77 | −1.82 | −1.25 |
| cg07411111 | TPD52L2 | tumor protein D52-like 2 | 289,518 | 998 | 1.04E-04 | 61.46 | 61.04 | 0.42 | 62.07 | 59.99 | 2.08 | 1.66 |
| cg09374353 | EHD1 | EH-domain containing 1 | 284,155 | 321 | 1.36E-04 | 20.73 | 20.54 | 0.19 | 18.44 | 19.61 | −1.18 | −1.37 |
| cg23670519 | 278,212 | 616 | 1.94E-04 | 27.86 | 27.27 | 0.59 | 27.09 | 24.35 | 2.74 | 2.15 | ||
| cg05307957 | ARID1A | AT rich interactive domain 1A (SWI-like) | 277,497 | 132 | 1.97E-04 | 2.70 | 2.78 | −0.08 | 2.99 | 3.38 | −0.39 | −0.31 |
| cg20305005 | SCN2A | sodium channel. voltage-gated. type II. alpha subunit | 273,040 | 240 | 2.54E-04 | 58.37 | 57.96 | 0.42 | 58.65 | 56.27 | 2.38 | 1.96 |
| cg13989999 | BCL2L1 | BCL2-like 1 | 268,258 | 3.290 | 3.89E-04 | 47.52 | 47.93 | −0.41 | 48.03 | 49.34 | −1.31 | −0.90 |
| cg06397161 | SYNGR1 | synaptogyrin 1 | 262,200 | 1.787 | 4.96E-04 | 42.48 | 42.79 | −0.31 | 44.56 | 46.19 | −1.63 | −1.32 |
| cg06959021 | TCHP | trichoplein. keratin filament binding | 255,728 | 173 | 6.39E-04 | 58.68 | 58.28 | 0.40 | 61.71 | 59.85 | 1.86 | 1.45 |
| cg01911191 | 253,223 | 1.609 | 7.82E-04 | 2.70 | 2.62 | 0.08 | 2.34 | 2.07 | 0.27 | 0.19 | ||
| cg25446789 | DTNB | dystrobrevin. beta | 150,180 | 257 | 8.25E-04 | 40.28 | 40.87 | −0.60 | 40.07 | 42.21 | −2.14 | −1.55 |
| cg14179401 | 249,065 | 983 | 9.35E-04 | 33.38 | 33.60 | −0.22 | 34.75 | 35.82 | −1.07 | −0.85 | ||
| cg08601457 | FYN | FYN proto-oncogene. Src family tyrosine kinase | 242,282 | 422 | 1.27E-03 | 7.11 | 7.28 | −0.17 | 5.98 | 6.68 | −0.70 | −0.53 |
| cg16702313 | C14orf43 | chromosome 14 open reading frame 43 | 239,378 | 153 | 1.45E-03 | 60.71 | 61.00 | −0.29 | 61.85 | 63.45 | −1.60 | −1.31 |
| cg13832372 | LHX6 | LIM homeobox 6 | 239,227 | 188 | 1.46E-03 | 10.96 | 11.42 | −0.45 | 10.32 | 12.60 | −2.28 | −1.83 |
| cg24242519 | FAM49A | family with sequence similarity 49. member A | 236,302 | 1.218 | 1.77E-03 | 13.47 | 13.75 | −0.28 | 13.51 | 14.61 | −1.10 | −0.82 |
| cg22638542 | SEC22C | SEC22 homolog C. vesicle trafficking protein | 227,163 | 329 | 2.60E-03 | 80.69 | 80.38 | 0.31 | 82.48 | 81.37 | 1.11 | 0.80 |
| cg01904243 | C14orf43 | chromosome 14 open reading frame 43 | 225,047 | 164 | 2.84E-03 | 13.97 | 14.22 | −0.25 | 12.62 | 13.82 | −1.20 | −0.95 |
| cg06338710 | GFI1 | growth factor independent 1 transcription repressor | 224,461 | 205 | 2.92E-03 | 76.24 | 76.90 | −0.66 | 77.50 | 79.53 | −2.03 | −1.38 |
| cg19048950 | LOC100188 | 219,307 | 463 | 3.72E-03 | 76.17 | 75.70 | 0.46 | 73.42 | 71.15 | 2.28 | 1.81 | |
| cg02104700 | S100P | S100 calcium binding protein P | 217,731 | 496 | 4.00E-03 | 2.42 | 2.49 | −0.07 | 2.45 | 2.67 | −0.22 | −0.15 |
| cg15417641 | CACNA1D | calcium channel. voltage-dependent. L type. alpha 1D subunit | 215,584 | 77 | 4.31E-03 | 50.18 | 48.86 | 1.32 | 53.39 | 46.64 | 6.76 | 5.44 |
| cg11222173 | RPTOR | regulatory associated protein of MTOR. complex 1 | 216,169 | 1.025 | 4.38E-03 | 62.03 | 61.49 | 0.54 | 61.00 | 58.74 | 2.26 | 1.71 |
| cg08062087 | C2orf66 | chromosome 2 open reading frame 66 | 215,666 | 587 | 4.39E-03 | 69.15 | 68.61 | 0.54 | 70.09 | 68.06 | 2.03 | 1.49 |
| cg14950321 | PLIN5 | perilipin 5 | 215,623 | 763 | 4.44E-03 | 39.87 | 40.42 | −0.55 | 41.31 | 43.13 | −1.82 | −1.27 |
| cg08778287 | IGF1R | insulin-like growth factor 1 receptor | 214,383 | 476 | 4.62E-03 | 33.41 | 32.78 | 0.64 | 31.37 | 28.84 | 2.54 | 1.90 |
| cg13698937 | C4orf46 | chromosome 4 open reading frame 46 | 210,788 | 694 | 5.45E-03 | 76.64 | 75.97 | 0.67 | 77.47 | 75.32 | 2.15 | 1.49 |
| cg00357551 | FAM196B | family with sequence similarity 196. member B | 207,522 | 1.016 | 6.33E-03 | 33.01 | 32.52 | 0.50 | 32.30 | 30.79 | 1.51 | 1.01 |
| cg04359840 | XYLT1 | xylosyltransferase I | 206,167 | 346 | 6.52E-03 | 47.27 | 47.85 | −0.58 | 47.80 | 49.99 | −2.19 | −1.61 |
| cg21698310 | PPP1R9B | protein phosphatase 1. regulatory subunit 15A | 203,847 | 822 | 7.32E-03 | 10.66 | 10.89 | −0.23 | 9.69 | 10.49 | −0.81 | −0.58 |
| cg00816037 | FAM38A | family with sequence similarity 38. member A | 197,944 | 281 | 9.10E-03 | 7.77 | 8.09 | −0.33 | 6.69 | 7.77 | −1.08 | −0.75 |
| cg24488469 | 192,915 | 314 | 1.11E-02 | 15.97 | 16.30 | −0.33 | 14.06 | 15.16 | −1.09 | −0.76 | ||
| cg12033822 | SLC35C2 | solute carrier family 35 (GDP-fucose transporter). member C2 | 189,184 | 1.519 | 1.34E-02 | 26.76 | 27.15 | −0.40 | 25.56 | 26.54 | −0.98 | −0.58 |
| cg22490254 | 186,085 | 396 | 1.45E-02 | 15.92 | 15.44 | 0.48 | 15.51 | 13.87 | 1.64 | 1.16 | ||
| cg26840970 | ZNF19 | zinc finger protein 19 | 315,231 | 309 | 1.54E-02 | 69.05 | 68.76 | 0.29 | 71.08 | 68.75 | 2.33 | 2.04 |
