

Abstract
Swertia mussotii Franch. is an important traditional Tibetan medicinal plant with pharmacological properties effective in the treatment of various ailments including hepatitis. Secoiridoids are the major bioactive compounds in S. mussotii. To better understand the secoiridoid biosynthesis pathway, we generated transcriptome sequences from the root, leaf, stem, and flower tissues, and performed de novo sequence assembly, yielding 98,613 unique transcripts with an N50 of 1,085 bp. Putative functions could be assigned to 35,029 transcripts (35.52%) based on BLAST searches against annotation databases including GO and KEGG. The expression profiles of 39 candidate transcripts encoding the key enzymes for secoiridoid biosynthesis were examined in different S. mussotii tissues, validated by qRT-PCR, and compared with the homologous genes from S. japonica, a species in the same family, unveiling the gene expression, regulation, and conservation of the pathway. The examination of the accumulated levels of three bioactive compounds, sweroside, swertiamarin, and gentiopicroside, revealed their considerable variations in different tissues, with no significant correlation with the expression profiles of key genes in the pathway, suggesting complex biological behaviours in the coordination of metabolite biosynthesis and accumulation. The genomic dataset and analyses presented here lay the foundation for further research on this important medicinal plant.
The herbaceous genus Swertia, in the family Gentianaceae, contains traditional medicinal plants that have been used in China, India, Korea, and Japan for thousands of years. There are approximately 170 species of Swertia, with approximately 79 occurring in China1. Several species have pharmacological properties and are used in the treatment of different ailments2,3,4,5. For example, Swertia chirata is traditionally used as a bitter tonic to stimulate appetite and as a febrifuge in India, whereas Swertia davidi has been widely used as a remedy for acute bacillary dysentery. Swertia mussotii Franch is well known as “Di-da” and “Zang Yin Chen” in China because of its long history of use as a curative for hepatitis6,7 and has been recognized as one of the eight most important Tibetan traditional medicinal plants. S. mussotii has been widely used as a crude drug in more than 10 types of complex Tibetan folk medicine for the treatment of hepatitis, along with other uses that exploit its anti-diabetic and anti-hyperlipidaemic effects.
Chemical and pharmacological studies on S. mussotii have identified several major active compounds, swertiamarin, mangiferin, gentiopicroside, sweroside, oleanolic acid and three xanthones8,9,10,11,12. These compounds are of considerable interest as theraputic metabolites, and have been studied in wide-ranging pharmacological applications. For example, swertiamarin, a secoiridoid, had significant hepatoprotective effects in a liver damage model induced by alpha-naphthylisothiocyanate13. Recent research has reported that swertiamarin attenuates liver injury, inflammation, and cholestasis in bile duct-ligated rats14. Swertiamarin can also significantly up-regulate the hepatic bile acid detoxification enzymes and efflux transporters in rats, resulting in an increase in the water solubility of hydrophobic bile acids and the elimination of conjugated bile acids15.
The natural population of S. mussotii is restricted to high altitudes (2,000 to 5,000 metres above sea level) in the Qinghai-Tibetan Plateau area in China. The natural resource of S. mussotii yields less than 50 tons every year, while the high demand of the crude drug market requires several times more than this yield1. With the increasing demand for the species for medicinal use and the harvesting of flowering plants before fruit maturation1, wild populations of S. mussotii are being exhausted because of over-collection16,17. S. mussotii has been listed as an endangered species by some local governments in China, and harvesting wild plants is now prohibited in some areas of its natural distribution18. Domestic cultivation of S. mussotii could potentially solve the problem of high demand, but its seeds have strong dormancy and very low germination efficiency. In addition, there are concerns regarding the possible changes in the bioactive compound contents through domestic cultivation. When seven pharmacologically active compounds were compared between plants from natural, high-altitude populations and cultivated plants at low altitudes, mangiferin, one of the most abundant and active compounds was significantly lower in the cultivated sample, although most of the compounds were not significantly different. If domestic cultivation of this medicinal crop is to be successful, we need a better understanding of the genetic underpinnings of the pathways producing these compounds.
The secoiridoid biosynthesis pathway is still largely unresolved in S. mussotii, although it is thought to be closely related to terpenoid backbone biosynthesis19,20. Only one full-length cDNA clone (SmG10H) encoding geraniol 10-hydroxylase has been isolated from S. mussotii using PCR with degenerate pairs of primers designed based on the G10H gene sequences from other species21. Geraniol 10-hydroxylase is thought to play an important role in iridoid monoterpenoid biosynthesis. Functional characterization by overexpressing SmG10H in transgenic S. mussotii calli increased the level of the SmG10H transcript and the swertiamarin content. In addition, treatment with methyl jasmonate (MeJA) also increased the transcription of this gene, with a concomitant increase in swertiamarin content21. Except for SmG10H, little is known about the functions and regulation of other genes associated with the secondary metabolic pathways in Swertia mussotii. Only eight Swertia mussotii sequences (matK, rpl16, ITS, SmG10H, and ITS1-5.8S-ITS2) have been deposited in the NCBI GenBank database21. Clearly, the lack of sequence information seriously impedes our understanding of the metabolic pathways through the molecular characterization of gene functions22. Therefore, a comprehensive research strategy is critical for improving our understanding the biology of this high-altitude plant for high yield and for elucidating the metabolic pathways leading to the biosynthesis of its active compounds for better pharmaceutical applications.
Transcriptomics using next-generation sequencing technology has proved to be a powerful and cost-effective approach that generates a large amount of transcribed sequence data useful for a wide range of research applications23,24. In this study, we established transcriptome databases from the root, stem, leaf, and flower tissues collected at the flowering stage of native-habitat-grown S. mussotii. Through comprehensive transcriptome data analyses, we identified thousands of genes and analysed a set of putative genes involved in the secoiridoid pathway. These transcriptome data represent the first genomic resource for S. mussotii, which not only lays the foundation for further functional genomics study, but will also aid in the identification, characterization, and genetic modification of secondary metabolic pathway genes in this important medicinal plant.
Result and Discussion
Illumina sequencing and read assembly
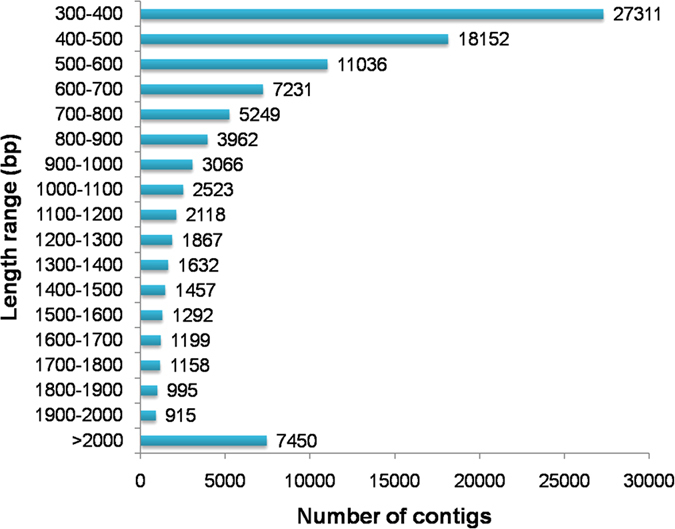
De novo transcriptome analysis is an excellent platform for generating comprehensive information of the gene space in an organism for the discovery of novel genes, development of molecular markers, and construction of gene expression networks for specific tissues/organs. This approach is particularly useful for non-model species lacking whole-genome sequencing data. To generate a transcriptome database for S. mussotii, twelve RNAseq libraries were constructed from the root, stem, leaf, and flower tissues collected from three individual plants. A total of 1,295,381,780 raw pair-end reads with a read length of 100 bp were generated. After a trimming process was performed to remove adaptors, primer sequences, poly-A tails, and short and low quality sequences, 1,195,207,248 (92.27%) high-quality reads were recovered, ranging from 240 to 338 million for each tissue (Supplementary Table S1). The filtered reads were de novo assembled using CLC Genomics Workbench (v8.0.2). The CD-HIT tool was used to obtain non-redundant contigs. The final assembly dataset contained 98,613 unique contigs (transcripts) with an N50 of 1,085 bp. The unique contigs were designated Swertia mussotii tentative consensus (SmTc) transcripts and assigned unique identifier numbers from SmTc_1 to SmTc_98613. The total accumulated size of the assembled transcripts was approximately 82 Mb, with sizes ranging from 300–13,875 bp and an average size of 829 bp. There were 76,007 transcripts (77.08%) in the size range of 300–1,000 bp, 15,156 (15.37%) at 1,001–2,000 bp, and 7,450 (7.55%) >2,000 bp (Fig. 1).
