

Abstract
Upregulation of antiapoptotic Bcl-2 proteins in certain tumors confers cancer cell resistance to chemotherapy or radiations. Members of the antiapoptotic Bcl-2 proteins, including Bcl-2, Mcl-1, Bcl-xL, Bcl-w, and Bfl-1, inhibit apoptosis by selectively binding to conserved α-helical regions, named BH3 domains, of pro-apoptotic proteins such as Bim, tBid, Bad, or NOXA. Five antiapoptotic proteins have been identified that interact with various selectivity with BH3 containing pro-apoptotic counterparts. Cancer cells present various and variable levels of these proteins, making the design of effective apoptosis based therapeutics challenging. Recently, BH3 profiling was introduced as a method to classify cancer cells based on their ability to resist apoptosis following exposure to selected BH3 peptides. However, these studies were based on binding affinities measured with model BH3 peptides and Bcl-2-proteins taken from mouse sequences. While the majority of these interactions are conserved between mice and humans, we found surprisingly that human NOXA binds to human Bfl-1 potently and covalently via conserved Cys residues, with over 2 orders of magnitude increased affinity over hMcl-1. Our data suggest that some assumptions of the original BH3 profiling need to be revisited and that perhaps further targeting efforts should be redirected toward Bfl-1, for which no suitable specific inhibitors or pharmacological tools have been reported. In this regard, we also describe the initial design and characterizations of novel covalent BH3-based agents that potently target Bfl-1. These molecules could provide a novel platform on which to design effective Bfl-1 targeting therapeutics.
Over the past two decades, efforts in the design of effective apoptosis based therapeutics targeting Bcl-2 family proteins have been based on detailed information on their expression and preferential affinity for pro-apoptotic BH3-containing proteins.1,2 Antiapoptotic Bcl-2 family proteins inhibit cancer cell death by forming stable heterodimers mediated by a conserved α-helical motif (termed BH3 domain) present on the surface of pro-apoptotic members of the same family. Five antiapoptotic Bcl-2 proteins, namely Bcl-2, Bcl-xL, Bcl-w, Mcl-1 and Bfl-1, interact with various specificity with several BH3 containing pro-apoptotic counterparts, including Bax, Bim, Bad, tBid, and NOXA. The latter can be divided functionally in activators, sensitizers, and effectors.3,4 The effectors, Bax and Bak, aided by the activators Bim and Bid, are believed to be the ultimate agents that cause MOMP (mitochondrial outer membrane permeabilization) by forming oligomers at the mitochondrial membrane, which result in the release of pro-apoptogenic molecules such as SMAC and cytochrome c.5 However, both the effectors and activators can be sequestered by antiapoptotic Bcl-2 proteins inhibiting cell death. Sensitizers, such as NOXA and Bad, function as cellular sentinels that respond to cellular damage, induced by chemotherapy, immunotherapy, or radiation in cancer cells, and by binding to antiapoptotic Bcl-2 family proteins, displace the bound activators and/or the effectors, resulting in apoptosis.6
While the activators and the effectors are generally promiscuous in binding the antiapoptotic Bcl-2 proteins, the sensitizers have different specificity. For example, Bad binds potently to Bcl-2, Bcl-xL, and Bcl-w, while NOXA has been reported to preferentially bind to Mcl-1 and in minor part to Bfl-1.7 Bcl-2 proteins expression has been highly studied in both leukemia and lymphoma cell lines and primary cell samples,8−10 and these studies culminated in 2016 with the approval by the FDA of Venetoclax,11−14 a potent and selective Bcl-2 antagonist that does not interact significantly with other antiapoptotic Bcl-2 proteins such as Bcl-xl, Mcl-1, or Bfl-1. Accordingly, increased Mcl-18,15 expression has been correlated with resistance to Bcl-2 antagonists in primary tumors and cancer cell lines, and this observation ignited intense research efforts aimed at developing novel Mcl-1 targeting agents in the past few years.15−26 The efficacy of these molecules as direct pro-apoptotic agents or as sensitizers for cancer cells to chemotherapy or immunotherapy, which act by activating cell death pathways in cancer cells, however, largely depends on the relative levels of antiapoptotic Bcl-2 proteins.27 Recently, BH3 profiling strategies have been reported and widely used as a way to classify tumors and cancer cell lines for their ability to respond to various BH3 peptides.28,29 In one implementation, cells that release cytochrome c after exposure to Bad derived BH3 peptides are classified as expressing Bcl-2, Bcl-xl, and/or Bcl-w, given the affinity of Bad for these three proteins. NOXA derived BH3 peptides have been used in similar assays to detect the expression of Mcl-1 in tumor cells, given that these peptides bound preferentially Mcl-1 over Bfl-1 and do not interact significantly with other antiapoptotic Bcl-2 proteins.7 While these and several other studies clearly point at Mcl-1 as a possible cause of resistance to Venetoclax and related antagonists in chronic lymphocytic leukemia (CLL), other studies suggested that Bfl-1 may have a more predominant role in resistance to both chemotherapy and Bcl-2 antagonists.23,30
In this paper, we analyzed the interactions between NOXA, ABT-199 (Venetoclax), and Bim with four antiapoptotic Bcl-2 proteins, namely Bcl-2, Bc-xL, Mcl-1, and Bfl-1, using biochemical, biophysical, and cellular assays. Our studies resulted in the surprising observation that human NOXA derived BH3 peptides, unlike the mouse derived peptides previously reported and commonly used for BH3 profiling, bound with high affinity to Bfl-1 with over 2 orders of magnitude increased affinity over Mcl-1. We also found that this affinity is owed to a specific disulfide bridge between NOXA and Bfl-1 that occurs only in the human proteins and not in mice. We identified a human derived NOXA peptide that is exquisitely selective for Bfl-1, which could be used in BH3 profiling applications and/or perhaps as a template for the design of novel Bfl-1 antagonists. On the basis of these findings, our studies suggest that the emphasis on Mcl-1 may need to be revisited and that perhaps further efforts in deriving Bfl-1 specific antagonists is warranted, given that to date no valid small molecule compounds nor pharmacological tools have been reported. In addition, we also report on initial Bim-BH3-based cell permeable agents that, by covalently inhibiting Bfl-1, can serve as suitable stepping stones to derive potent Bfl-1 targeting compounds. Given the recent report by scientists at Genentech,31 we also report a comparison between a cell-penetrating linear and a stapled version of these covalent peptides aimed at deriving cell active covalent Bfl-1 antagonists. In summary, our findings should facilitate further studies probing the role of Bfl-1 in the development of resistance in CLL, melanomas, and other tumors and for the development of novel apoptosis-based therapeutics.
Results and Discussion
Human NOXA Interacted with Human Bfl-1 Covalently and Potently, and with Higher Affinity Compared to Human Mcl-1
The interaction between pro- and antiapoptotic Bcl-2 proteins has been proven in several instances to be mediated by a conserved α-helical BH3 peptide on the surface of pro-apoptotic proteins that bound with various affinities and specificities to a BH3-binding groove present on the surface of antiapoptotic Bcl-2 proteins. Hence, biophysical studies on several BH3 peptides in complex with various antiapoptotic Bcl-2 proteins have been reported over the past two decades.32
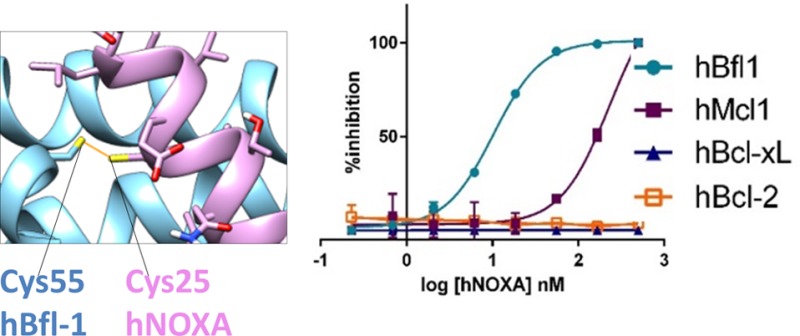
The X-ray structure of human Bfl-1 in complex with a BH3 peptide from NOXA (Figure 1A) was recently deposited in the protein data bank (PDB ID 3MQP). While the general positioning of the peptide into the BH3 binding groove of Bfl-1 largely resembled that of the previously reported complexes between other BH3 peptides and other antiapoptotic Bcl-2 family proteins (for example, mNOXA B/mMcl-1 complex in Figure 1B; PDB ID 2JM6(33)), we noticed that hBfl-1 presented a unique and peculiar structural feature. The conserved residue Cys 25 in hNOXA (Figure 1A,C) was precisely juxtaposed to the conserved residue Cys 55 in human Bfl-1 (Figure 1A,C), to possibly form a disulfide bond. Accordingly, we found that a hNOXA BH3 peptide (Table 1) and a human Bfl-1 recombinant construct that lacked the C-terminal transmembrane domain (hBfl-1ΔTm(1–149)) formed a covalent complex, as we could observe by SDS gel electrophoresis (Figure 1D). Moreover, further confirmation of covalent modification of hBfl-1 was obtained using MALDI TOF mass spectrometry, which demonstrated the expected mass increase when hNOXA was incubated with hBfl-1. Specifically, we observed the following masses: hBfl1ΔTM(1–149) = 19673.8 Da; hNOXA = 2463.0 Da; complex hBfl-1/hNOXA = 22134.4 Da, which is nearly identical (well within the accuracy of the method) to the exact expected theoretical mass of the covalent adduct, 22134.8 Da (Figure 1E).