| cg09608073 | CHSY3 | chondroitin sulfate synthase 3 | 184,657 | 712 | 1.55E-02 | 9.31 | 9.50 | −0.19 | 7.99 | 8.47 | −0.48 | −0.29 |
| cg19925780 | 179,222 | 134 | 1.85E-02 | 57.36 | 56.05 | 1.30 | 59.58 | 56.04 | 3.54 | 2.24 | ||
| cg25223634 | C10orf26 | chromosome 10 open reading frame 26 | 172,484 | 368 | 1.90E-02 | 37.56 | 38.21 | −0.64 | 35.14 | 37.58 | −2.43 | −1.79 |
| cg00026474 | ST3GAL1 | ST3 beta-galactoside alpha-2.3-sialyltransferase 1 | 178,373 | 842 | 1.96E-02 | 19.10 | 19.54 | −0.44 | 19.23 | 20.63 | −1.40 | −0.96 |
| cg25114611 | FKBP5 | FK506 binding protein 5 | 362,777 | 198 | 1.97E-02 | 30.27 | 30.36 | −0.08 | 30.44 | 31.81 | −1.37 | −1.28 |
| cg01609214 | MIR30D | microRNA 30d | 174,169 | 418 | 2.24E-02 | 89.84 | 89.03 | 0.81 | 90.88 | 87.38 | 3.50 | 2.69 |
| cg21188533 | CACNA1D | calcium channel. voltage-dependent. L type. alpha 1D subunit | 173,743 | 61 | 2.24E-02 | 43.74 | 41.82 | 1.91 | 52.19 | 43.74 | 8.45 | 6.54 |
| cg06627354 | TRPM8 | transient receptor potential cation channel. subfamily M. member 8 | 173,046 | 285 | 2.32E-02 | 65.91 | 65.21 | 0.70 | 64.45 | 61.66 | 2.79 | 2.10 |
| cg25722983 | STK40 | serine/threonine kinase 40 | 290,462 | 740 | 2.36E-02 | 45.26 | 45.55 | −0.29 | 46.40 | 47.91 | −1.52 | −1.22 |
| cg25233339 | ATP1B3 | ATPase. Na+/K+ transporting. beta 3 polypeptide | 250,770 | 212 | 2.37E-02 | 3.69 | 3.79 | −0.10 | 3.03 | 3.48 | −0.45 | −0.35 |
| cg00449189 | 171,418 | 702 | 2.49E-02 | 12.06 | 12.39 | −0.33 | 10.75 | 11.51 | −0.77 | −0.44 | ||
| cg08287903 | UGT8 | UDP glycosyltransferase 8 | 169,875 | 180 | 2.58E-02 | 48.84 | 48.15 | 0.69 | 51.00 | 48.24 | 2.76 | 2.06 |
| cg05055821 | 168,762 | 322 | 2.69E-02 | 9.90 | 9.65 | 0.26 | 9.44 | 8.67 | 0.77 | 0.52 | ||
| cg05525812 | 171,414 | 3.675 | 2.76E-02 | 16.95 | 17.39 | −0.44 | 15.71 | 16.55 | −0.84 | −0.40 | ||
| cg20530056 | IKBKE | inhibitor of kappa light polypeptide gene enhancer in B-cells. kinase epsilon | 168,953 | 1.699 | 2.80E-02 | 63.75 | 64.26 | −0.51 | 64.64 | 65.97 | −1.33 | −0.82 |
| cg01963224 | 166,963 | 102 | 2.84E-02 | 12.83 | 13.10 | −0.28 | 11.88 | 12.94 | −1.06 | −0.78 | ||
| cg05548393 | SLC30A8 | solute carrier family 30 (zinc transporter). member 8 | 165,967 | 483 | 2.98E-02 | 77.09 | 76.22 | 0.87 | 78.67 | 76.30 | 2.37 | 1.50 |
| cg24715767 | PRDM2 | PR domain containing 2. with ZNF domain | 165,652 | 337 | 3.00E-02 | 57.80 | 57.13 | 0.67 | 61.48 | 59.19 | 2.29 | 1.62 |
| cg05603910 | ANO9 | anoctamin 9 | 167,628 | 3.304 | 3.10E-02 | 78.58 | 77.55 | 1.03 | 75.77 | 73.98 | 1.79 | 0.77 |
| cg16660971 | RPTOR | regulatory associated protein of MTOR. complex 1 | 163,969 | 323 | 3.17E-02 | 72.09 | 71.01 | 1.08 | 71.01 | 66.95 | 4.06 | 2.98 |
| cg06361984 | NDE1 | nudE neurodevelopment protein 1 | 159,841 | 809 | 3.69E-02 | 82.89 | 82.22 | 0.67 | 81.19 | 79.42 | 1.77 | 1.10 |
| cg17873451 | LOC440925 | 157,993 | 231 | 3.84E-02 | 4.11 | 4.26 | −0.15 | 3.54 | 3.90 | −0.37 | −0.22 | |
| cg03220447 | NAV2 | neuron navigator 2 | 159,803 | 2.277 | 3.87E-02 | 12.43 | 12.70 | −0.27 | 12.44 | 12.97 | −0.53 | −0.26 |
| cg05756780 | IL6R | interleukin 6 receptor | 156,614 | 1.147 | 4.13E-02 | 23.32 | 23.63 | −0.31 | 22.65 | 23.65 | −1.00 | −0.69 |
| cg02104644 | SYT7 | synaptotagmin VII | 158,677 | 4.255 | 4.27E-02 | 19.47 | 20.07 | −0.59 | 18.63 | 19.87 | −1.24 | −0.64 |
| cg08242636 | CBFB | core-binding factor. beta subunit | 155,174 | 1.149 | 4.33E-02 | 5.10 | 5.25 | −0.15 | 5.00 | 5.37 | −0.37 | −0.22 |
| cg22006825 | HNRNPUL1 | heterogeneous nuclear ribonucleoprotein U-like 1 | 171,009 | 17.624 | 4.41E-02 | 19.13 | 19.57 | −0.45 | 19.20 | 19.75 | −0.55 | −0.10 |
| cg07201017 | FLJ41350 | LBX1 antisense RNA 1 (head to head) | 159,918 | 6.965 | 4.48E-02 | 16.26 | 16.59 | −0.33 | 15.82 | 16.34 | −0.52 | −0.19 |
| cg02571448 | PCBP3 | poly(rC) binding protein 3 | 151,595 | 974 | 4.81E-02 | 41.12 | 41.91 | −0.79 | 41.66 | 43.59 | −1.93 | −1.14 |
| cg24968629 | CELSR1 | cadherin. EGF LAG seven-pass G-type receptor 1 | 177,517 | 192 | 4.85E-02 | 74.90 | 73.75 | 1.15 | 70.52 | 65.45 | 5.07 | 3.92 |
| cg25474070 | IL3 | interleukin 3 | 173,116 | 895 | 4.92E-02 | 62.86 | 63.34 | −0.47 | 64.15 | 65.74 | −1.59 | −1.12 |
| cg19859980 | C1orf97 | chromosome 1 open reading frame 97 | 154,094 | 4.225 | 4.92E-02 | 2.51 | 2.60 | −0.09 | 2.59 | 2.74 | −0.15 | −0.06 |
| cg03699074 | FAM38A | family with sequence similarity 38. member A | 150,341 | 568 | 4.94E-02 | 14.23 | 14.69 | −0.46 | 12.81 | 14.07 | −1.26 | −0.79 |
*Methylation level (%); #Δβ = difference in % methylation of current-never smokers.