Figure 1. Overview of transcriptome assembly data showing the size distribution of transcripts.
Functional annotation
Functional annotation of assembled transcripts provides insight into their molecular functions and biological processes in an organism. The S. mussotii transcripts were annotated based on protein similarity using BLAST searches against several public databases. The statistical summary for this annotation is shown in Supplementary Table S2. Among the 98,613 transcripts, 30,489 (30.92%) could be annotated in TrEMBL, 11,593 (11.76%) in Swiss-Prot, 11,175 (11.33%) in Clusters of Orthologous Groups (COG), 22,288 (22.60%) in Gene Ontology (GO), and 7,654 (7.76%) in the Kyoto Encyclopedia of Genes and Genomes (KEGG). Taken together, 35,029 transcripts (35.52%) could be assigned at least one putative function from one of these databases (Supplementary Table S2), while 64.48% of the transcripts had no significant protein matches (Supplementary Table S3). These transcripts may represent novel or diverse proteins and long non-coding RNAs in S. mussotii, or could be derived from less conserved 3′ or 5′ untranslated regions of the genes25,26.
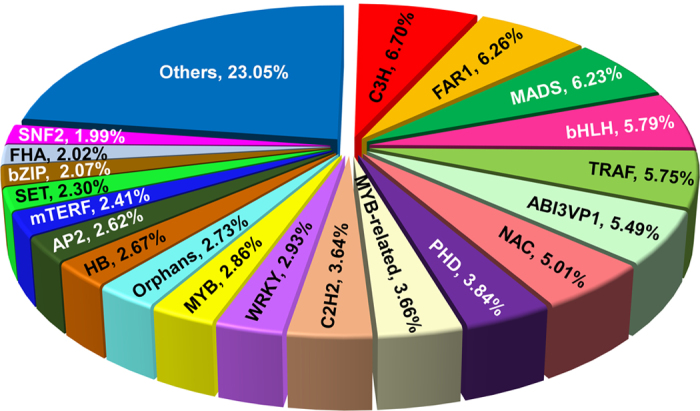
Transcription factors (TFs) are diverse groups of gene families that play key regulatory roles in plant growth and development via controlling the expression of genes through specific binding to cis-regulatory elements present in the promoter regions of targeted genes27. Many TF families, including MYB, MYB-related, WRKY and bHLH, are known to regulate secondary metabolism biosynthesis in plants28,29,30. The number of genes encoding different TF families varies in different plant species and they often perform species-/tissue-specific or developmental stage-specific function(s)31. Our analysis of the S. mussotii transcriptome data revealed that 7,481 transcripts (7.59%) encode putative TFs that can be classified into 82 TF families (Supplementary Table S4). Members of the C3H transcription factor family were the most abundant (501, 6.70%) followed by FAR1 (468, 6.26%), MADS (466, 6.23%), bHLH (433, 5.79%) and TRAF (430, 5.75%) (Fig. 2). The identification of this large set of TFs, along with their expression profiling in individual tissues provides a rich resource for future characterization of specific TFs in various biochemical pathways in S. mussotii.
Figure 2. Distribution of transcription factor families.
When GO was used to classify gene functions, 22,288 transcripts could be assigned to one or more GO terms within the three domains and 22 functional categories (Fig. 3, Supplementary Table S5). Within the cellular component domain, the three most enriched categories were “cell” (11,559, 51.86%), “cell part” (11,422, 51.25%) and “organelle” (4,413, 19.80%). In the molecular function domain, the three most matched categories were “binding” (11,095, 49.78%), “catalytic activity” (10,891, 48.86%), and “transporter activity” (1,213, 5.44%). In the biological process domain, the three most common categories were “cellular process” (8,846, 39.69%), “metabolic process” (8,058, 36.15%), and “biological regulation” (1,904, 8.54%). The most commonly assigned functional categories in each domain were consistent with the results from other Gentianaceae species32,33.
Figure 3. Frequencies and mean expression levels of transcripts matching GO terms.

The percentage of transcripts matching GO terms is shown for each category as red bars.
The KEGG pathway database is a collection of maps representing the experimental knowledge concerning metabolism and provides various functions for genes in cells and organisms. We assigned gene functions to the biological pathways described in KEGG to better understand the biological functions of the genes in specific metabolic pathways (Fig. 4). To systematically analyse inner cell metabolic pathways and complex biological behaviours, we classified transcripts by mapping the annotated coding region sequences to the reference canonical pathways in the KEGG pathway database. From this analysis, we identified that 7,654 transcripts (7.76%) could be assigned to five main categories: “metabolism”, “genetic information processing”, “environmental information processing”, “cellular process”, and “organismal systems” with 33 sub-categories and 280 pathways (Fig. 4, Supplementary Table S6). The top 3 pathways with the largest numbers of mapped transcripts were “translations” (865 transcripts, 11.30%), “signal transduction” (697 transcripts, 9.11%), and “carbohydrate metabolism” (630 transcripts, 8.23%). Furthermore, we identified 643 protein-coding transcripts that have significant matches to 333 enzymes. These enzymes have assigned functions in 23 secondary metabolic pathways in KEGG (Table 1). Among them, 86 transcripts encode key enzymes involved in the pathways for terpenoid biosynthesis, including the synthesis of terpenoid backbone (54 transcripts), monoterpenoids (4 transcripts), diterpenoids (14 transcripts), and sesquiterpenoid and triterpenoid (14 transcripts). There were 123 transcripts found related to the flavonoid biosynthesis pathway including the phenylpropanoid (101 transcripts), flavonoid (18 transcripts), and flavone and flavonol biosynthesis pathways (4 transcripts). Only 35 transcripts were associated with alkaloid biosynthesis. Identification and future characterization of transcripts involved in different metabolic pathways will help us better understand their functions in the biosynthesis of active compounds in Gentianaceae plants.
Figure 4. KEGG pathway classification map.
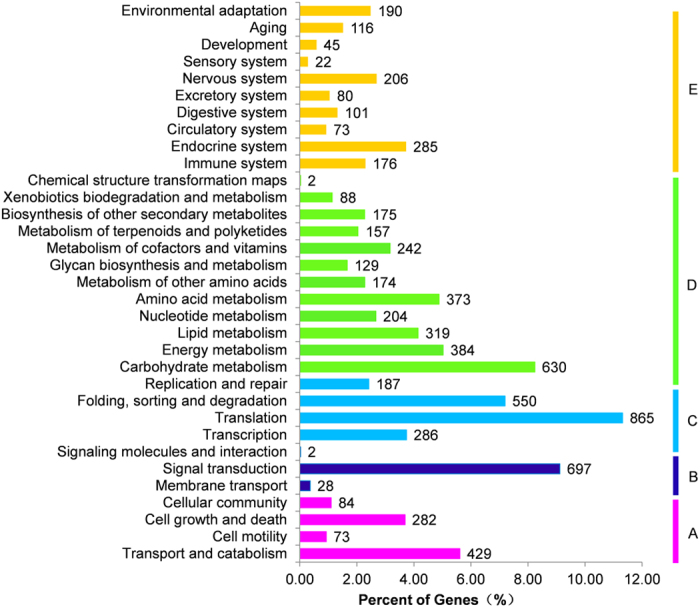
Genes were divided into five branches according to the biological pathways they participated in: (A), Cellular Processes; (B), Environmental Information Processing; (C), Genetic Information Processing; (D), Metabolism; (E), Organismal Systems.