Figure 1.

Human NOXA interacted with human Bfl-1 covalently and potently, and with higher affinity compared to human Mcl-1. Panel A: Structure of hBfl-1 and hNOXA complex with a close-up view of the covalent bond between the Cys 55 of hBfl-1 and the N-terminal Cys of a hNOXA derived peptide (see sequence in Table 1) modeled on the hNOXA BH3 peptide cocrystallized with hBfl-1 (PDB ID: 3MQP). The newly formed bond is colored in orange, while hBfl-1 is represented by light blue ribbons and hNOXA peptide by pink sticks and ribbons. Panel B: Binding pose of a hNOXA derived peptide modeled on the hNOXA BH3 peptide cocrystallized with mMcl-1 (PDB ID: 2JM6)2. The close-up view shows the absence of a covalent interacting Cys residue in the BH3 hydrophobic cleft of mMcl-1, here represented by gray ribbons while hNOXA peptide by pink sticks and ribbons. Panel C: Sequence alignment of selected pro-apoptotic Bcl-2 proteins hBIK, hHRK, hBIM, hBAD, hBID, hBMF, hNOXA, and the hBAX subfamily proteins hBAX and hBAK (upper panel). Structural alignment (lower panel) of selected antiapoptotic Bcl-2 proteins hBfl-1, hMcl-1, hBcl-2, and hBcl-xL. Bcl-2 Homology (BH) regions belonging to pro-apoptotic and antiapoptotic proteins are included in pink and blue rectangular boxes, respectively. Interacting Cys residues present in both hBfl-1 (Cys 55) and hNOXA (Cys 25) are highlighted in yellow, while red-colored residues indicate strictly conserved residues of pro-apoptotic proteins. Panel D: SDS-PAGE gel electrophoresis followed by Coomassie staining of the hBfl-1 protein (10 μM) in the absence and presence of equimolar concentrations of a hNOXA derived peptide (Table 1) at different time points (15 min, 30 min, 1h, 2h). Panel E: MALDI-TOF MS spectra of hBfl-1 collected in absence (blue) and presence (pink) of a hNOXA derived peptide (Table 1) after 2 h incubation at RT and at a protein–ligand ratio of 1:2. Panel F: Cartoon representing the principle of the DELFIA assays. These assays were used to quantify the ability of our test peptides to displace a biotinylated BID BH3 peptide from hBfl-1, hMcl-1, hBcl-xL, and hBcl-2. Representative DELFIA dose–response curves relative to the hNOXA derived peptide (Table 1) against these proteins are reported.
Table 1. NOXA Derived Peptide Sequences and Relative IC50 Values in a DELFIA Assay against the Bcl-2 Family Proteins hBfl-1, hMcl-1, hBcl-xL, and hBcl-2a.
| DELFIA IC50 (nM) |
|||||
|---|---|---|---|---|---|
| compd ID | aa sequence | hBfl-1 | hMcl-1 | hBcl-xL | hBcl-2 |
| hNOXA | Ac-CATQLRRFGDKLNFRQKLLN-NH2 | 7.5 ± 2 | 228 ± 1 | >1 μM | >1 μM |
| homo_hNOXA | Ac-hCATQLRRFGDKLNFRQKLLN-NH2 | 83 ± 11 | 266 ± 31 | >1 μM | >1 μM |
| λ_hNOXA | Ac-λATQLRRFGDKLNFRQKLLN-NH2 | 287 ± 69 | 233 ± 25 | >1 μM | >1 μM |
| hNOXAs | Ac-CATQLRRFGDKLN-NH2 | 15 ± 2 | >1 μM | >1 μM | >1 μM |
| NOXA A | Ac-AELPPEFAAQLRKIGDKVYC-NH2 | >1 μM | 450 ± 1 | >1 μM | >1 μM |
| NOXA B | Ac-PADLKDECAQLRRIGDKVNL-NH2 | 431 ± 35 | 479 ± 28 | >1 μM | >1 μM |
| C1A_hNOXAs | Ac-AATQLRRFGDKLN-NH2 | >1 μM | >1 μM | n.t. | n.t. |
| cyc_hNOXA | Ac-CATQLRRFGDKLNFRQKLLNLISKLFC-NH2 | 86 ± 13 | 344 ± 41 | n.t. | n.t. |
hC = homocysteine; λ = Dap-2-chloroacetamide; C1–C27 in cyc-NOXA = disulfide bridge. n.t.: not tested. Values are the results of multiple measurements run in duplicates and represent the mean ± SE.
Over the past several years, most laboratories have relayed on a fluorescence polarization assay to quantify the ability of Bcl-2 antagonists to displace FITC-labeled BH3 peptides. While this assay has been used with some success, we found that it is prone to producing false positives, owed to the indirect nature of the readout and the relatively narrow assay window. In fact, the assay relied on indirect measurements of the overall rotational diffusion times of the peptide in the free and bound state. Compounds with low solubility may have an effect on the solubility of the peptide, usually present in the assay well at relatively low (nanomolar) concentrations relative to test agents that could result in false positives, or artificially low or otherwise inaccurate inhibition constants. For these reasons, we opted to derive a more robust assay, as we recently reported.34 Hence, we developed a panel of assays based on the DELFIA (Dissociation Enhanced Lanthanide Fluorescence Immunoassay) platform34 for hBcl-2ΔTm, hMcl-1ΔTm, hBcl-xLΔTm, and hBfl-1ΔTm (Figure 1F). In these assays, a biotinylated Bid BH3 peptide was captured on DELFIA streptavidin coated plates (see Methods), while histidine tagged recombinant proteins were conjugated with a highly fluorescent Eu-tagged anti-His antibody (PerkinElmer, see Methods). After washing steps, the displacement of the immuno-reaction complex from the assay well by a test molecule was detected by a decrease in fluorescence signal (Figure 1F).34
Using this robust biochemical assay, we tested the ability of hNOXA BH3 peptide (Table 1) to displace the binding of Bid-BH3 from selected antiapoptotic proteins. Not surprisingly, hNOXA did not show any appreciable affinity for both Bcl-2 and Bcl-xL, as previously reported,7 while it displayed a greater affinity for both hMcl-1 and hBfl-1. Mouse derived NOXA A peptide, commonly used to detect Mcl-1 in BH3 profiling,29,35−39 indeed interacted more potently, albeit with triple digit nanomolar affinity with hMcl-1 compared to hBfl-1 (Table 1). NOXA B on the contrary had similar relatively weak affinities for both human proteins (Table 1). This affinity trend followed what was reported previously using these mouse-derived NOXA peptides tested against mouse Mcl-1 and mouse Bfl-1. These previous studies originated the common knowledge that Mcl-1 has a greater affinity for NOXA than Bfl-1.7,29,35−39 However, contrary to these observations, here we found that human NOXA displayed a dramatically greater affinity for human Bfl-1 than human Mcl-1. The increased affinity of human NOXA for human Bfl-1 versus human Mcl-1 could be attributable to a covalent interaction between hBfl-1 and hNOXA. Such interaction was structurally unique and not present in other BH3 peptides (including mouse derived NOXA A peptide, for example) or in other antiapoptotic Bcl-2 proteins (Figure 1C). The mouse derived NOXA B peptide did contain a Cys residue, but this did not appear to be at the proper position in the BH3 sequence for covalent interaction (Table 1).