Expression profile
All 26 expression probes in Table 2 show large ranking differences between the sexes, with 23 exhibiting higher significance in females (median ranks: 111 in females and 19,775 in males), while the remaining 3 probes show higher significance in males. All probes are underexpressed in smokers with the exception of 2 which are overexpressed particularly in male smokers. Owing to population size limitations, many of the identified signals do not reach statistical significance in the sex-stratified analyses. However, for the 23 female-specific probes, the median FDR value with regard to the current-versus-never smoker comparisons is 0.40 (range 0.01–0.91) in females while all 23 have FDR > 0.80 in males (not shown), suggesting that the effects of smoking are largely limited to females. Importantly, as shown in Table 2 these significance ranking differences are accompanied by a corresponding difference in effect sizes for all the signals identified, with the median effect size (ratio of expression in current divided by never smokers) observed being 0.80 (range 0.72–0.93) and 0.96 (range 0.93–1.00) in females and males, respectively. No expression probe shows opposite effects of smoking in the two sexes.
Epigenetic profile
Turning to the epigenetic profile, the 92 CpG sites (associated with 72 genes) thus identified (Table 3) show both under- and overmethylation in smokers while their ranking differences between the sexes are also substantial, with the median rank values being 432 and 215,917 in females and males, respectively. The median FDR value of these CpG sites is 0.12 (range 0.0002–0.68) in females, while all 92 sites have FDR > 0.9 in males. In complete analogy to what is observed for the transcriptomic profile, for all CpG sites the impact of smoking (Δβ = βsmokers − βnon-smokers) is greater in females than in males, with the median absolute Δβ values being 1.55% (range 0.15–8.45%) and 0.33% (range 0.00–8.45%), in females and males, respectively. No CpG sites exhibit opposite effects of smoking in the two sexes. There was no statistically significant overabundance in the distribution of the CpG sites in relation to their locations (TSS200, TSS1500, body, 3′UTR, 5′UTR, 1st exon, intragenic) or to their occurrence in CpG islands and their regions (island, non-island S-shore, S-shelf, N-shore, N-shelf). Finally, we note that one gene, PLIN5, appears to be more overexpressed in females but more undermethylated in males, implying a possibility of differential epigenetic regulation in the two sexes.
Consistency and stability of observed sex effects
Owing to the limited population size no direct replication between the two cohorts was conducted. However, cohort-stratified analyses show that the features listed in Tables 3 and 4 tend to be among the most significant features, in terms of sex differences, also in the individual cohorts. Thus, the median rank value of the 92 CpG sites of Table 4 was 3,048 (out of a total of 410,987 sites examined; top 0.7%) in females and 195,213 in males in NSHDS, and 9,098 (top 2%) and 218,704, respectively, in EPIC Italy, thus demonstrating a clear trend of higher female sensitivity in both cohorts. A similar trend was seen with the corresponding transcriptomic signals of Table 2, with the 23 female-specific signals having median ranks of 585 (out of a total of 29,667 probes examined; top 2%) in females and 25,742 in males in NSHDS, and 1,708 (top 6%) and 12,338 in females and males, respectively, in EPIC Italy. As regards the 3 male-specific transcriptomic signals, all 3 in NSHDS and 2 out of 3 in EPIC ranked higher in males.
Table 4. Female-specific DEGs/DMGs of potential relevance to CVD. and their expression/methylation changes observed in present study or reported clinical studies (where available).
| Gene | Direct evidence of links with CVD pathogenesis | Mechanistic basis of links with CVD | Current study |
Vasclular inflam-mation28 | Chronic artery occlussion48 | Myocardial infarction63 | Coronary heart disease64 | Coronary artery disease65 | Peripheral arterial disease66 | Myocardial infarction67 | Venous thrombo-embolism68 | Atherosus-ceptibility69 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| expr* | meth# | expr* | expr* | expr* | expr* | expr* | expr* | expr* | expr* | meth# | |||
| F2R (PAR1) | polymorphisms in human CVD14 | thrombin signaling/vascular haemostasis | down | down | down | ||||||||
| GP5 | up | ||||||||||||
| FYN | transgenic mice19 | down | up | ||||||||||
| IGF1R | up | up | |||||||||||
| EGF | up | ||||||||||||
| EPS8 | transgenic mice24 | up | |||||||||||
| RPTOR | transgenic mice26 | up | |||||||||||
| HMOX1 | polymorphisms and differential expression in human CVD28; transgenic mice28 | anti-inflammatory | down | down | |||||||||
| HDAC4 | regulation of hypertrophy | ||||||||||||
| BCL2L1 | anti-apoptotic | down | up | ||||||||||
| CACNA1D | polymorphisms in human CVD37; transgenic mice35,36 | calcium signalling | up | ||||||||||
| TANGLN | polymorphisms in human CVD70; transgenic mice42 | VSMC/endothelial cell function | down | ||||||||||
| SYNE1 | polymorphisms in human CVD37; transgenic mice35,36 | down | down | ||||||||||
| IL32 | differential expression in human CVD45; transgenic mice45 | pro-inflammatory | down | ||||||||||
| PLIN5 | transgenic mice46 | oxidative stress | down | ||||||||||
| HNRNPUL1 | polymorphisms in human CVD47 | unknown | down | ||||||||||
| miR-30d | differential expression in human CVD48 | regulation of cardiomyocyte apoptosis71 | up | up | down | ||||||||
| C14orf43 | differential expression in human CVD61 | unknown | down | down | |||||||||
| C1orf21 | differential expression in human CVD59 | unknown | down | down | |||||||||
| ST3GAL1 | differential expression in human CVD59 | unknown | down | up | |||||||||
| ZNF19 | differential expression in human CVD59 | unknown | up | down | |||||||||
The genes are listed in the order in which they are discussed in the main text. Col. 1 presents evidence based on human population and transgenic animal studies.
*Expression; #methylation.