Table 1. Secondary metabolism pathways in S. mussotii.
| Pathway ID | Pathways | Function categories | SmTc numbers |
|---|---|---|---|
| ko00100 | Steroid biosynthesis | 18 | 31 |
| ko00130 | Ubiquinone and other terpenoid-quinone biosynthesis | 21 | 36 |
| ko00230 | Purine metabolism | 91 | 160 |
| ko00232 | Caffeine metabolism | 2 | 2 |
| ko00400 | Phenylalanine, tyrosine and tryptophan biosynthesis | 23 | 41 |
| ko00860 | Porphyrin and chlorophyll metabolism | 32 | 48 |
| ko00900 | Terpenoid backbone biosynthesis | 28 | 54 |
| ko00901 | Indole alkaloid biosynthesis | 1 | 1 |
| ko00902 | Monoterpenoid biosynthesis | 2 | 4 |
| ko00903 | Limonene and pinene degradation | 2 | 11 |
| ko00904 | Diterpenoid biosynthesis | 8 | 14 |
| ko00905 | Brassinosteroid biosynthesis | 7 | 7 |
| ko00906 | Carotenoid biosynthesis | 21 | 30 |
| ko00908 | Zeatin biosynthesis | 5 | 13 |
| ko00909 | Sesquiterpenoid and triterpenoid biosynthesis | 8 | 14 |
| ko00940 | Phenylpropanoid biosynthesis | 19 | 101 |
| ko00941 | Flavonoid biosynthesis | 14 | 18 |
| ko00942 | Anthocyanin biosynthesis | 3 | 5 |
| ko00944 | Flavone and flavonol biosynthesis | 4 | 4 |
| ko00945 | Stilbenoid, diarylheptanoid and gingerol biosynthesis | 5 | 14 |
| ko00950 | Isoquinoline alkaloid biosynthesis | 9 | 15 |
| ko00960 | Tropane, piperidine and pyridine alkaloid biosynthesis | 8 | 18 |
| ko00965 | Betalain biosynthesis | 2 | 2 |
Simple sequence repeat (SSR) analysis
SSRs have been extensively used as molecular markers for various genotyping applications because they are abundant, easy to develop and detect, and highly polymorphic among species/accessions34,35. We screened the assembled transcript sequences using the MISA search tool and identified 33,529 SSRs (Supplementary Table S7). Among them, 8,778 SSRs (2–6 nt) were of the minimum length of 12 bp, with a frequency of one SSR per 9.3 kb of transcriptome sequences.
The identified SSRs were dominated by di and tri-nucleotide repeats representing approximately 43.23% (3,795) and 47.30% (4,152), respectively, of the SSRs with 2–6 nt (Fig. 5a). Among the di-nucleotide repeats, AT/AT was present at the highest (23.06%) frequency, followed by AG/CT (10.06%) and AC/GT (9.97%) (Fig. 5b). For tri-nucleotide repeats, AAG/CTT showed the highest occurrence (9.75%), followed by AAT/ATT (8.34%) and ACC/GGT (6.33%). AAC/GTT was the least abundant (2.78%) (Fig. 5b). This distribution is similar to those of other Gentianaceae family members, such as Gentiana straminea33 and Veratrilla ballonii36. Small fractions of tetra- (4.60%), penta- (2.65%) and hexa-nucleotide (2.21%) SSRs were also identified in S. mussotii (Fig. 5a). Given that the identified SSRs are present in transcripts, short repeat sequences could have played roles in gene evolution37,38. The availability of this large number of SSRs can greatly enhance large-scale genotyping studies for various applications in S. mussotii, such as genetic diversity assessment and association mapping for important traits.
Figure 5. Simple sequence repeats (SSRs) in the S. mussotii transcriptome.
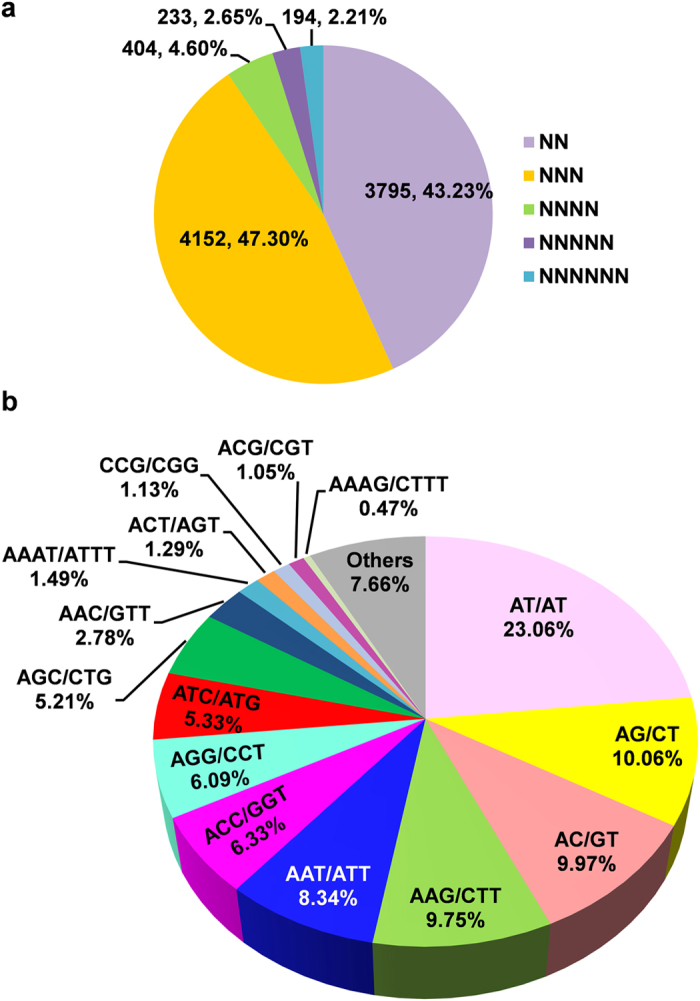
(a) Distribution of different classes of SSRs. (b) Frequency of most abundant SSR motifs.
Differential gene expression analysis
RNA-seq has emerged as a powerful technology to measure gene expression and tissue specificity at the whole-genome level39,40. To investigate the differential gene expression among different tissues, we mapped the high-quality reads from individual samples onto the S. mussotii transcriptome using CLC Genomics Workbench. Approximately 85–91% of the total reads were successfully mapped to the S. mussotii transcriptome (Supplementary Table S1). The differentially expressed transcripts (DETs) were identified based on the normalized RPKM (reads per kilobase of transcript per million mapped reads) value for each transcript in individual tissue (Supplementary Table S8) with Expander software (http://acgt.cs.tau.ac.il/expander/) using a t-test with a p-value cut-off ≤ 0.05 and at least a 2-fold expression change among different tissues41. Substantial transcriptional differences were seen in the pairwise comparisons between different tissues (Fig. 6a). The number of DETs was highest betweenthe flower and leaf and lowest between the stem and root (Fig. 6a). We further investigated DETs in one tissue in respect to the other three tissues. In this case, a DET was identified by testing its RPKM value from one tissue against the values from the three other tissues with the same statistical parameter. In the flowers, 9,344 transcripts were up-regulated and 1,914 transcripts were down-regulated relative to their expression in the three other tissues (Fig. 6b). Leaves had the largest number of up-regulated transcripts (14,707). In roots, 9,800 transcripts were up-regulated, while 3,822 transcripts were down-regulated. Stems had the smallest number of DETs, with 5,884 up-regulated and 613 down-regulated (Fig. 6b). In addition, we defined a transcript as having tissue-specific expression if its reads only came from a single tissue. A total of 11,560 transcripts exhibited tissue-specific expression, and 2,109, 350, 6,675 and 2,426 transcripts came from root, stem, leaf, and flower, respectively (Fig. 6c).
Figure 6. Differential expression analysis of S. mussotii transcripts.