While submitting this manuscript, Huhn et al. reported on covalent stapled (but not the corresponding linear) NOXA and BIM peptides.40 However, we want to emphasize that a lack of quantitative information in these recent studies precluded the authors from making these critical observations on the relative affinities of hNOXA for hBfl-1 and hMcl-1. Because the binding of hNOXA for hBfl-1 was likely driven by the disulfide bridge formation, we derived a minimal peptide region (hNOXAs, Table 1) that retained a low nanomolar affinity for hBfl-1 (IC50 = 15 nM) while no displacement was observed for this agent when tested against other antiapoptotic Bcl-2 family proteins including hMcl-1 (Table 1) up to 1 μM. In agreement, mutating the Cys residue in hNOXA peptide resulted in an inactive molecule (Table 1, Supporting Information Figure S1), further demonstrating the dependence of the disulfide bridge for the interaction between hBfl-1 and hNOXA. The striking differences between mouse NOXA, possessing two BH3 domains (namely NOXA A and NOXA B), and human NOXA, having only one BH3 domain whose activity seemed dependent on a specific disulfide bridge formation with hBfl-1, may be due to different activation and regulation mechanisms of these proteins. While detailed structural studies on mouse or human NOXA have remained elusive, a recent report suggested that human NOXA was regulated by a specific phosphorylation event at Ser 13.41 A current hypothesis based on modeling studies suggested the presence of a phosphorylated, cytosolic, inactive version of NOXA that could bring the BH3 helix and the transmembrane helix together to form an α-hairpin.41 On the basis of such a model, we hypothesized that this conformation may be stabilized/regulated by an intramolecular disulfide bridge between Cys 25 in the BH3 domain and Cys 51 in the TM helix of hNOXA, also unique to this protein among the Bcl-2 family proteins. This structural motif was previously hypothesized, and it is present in nature in other proteins.42 To preliminarily test this hypothesis, we synthesized a truncated cyclic NOXA peptide (cyc_hNOXA, Table 1), constituted by the BH3 helix from Cys 25 and the TM helix up to Cys 51 that are disulfide bridged. This “closed” conformation of the protein resulted in a significantly reduced affinity for hBfl-1 (IC50 = 85.8 nM, Table 1) when tested side by side with the linear version. While still speculative, these preliminary studies may suggest that, in addition to phosphorylation of Ser13, a possible redox mechanism of regulation of hNOXA would involve the reduction of an intramolecular disulfide bridge that would expose the TM domain for translocation to the mitochondrial membrane. These observations would also explain the activation of NOXA (but not other pro-apoptotic factors) by UV radiation,43 given that UV may catalyze the opening of the disulfide bridge. In tumors cells, dysfunctional mitochondria can cause a shift from oxidative phosphorylation to active glycolysis that in turn increases ROS (reactive oxygen species) generation. Hence, under our hypotheses and observations, such altered redox potential in cancer cells could be a target for both hNOXA activation and/or hNOXA/hBfl-1 interactions.
In conclusion, our observations that hNOXA had greater affinity for hBfl-1 may have tremendous implications on defining the role of hBfl-1 in cancer cell resistance that thus far have been overlooked, despite several reports.15,44,45 Moreover, we have identified that hNOXA derived peptides can serve as hBfl-1 potent BH3 agents that can be generally used for profiling, perhaps complementing the recent work by Butterworth reporting an elegant application of molecules in BH3 profiling,46 or to generate possibly cell permeable therapeutics such as stapled peptides40 or using other cell permeabilization techniques. Finally, albeit speculative, our findings provide a structural framework for the possible regulation and activation of hNOXA via intra- and intermolecular disulfide bond formations that will require further investigations.
Introducing a Mild Michael Acceptor in Bim BH3 Peptide Produced a Potent, Covalent hBfl-1 Antagonist
Analysis of the crystal structure of hBfl-1 in complex with a hNOXA peptide revealed that the distance between the sulfur atoms of hNOXA Cys 25 and hBfl-1 Cys 55 was approximately 3.7 Å (Figure 1A), although a second more distant rotamer for the side chain of Cys 55 in Bfl-1 was also deposited in the PDB ID 3MQP. Because of the presence of this second conformation in which Cys 55 is more distant from Cys 25 in hNOXA, we synthesized and tested two additional hNOXA derived BH3 peptides (Table 1) in which the conserved Cys 25 was replaced by either a homoCys (hence one carbon longer) or a mild Michael acceptor (Dap-2-chloroacetamide, hence two atoms longer than the Cys; Table 1). By using the DELFIA displacement assay, hNOXA remained the most active against hBfl-1 (Table 1), although all peptides were able to covalently interact with hBfl-1 by SDS gel electrophoresis (Supporting Information Figure S2).
On the basis of these observations, we further analyzed the structure of the complex between BIM-BH3 and hBfl-1 (PDB ID 2VM6; Figure 2A).47 We chose the BIM residue Trp147 as a possible target for the introduction of a mild Michael acceptor to react covalently with hBfl-1, thus mimicking what we observed with hNOXA. The distance between the γ-carbon of the side chain of Trp147 and the sulfur atom of hBfl-1 Cys 55 is 3.8 Å from the X-ray structure (PDB ID 2VM6;47Figure 2A) and therefore represented an ideal place for the placement of a mild Michael acceptor. Our considerations in designing covalent peptides followed the general strategies as we recently reported while targeting the ubiquitin ligase SIAH interactions with a peptide derived by the protein Sip.48 On the basis of this experience, we introduced a Dap-2-chloroacetamide in lieu of Trp 147 in a BIM BH3 peptide (Table 2). The resulting agent, 130E7 (Table 2), was tested against the four antiapoptotic Bcl-2 proteins using the DELFIA assays (Figure 2C, Table 2), side by side with the wild type BIM BH3 corresponding peptide (Table 1) and the Bcl-2 selective antagonist ABT-199 (Venetoclax;14Table 2). The BIM BH3 peptide was cross-reactive with all four Bcl-2 proteins as previously reported, with IC50’s in the nanomolar range (Figure 2C, Table 2). However, the introduction of the Dap-2-chloroacetamide in the peptide resulted in an agent (130E7) that was more potent against hBfl-1 compared to the other Bcl-2 proteins tested, again owed to the covalent nature of these interactions. In comparison, ABT-199 resulted as the most potent against Bcl-2 and inactive against Bcl-xL, Mcl-1, and Bfl-1 (Table 2), again in agreement with what was recently reported for this agent.14 To further characterize the binding mode of 130E7 for hBfl-1, we tested its ability to form a covalent adduct in vitro (Figure 2D,E). The molecule interacted with hBfl-1 covalently as demonstrated by SDS gel electrophoresis (Figure 2D). In addition, to further confirm the covalent modification of hBfl-1, we used MALDI TOF mass spectrometry, which demonstrated the expected mass increase when 130E7 was incubated with hBfl-1 (Figure 2E). Specifically, we observed the following masses: hBfl1ΔTM(1–149) = 19673.8 Da; 130E7 = 2545.5 Da; complex hBfl-1/130E7 = 22182.6 Da (theoretical expected mass for the covalent adduct is 22183.3 Da). Therefore, these data identified that 130E7 is a potent, covalent hBfl-1 antagonist.
Figure 2.