The observation that almost all sex-specific features identified show higher sensitivity in females is striking. For comparison it is noted that only 116 of the 350 transcripts and 1,009 of the 1,273 CpGs differentially modified by smoking in the mixed population have lower FDR values in females. Although, as stated above, the rank-based comparison we employed is not expected to be significantly affected by group size, in view of the larger number of females in our study (320 female versus 134 male current and never smokers), we ran the same analysis as described above 10 times, in each case using all 134 male subjects and an equal number of females sampled randomly from our population while maintaining constant the proportions of subjects coming from each of the two cohorts and with the different types of smoking status (see Fig. 1, right). In each such resampling analysis, the resulting sex-specific epigenetic or transcriptomic signals included on average 50.3% (S.D. 7.3%) and 50.8% (S.D. 14.2%) of the signals shown in Tables 2 and 3, respectively, while the cumulative % overlap (average of each successive resampling round plus all preceding ones) for both lists tended towards approx. 50%, reflecting the similar loss of statistical power of the smaller subpopulations employed (Suppl. Table S3 and Suppl. Fig. S2). These observations provide confirmation that the identification of the sex-specific signals was not subject to bias by the group size of each sex class.
Discussion
A number of recent studies on smoking-induced changes in methylation profiles, which employed effect size as the response classification parameter, failed to detect any sex-specific responses11,12, possibly because of residual confounding arising from insufficiently accurate adjustment for tobacco smoke exposure. By comparing in sex-stratified analyses the significance ranking of transcriptomic and epigenetic signals previously shown to differ between current and never smokers in a mixed-sex population, we identified a number of features which exhibit significantly different responses in the two sexes. Because the highly stringent nature of our non-parametric, rank-based statistical methodology inevitably attenuates sensitivity, it is possible that additional features with sex-specific behaviour may exist. On the other hand this approach has the advantage that it minimizes false positive findings and maximizes specificity.
Our observation that almost all features identified show stronger responses in females implies the possibility of higher female susceptibility to diseases related to the corresponding genes. The most notable observation regarding the list of sex-specific DEGs and DMGs relates to the presence of multiple genes related to CVD, especially genes involved in thrombin signaling and vascular and endothelial cell function. Table 4 summarises the relevant evidence (discussed further below) and compares the changes observed in the present study with those reported in clinical studies, where such information is available. It can be seen that the direction of change reported in these studies is in concordance with that which we have observed in apparently healthy female smokers, supporting the relevance of these changes to disease pathogenesis.
Genes involved in thrombin signaling
Thrombin, a serine protease, has an essential role in coagulation and haemostasis mediated by platelets, while in addition it elicits important effects in endothelial and vascular smooth muscle cells (VSMC). For these reasons thrombin-mediated effects are of great importance in the pathogenesis of CVD. Most of the cellular effects of thrombin are initiated via the activation of a family of G-protein-coupled receptors called protease-activated receptors (PARs), which are transmembrane proteins expressed on different types of cells including platelets, endothelial cells and VSMC. The main thrombin receptor on platelets and blood vessel cells is PAR1, also known as coagulation factor II (thrombin) receptor (F2R). This gene plays a key role in vascular function and CVD13 and its genetic variants are known to influence platelet function14,15. In our previously published analysis of the effects of smoking on transcriptomic and epigenetic profiles10 we found F2R to be differentially underexpressed in current smokers with a statistical significance of FDR = 0.15 which just fails to reach the threshold adopted in the present analysis (FDR < 0.10). Inclusion of the F2R-related expression probe together with the 350 probes with FDR < 0.10 in the sex-stratified analysis described above reveals a highly significant female specificity of the effect of smoking on this gene. Thus, in male- and female-stratified analyses, respectively, the significance rankings for current-versus-never-smoker comparisons were 29,197 and 1,283 (p = 1.08 × 10−4), the FDR values 0.98 and 0.040 and the effect sizes 1.00 and 0.88.
The interaction of thrombin with PAR1 in platelets is facilitated by its initial binding to the GPIb-IX-V complex which plays a critical role in thrombosis, atherogenesis and inflammation16. This complex includes the glycoprotein GP5, which is associated with a CpG site we found to be differentially overmethylated in female smokers (Table 3). Following its activation via the cleavage of its N-terminal domain by thrombin, PAR1 initiates multiple kinase signaling pathways which lead to different effects depending on the cells concerned (Fig. 2). Such effects include hemostasis and thrombosis in the case of platelets, induction of pro-inflammatory phenotype in the case of endothelial cells, increase of vascular permeability, proliferation, migration and hypertrophy in the case of VSMCs, thus contributing to the pathogenesis of different types of CVD.
Figure 2. Schematic representation of some of the interactions involved in PAR1 signalling initiated by thrombin.
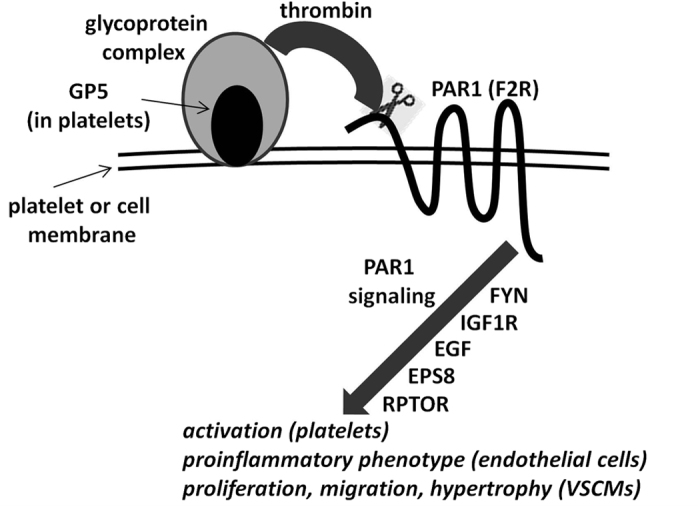
All genes shown, are preferentially modified in female smokers, PAR1 being differentially expressed and the others differentially methylated. The genes shown near the arrow participate in signalling downstream of PAR1 and the order in which they are shown does not imply a sequential interaction.
Signaling by the activated PAR1 receptor is controlled by, among other factors, Src kinases17, including FYN (FYN proto-oncogene, Src family tyrosine kinase) which is associated with a CpG site differentially undermethylated in female smokers. Following initiation of GPIb-IX-V/PAR1 signalling, FYN phosphorylates PKCδ (protein kinase Cδ) which subsequently negatively modulates platelet activation18. The importance of FYN for haemostasis-related disease is underlined by the report that FYN-deficient mice show an altered haemostatic response19.
Another gene which influences platelet function is IGF1R (insulin-like growth factor 1 receptor), which is associated with a CpG site differentially overmethylated in female smokers. The IFG1R protein is expressed at high levels on the plasma membrane of platelets while its ligand, IGF1, is a growth factor found in the α granules in platelets. Stimulation of platelets with IGF1 results in rapid phosphorylation of IGF1R and potentiation of PAR1-induced platelet aggregation20.