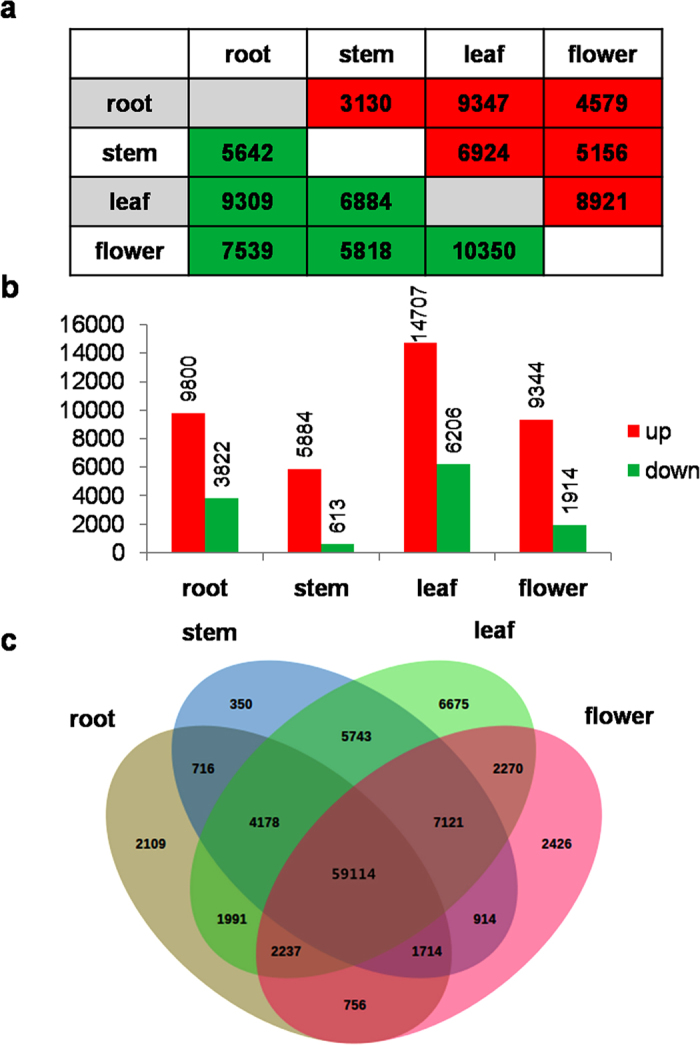
(a) Pair-wise comparisons of different tissues showing DETs. (b) The number of significantly (P-value ≤ 0.05 and at least two-fold change) up- and down- regulated transcripts in each tissue as compared to all three other tissues. (c) Venn diagram representing the number of DETs among S. mussotii tissues.
Among the identified DETs, 2,946 encoded transcription factors, and 565, 203, 1,046, and 1,132 came from the root, stem, leaf, and flower, respectively (Supplementary Table S4). Future characterization of specific transcription factors is required for a better understanding of the gene expression profiles and regulation in various biochemical pathways in S. mussotii. The DETs were further analysed using the KEGG database to explore their functional categories. Among 2,428 DETs with assigned functions in the biochemical pathways, 562, 114, 1,019, and 733 were from root, stem, leaf, and flower tissues, respectively, and these DETs were associated with 193, 60, 243, and 216 pathways in the corresponding tissues (Supplementary Table S9). These DETs had enrichment for 88, 23, 99, and 47 pathways in the root, stem, leaf, and flower tissues, respectively, while fifteen pathways were commonly enriched in all tissues, including the biosynthesis of secondary metabolites, microbial metabolism in diverse environments, starch and sucrose metabolism, amino sugar and nucleotide sugar metabolism in carbohydrate metabolism, the PI3K-Akt signalling pathway, the AMPK signalling pathway, and plant hormone signal transduction in the signal transduction group. We found that certain pathways were specifically enriched in particular tissues. The 25 pathways only enriched in the leaf were mainly related to energy metabolisms and signalling transductions. The 12 root enriched pathways were involved in the phosphonate and phosphinate metabolism pathways. In the flowers, specifically enriched pathways were primarily associated with secondary metabolism groups, including the flavonoid, sesquiterpenoid, and triterpenoid biosynthesis pathways. Pathways specifically enriched in the stem were not observed (Supplementary Table S9).
Putative genes involved in the secoiridoids biosynthesis pathway
In the proposed secoiridoid biosynthesis pathway (Fig. 7a), isopentenyl diphosphate (IPP) and its allylic isomer, dimethylallyldiphosphate (DMAPP), are the universal precursors in the first enzymatic step. These chemicals can be derived from both the cytosolic mevalonate (MVA) and/or plastidial methylerythritol phosphate (MEP) pathways. The prenyl-transfer of IPP on DMAPP catalysed by geranyl diphosphate synthase (GPPS)42 forms geranyl diphosphate (GPP), which is followed by the formation of the monoterpene geraniol by geraniol synthase (GES)43. It has been proposed that geraniol is the start of the secoiridoid pathway and that it is hydroxylated into 10-hydroxygeraniol by geraniol 10-hydroxylase21, leading to the formation of secologanin. Ultimately, secologanin is converted into swertiamarin, gentiopicroside, and other secoiridoid compounds through several currently unknown steps44. When examining the annotation of transcriptome contigs against the KEGG database, we identified 39 transcripts that could be classified into 24 enzyme categories associated with three metabolic pathways leading to secoiridoid biosynthesis (Table 2). It appeared that the genes encoding all of the enzymes in these pathways were identified in the transcriptome pool of S. mussotii. The analysis of the metabolic pathways with transcriptomic data help us identify the coding sequences of the genes involved in specific processes of the pathway in targeted plant species and cane aid in uncovering their family members and expression patterns in different tissues. For example, the reaction catalysed by the enzyme 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) is an important step in the MVA pathway45. The HMGR protein contains an N-terminal transmembrane domain, a highly divergent linker domain, and a C-terminal conserved catalytic domain46. The upregulation of HMGR genes could improve the yield of terpenes in transgenic plants47,48,49,50,51. The HMGR genes from a number of medicinal plants, including Panax gingseng, Salvia miltiorrhiza, Panax notoginseng, Siraitia grosvenorii, Aquilaria sinensis, and Cyclocarya paliurus, have been cloned and characterized. The HMGR gene family members range from 1 to 9 in different species, although most have fewer than three. In the Gentianaceae family, this gene has two copies in each species examined thus far, including Gentiana rigescens, G. macrophylla and G. lutea. In S. mussotii, we identified 4 full-length HMGR transcripts (SmHMGR1, SmHMGR2, SmHMGR3, and SmHMGR4) (Supplementary Figure S1). A sequence alignment revealed that these transcripts were conserved in the C-terminus region and diverged in the N-terminus region (Supplementary Figure S1). Transcriptome profiling data showed that SmHMGR1 was constitutively highly expressed in all four tissues, while the other three genes all displayed differential expression patterns (Fig. 7b). Although the exact function of each HMGR gene is not clear in S. mussotii, we can speculate that HMGR1 might play an important role in the pathway given its high expression level in all of the tissues.
Figure 7. Schematic representation of the proposed swertiamatin biosynthesis pathway and the differential expression of genes involved in the pathway.

(a) Proposed pathway of swertiamatin biosynthesis. (b) The expression of genes involved in the MVA pathway. (c) The expression of genes involved in the MEP pathway. (d) The expression of genes involved in the secoiridoid pathway.