Characterization of covalent hBIM derived peptides. Panel A: Structure of hBfl-1 in complex with hBIM (PDB ID: 2VM6). The close-up view shows the presence of a Trp residue in hBIM (Trp 147) that corresponds to Cys 25 in hNOXA (see Figure 1). hBfl-1 is represented by light-blue ribbons, while hBIM peptide is represented by purple sticks and ribbons. Panel B: Structure of the complex between hBfl-1 and a covalent hBIM derived peptide named 130E7 (Table 1) with a close-up view of the covalent bond between the Cys 55 of hBfl-1 and the Dap-2-chloroacetamide in 130E7 modeled on the hBIM BH3 peptide from the PDB ID 2VM6. The newly formed bond is colored in orange, while hBfl-1 is represented by light blue ribbons and 130E7 is depicted as green sticks and ribbons. Panel C: Dose–response DELFIA curves for the displacement of a BID peptide from the Bcl-2 family proteins hBfl-1, hMcl-1, hBcl-xL, and hBcl-2 by compounds 130E7, 130E8 (hBIM), and ABT-199. The arrows emphasize differences in affinities against each of the tested proteins between 130E7, ABT-199, and 130E8. The covalent agent 130E7 showed an increase in affinity only against Bfl-1, owed to the covalent interaction with Cys 55, while reduced affinities are observed between this agent and other Bcl-2 proteins lacking such residues in the BH3 binding groove. Panel D: SDS-PAGE gel electrophoresis followed by Coomassie staining of the hBfl-1 protein (10 μM) in absence and presence of equimolar concentrations of 130E7 at different time points (15 min, 30 min, 1 h, 2 h). Panel E: MALDI-TOF MS spectra of hBfl1 collected in the absence (blue) and presence (green) of 130E7 after 2 h incubation at RT and at a protein–ligand ratio of 1:2.
Table 2. BIM Derived Peptide Sequences and Relative IC50 Values in a DELFIA Assay against the Bcl-2 Family Proteins hBfl-1, hMcl-1, hBcl-xL, and hBcl-2a.

λ = Dap-2-chloroacetamide, Xi and Xi+4 = (S)-2-(4′-pentenyl)alanine
Cell Permeable Covalent Bim-BH3 Mimetics Targeting Bfl-1: Biochemical and Biophysical Validation
To study the effect of hBfl-1 inhibition in cells, cell penetrating versions of 130E7 were designed and tested. Two parallel strategies were adopted either using an N-terminal cell penetrating sequence (130G4, Figure 3A) or by synthesizing a hydrocarbon stapled version of the agent (130D11, Figure 3B), similar to what was reported for the linear BIM BH3 peptide.49−52 A similar covalent stapled peptide was reported while this manuscript was submitted,40 using an acrylamide instead of a Dap-2-chloroacetamide as we report here. However, characterization of such an agent with respect to affinity and selectivity for Bcl-2 proteins has not been reported in detail, and the manuscript relied only on SDS page to prove the covalent interaction.40 Here, we carefully tested each molecule in DELFIA displacement assays against the selected antiapoptotic Bcl-2 proteins, in addition to SDS gel electrophoresis, MALDI TOF spectrometry, and isothermal titration calorimetry. Introducing a cell penetrating sequence at the N-terminus of 130E7 (resulting in agent 130G4; Figure 3A, Table 2), or introducing a hydrocarbon staple in 130E7 (resulting in the stapled peptide 130D11; Figure 3B, Table 2), did not significantly alter their respective binding affinities for hBfl-1, likely driven by the covalent interactions (Table 2). However, the binding of the stapled 130D11 to other Bcl-2 proteins got somewhat reduced, in agreement with recent observations about the stapled version of BH3 peptides31 (Figure 3C, Table 2). Surprisingly, these comparative data between linear and stapled versions of these covalent agents have not been reported by Huhn et al.(40) The selectivity of the covalent interaction against hBfl-1 for both agents was further probed by SDS gel electrophoresis by incubating each agent with hBfl-1, hMcl-1, hBcl-2, or hBcl-xL as shown in Figure 3D for 130G4 and in Figure 3E for 130D11. A new band for the adduct was observed only when hBfl-1, and not the other proteins, was exposed to the covalent agents (Figure 3E,F). The new bands appeared at approximately the expected molecular weights: expected MW for hBfl-1/130G4 adduct, 24265 Da; expected MW for hBfl-1/130D11 adduct, 22183 Da (Figure 3 E,F). In addition, and again in agreement with previous recent studies,31 introduction of the hydrocarbon staple significantly reduced the solubility of the resulted agent, 130D11, as evident by 1D 1H NMR (Figure 3F). To further characterize the direct dissociation constants and the thermodynamics of binding of these agents to hBfl-1, we tested them using isothermal titration calorimetry (ITC; Figure 3G), resulting in Kd values in the low nanomolar range against hBfl-1 (2.6 and 23.0 nM, for 130E7 and 130G4, respectively; Figure 3G and Table S2). However, likely due to its limited solubility, the binding of the hydrocarbon stapled version (130D11) to hBfl-1 was not detected under our experimental conditions (Figure 3G). In addition, the affinity for Bcl-2 and Bcl-xl also was significantly compromised in the stapled peptide, compared to the linear peptide, similar to what was recently reported.31 As mentioned, covalent stapled versions of NOXA and BIM derived peptides were reported while this manuscript was submitted; however, and surprisingly, no detailed quantitative information on the relative binding affinities between linear and stapled agents nor between covalent and noncovalent agents was reported.40
Figure 3.

Selectivity profiling and in vitro evaluation of two covalent cell penetrating peptides. Panel A: Cartoon representation of 130G4, derived by linking the N-terminus of 130E7 to a cell penetrating sequence constituted by five PRR repeats (Table 1). Panel B: Cartoon representation of 130D11, a hydrocarbon stapled derivative of 130E7 (Table 1). In the sequences of the peptides reported in panels A and B, we used the same abbreviations as in Table 1: λ = Dap-2-chloroacetamide, Xi and Xi+4 = (S)-2-(4′-pentenyl)alanine. Panel C: Dose–response curves for the displacement of a BID peptide from the Bcl-2 family proteins hBfl-1, hMcl-1, hBcl-xL, and hBcl-2 by compounds 130G4 and 130D11 in DELFIA assays. Panels D and E: NUPAGE gel electrophoresis followed by Coomassie staining of 130G4 and 130D11 against selected Bcl-2 family proteins (hBfl-1, hBcl-xL, hBcl-2, hMcl-1) after 2 h incubation at RT and at a protein–ligand ratio of 1:2. The noncovalent peptide 130E8 (Table 1) was used as negative control. Panel F: Superimposition of the amide region (6–9 ppm) in the 1D 1H NMR spectra of 100 μM 130D11 (blue), 130G4 (red), and 130E7 (black) to address compounds’ solubility. The spectra were collected in PBS buffer at pH = 7.4 containing 10% D2O + 1% d6-DMSO. 3-(Trimethylsilyl) propionic-2,2,3,3-d4 acid (TMSP) at 11.1 μM has been used as an internal reference (0.0 ppm). Panel G: Isothermal titration calorimetry experiments of 130G4, 130D11, and 130E7 against hBfl-1. Kd values in the low nanomolar range were observed for 130G4 and 130E7 (Kd = 23.0 nM and 2.6 nM, respectively) while, likely due to its limited solubility, no significant binding was observed for 130D11 under our experimental conditions. Similar data were obtained against other Bcl-2 family proteins (Supporting Information Table S2).