As already mentioned, thrombin-mediated PAR (including PAR1) signaling also operates in VSMC and endothelial cells, thereby playing an important role in diverse cellular activities related to inflammation, CVD, tumor growth and other conditions. In this context the genes discussed above (with the exception of GP5 which is expressed only in platelets) can be anticipated to affect by analogous mechanisms the pathogenesis of such diseases. Moreover, a number of additional genes of relevance to thrombin signaling in VSMC and endothelial cells is included in the list of genes found to be differentially modified in female smokers. One of these genes is EGF (epidermal growth factor), differentially overmethylated in female smokers, a potent mitogenic factor in many cell types acting through its receptor EGFR. Activation of EGFR promotes thrombin-induced proliferation of VSMC21, while its inhibition attenuates thrombin-stimulated signalling along the PI3K-Akt-mTOR-S6K1 axis, leading to effects on cell proliferation and motility22. Importantly, EGFR signalling is coordinated by EPS8 (epidermal growth factor receptor pathway substrate 8)23, which is underexpressed in female smokers. In support of a probable role of EPS8 in vascular disease is the report that EPS8-null mice show increased vascular permeability24. Finally, RPTOR (regulatory associated protein of MTOR, complex 1), overmethylated at 2 CpG sites in female smokers, negatively regulates mTOR kinase25 which, as mentioned above, is involved in thrombin signaling in VSMC. It is noted that mice with an RPTOR deletion targeted on the myocardium have been reported to develop dilated cardiomyopathy26.
Other genes related to CVD pathogenesis
The list of genes exhibiting sex-specific response to tobacco smoking includes a number of additional members for which there is significant clinical or mechanistic evidence, including evidence from transgenic animal studies, that they are linked with CVD pathogenesis. The gene with the largest expression change in female smokers is HMOX1 (haem oxygenase 1), well known for its antioxidant and anti-inflammatory properties as well as for its protective role against CVD27,28,29. HMOX1 is also known to protect against smoking-induced COPD30, a disease for which there is strong evidence of differentially higher susceptibility in female smokers1,5,6. Another gene of interest is HDAC4 (histone deacetylase 4), differentially overmethylated in female smokers, which plays a global role in the epigenetic control of gene expression by modifying histones as well as non-histone proteins31 and plays an important role in regulating hypertrophic responses32. Finally of particular note is the female-specific demethylation of the well known anti-apoptotic gene BCL2L1 (BCL2-like 1), which plays a key role in the regulation of platelet activation and apoptosis33. Among the consequences of platelet apoptosis is the production of microparticles, which are recognised to play an important role in inflammation, CVD, coagulation and angiogenesis34.
The largest sex-related difference in the impact of smoking (absΔΔβ, last column in Table 4) is observed at 3 CpG sites associated with CACNA1D (calcium channel, voltage-dependent, L type, alpha 1D subunit). This gene encodes for the cav1.3 subunit of a voltage-gated, L-type calcium channel and human and animal studies strongly support its association with various pathological conditions, including cardiovascular and neurological disorders35,36,37,38. On a side note, it is of interest that cav1.3 physically interacts with the receptor of GABAB, with activation of the latter leading to an increase in the L-type calcium channel currents39. Given the key role of this receptor in the mechanism of addiction, it is possible that any sex-related variation in CACNA1D expression may be reflected in corresponding differences in susceptibility to nicotine addiction. In support of this idea, several lines of evidence indicate that females have a higher susceptibility to nicotine dependence, including faster progression to dependence, shorter and less frequent abstinence periods, greater difficulty to quit, and poorer response to smoking cessation treatments38,40,41.
Other genes listed in Table 4, for which there is evidence of varying strength of links with different types of CVD, include TAGLN (transgelin42), SYNE1 (spectrin repeat containing, nuclear envelope 1)43,44, IL32 (interleukin 32)45, PLIN5 (perilipin 5)46, HNRNPUL1 (Heterogeneous Nuclear Ribonucleoprotein U-Like 1)47 and miR-30d48. Finally, Table 4 shows a number of genes (C14orf43, C1orf21, ST3GAL1 and ZNF19) which do not have any function related to CVD pathogenesis, however they have been reported to be differentially expressed in patients with different types of CVD.
Molecular basis of sex-specific effects of tobacco smoke exposure
The previous discussion shows that numerous genes among those found to be differentially altered in female smokers interact closely in the context of thrombin signaling in platelets and vascular/endothelial cells. While the molecular basis for such differential female susceptibility to tobacco smoke is not currently understood, there is strong evidence that thrombin signaling and hemostasis are subject to hormonal influences and it is possible that such influences may also modify the responses to tobacco smoking. Megakaryocytes and platelets express the estrogen and androgen receptors, and the coagulation cascade is known to be influenced by variations in the levels of female sex steroids49,50,51, while it has been suggested that females have greater baseline platelet reactivity which may be attenuated by estrogens51,52. Furthermore, it has been reported that platelets from women are more responsive than those of men to thrombin agonists51, and that females with atherosclerosis show higher PAR1-mediated platelet activation53. Sex hormones contribute to the modulation of additional genes related to CVD and may therefore also modify the impact of smoking. For example, the expression of HMOX1 under conditions of oxidative stress is modulated by estrogen receptor alpha54, while estrogen receptor beta modulates the expression of HDAC4 under the influence of hypertrophic factors in rat cardiomyocytes55.
Summarising the preceding discussion, a large number of genes which are known or suspected to play a role in the mechanism of CVD, and to modulate corresponding disease risks, have their expression or methylation in WBCs modified by smoking significantly more in females than in males. It is not known whether similar changes occur in other tissues of smokers. However we have recently reported that tobacco smoking causes similar changes in expression and CpG methylation in the Ah receptor repressor gene in WBCs and lung cells10. We have also shown that the genes which are differentially expressed or methylated in WBCs of smokers are closely related to many smoking-induced diseases regardless of their target tissue, implying that changes observed in blood cells may reflect more global effects. This conclusion is further supported by the concordance of the results presented in the present study with the conclusions of epidemiological studies which consistently point to a higher female susceptibility to tobacco-induced CVD. Furthermore, we also report analogous, although more limited, findings supporting higher female susceptibility to tobacco-induced COPD and nicotine addiction. Although our evaluation of the disease-relevance of the sex-specific DEGs and DMGs presented above did not include cancer, many of the signaling pathways discussed are also highly relevant to carcinogenesis56. It is noted that the current evidence regarding sex susceptibility to tobacco-related cancer is mixed7,8,9,57.
In conclusion, the results presented here underline the utility of blood-based omics profiling for identifying health hazards associated with environmental exposures and suggest a potential for use of such data in identification of susceptible sub-groups.
Materials and Methods
The present report is based on data from the Envirogenomarkers project (www.envirogenomarkers.net). Envirogenomarkers is a prospective case-control study nested within the European Prospective Investigation into Cancer and Nutrition study (EPIC-ITALY) and the Northern Sweden Health and Disease Study (NSHDS)58,59, in which subjects asymptomatic at the time of enrolment provided a blood sample and information on dietary habits, lifestyle, health history etc. The EnviroGenomarkers project and its associated studies and experimental protocols were approved by the Regional Ethical Review Board of the Umea Division of Medical Research, for the Swedish cohort, and the Florence Health Unit Local Ethical Committee, for the Italian cohort, and all participants gave written informed consent. All methods were carried out in accordance with the approved guidelines.