Table 2. Summary of transcripts in S. mussotii encoding enzymes involved in the secoiridoid pathway.
| Pathway | Gene | Gene name | E.C. number | KO | SmTc |
|---|---|---|---|---|---|
| MVA | ACCT1 | acetyl-CoA C-acetyltransferase | 2.3.1.9 | K00626 | SmTc_2083 |
| ACCT2 | acetyl-CoA C-acetyltransferase | 2.3.1.9 | K00626 | SmTc_5029 | |
| ACCT3 | acetyl-CoA C-acetyltransferase | 2.3.1.9 | K00626 | SmTc_33675 | |
| HMGS1 | hydroxymethylglutaryl-CoA synthase | 2.3.3.10 | K01641 | SmTc_5929 | |
| HMGS2 | hydroxymethylglutaryl-CoA synthase | 2.3.3.10 | K01641 | SmTc_7106 | |
| HMGS3 | hydroxymethylglutaryl-CoA synthase | 2.3.3.10 | K01641 | SmTc_8664 | |
| HMGS4 | hydroxymethylglutaryl-CoA synthase | 2.3.3.10 | K01641 | SmTc_81097 | |
| HMGR1 | hydroxymethylglutaryl-CoA reductase | 1.1.1.34 | K00021 | SmTc_1585 | |
| HMGR2 | hydroxymethylglutaryl-CoA reductase | 1.1.1.34 | K00021 | SmTc_10138 | |
| HMGR3 | hydroxymethylglutaryl-CoA reductase | 1.1.1.34 | K00021 | SmTc_15910 | |
| HMGR4 | hydroxymethylglutaryl-CoA reductase | 1.1.1.34 | K00021 | SmTc_22303 | |
| MVK | mevalonate kinase | 2.7.1.36 | K00869 | SmTc_7721 | |
| PMK | phosphomevalonate kinase | 2.7.4.2 | K00938 | SmTc_5121 | |
| MVD1 | diphosphomevalonate decarboxylase | 4.1.1.33 | K01597 | SmTc_2684 | |
| MVD2 | diphosphomevalonate decarboxylase | 4.1.1.33 | K01597 | SmTc_2834 | |
| MEP | DXS1 | 1-deoxy-D-xylulose-5-phosphate synthase | 2.2.1.7 | K01662 | SmTc_5909 |
| DXS2 | 1-deoxy-D-xylulose-5-phosphate synthase | 2.2.1.7 | K01662 | SmTc_7421 | |
| DXS3 | 1-deoxy-D-xylulose-5-phosphate synthase | 2.2.1.7 | K01662 | SmTc_10119 | |
| DXS4 | 1-deoxy-D-xylulose-5-phosphate synthase | 2.2.1.7 | K01662 | SmTc_13486 | |
| DXR | 1-deoxy-D-xylulose-5-phosphate reductoisomerase | 1.1.1.267 | K00099 | SmTc_8142 | |
| ISPD | 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase | 2.7.7.60 | K00991 | SmTc_23281 | |
| ISPE | 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase | 2.7.1.148 | K00919 | SmTc_19780 | |
| ISPF | 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase | 4.6.1.12 | K01770 | SmTc_11729 | |
| GCPE | (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase | 1.17.7.1 | K03526 | SmTc_845 | |
| ISPH | 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase | 1.17.7.4 | K03527 | SmTc_757 | |
| IDI1 | isopentenyl-diphosphate delta-isomerase | 5.3.3.2 | K01823 | SmTc_744 | |
| IDI2 | isopentenyl-diphosphate delta-isomerase | 5.3.3.2 | K01823 | SmTc_1157 | |
| GPPS1 | geranyl diphosphate synthase | 2.5.1.1 | K14066 | SmTc_17467 | |
| GPPS2 | geranyl diphosphate synthase | 2.5.1.1 | K14066 | SmTc_6570 | |
| Secoiridoid | GES | geranyl diphosphate diphosphatase | 3.1.7.11 | K20979 | SmTc_70847 |
| G10H | geraniol 10-hydroxylase | 1.14.13.152 | K15099 | SmTc_41455 | |
| 8HGO | 8-hydroxygeraniol oxidoreductase | 1.1.1.324 | SmTc_81115 | ||
| IS | iridoid synthase | 1.3.1.99 | K20144 | SmTc_35128 | |
| IO | iridoid oxidase | SmTc_29298 | |||
| 7-DLGT | 7-deoxyloganetic acid glucosyl transferase | 2.4.1.323 | SmTc_15909 | ||
| DL7H | 7-deoxyloganic acid hydroxylase | SmTc_8300 | |||
| LAMT | loganic acid O-methyltransferase | 2.1.1.50 | SmTc_23275 | ||
| SLS1 | secologanin synthase | 1.3.3.9 | K13400 | SmTc_4315 | |
| SLS2 | secologanin synthase | 1.3.3.9 | K13400 | SmTc_16024 |
Compared to the MVA and MEP pathways, the secoiridoid pathway is not been well understood. Recently, it has been shown that the CrDXS and CrG10H genes isolated from Catharanthus roseus are important genes in regulating terpenoid indole alkaloid biosynthesis52, filling important knowledge gaps in understanding the pathway. When we examined the expression profiles of the genes in the three pathways using the S. mussotii transcriptome data, it was noted that the transcripts of the MVA and MEP pathway genes, such as hydroxymethylglutaryl-CoA reductase (SmHMGR), diphosphomevalonate decarboxylase (SmMVD), hydroxymethylglutaryl-CoA synthase (SmHMGS), and 1-deoxy-D-xylulose-5-phosphate synthase (SmDXS), were more abundant in the flower tissues than in other tissues (Fig. 7b,c). However, in the secoiridoid pathway, five out of seven transcripts had high expression level in the stem, leaf and flower tissues but much lower expression in the root (Fig. 7c). Only the Sm7DLGT gene encoding 7-deoxyloganetic acid glucosyl transferase showed a higher expression in the root tissue than in the other three tissues (Fig. 7d). Two transcripts (SmSLS1 and SmSLS2) both encode a secologanin synthase enzyme. The expression of the SmSLS1 gene was constantly low in all of the tissues, while the expression of SmSLS2 was higher in these tissues, particularity in the flower tissue (Fig. 7d).
Recently, a transcriptome study on a species (Swertia japonica) belonging to the same Swertia family was reported53. By comparing the transcriptome data from closely related species, we can identify transcripts that are shared and different in the two species and predict the evolutionary relationships of the key enzymes in the metabolic pathways. When the S. mussotii genes involved in the secoiridoid biosynthesis pathway were compared with those from S. japonica, we found that all of the genes identified in the pathway had good matches in the S. japonica transcriptome dataset, suggesting a conserved gene set in the pathway. The genes in the two species shared high sequence identity at the nucleotide level (Supplementary Table S10). Among these genes, only two had sequence identity between 80 to 85%; the rest (37) were over 90% identity. We further compared their expression patterns by examining the expression ratios of these pathway genes from leaf and root, the two tissues with transcriptome data available from both species (Supplementary Table S10). In the three pathways that are associated with the biosynthesis of secoiridoids, genes from the MVA pathway appeared to have similar ratios. The highest ratio difference was no more than four-fold between the two species. In both the secoiridoid and MEP pathways, genes with ratio differences more than 20-fold were noted, although many genes shared similar ratios. A better understanding of their differences in terms of gene expression and evolutionary relationship could be useful in the future comparison of metabolomics data to elucidate variations of the bioactive compounds in the two species.
qRT-PCR validation of different expression patterns
To validate the transcriptome analysis data and to provide a better understanding of the molecular basis of the metabolic pathways involved in swertiamarin biosynthesis, we selected 7 transcripts encoding key enzymes in the secoiridoid pathway to examine their expression patterns using qRT-PCR (Fig. 8). One of these transcripts, SmG10H, which encodes geraniol 10-hydroxylase, has been cloned in S. mussotii and has shown a potential role in swertiamarin biosynthesis21. Previous expression analysis of the SmG10H gene in leaf, stem, and root tissues indicated its highest expression level was in leaf tissue21. Our qRT-PCR data were consistent with this observation and showed that SmG10H expression in the flower was equivalent to that in the stem (Fig. 8). The higher expression of G10H in leaf tissue than root tissue has also been reported for Gentiana rigescens and G. macrophylla32,54. However, in Swertia chirayita, the ScG10H gene was most highly expressed in the roots55. Their different expression patterns could suggest that the regulation of G10H genes in the secoiridoid pathway might be different, even among closely related species.
Figure 8. The expression pattern of seven genes in swertiamarin biosynthesis pathway across different tissues.

R, root; S, stem; L, leaf; F, flower.