Bfl-1 Inhibition Reverted Resistance to Apoptosis in Cell Lines and Primary CLL Cells
To further assess the ability of our hBfl-1 covalent antagonists to penetrate live cells, we tested them against HEK293 cells that were transfected with HA-tagged-wt-hBfl-1 (Sino biologicals). In this assay, cell penetration by the agents and formation of the covalent adduct was visible by simple Western blot analysis owed to the increased molecular weight of the covalent complex. 130G4 was able to penetrate the cell membrane and form a covalent adduct with hBfl-1 in a dose dependent manner after exposure of transfected cells with the agents for 6 h, as suggested by the new band that has approximately an increase of 5 kDa in MW (Figure 4A). On the contrary, reliable cell permeability data with the stapled peptide were difficult to obtain due to the limited solubility of the agent. Hence, for further cellular studies, we preferred 130G4 to the less soluble hydrocarbon stapled 130D11. Indeed, a similarly derived covalent BIM staple peptide recently reported was tested only at relatively high concentrations (40 μM) to detect cellular activity.40 In agreement, when tested against a melanoma cell line that expressed higher levels of both NOXA and Bfl-1 SKMEL-5,9,53 we observed that while 130G4 was active in the low micromolar range, 130D11 was inactive up to 10 μM. Indeed, cell activities for a similar stapled covalent peptide recently reported were obtained only at 40 μM.40 Hence, we subsequently tested 130G4 against the lymphoma cell line Raji, a line that expresses Bfl-1,53 and measured the specific induced apoptosis (SIA) by the agent when used alone or in combination with fludarabine or etoposide at different concentrations (Figure 4C). In these experiments, 130G4 had single agent activity with EC50 values <5 μM (Figure 4C). However, testing etoposide or fludarabine in the presence of 5 μM of 130G4 dramatically sensitized this cell line to the agents (Figure 4C). Similarly, we preliminarily tested the agent against primary cells isolated from patients affected by chronic lymphocytic leukemia (CLL) that are resistant to fludarabine or etoposide (Figure 4D). These experiments were conducted against the CLL cells alone or cocultured with stroma-NK-term cells to best reproduce the tumor environment. In both experiments 130G4 had similar single agent activity (EC50 < 5 μM), while both fludarabine and etoposide had limited activity. However, at the least, an additive effect was observed when the agents were used in combination, suggesting that 130G4 or agents with similar activity against Bfl-1 could be used in fludarabine resistant patients.
Figure 4.

Cellular validation of compound 130G4. Panel A: Western-blot analysis of HEK293 cells transfected with N-HA-tagged hBfl-1 and treated for 6 h with increasing concentrations (10, 20, 30, 50 μM) of 130G4. Cells treated with DMSO only were loaded as negative controls. Samples were probed with an anti-HA antibody (see Methods). Molecular weight shifts corresponding to the covalent adducts of hBfl-1 are indicated. Panel B: Viability dose–response curve of human malignant melanoma SKMEL-5 cells exposed for 24 h to different concentrations (from 0.4 to 100 μM) of the test compound 130G4. Antiproliferative activity was assessed using the ATPlite assay from PerkinElmer and normalized to control cells which were treated with the vehicle, DMSO. The reported IC50 value was calculated by Prism5 (GraphPad). Panel C: Synergistic assay on Raji cell line derived cells incubated with the hBfl-1 inhibitor 130G4 alone (1, 5, 10, 30 μM) or in combination with different concentrations of Fludarabine (F-ara-A, 1, 3, 10 μM) or Etoposide (10, 30, 90 μM) for 48 h at 37 °C in a CO2 incubator. The in vitro cytotoxicity was measured using PI/DiOC6 followed by flow cytometry data analysis, and the data were reported as % SIA (specific induced apoptosis, see Methods). Panel D: Evaluation of a synergistic effect of 130G4 with etoposide and fludarabine (F-ara-A) in CLL cells and CLL cells cocultured with stromal cell support. In the washout experiment, CLL cells alone or cocultured with stroma-NK-tert cells were incubated with the Bfl-1 inhibitor 130G4 alone or in combination with different concentrations of fludarabine (F-ara-A) or etoposide for 48 h at 37 °C in CO2 incubator. The in vitro cytotoxicity was measured in all the samples after 48 h using CD19/CD5/DioC6 staining followed by flow cytometry data analysis, and the data were presented as % SIA (specific induced apoptosis). The two graphs in the panel represent data collected from two different CLL patients.
Previous cellular and genetic studies implicated Bfl-1 as a critical Bcl-2 family protein responsible for the resistance to both chemotherapy and Bcl-2 antagonists treatment in CLL and in melanomas. However, misleading binding data using mouse NOXA derived BH3 peptides (NOXA A and NOXA B, Table 1), which bound more potently to Mcl-1, out-shadowed the role of Bfl-1 in cancer cells. Our studies also revealed a possible mechanism of interaction between hNOXA and hBfl-1 that we hypothesized being regulated by the formation of an intermolecular disulfide bridge with a Cys in the TM domain of the protein, in a cytosolic, inactive state of the protein. These observations, if confirmed, could have far reaching ramifications in deciphering a possible mechanism of redox-regulation of apoptosis in cells and may stimulate the design of novel covalent inhibitors that exploit the conserved Cys 55 in hBfl-1. To this end, our studies with our preliminarily derived cell-permeable covalent agent provide the necessary proof-of-concept data and represent a valuable stepping stone for the design of more drug like agents.
Methods
Protein Expression and Purification
hBfl-1 Protein Expression
A modified pET21a vector encoding a human Bfl-1 fragment (residues 1–149) and an N-terminal His tag was used for these studies. Transformed Rosetta-gami (DE3) competent cells (Novagen) were transferred to Luria broth (LB) supplemented with the appropriate antibiotic and allowed to grow at 37 °C under shaking conditions. When the optical density reached 0.7, the culture was induced with 0.1 mM IPTG (isopropyl 1-thio-d-galactopyranoside) and the growing cells were left shaking overnight at 15 °C. Bacteria were collected and lysed at 4 °C. The overexpressed protein was purified using Ni2+ affinity chromatography with a linear gradient of imidazole. The eluted fractions were dialyzed against a buffer containing 50 mM phosphate and 150 mM NaCl, at pH = 6.5. Typical yields were 0.8 g/L. The final protein sequence was MHHHHHHSSGVDLGTENLYFQSMTDCEFGYIYRLAQDYLQCVLQIPQPGSGPSKTSRVLQNVAFSVQKEVEKNLKSCLDNVNVVSVDTARTLFNQVMEKEFEDGIINWGRIVTIFAFEGILIKKLLRQQIAPDVDTYKEISYFVAEFIMNNTGEWIRQNGGWENGFVKKFE. Similarly, hBcl-xL samples were prepared and purified as described previously.54 Briefly, E. coli strain BL21 was transformed with the pET-21b plasmid (Novagen) carrying the gene coding for Bcl-xL ΔTM (Bcl-xL deletion mutant lacking the transmembrane domain). Bacteria were grown at 37 °C, and induction of protein expression was carried out when the OD600 reached 0.6 with 1 mM IPTG for 4 h at 37 °C. Following cell harvest and lysis by sonication, hBcl-xL was purified using a Ni-affinity column (Amersham). The eluate was dialyzed against 40 mM phosphate buffer (pH = 7.5) and 150 mM NaCl. Likewise, the hMcl-1 (172–323) gene was inserted into a pET15b vector for expression in E. coli. Bacteria cultures were induced when OD600 reached 0.6 with 1 mM IPTG and left shaking overnight at 20 °C. Similarly to hBfl-1 and hBcl-Xl, hMcl-1 protein was purified using Ni2+ affinity chromatography with a linear gradient of imidazole. Recombinant hBcl-2 protein was purchased from Sino Biologicals.
Agents and Peptides
Linear peptides were synthesized on a Liberty Blue microwave peptide synthesizer (CEM Corporation, NC) and a PS3 automated peptide synthesizer using Fmoc protocols. Stapled and covalent peptides reported in Tables 1 and 2 were synthesized by InnoPep (San Diego, CA) following standard solid phase peptide synthetic procedures. Fmoc-Dap-2-chloroacetamide–OH was prepared and incorporated as a building block at the N-terminus of selected peptides in solid phase conditions.
Mass Spectrometry Analysis
Molecular weight analysis was done using an Autoflex II MALDI TOF/TOF (Bruker Daltonics).
hBfl-1 was incubated in the absence and in the presence of hNOXA and 130E7 in a 1:2 ratio for 2 h. For mass spectroscopy analysis, equal volumes (2 μL) of each sample dissolved in the reaction buffer (16 mM phosphate, 50 mM NaCl, pH = 6.5) and MALDI Matrix solution (sinapic acid; 20 mg mL–1 in 50% acetonitrile -0.1% trifluoroacetic acid solution) were cocrystallized on the MALDI target plate and allowed to air-dry for 10 min. Mass spectra were acquired and processed with FlexAnalysis 2.4. The control protein molecular weight spectra as well as the protein and peptide conjugated spectra are shown. The protein mass shift correlating to peptide molecular weight was observed in a conjugated sample. The data were further processed using the FlexAnalysis peak picking method. With our current instrumentation and these experimental conditions, we expect on average a variation in the observed molecular mass of about ±50 Da, depending on the presence of slightly different ionization states of side chains, the presence of different numbers of nature of counterions, etc.