Owing to the Envirogenomarkers project’s design, some of the participating subjects had been selected on the basis of the fact that they went on to develop breast cancer or B-cell lymphoma 2–16 years after recruitment, however for the purposes of the present study they were all treated as apparently healthy. We have previously shown that inclusion of such subjects did not significantly affect the list of smoking-modified transcriptomic and epigenetic features10. Anthropometric measurements and lifestyle parameters had been collected through questionnaires at recruitment (1993–1998 for EPIC-ITALY; 1990–2006 for NSHDS). Information on smoking was obtained through questionnaires and included data on duration, number of cigarettes smoked per day and (only in Italy) pack-years. In addition, for a fraction of the subjects data on plasma cotinine concentration were also available. Details of the subjects involved in the present study are shown in Table 1. Sample collection, storage and processing procedures have been described elsewhere58,59. Based on the conclusions of a previously published pilot study60, subjects were included in the study only if the processing of their blood samples and placement of their buffy coats in cold storage had been completed within 2 hours of collection so as to minimize effects on the transcriptomic profile.
RNA and DNA extraction from buffy coats, genome-wide analysis of gene expression (Agilent 4 × 44 K human whole genome microarray platform) and CpG methylation (Illumina Infinium HumanMethylation450 platform) and the corresponding data quality assessment and preprocessing, were conducted as described previously60. Cotinine levels (AUC) in plasma were measured by reverse-phase chromatography on an Acquity UPLC system (Waters Corporation, Milford, MA, USA) with a Acquity HSS T3 C18 10 mm × 2.1 mm, 1.8 μm, column (Waters) and a binary gradient elution comprising of water 0.1% formic acid and acetonitrile 0.1% formic acid for 19 min. Online analysis of the eluent was performed using a quadrupole time-of-flight mass spectrometer (QTOF-Ultima-MS; Waters) in the positive ion mode. Data were processed using Databridge and XCMS software (Waters). We confirmed the identity of cotinine with authentic standard and accurate mass.
Data analysis and the derivation of lists of expression probes and CpG sites which differed significantly between current and never smokers has been described in detail previously10. Briefly, linear mixed models were ran, using M values for DNA methylation or log2 intensities of mRNA expression as dependent variables, plus date of isolation, labeling, and hybridization for RNA expression, or date of analysis for methylation, as random variables. All analyses additionally adjusted for sex, age, BMI and cohort. Owing to the design of the EnviroGenomarkers project, future disease (breast cancer, B-cell lymphoma) and case-control status were also included as fixed variables. In the case of DNA methylation data, the models were also adjusted for blood cell composition estimated with a published algorithm61. Multiple testing was accounted for with high stringency by using Bonferroni or FDR Benjamini-Hochberg correction. This procedure led to the identification of lists, recently published10, of 1,273 CpG sites (FDR < 0.05) and 350 transcripts (FDR < 0.10) which differ significantly in current relative to never smokers (Suppl. Tables 1 and 2).
We looked for sex-related differences among the above mentioned expression and DNA methylation features by employing rank-based, non-parametric, statistical testing methodology based on the evaluation of the differences in the significance ranks of the probes in the two classes (Fig. 2). Towards this end we first conducted current-versus-never smoker comparisons, using the same statistical models as previously, for all transcriptomic and epigenetic features (29,667 expression probes and 410,987 methylation probes, respectively) separately in males and females, ranking all features by the corresponding significance (p-value). Subsequently we extracted from these lists the rank values of the features which we had previously found to be significant in the mixed population (350 expression probes significant at FDR < 0.10 and 1,273 CpGs significant at Bonferoni-corrected p < 0.05) and calculated their differences between the two sexes. We thus derived a distribution of differences which conforms to the normality assumption (steep unimodal) and was used as basis in order search for differences that violate the null hypothesis that only non-sex-related differences are observed, i.e. signal rankings in sex-stratified analyses are equal. The statistic was calculated from the corresponding complementary cumulative distribution function (Survival Function)62 which describes the probability that a variate takes a value greater than a particular number, taking as a significance threshold the value of p < 0.05. The workflow of this analysis is described diagrammatically in Fig. 1. The tool for the implementation of the statistical evaluation of sex-specific rank differences described above is publically available, under the name RIPOSTE (“Rank DrIven POpulation STatistical Evaluation”), on the Galaxy platform at http://mebioinfo.ekt.gr/galaxy, where instructions for its use are also given.
In order to check for any bias introduced by the difference in male and female population sizes on the selection of sex-specific signals, we implemented a permutation probabilistic approach (also illustrated in Fig. 1, right) by randomly resampling 10 times the full population so as to extract subpopulations even with respect to sex and smoking status, subsequently applying to all subpopulations thus selected the same analytical workflow as described above. In each subpopulation we used all available male subjects (134) and an equal number of females selected randomly while maintaining unaltered the proportions of females from the different cohorts and with different smoking status. For each resampling we counted the number of significant (p < 0.05 for rank difference) signals that came from among those obtained with the full male and female populations (shown in Tables 2 and 3).
Additional Information
How to cite this article: Chatziioannou, A. et al. Blood-based omic profiling supports female susceptibility to tobacco smoke-induced cardiovascular diseases. Sci. Rep. 7, 42870; doi: 10.1038/srep42870 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
Research support by the European Union (Grants number 226756 and 308610). Epigenomics sample analyses were conducted under contract by CBM (Cluster in Biomedicine) S.c.r.l., Trieste, Italy, an Illumina Certified Service Provider. The authors wish to thank Margarita Bekyrou and Stella Kaila for their technical contributions.
Footnotes
The authors declare no competing financial interests.
Author Contributions S.A.K., P.G. and A.C. designed the research. I.A.B., A.J. and D.P. organised the epidemiologic part of the work. A.C., P.G., D.G.H., I.L., I.V., A.P.S., A.E.P., M.B. and M.C.-H. performed the experiments and analysed the data. The EnviroGenomarkers consortium contributed to the design and conduct of the research and the evaluation of the results. S.A.K., A.C., H.K., T.M.C.M.K., J.C.S.K. and P.V. supervised the research. S.A.K. wrote and all authors commented on the manuscript.
Contributor Information
The EnviroGenomarkers project consortium:
Ralph Gottschalk, Danitsja van Leeuwen, Leen Timmermans, Benedetta Bendinelli, Rachel Kelly, Roel Vermeulen, Lutzen Portengen, Fatemeh Saberi-Hosnijeh, Beatrice Melin, Göran Hallmans, Per Lenner, Toby J. Athersuch, Manolis Kogevinas, Euripides G. Stephanou, Antonis Myridakis, Lucia Fazzo, Marco De Santis, Pietro Comba, Hannu Kiviranta, Panu Rantakokko, Riikka Airaksinen, Päivi Ruokojärvi, Mark Gilthorpe, Sarah Fleming, Thomas Fleming, Yu-Kang Tu, Bo Jonsson, Thomas Lundh, Wei J. Chen, Wen-Chung Lee, Chuhsing Kate Hsiao, Kuo-Liong Chien, Po-Hsiu Kuo, Hung Hung, and Shu-Fen Liao
References
- US Department of Health and Human Services, The Health Consequences of Smoking–50 Years of Progress. A Report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, P1–978 (2014).