Another well-characterized gene in the secoiridoid pathway is 7DLGT, which encodes 7-deoxyloganetic acid glucosyl transferase. Previous reports have shown that the 7DLGT enzyme possesses a high catalytic efficiency towards 7-deoxyloganetic acid, and when its expression was reduced via virus-induced silencing in C. roseus, the secologanin levels declined significantly56. Secologanin is a key building block in the biosynthesis of many monoterpene indole alkaloids, including swertiamarin. Our qRT-PCR results showed that the expression pattern of Sm7DLGT was quite different from the other transcripts in the pathway. First, its expression level was low in all of the tissues compared to the expression levels of other transcripts in this pathway. Second, consistent with the transcriptome analysis data (Fig. 7c), Sm7DLGT was the only gene with its highest expression in the root tissue; similar to what was reported for Swertia chirayita55. The low expression of Sm7DLGT supports the hypothesis that it may be the rate-limiting enzyme in the secoiridoids biosynthesis pathway. Therefore, this gene could be a potential target for a biotechnological approach to engineer an improved the pathway for increasing swertiamarin yield in S. mussotii in the future.
Secologanin synthases (SLS) are P450 enzymes that catalyse an unusual ring-opening reaction using loganin in the biosynthesis of secologanin in the secoiridoid pathway. In C. roseus, there are two isoforms of SLS, CrSLS1(GenBank L10081) and CrSLS2 (KF415117), with 94.0% nucleotide identity and the same enzymatic function57. However, the two SLS genes (SmSLS1 and SmSLS2) from S. mussotii shared only 63.0% nucleotide identity, suggesting they have diverged extensively. While SmSLS2 (SmTc_16024) and CrSLS2 shared 74.4% amino acid identity, SmSLS1 (SmTc_4315) had the highest amino acid identity (49.8%) with the SLS protein from Nothapodytes nimmoniana based on the BLAST search result (Supplementary Figure S2)58. qRT-PCR indicated that the expression patterns of the two SmSLS genes were quite different. The highest expression of SLS1 was in the leaf and lowest in the flower, while the highest expression of SLS2 was in the flower and lowest in the root. Given the large differences at the sequence level and in their expression patterns, it is uncertain if SmSLS1 and SmSLS2 have similar biological functions in secologanin biosynthesis.
Bioactive compound biosynthesis and accumulation
To examine the possible associations of gene expression in the secoiridoid pathway with the metabolite products, we determined the contents of three bioactive compounds of the secoiridoid pathway, sweroside, swertiamarin, and gentiopicroside, in four tissues (root, leaf, stem, and flower). These compounds have similar chemical structures. Compared with gentiopicroside, on C-5, swertiamarin has a hydroxy group, while sweroside has a proton. Among the three compounds, gentiopicroside has the highest content in all of the tissues (Fig. 9). These compounds had similar accumulation patterns with the lowest content in the root and highest in the flower tissue. The high level of these metabolites in the flower tissue may have an ecological advantage for the plants, since they are bitter in taste. Thus, its high content may serve as a defence mechanism against predators. The low content in the root tissue showed a correlation with the relatively low expression of most biosynthesis genes except 7DLGT. The highest content in the flower tissue did not correlate with the moderate relative expression of these genes. We also observed that the leaf tissue, which had the highest gene expression level, had a compound content level below that of the flower tissue. Taken together, the expression profiles of the metabolic biosynthesis genes in different tissues did not show significant correlations in relation to the metabolite contents. This result agrees with several previous reports, including the study in which the expression levels of the genes involved in the phenylpropanoid pathway did not show significant correlations with the accumulation pattern of shikonin59. An explanation for these observations could be that the highly expressed genes are involved in the biosynthesis of other secondary metabolites or that the biosynthesis is primarily controlled at the protein or enzyme levels. Alternatively, considering that the measured content reflects the accumulated level of the product, a transport mechanism wherein the metabolites are synthesized in the source (leaf) and rapidly transported into the sink (flower) might be involved.
Figure 9. The content of sweroside, swertiamarin and gentiopicroside in different tissues.

R, root; S, stem; L, leaf; F, flower.
In conclusion, we generated a comprehensive transcriptome assembly representing the gene space in S. mussotii. The deep transcriptome sequence data allowed us to identify and characterize the expressions levels of candidate transcripts encoding key enzymes involved in terpenoid metabolic pathways, providing insight into the biosynthesis of bioactive compounds in S. mussotii. The quantitative analyses of gene expression in relation to the bioactive compound contents across different tissues revealed the complexity of gene expression and metabolite accumulation in the secoiridoid biosynthesis pathway. Clearly, further corroboration through molecular, biochemical, enzymatic, and physiological studies will be needed to gain a better understanding of the underlying regulatory mechanisms involved. Nevertheless, the transcriptome data represent the first genomics resource for S. mussotii and lay the foundation for future research towards the improvement of this ethnomedicinal crop through genetics, genomics, and biotechnological approaches.
Methods
Plant material
Fresh roots, stems, leaves and flowers were collected from three individual S. mussotii plants at the full-bloom stage in their wild habitat located in Zhongda County in Yushu Tibetan Autonomous Prefecture of Qinghai Province. The samples were immediately frozen in liquid nitrogen and stored at −80 °C prior to RNA extraction.
RNA isolation and transcriptome sequencing
Total RNA from the four tissues from three individual plants was extracted and processed separately as three replicates. The quality of the RNA samples was evaluated using a Bioanalyzer (Agilent Technologies, USA). RNAseq libraries were constructed with insert sizes of 200 to 500 bp, and were sequenced using the Illumina HiSeq 2000 platform with paired-end (PE) reads of 100 bp.
De novo assembly and sequence processing
The raw data of the twelve RNA-seq reads were quality trimmed and then used for sequence assembly using CLC Genomics Workbench (v8.0.2) with the default parameters. For the removal of the redundant sequences, the CD-HIT60 package with an identity parameter of 95% was employed to generate a set of non-redundant contig sequence files. Contigs (transcripts) shorter than 300 bp were not included in the final S. mussotii transcriptome assembly dataset for further analyses.
Gene function annotation
The consensus contig sequence files were annotated against the Nr (NCBI non-redundant protein sequences), PFAM (protein family), and Swiss-Prot (a manually annotated and reviewed protein sequence database) databases using local BLASTX program with an E-value threshold of 1e−10. GO analysis was performed using Blast2GO61 with the same E-value cutoff. To identify the transcription factors in the S. mussotii transcript dataset, we also applied the BLASTX search program against the plant transcription factor database62. Annotated sequences were further characterized based on KOG/COG classifications. Metabolic pathway mapping of the transcripts in the Nr Unigene set was performed using the KEGG Automatic Annotation Server63.
Identification of SSRs
The transcripts sequences were scanned for the presence of SSRs using MISA (MIcroSAtellite) with default parameters64. The minimum number of repeat units for di-nucleotide was six, whereas for tri-, tetra-, penta- and hexa-nucleotide repeats, the minimum number of repeat units was greater than five in the search criteria.
Differential expression analysis
To estimate the expression pattern of each transcript in different tissue samples, high-quality reads from each sample were mapped on the final transcriptome assembly using CLC Genomics Workbench (v8.0.2). A maximum of two mismatches was allowed for mapping. The read counts were normalized by calculating the RPKM for each transcript in an individual tissue. Differential gene expression analysis was performed using Expander (v7.1)41 software based on t-test. A P-value cut-off ≤0.05 (multiple tests correction: FDR) and a change of at least a two-fold change were used to identify significantly different transcript expressions levels.
qRT-PCR analysis
Quantitative real-time PCR was performed using a QuantStudio Flex 6 system (Applied Biosystems, Foster City, CA, USA) with the Brilliant II SYBR Green QRT-PCR Master Mix Kit, 1-Step (Agilent Technologies, Cedar Creek, TX, USA). Gene-specific primers for the seven selected genes in the secoiridoid pathway were designed using Primer 3, and the primer sequences are listed in Supplementary Table S11. Real time PCR for each gene was performed using three biological replicates and three technical replicates. The tubulin transcript served as an internal control for normalization. The relative gene expression levels were calculated using the 2−ΔΔCT method65.