DELFIA (Dissociation Enhanced Lanthanide Fluorescence Immunoassay)
One hundred microliters of a 600 ng/mL solution of biotin-BID (Biotin-lc-EDIIRNIARHLAQVGDSMDR-NH2, where lc indicates a hydrocarbon chain of six methylene groups, aminohexanoic acid) was added to each well of 96-well streptavidin-coated plates (PerkinElmer). After 2 h of incubation and elimination by three washing steps of the unbound biotin-BID peptide, 11 μL of a preincubated solution of the protein and a serial dilution of the test peptide were added to the assay plate, followed by the addition of 89 μL of a 1.56 nM solution of Eu–N1-labeled anti-6xHis Antibody (PerkinElmer). After 1 h of incubation at RT to allow the immunoreaction to complete, a second washing step allowed removal of the unbound protein in complex with Eu antibodies if displaced by a test compound. Subsequently, 200 μL of enhancement solution (PerkinElmer) was added to each well and fluorescence measured after 10 min of incubation (excitation wavelength, 340 nm; emission wavelength, 615 nm). The protein final concentrations, determined by titrations, were 15 nM for hBfl-1, 16 nM for hMcl-1, 7 nM for hBcl-2, and 8.5 nM for hBcl-xL, while the antibody final concentration was 22.2 ng/well when compounds were tested against hBfl-1, hMcl-1, and hBcl-xL and 14.8 ng/well in the presence of hBcl-2. Each well received a final DMSO concentration equal to 1%. Protein, peptide, and antibody solutions were prepared in DELFIA assay buffer (PerkinElmer). Counts were normalized to control wells, which were treated with the vehicle, DMSO, and reported as % of inhibition. The reported IC50 values were calculated by Prism5 (GraphPad).
Gel Electrophoresis
Synthetic peptides were incubated for 2 h and at equimolar concentration (10 μM) with human Bcl-2 family proteins (hBfl-1, hMcl-1, hBcl-2, hBcl-xL). Note that the paper from Huhn et al. used 40 μM protein and 120 μM peptides.40 Samples were subjected to gel electrophoresis with SDS-PAGE gel followed by treatment with Coomassie dye as a staining protocol. For protein band detection, the NuPAGE 12% bis-tris mini gels (Life Technologies) were stained with SymplyBlue SafeStain (Life Technologies) according to the manufacture’s protocol.
NMR Spectroscopy
NMR spectra were acquired on a 600 MHz Bruker Avance spectrometer equipped with a TCI cryoprobe. All NMR data were processed and analyzed using TOPSPIN2.1 (Bruker Biospin, Billerica, MA, USA) and SPARKY3.1 (University of California, San Francisco, CA, USA). Solubility of test compounds was evaluated at 100 μM and in the following buffer conditions: PBS at a pH = 7.4 containing 10% D2O + 1% d6-DMSO. 3-(Trimethylsilyl) propionic-2,2,3,3-d4 acid (TMSP) at 11.1 μM was used as an internal reference (0.0 ppm).
Isothermal Titration Calorimetry and Molecular Modeling
Isothermal titration calorimetry was performed on a VP-ITC calorimeter from Microcal (Northampton, MA, USA). When indicated, measurements were performed in a reverse fashion (i.e., the protein was titrated into the compound solution). All titrations were performed at 25 °C in PBS buffer supplemented with 10% DMSO. Experimental data were analyzed using Microcal Origin software provided by the ITC manufacturer (Microcal). Modeling studies and figures were prepared using Chimera (http://www.cgl.ucsf.edu/chimera).
Cell Based Assays
Antiproliferative Activity of SKMEL-5 Cells
Human malignant melanoma SKMEL-5 cells were obtained from the American Type Culture Collection (ATCC HTB-70, Manassas, VA, USA) and were maintained in 5% CO2 at 37 °C and cultured in Eagle’s Minimum Essential Medium (EMEM; Cellgro) plus GlutaMAX supplemented with 10% fetal bovine serum (FBS; Omega Scientific) and 1% penicillin/streptomycin (Omega Scientific).
Approximately 30 000 cells were seeded into individual wells of a 96-well tissue culture plate and incubated for 24 h. Cells were replenished with fresh medium (0.1 mL/well; 5% FBS, no antibiotics) and exposed to triplicates of different concentration solutions (from 0.4 to 100 μM) of test compounds. The analyzed inhibitors were dissolved in DMSO, reaching a final DMSO concentration of 1%. After incubation for 24 h at 37 °C and 5% CO2, cell viability was assessed using ATPlite assay from PerkinElmer (Waltham, MA). Viability was normalized to control cells which were treated with the vehicle, DMSO. The reported IC50 values were calculated by Prism5 (GraphPad).
Western Blot
Twenty-four hours before the transfection, HEK293T was plated into a six-well plate at a seeding density of 500 000 cells per well. At 90% confluence, cells were transfected with N-HA-tagged Bfl1 (Bcl-2-related protein A1, full length; organism, Homo sapiens; catalog number, HG10562-NY, Sino Biological Inc.), by using the jetPRIME transfection reagent (Polyplus). After 16 h, different concentrations of Bfl1 inhibitors were added to the cells at a final DMSO concentration of 1% and incubated for 6 h. Cells were then washed with cold PBS and lysed with lysis buffer (50 mM Tris-HCl, at a pH of 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA) for 30 min on ice. Cell lysates were centrifuged at 13 000g for 30 min at 4 °C. The concentration of the protein content was determined using Bio-Rad protein assay solution. Equal amounts of cell lysate (40 μg) were resolved by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (PerkinElmer Life Sciences, Waltham, MA). Membranes were blocked with 5% BSA/PBST for 2 h and incubated with primary antibodies (rabbit polyclonal anti-HA antibody, Santa Cruz cat#: sc-805, dilution 1:1000) overnight at 4 °C with shaking. Following three washes with PBST (PBS containing Tween 20, 0.02% v/v), membranes were incubated with the secondary antibody (IRDye 800CW Donkey anti-Rabbit, 1:10 000) for 1 h shaking at RT. Bound antibodies were detected with LICOR.
Raji Cell Culture
In the washout experiment, Raji cell line derived (20 000) cells were placed per well in a 96-well plate. The cells were incubated with the Bfl-1 inhibitor 130G4 alone or in combination with different concentrations of Fludarabine (F-ara-A) or etoposide for 48 h at 37 °C in CO2 incubator. The in vitro cytotoxicity was measured in all the samples after 48 h using PI/DiOC6 followed by flow cytometry data analysis, and the data were reported as % SIA (specific induced apoptosis, see below).
Chronic Lymphocytic Leukemia (CLL) Sampling and Cell Culture
Peripheral blood mononuclear cells (PBMC) from patients with CLL were obtained from the CLL Research Consortium tissue bank. After CLL diagnosis was confirmed,55 patients provided written informed consent for blood sample collection. The institutional review board of the University of California, San Diego, approved the protocol. PBMCs were isolated by Ficoll-Hypaque gradient density centrifugation (Cat# 17-1440-03, GE Healthcare Life Science) and used fresh or viably frozen and stored in liquid nitrogen for later use.