- Huxley R. R. & Woodward M. Cigarette smoking as a risk factor for coronary heart disease in women compared with men: a systematic review and meta-analysis of prospective cohort studies. Lancet 378, 1297–305 (2011). [DOI] [PubMed] [Google Scholar]
- Appelman Y., van Rijn B. B., Ten Haaf M. E., Boersma E. & Peters S. A. Sex differences in cardiovascular risk factors and disease prevention. Atherosclerosis 241, 211–8 (2015). [DOI] [PubMed] [Google Scholar]
- Peters S. A., Huxley R. R. & Woodward M. Smoking as a risk factor for stroke in women compared with men: a systematic review and meta-analysis of 81 cohorts, including 3,980,359 individuals and 42,401 strokes. Stroke 44, 2821–8 (2013). [DOI] [PubMed] [Google Scholar]
- Aryal S., Diaz-Guzman E. & Mannino D. M. Influence of sex on chronic obstructive pulmonary disease risk and treatment outcomes. Int. J. Chron. Obstruct. Pulmon. Dis. 9, 1145–54 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forey B. A., Thornton A. J. & Lee P. N. Systematic review with meta-analysis of the epidemiological evidence relating smoking to COPD, chronic bronchitis and emphysema. BMC Pulm. Med. Jun 14, doi: 10.1186/1471-2466-11-36 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyohara C. & Ohno Y. Sex differences in lung cancer susceptibility: a review. Gend. Med. 7, 381–401 (2010). [DOI] [PubMed] [Google Scholar]
- Papadopoulos A. et al. Heavy smoking and lung cancer: are women at higher risk? Result of the ICARE study. Br. J. Cancer 110, 1385–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell H. A., Iyen-Omofoman B., Hubbard R. B., Baldwin D. R. & Tata L. J. The association between smoking quantity and lung cancer in men and women. Chest 143, 123–9 (2013). [DOI] [PubMed] [Google Scholar]
- Georgiadis P. et al. Omics for prediction of environmental health effects: Blood leukocyte-based cross-omic profiling reliably predicts diseases associated with tobacco smoking. Sci. Rep. Feb 3, doi: 10.1038/srep20544 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilinger S. et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One 8, e638l2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenker I. M. S. et al. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with Smoking. Hum. Mol. Genet. 22, 843–851 (2013). [DOI] [PubMed] [Google Scholar]
- Leger A. J., Covic L. & Kuliopulos A. Protease-activated receptors in cardiovascular diseases. Circulation 114, 1070–7 (2006). [DOI] [PubMed] [Google Scholar]
- Smith S. M. et al. PAR-1 genotype influences platelet aggregation and procoagulant responses in patients with coronary artery disease prior to and during clopidogrel therapy. Platelets 16, 340–5 (2005). [DOI] [PubMed] [Google Scholar]
- Ramström S. et al. Platelet PAR1 receptor density - correlation to platelet activation response and changes in exposure after platelet activation. Thromb. Res. 121, 681–8 (2007). [DOI] [PubMed] [Google Scholar]
- Li R. & Emsley J. The organizing principle of the platelet glycoprotein Ib-IX-V complex. J. Thromb. Haemost. 11, 605–14 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senis Y. A., Mazharian A. & Mori J. Src family kinases: at the forefront of platelet activation. Blood 124, 2013–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby D. & Poole A. W. Physical and Functional Interaction between Protein Kinase Cδ and Fyn Tyrosine Kinase in Human Platelets. J. Biol. Chem. 278, 24533–24541 (2003). [DOI] [PubMed] [Google Scholar]
- Reddy K. B., Smith D. M. & Plow E. F. Analysis of Fyn function in hemostasis and alphaIIbbeta3-integrin signaling. J. Cell Sci. 121, 1641–8 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hers I. Insulin-like growth factor-1 potentiates platelet activation via the IRS/PI3Kalpha pathway. Blood 110, 4243–52 (2007). [DOI] [PubMed] [Google Scholar]
- Hsieh H. L. et al. Thrombin induces EGF receptor expression and cell proliferation via a PKC(delta)/c-Src-dependent pathway in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 29, 1594–601 (2009). [DOI] [PubMed] [Google Scholar]
- Cao H., Dronadula N. & Rao G. N. Thrombin induces expression of FGF-2 via activation of PI3K-Akt-Fra-1 signaling axis leading to DNA synthesis and motility in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 290, C172–C182 (2006). [DOI] [PubMed] [Google Scholar]
- Lanzetti L. et al. The Eps8 protein coordinates EGF receptor signalling through Rac and trafficking through Rab5. Nature 408, 374–7 (2000). [DOI] [PubMed] [Google Scholar]
- Giampietro C. et al. The actin-binding protein EPS8 binds VE-cadherin and modulates YAP localization and signaling. J. Cell Biol. 211, 1177–92 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrales A., López E., Lee-Rivera I. & López-Colomé A. M. ERK1/2-dependent activation of mTOR/mTORC1/p70S6K regulates thrombin-induced RPE cell proliferation. Cell Signal. 25, 829–38 (2013). [DOI] [PubMed] [Google Scholar]
- Shende P. et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation 123, 1073–82 (2011). [DOI] [PubMed] [Google Scholar]
- Haines D. D., Lekli I., Teissier P., Bak I. & Tosaki A. Role of haeme oxygenase-1 in resolution of oxidative stress-related pathologies: focus on cardiovascular, lung, neurological and kidney disorders. Acta Physiol. (Oxf) 204, 487–501 (2012). [DOI] [PubMed] [Google Scholar]
- Wenzel P. et al. Heme oxygenase-1 suppresses a pro-inflammatory phenotype in monocytes and determines endothelial function and arterial hypertension in mice and humans. Eur. Heart J. 36, 3437–46 (2015). [DOI] [PubMed] [Google Scholar]
- Chan K. H., Ng M. K. & Stocker R. Haem oxygenase-1 and cardiovascular disease: mechanisms and therapeutic potential. Clin. Sci. (Lond.) 120, 493–504 (2011). [DOI] [PubMed] [Google Scholar]
- Dolinay T., Choi A. M. & Ryter S. W. Heme Oxygenase-1/CO as protective mediators in cigarette smoke- induced lung cell injury and chronic obstructive pulmonary disease. Curr. Pharm/Biotechnol. 13, 769–76 (2012). [DOI] [PubMed] [Google Scholar]
- Wang Z., Qin G. & Zhao T. C. HDAC4: mechanism of regulation and biological functions. Epigenomics 6, 139–50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra M. Class IIa HDACs - new insights into their functions in physiology and pathology. FEBS J. 282, 1736–44 (2015). [DOI] [PubMed] [Google Scholar]
- Vogler M. et al. BCL2/BCL-X(L) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood 117, 7145–54 (2011). [DOI] [PubMed] [Google Scholar]
- Durante A. & Zalewski J. Microparticles: A novel player in cardiovascular diseases. Cardiology 132, 249–51 (2015). [DOI] [PubMed] [Google Scholar]
- Vandael D. H. et al. Ca(v)1.3 and BK channels for timing and regulating cell firing. Mol. Neurobiol. 42, 185–98 (2010). [DOI] [PubMed] [Google Scholar]
- Mangoni M. E. et al. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc. Natl. Acad. Sci. USA 100, 5543–8 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig S. M. et al. Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat. Neurosci. 14, 77–84 (2011). [DOI] [PubMed] [Google Scholar]