Extraction and estimations of sweroside, swertiamarin and gentiopicroside
Individual tissues (root, stem, leaf, and flower) were pooled from three plants. The dry materials were ground into fine powder. Approximately 50 mg of powder was extracted in 1 mL of methanol by ultrasonication at room temperature for 1 h, followed by centrifugation. The supernatant was then diluted to an appropriate concentration with methanol. Samples were separated using ultra high-performance liquid chromatography (UHPLC) with a HSS T3 column (2.1 × 100 mm, 1.7 μm, Waters) and a Thermo Scientific™ Dionex™ UltiMate™ 3000 Rapid Separation LC (RSLC) system. The mobile phase consisted of solvents (A), 0.10% aqueous formic acid, and (B), acetonitrile containing 0.10% formic acid. The gradient conditions were applied as follows: from 5.00% to 10.00% B in 1 min, from 10.00% to 60.00% B until 6 min, from 60.00% to 100.00% B in 6.5 min, maintained at 100.00% B for 3.5 min, from 10.1 min to 13 min, and maintained at 5.00% B. The flow rate was 300 μL/min, and the injection volume was 1 μL. Liquid chromatography-mass spectrometry (LC-MS) was performed using a Thermo Scientific™ Q Exactive™ Hybrid Quadrupole Orbitrap mass spectrometer equipped with a HESI-II probe and controlled with Xcalibur 2.2 SP1.48 software (Thermo Fisher Scientific)66. The sweroside, swertiamarin, and gentiopicroside content of each tissue was identified by the retention time and mass spectra data compared with standards, and their quantity was estimated by linear regression. The quantification was estimated in triplicate and the data were interpreted in terms of % of dry weight (DWT).
Data availability
The raw Illumina data generated in this study were deposited in the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA320459.
Additional Information
Accession Codes: The datasets of raw read sequences from each tissue were deposited in the NCBI Short Read Archive (SRA) database under the BioProject accession number PRJNA320459.
How to cite this article: Liu, Y. et al. Deep sequencing and transcriptome analyses to identify genes involved in secoiridoid biosynthesis in the Tibetan medicinal plant Swertia mussotii. Sci. Rep. 7, 43108; doi: 10.1038/srep43108 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This research was funded by the National Natural Science Foundation of China [81274158, 81373765 and 31161140345], New Century Excellent Talents in University of China [NCET-12-0578 and NCET-13-0624], the Programme of Introducing Talents of Discipline to University of China [B08044], China Postdoctoral Science Foundation [20110490556], the Fundamental Research Funds for the Central Universities [2016SHXY04], and Minzu University of China [2015MDTD16C and ydzxxk201618].
Footnotes
The authors declare no competing financial interests.
Author Contributions Y.L., Y.Q.G., C.L. and L.H. conceived and designed the experiments. Y.L. and L.T. collected the materials. Y.L., F.G., L.Z., T.M., P.C., N.H., R.G., D.P., and J.S. performed the experiments. Y.W. and Y.L. compiled and interpreted the data. Y.L., Y.W., Y.Q.G. wrote the manuscript. All authors reviewed and discussed the manuscript.
References
- Yang Y. C. Book of Tibetan Medicine. Qinghai People’s Press: Xining 111–112 (1991). [Google Scholar]
- Negi J. S., Singh P. & Rawat B. Chemical constituents and biological importance of swertia: a review. Curr. Res. Chem. 3, 1–15 (2011). [Google Scholar]
- Kakiuchi N. et al. Phylogenetic examination of crude drugs derived from Yunnanese Swertia plants. J Nat Med 68, 206–10 (2014). [DOI] [PubMed] [Google Scholar]
- Dutt B., Srivastava L. J. & Singh J. M. Swertia spp: a source of bitter compounds for medicinal use. Anc Sci Life 15, 226–9 (1996). [PMC free article] [PubMed] [Google Scholar]
- Brahmachari G. et al. Swertia (Gentianaceae): chemical and pharmacological aspects. Chem Biodivers 1, 1627–51 (2004). [DOI] [PubMed] [Google Scholar]
- Yamahara J., Konoshima T., Sawada T. & Fujimura H. Biologically active principles of crude drugs: pharmacological actions of Swertia japonica extracts, swertiamarin and gentianine (author’s transl). Yakugaku Zasshi 98, 1446–51 (1978). [DOI] [PubMed] [Google Scholar]
- Kikuzaki H., Kawasaki Y., Kitamura S. & Nakatani N. Secoiridoid glucosides from Swertia mileensis. Planta Med 62, 35–8 (1996). [DOI] [PubMed] [Google Scholar]
- Zhang J. S., Wang X. M., Dong X. H., Yang H. Y. & Li G. P. Studies on chemical constituents of Swertia mussotii. Journal of Chinese Medicinal Materials 32, 511–514 (2009). [PubMed] [Google Scholar]
- Sun H. F. & Ding J. Y. Seperation and identification of xanthones of Swertia mussotii. Acta Botanica Sinica 23, 464–469 (1981). [Google Scholar]
- Sun H. F., Hu B. L., Ding J. Y. & Fan S. F. The glucosides from Swertia mussotii Franch. Acta Botanica Sinica 33, 31–37 (1991). [Google Scholar]
- Ding J. Y. & Sun H. F. Effective antihepatitic component of Swertia mussotii: separation and identification of mantgiferin and leanolic acid. Chineses Traditional Herbage Drugs 9, 391–392 (1980). [Google Scholar]
- Shang J., Zhang G. Y., Yang C. B. & Chen Z. Studies on chemical constituents of Swertia mussotii Franch. J Qinghai Normal Univ (Natural Science Edition), 66–67 (2008). [Google Scholar]
- Tian C., Zhang T., Wang L., Shan Q. & Jiang L. The hepatoprotective effect and chemical constituents of total iridoids and xanthones extracted from Swertia mussotii Franch. J Ethnopharmacol 154, 259–66 (2014). [DOI] [PubMed] [Google Scholar]
- Zhang L. et al. Swertianlarin, an herbal agent derived from Swertia mussotii Franch., attenuates liver injury, inflammation, and cholestasis in common bile duct-ligated rats. Evid Based Complement Alternat Med 2015, 948376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X. C. et al. Swertianlarin, isolated from Swertia mussotii Franch., increases detoxification enzymes and efflux transporters expression in rats. Int J Clin Exp Pathol 8, 184–95 (2015). [PMC free article] [PubMed] [Google Scholar]
- Yin H., Zhao Q., Sun F. M. & An T. Gentiopicrin-producing endophytic fungus isolated from Gentiana macrophylla. Phytomedicine 16, 793–7 (2009). [DOI] [PubMed] [Google Scholar]
- Cao J. P., Liu X., Hao J. G. & Zhang X. Q. Tissue culture and plantlet regeneration of Gentiana macrophylla. Acta Botanica Boreali-occidentalia Sinica 25, 1101–1106 (2005). [Google Scholar]
- Liu J. Q., Chen Z. D., Liao Z. X. & Lu A. M. A comparision of the ITS sequences of the Tibetan medicine Zang Yin Chen Swertia mussotii and its adulterant species. Acta Pharmaceutica Sinica 36, 67–70 (2001). [PubMed] [Google Scholar]
- Zheng X., Xu H., Ma X., Zhan R. & Chen W. Triterpenoid saponin biosynthetic pathway profiling and candidate gene mining of the Ilex asprella root using RNA-Seq. Int J Mol Sci 15, 5970–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun P. et al. Transcriptome analysis reveals putative genes involved in iridoid biosynthesis in Rehmannia glutinosa. Int J Mol Sci 13, 13748–63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. et al. Cloning and functional analysis of geraniol 10-hydroxylase, a cytochrome P450 from Swertia mussotii Franch. Biosci Biotechnol Biochem 74, 1583–90 (2010). [DOI] [PubMed] [Google Scholar]
- Wang J., Zhao C., Liu C., Xia G. & Xiang F. Introgression of Swertia mussotii gene into Bupleurum scorzonerifolium via somatic hybridization. BMC Plant Biol 11, 71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Gerstein M. & Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10, 57–63 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. et al. A low-cost library construction protocol and data analysis pipeline for Illumina-based strand-specific multiplex RNA-seq. PLoS ONE 6, e26426 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLay B. et al. Transcriptome analysis of the salivary glands of potato leafhopper, Empoasca fabae. J Insect Physiol 58, 1626–34 (2012). [DOI] [PubMed] [Google Scholar]
- Stafford-Banks C. A., Rotenberg D., Johnson B. R., Whitfield A. E. & Ullman D. E. Analysis of the salivary gland transcriptome of Frankliniella occidentalis. PLoS One 9, e94447 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman D. S. Transcription factors: an overview. Int J Biochem Cell Biol 29, 1305–12 (1997). [DOI] [PubMed] [Google Scholar]