PBMCs were separated from heparinized venous blood by density gradient centrifugation using Ficoll-Hypaque media (GE Healthcare). Samples with >95% double positive cells for CD5 and CD19, as assessed by flow cytometry, were selected and used. Briefly, primary CLL-B leukemia cells derived from CLL patients were cultured in RPMI supplemented with 10% heat inactivated FBS (fetal bovine serum, catalog # FB-02, Omega Scientific, Tarzana, CA) and 1% antibiotic at a density of 3 × 105 cells per well in a total volume of 100 μL at 37 °C and 5% CO2. Primary CLL cells were cultured alone in 96-well round-bottom plates (catalog # 3596, Corning, NY) or cocultured with NK-tert stromal cells in 96-well flat bottom plates (catalog # 3799, Corning, NY) with a ratio of 20:1 (NK-tert/CLL) by seeding 300 000 cells/well.56 The human mesenchymal NK-tert stromal cell line was derived from bone marrow. Stroma-NK-tert cells are fibroblast derived from human bone marrow and immortalized with human telomerase reverse transcriptase (hTERT) bearing exogene MFG-tsT-IRES-neo (RCB2350- RIKEN Bioresource Center, Japan). These cells were obtained from the RIKEN Cell Bank (RIKEN, Yokohama, Japan). NK-tert cells alone were maintained in DMEM (catalog # 10-017-CV, Corning Cellgrov, Manassas, VA 20109) supplemented with 10% FBS and 1% antibiotic.
Flow Cytometry
The flow cytometric data collection and analysis was carried out using FACScalibur (BD Biosciences, San Jose, CA) and FlowJo software (version 9, TreeStar Inc., Ashland, OR).
Detection of Apoptosis
For assessment of apoptotic cells, first cells alone or cocultured with stroma cell support were stained to select CLL-B with CD19/CD5 antibodies to gate on CLL-B cells. The CLL-B cell specific staining was done using a 1:40 dilution of CD19-PerCP-Cy5.5 (catalog # 8045-0198, clone SJ25C1, eBioscience, San Diego) and a 1:50 dilution of APC mouse antihuman CD5 (catalog # 555355, clone UCHT2, BD Biosciences) specific antibodies. The staining was done at 4 °C for 30 min followed by washing using FACS buffer two times. Following that, apoptotic and viable cells were discriminated by staining the cells with a 1:1000 dilution of 40uM 3,3′-dihexyloxacarbocyanine iodide (DiOC6; catalog #D-273, Molecular Probes, Eugene, OR, USA) for 30 min at 37 °C. Following DiOC6 staining, samples were subjected to flow cytometry analysis.
Calculation of Specific Induced Apoptosis (SIA)
In order to discriminate the compound specific induced apoptosis vs background spontaneous cell death from in vitro culture conditions, we calculated the percentage of specific induced apoptosis (% SIA) using the following formula: % SIA = [(compound induced apoptosis – media only spontaneous apoptosis)/(100 – media only spontaneous apoptosis)] × 100.
Statistical Analysis
The data sets were analyzed using GraphPad Prism software (v. 5.0c; San Diego, CA). The statistical significance was determined by using paired or unpaired Student’s t test or one-way ANOVA followed by Bonferroni correction’s multiple comparisons test. Statistical differences for the mean values are indicated as follows: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. The IC50 value was defined as the drug concentration that inhibits 50% cell growth compared with untreated controls and calculated by Graphpad Prism 6.0 software. Unless indicated, data are presented as the mean ± SEM.
Acknowledgments
This work was supported in part by National Institutes of Health grant CA168517 (to M.P.), grant CA P01081534 Chronic Lymphocytic Leukemia (CLL) Research Consortium (to T.J.K., M.P., and J.E.C.), the UC San Diego Foundation Blood Cancer Research Fund (to T.J.K.), and the Bennett Family Foundation (to J.E.C.). M.P. holds the Daniel Hays Chair in Cancer Research at the School of Medicine at UCR. Molecular graphics and analyses were performed with the UCSF Chimera package (http://www.cgl.ucsf.edu/chimera). Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). We also thank the Sanford-Burnham-Prebys Medical Discovery Institute (La Jolla, CA) Protein Analysis Facility supported by NIH grant CA030199 for support with earlier ITC measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.6b00962.
Molecular weights, SDS-PAGE gel electrophoresis data, DELFIA assay curves, and Kd values derived by ITC experiments for selected peptides reported in the manuscript (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fesik S. W. (2005) Promoting apoptosis as a strategy for cancer drug discovery. Nat. Rev. Cancer 5, 876–885. 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
- Opferman J. T. (2016) Attacking cancer’s Achilles heel: antagonism of anti-apoptotic BCL-2 family members. FEBS J. 283, 2661–2675. 10.1111/febs.13472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J. M.; Cory S. (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281, 1322–1326. 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- Czabotar P. E.; Lessene G.; Strasser A.; Adams J. M. (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63. 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- Wei M. C.; Zong W. X.; Cheng E. H.; Lindsten T.; Panoutsakopoulou V.; Ross A. J.; Roth K. A.; MacGregor G. R.; Thompson C. B.; Korsmeyer S. J. (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730. 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce C. M.; Reed J. C. (2016) Finally, An Apoptosis-Targeting Therapeutic for Cancer. Cancer Res. 76, 5914–5920. 10.1158/0008-5472.CAN-16-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Willis S. N.; Wei A.; Smith B. J.; Fletcher J. I.; Hinds M. G.; Colman P. M.; Day C. L.; Adams J. M.; Huang D. C. (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17, 393–403. 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Kitada S.; Reed J. C. (2004) MCL-1 promoter insertions dial-up aggressiveness of chronic leukemia. J. Natl. Cancer Inst 96, 642–643. 10.1093/jnci/djh153. [DOI] [PubMed] [Google Scholar]
- Placzek W. J.; Wei J.; Kitada S.; Zhai D.; Reed J. C.; Pellecchia M. (2010) A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 1, e40. 10.1038/cddis.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalinska M.; Kilianska Z. M. (2012) Targeting Bcl-2 in CLL. Curr. Med. Chem. 19, 5109–5115. 10.2174/092986712803530566. [DOI] [PubMed] [Google Scholar]
- Del Poeta G.; Postorino M.; Pupo L.; Del Principe M. I.; Dal Bo M.; Bittolo T.; Buccisano F.; Mariotti B.; Iannella E.; Maurillo L.; Venditti A.; Gattei V.; de Fabritiis P.; Cantonetti M.; Amadori S. (2016) Venetoclax: Bcl-2 inhibition for the treatment of chronic lymphocytic leukemia. Drugs Today (Barc) 52, 249–260. 10.1358/dot.2016.52.4.2470954. [DOI] [PubMed] [Google Scholar]
- Starr P. (2016) Venetoclax Shows Strong Activity in CLL. Am. Health Drug Benefits 9, 21. [PMC free article] [PubMed] [Google Scholar]
- Stilgenbauer S.; Eichhorst B.; Schetelig J.; Coutre S.; Seymour J. F.; Munir T.; Puvvada S. D.; Wendtner C. M.; Roberts A. W.; Jurczak W.; Mulligan S. P.; Bottcher S.; Mobasher M.; Zhu M.; Desai M.; Chyla B.; Verdugo M.; Enschede S. H.; Cerri E.; Humerickhouse R.; Gordon G.; Hallek M.; Wierda W. G. (2016) Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 17, 768–778. 10.1016/S1470-2045(16)30019-5. [DOI] [PubMed] [Google Scholar]
- Souers A. J.; Leverson J. D.; Boghaert E. R.; Ackler S. L.; Catron N. D.; Chen J.; Dayton B. D.; Ding H.; Enschede S. H.; Fairbrother W. J.; Huang D. C.; Hymowitz S. G.; Jin S.; Khaw S. L.; Kovar P. J.; Lam L. T.; Lee J.; Maecker H. L.; Marsh K. C.; Mason K. D.; Mitten M. J.; Nimmer P. M.; Oleksijew A.; Park C. H.; Park C. M.; Phillips D. C.; Roberts A. W.; Sampath D.; Seymour J. F.; Smith M. L.; Sullivan G. M.; Tahir S. K.; Tse C.; Wendt M. D.; Xiao Y.; Xue J. C.; Zhang H.; Humerickhouse R. A.; Rosenberg S. H.; Elmore S. W. (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208. 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Vogler M.; Butterworth M.; Majid A.; Walewska R. J.; Sun X. M.; Dyer M. J.; Cohen G. M. (2009) Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood 113, 4403–4413. 10.1182/blood-2008-08-173310. [DOI] [PubMed] [Google Scholar]