- Zamponi G. W. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug. Discov. 15, 19–3 (2016). [DOI] [PubMed] [Google Scholar]
- Im B. H. & Rhim H. GABA(B) receptor-mediated ERK1/2 phosphorylation via a direct interaction with Ca(V)1.3 channels. Neurosci. Lett. 513, 89–94 (2012). [DOI] [PubMed] [Google Scholar]
- Cepeda-Benito A., Reynoso J. T. & Erath S. Meta-analysis of the efficacy of nicotine replacement therapy for smoking cessation: Differences between men and women. Journal of Consulting and Clinical Psychology 72, 712–722 (2004). [DOI] [PubMed] [Google Scholar]
- Scharf D. & Shiffman S. Are there gender differences in smoking cessation, with and without bupropion? Pooled- and meta-analyses of clinical trials of Bupropion SR. Addiction 99, 1462–1469 (2004). [DOI] [PubMed] [Google Scholar]
- Chen R. et al. Transcriptome profiling reveals that the SM22α-regulated molecular pathways contribute to vascular pathology. J. Mol. Cell Cardiol. 72, 263–72 (2014). [DOI] [PubMed] [Google Scholar]
- Banerjee I. et al. Targeted ablation of nesprin 1 and nesprin 2 from murine myocardium results in cardiomyopathy, altered nuclear morphology and inhibition of the biomechanical gene response. PLoS Genet. Feb 20, doi: 10.1371/journal.pgen.1004114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puckelwartz M. J. Nesprin-1 mutations in human and murine cardiomyopathy. J. Mol. Cell Cardiol. 48, 600–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinhuis B. et al. Towards a role of interleukin-32 in atherosclerosis. Cytokine 64, 433–40 (2013). [DOI] [PubMed] [Google Scholar]
- Wang H. et al. Cardiomyocyte-specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction. J. Lipid Res. 54, 953–65 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiffman D. et al. Gene Variants of VAMP8 and HNRPUL1 are associated with early-onset myocardial infarction. Arteriosclerosis, Thrombosis, and Vascular Biology 26, 1613–1618 (2006). [DOI] [PubMed] [Google Scholar]
- Hakimzadeh N. et al. Circulating MicroRNAs Characterizing Patients with Insufficient Coronary Collateral Artery Function. PLoS One Sep 2, doi: 10.1371/journal.pone.0137035 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaireti R., Gustafsson K. M., Byström B., Bremme K. & Lindahl T. L. Endogenous thrombin potential is higher during the luteal phase than during the follicular phase of a normal menstrual cycle. Hum Reprod. 28, 1846–52 (2013). [DOI] [PubMed] [Google Scholar]
- Farré A. L., Modrego J. & Zamorano-León J. J. Effects of hormones on platelet aggregation. Horm. Mol. Biol. Clin. Investig. 18, 27–36 (2014). [DOI] [PubMed] [Google Scholar]
- Wang T. Y. et al. Platelet biology and response to antiplatelet therapy in women: implications for the development and use of antiplatelet pharmacotherapies for cardiovascular disease. J. Am. Coll. Cardiol. 59, 891–900 (2012). [DOI] [PubMed] [Google Scholar]
- Roy-O’Reilly M. & McCullough L. D. Sex differences in stroke: the contribution of coagulation. Exp. Neurol. 259, 16–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremmel T., Kopp C. W., Eichelberger B., Koppensteiner R. & Panzer S. Sex differences of leukocyte-platelet interactions and on-treatment platelet reactivity in patients with atherosclerosis. Atherosclerosis 237, 692–5 (2014). [DOI] [PubMed] [Google Scholar]
- Marcantoni E., Di Francesco L., Dovizio M., Bruno A. & Patrignani P. Novel insights into the vasoprotective role of heme oxygenase-1. Int. J. Hypertens. 2012, 127910, doi: 10.1155/2012/127910 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedram A. et al. Estrogen regulates histone deacetylases to prevent cardiac hypertrophy. Mol. Biol. Cell. 24, 3805–18 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco A. T., Corken A. & Ware J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood 126, 582–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. et al. Gender susceptibility for cigarette smoking-attributable lung cancer: A systematic review and meta-analysis. Lung Cancer 85, 351–360 (2014). [DOI] [PubMed] [Google Scholar]
- Bingham S. & Riboli E. Diet and cancer - the European Prospective Investigation into Cancer and Nutrition. Nat. Rev. Cancer 4, 206–15 (2004). [DOI] [PubMed] [Google Scholar]
- Hallmans G. et al. Cardiovascular disease and diabetes in the Northern Sweden Health and Disease Study Cohort - evaluation of risk factors and their interactions. Scand. J. Public Health Suppl. 61, 18–24 (2003). [DOI] [PubMed] [Google Scholar]
- Hebels D. G. et al. Performance in omics analyses of blood samples in long-term storage: opportunities for the exploitation of existing biobanks in environmental health research. Environ Health Perspect. 121, 480–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics May 8, doi: 10.1186/1471-2105-13-86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M., Hastings N. & Peacock B. Statistical Distributions, 3rd ed. New York: Wiley, p. 13 (2000). [Google Scholar]
- Ward J. A. et al. Circulating Cell and Plasma microRNA Profiles Differ between Non-ST-Segment and ST-Segment-Elevation Myocardial Infarction. Fam. Med. Med. Sci. Res. October 1, doi: 10.4172/2327-4972.1000108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joehanes R. et al. Gene expression signatures of coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 33, 1418–1426 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nührenberg T. G. et al. Transcriptome analysis in patients with progressive coronary artery disease: identification of differential gene expression in peripheral blood. J. Cardiovasc. Transl. Res. 6, 81–93 (2013). [DOI] [PubMed] [Google Scholar]
- Masud R., Shameer K., Dhar A., Ding K. & Kullo I. J. Gene expression profiling of peripheral blood mononuclear cells in the setting of peripheral arterial disease. J. Clin. Bioinforma. Mar 12, doi: 10.1186/2043-9113-2-6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al. Dysregulated expression of microRNAs and mRNAs in myocardial infarction. Am. J. Transl. Res. 7, 2291–304 (2015). [PMC free article] [PubMed] [Google Scholar]
- Lewis D. A. et al. Whole blood gene expression profiles distinguish clinical phenotypes of venous thromboembolism. Thromb. Res. 135, 659–65 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. Z., Manduchi E., Stoeckert C. J. Jr. & Davies P. F. Arterial endothelial methylome: differential DNA methylation in athero-susceptible disturbed flow regions in vivo. BMC Genomics Jul 7, doi: 10.1186/s12864-015-1656-4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai K. et al. Highly multiplexed genotyping of coronary artery disease-associated SNPs using MALDI-TOF mass spectrometry. Hum. Mutat. 20, 133–8 (2002). [DOI] [PubMed] [Google Scholar]
- Melman Y. F. et al. Circulating MicroRNA-30d Is Associated With Response to Cardiac Resynchronization Therapy in Heart Failure and Regulates Cardiomyocyte Apoptosis: A Translational Pilot Study. Circulation 131, 2202–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.