- Schluttenhofer C. & Yuan L. Regulation of specialized metabolism by WRKY transcription factors. Plant Physiol 167, 295–306 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano Y., Yamaguchi M., Endo H., Rejab N. A. & Ohtani M. NAC-MYB-based transcriptional regulation of secondary cell wall biosynthesis in land plants. Front Plant Sci 6, 288 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens J. et al. The bHLH transcription factors TSAR1 and TSAR2 regulate triterpene saponin biosynthesis in Medicago truncatula. Plant Physiol 170, 194–210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K. B. Transcriptional regulation in plants: the importance of combinatorial control. Plant Physiol 118, 1111–20 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Allan A. C., Li C., Wang Y. & Yao Q. De novo assembly and characterization of the transcriptome of the Chinese medicinal herb, Gentiana rigescens. Int J Mol Sci 16, 11550–73 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D. et al. De novo sequencing transcriptome of endemic Gentiana straminea (Gentianaceae) to identify genes involved in the biosynthesis of active ingredients. Gene 575, 160–70 (2016). [DOI] [PubMed] [Google Scholar]
- Varshney R. K., Graner A. & Sorrells M. E. Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23, 48–55 (2005). [DOI] [PubMed] [Google Scholar]
- Hancock J. M. Simple sequences in a “minimal’ genome. Nat Genet 14, 14–5 (1996). [DOI] [PubMed] [Google Scholar]
- Wang L. et al. De novo transcriptome assembly and development of novel microsatellite markers for the traditional Chinese medicinal herb, Veratrilla baillonii Franch. (Gentianaceae). Evol Bioinform Online 11, 39–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr B. & Schlotterer C. Long microsatellite alleles in Drosophila melanogaster have a downward mutation bias and short persistence times, which cause their genome-wide underrepresentation. Genetics 155, 1213–20 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth G., Gaspari Z. & Jurka J. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res 10, 967–81 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M. Next-generation sequencing technologies for gene expression profiling in plants. Brief Funct Genomics 11, 63–70 (2012). [DOI] [PubMed] [Google Scholar]
- Ozsolak F. & Milos P. M. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet 12, 87–98 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I. et al. Expander: from expression microarrays to networks and functions. Nat Protoc 5, 303–22 (2010). [DOI] [PubMed] [Google Scholar]
- Rai A., Smita S. S., Singh A. K., Shanker K. & Nagegowda D. A. Heteromeric and homomeric geranyl diphosphate synthases from Catharanthus roseus and their role in monoterpene indole alkaloid biosynthesis. Mol Plant 6, 1531–49 (2013). [DOI] [PubMed] [Google Scholar]
- Simkin A. J. et al. Characterization of the plastidial geraniol synthase from Madagascar periwinkle which initiates the monoterpenoid branch of the alkaloid pathway in internal phloem associated parenchyma. Phytochemistry 85, 36–43 (2013). [DOI] [PubMed] [Google Scholar]
- Tai H. M., Huang M. H. & Yang C. C. Formal Total Synthesis of (±)-Dimethyl Secologanoside. Journal of the Chinese Chemical Society 50, 441–444 (2003). [Google Scholar]
- Bach T. J. Some new aspects of isoprenoid biosynthesis in plants - a review. Lipids 30, 191–202 (1995). [DOI] [PubMed] [Google Scholar]
- Li W. et al. Species-specific expansion and molecular evolution of the 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) gene family in plants. Plos ONE 9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harker M. et al. Enhancement of seed phytosterol levels by expression of an N-terminal truncated Hevea brasiliensis (rubber tree) 3-hydroxy-3-methylglutaryl-CoA reductase. Plant Biotechnology Journal 1, 113–121 (2003). [DOI] [PubMed] [Google Scholar]
- Chappell J., Wolf F., Proulx J., Cuellar R. & Saunders C. Is the reaction catalyzed by 3-hydroxy-3-methylglutaryl coenzyme-a reductase a rate-limiting step for isoprenoid biosynthesis in plants. Plant Physiology 109, 1337–1343 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller H. et al. Expression of the Hevea brasiliensis (Hbk) Mull. Arg. 3-hydroxy-3-methylglutaryl-coenzyme a reductase-1 in tobacco results in sterol overproduction. Plant Physiology 109, 761–770 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey S. J. et al. Enhanced seed phytosterol accumulation through expression of a modified HMG-CoA reductase. Plant Biotechnology Journal 4, 219–229 (2006). [DOI] [PubMed] [Google Scholar]
- Munoz-Bertomeu J., Sales E., Ros R., Arrillaga I. & Segura J. Up-regulation of an N-terminal truncated 3-hydroxy-3-methylglutaryl CoA reductase enhances production of essential oils and sterols in transgenic Lavandula latifolia. Plant Biotechnology Journal 5, 746–758 (2007). [DOI] [PubMed] [Google Scholar]
- Wang C. T., Liu H., Gao X. S. & Zhang H. X. Overexpression of G10H and ORCA3 in the hairy roots of Catharanthus roseus improves catharanthine production. Plant Cell Reports 29, 887–894 (2010). [DOI] [PubMed] [Google Scholar]
- Rai A. et al. High-throughput sequencing and de novo transcriptome assembly of Swertia japonica to identify genes involved in the biosynthesis of therapeutic metabolites. Plant Cell Rep 35, 2091–111 (2016). [DOI] [PubMed] [Google Scholar]
- Hua W. et al. An insight into the genes involved in secoiridoid biosynthesis in Gentiana macrophylla by RNA-seq. Mol Biol Rep 41, 4817–25 (2014). [DOI] [PubMed] [Google Scholar]
- Padhan J. K., Kumar V., Sood H., Singh T. R. & Chauhan R. S. Contents of therapeutic metabolites in Swertia chirayita correlate with the expression profiles of multiple genes in corresponding biosynthesis pathways. Phytochemistry 116, 38–47 (2015). [DOI] [PubMed] [Google Scholar]
- Asada K. et al. A 7-deoxyloganetic acid glucosyltransferase contributes a key step in secologanin biosynthesis in Madagascar periwinkle. Plant Cell 25, 4123–34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duge de Bernonville T. et al. Characterization of a second secologanin synthase isoform producing both secologanin and secoxyloganin allows enhanced de novo assembly of a Catharanthus roseus transcriptome. BMC Genomics 16, 619 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha B. L. et al. Transcriptome analysis of stem wood of Nothapodytes nimmoniana (Graham) Mabb. identifies genes associated with biosynthesis of camptothecin, an anti-carcinogenic molecule. J Biosci 41, 119–31 (2016). [DOI] [PubMed] [Google Scholar]
- Yamamura Y., Sahin F. P., Nagatsu A. & Mizukami H. Molecular cloning and characterization of a cDNA encoding a novel apoplastic protein preferentially expressed in a shikonin-producing callus strain of Lithospermum erythrorhizon. Plant Cell Physiol 44, 437–46 (2003). [DOI] [PubMed] [Google Scholar]
- Fu L., Niu B., Zhu Z., Wu S. & Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A. & Gotz S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008, 619832 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Rodriguez P. et al. PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res 38, D822–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya Y., Itoh M., Okuda S., Yoshizawa A. C. & Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35, W182–5 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel T., Michalek W., Varshney R. K. & Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106, 411–22 (2003). [DOI] [PubMed] [Google Scholar]
- Pfaffl M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research 29 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao G. et al. Characterization of Shrimp Oil from Pandalus borealis by High Performance Liquid Chromatography and High Resolution Mass Spectrometry. Mar Drugs 13, 3849–76 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw Illumina data generated in this study were deposited in the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA320459.