- Albershardt T. C.; Salerni B. L.; Soderquist R. S.; Bates D. J.; Pletnev A. A.; Kisselev A. F.; Eastman A. (2011) Multiple BH3 mimetics antagonize antiapoptotic MCL1 protein by inducing the endoplasmic reticulum stress response and up-regulating BH3-only protein NOXA. J. Biol. Chem. 286, 24882–24895. 10.1074/jbc.M111.255828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruncko M.; Wang L.; Sheppard G. S.; Phillips D. C.; Tahir S. K.; Xue J.; Erickson S.; Fidanze S.; Fry E.; Hasvold L.; Jenkins G. J.; Jin S.; Judge R. A.; Kovar P. J.; Madar D.; Nimmer P.; Park C.; Petros A. M.; Rosenberg S. H.; Smith M. L.; Song X.; Sun C.; Tao Z. F.; Wang X.; Xiao Y.; Zhang H.; Tse C.; Leverson J. D.; Elmore S. W.; Souers A. J. (2015) Structure-guided design of a series of MCL-1 inhibitors with high affinity and selectivity. J. Med. Chem. 58, 2180–2194. 10.1021/jm501258m. [DOI] [PubMed] [Google Scholar]
- Cohen N. A.; Stewart M. L.; Gavathiotis E.; Tepper J. L.; Bruekner S. R.; Koss B.; Opferman J. T.; Walensky L. D. (2012) A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem. Biol. 19, 1175–1186. 10.1016/j.chembiol.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M.; Marcellus R. C.; Roulston A.; Watson M.; Serfass L.; Murthy Madiraju S. R.; Goulet D.; Viallet J.; Belec L.; Billot X.; Acoca S.; Purisima E.; Wiegmans A.; Cluse L.; Johnstone R. W.; Beauparlant P.; Shore G. C. (2007) Small molecule obatoclax (GX15–070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. U. S. A. 104, 19512–19517. 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard D. J.; Lena R.; Bannister T.; Blake N.; Pierceall W. E.; Carlson N. E.; Keller C. E.; Koenig M.; He Y.; Minond D.; Mishra J.; Cameron M.; Spicer T.; Hodder P.; Cardone M. H. (2013) Hydroxyquinoline-derived compounds and analoguing of selective Mcl-1 inhibitors using a functional biomarker. Bioorg. Med. Chem. 21, 6642–6649. 10.1016/j.bmc.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadarajan S.; Vogler M.; Butterworth M.; Dinsdale D.; Walensky L. D.; Cohen G. M. (2013) Evaluation and critical assessment of putative MCL-1 inhibitors. Cell Death Differ. 20, 1475–1484. 10.1038/cdd.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J.; Stebbins J. L.; Kitada S.; Dash R.; Placzek W.; Rega M. F.; Wu B.; Cellitti J.; Zhai D.; Yang L.; Dahl R.; Fisher P. B.; Reed J. C.; Pellecchia M. (2010) BI-97C1, an optically pure Apogossypol derivative as pan-active inhibitor of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J. Med. Chem. 53, 4166–4176. 10.1021/jm1001265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yecies D.; Carlson N. E.; Deng J.; Letai A. (2010) Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 115, 3304–3313. 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abulwerdi F.; Liao C.; Liu M.; Azmi A. S.; Aboukameel A.; Mady A. S.; Gulappa T.; Cierpicki T.; Owens S.; Zhang T.; Sun D.; Stuckey J. A.; Mohammad R. M.; Nikolovska-Coleska Z. (2014) A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 13, 565–575. 10.1158/1535-7163.MCT-12-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abulwerdi F. A.; Liao C.; Mady A. S.; Gavin J.; Shen C.; Cierpicki T.; Stuckey J. A.; Showalter H. D.; Nikolovska-Coleska Z. (2014) 3-Substituted-N-(4-hydroxynaphthalen-1-yl)arylsulfonamides as a novel class of selective Mcl-1 inhibitors: structure-based design, synthesis, SAR, and biological evaluation. J. Med. Chem. 57, 4111–4133. 10.1021/jm500010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D.; Zhang Q.; Schreiber J. S.; Parsels L. A.; Abulwerdi F. A.; Kausar T.; Lawrence T. S.; Sun Y.; Nikolovska-Coleska Z.; Morgan M. A. (2015) Targeting mcl-1 for radiosensitization of pancreatic cancers. Transl Oncol 8, 47–54. 10.1016/j.tranon.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner E. S.; Holmgren A. (2006) The thioredoxin system in cancer-introduction to a thematic volume of Seminars in Cancer Biology. Semin. Cancer Biol. 16, 419. 10.1016/j.semcancer.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Certo M.; Del Gaizo Moore V.; Nishino M.; Wei G.; Korsmeyer S.; Armstrong S. A.; Letai A. (2006) Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365. 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Del Gaizo Moore V.; Letai A. (2013) BH3 profiling--measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer Lett. 332, 202–205. 10.1016/j.canlet.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A.; Norberg M.; Okvist A.; Derkow K.; Choudhury A.; Tobin G.; Celsing F.; Osterborg A.; Rosenquist R.; Jondal M.; Osorio L. M. (2007) Upregulation of bfl-1 is a potential mechanism of chemoresistance in B-cell chronic lymphocytic leukaemia. Br. J. Cancer 97, 769–777. 10.1038/sj.bjc.6603951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T.; Zobel K.; Fedorova A.; Quan C.; Yang H.; Fairbrother W. J.; Huang D. C.; Smith B. J.; Deshayes K.; Czabotar P. E. (2013) Stabilizing the pro-apoptotic BimBH3 helix (BimSAHB) does not necessarily enhance affinity or biological activity. ACS Chem. Biol. 8, 297–302. 10.1021/cb3005403. [DOI] [PubMed] [Google Scholar]
- Sivaraman T.; Sivakumar D. (2016) A Review on Structures and Functions of Bcl-2 Family Proteins from Homo sapiens. Protein Pept. Lett. 23, 932. 10.2174/0929866523666160719094636. [DOI] [PubMed] [Google Scholar]
- Czabotar P. E.; Lee E. F.; van Delft M. F.; Day C. L.; Smith B. J.; Huang D. C.; Fairlie W. D.; Hinds M. G.; Colman P. M. (2007) Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc. Natl. Acad. Sci. U. S. A. 104, 6217–6222. 10.1073/pnas.0701297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rega M. F.; Reed J. C.; Pellecchia M. (2007) Robust lanthanide-based assays for the detection of anti-apoptotic Bcl-2-family protein antagonists. Bioorg. Chem. 35, 113–120. 10.1016/j.bioorg.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Deng J.; Carlson N.; Takeyama K.; Dal Cin P.; Shipp M.; Letai A. (2007) BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 12, 171–185. 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Montero J.; Letai A. (2016) Dynamic BH3 profiling-poking cancer cells with a stick. Mol. Cell Oncol 3, e1040144. 10.1080/23723556.2015.1040144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan J.; Letai A. (2013) BH3 profiling in whole cells by fluorimeter or FACS. Methods 61, 156–164. 10.1016/j.ymeth.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan J.; Montero J.; Rocco J.; Letai A. (2016) iBH3: simple, fixable BH3 profiling to determine apoptotic priming in primary tissue by flow cytometry. Biol. Chem. 397, 671–678. 10.1515/hsz-2016-0107. [DOI] [PubMed] [Google Scholar]
- Touzeau C.; Ryan J.; Guerriero J.; Moreau P.; Chonghaile T. N.; Le Gouill S.; Richardson P.; Anderson K.; Amiot M.; Letai A. (2016) BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 30, 761–764. 10.1038/leu.2015.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huhn A. J.; Guerra R. M.; Harvey E. P.; Bird G. H.; Walensky L. D. (2016) Selective Covalent Targeting of Anti-Apoptotic BFL-1 by Cysteine-Reactive Stapled Peptide Inhibitors. Cell Chem. Biol. 23, 1123–1134. 10.1016/j.chembiol.2016.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowman X. H.; McDonnell M. A.; Kosloske A.; Odumade O. A.; Jenness C.; Karim C. B.; Jemmerson R.; Kelekar A. (2010) The proapoptotic function of Noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Mol. Cell 40, 823–833. 10.1016/j.molcel.2010.11.035. [DOI] [PubMed] [Google Scholar]
- Starovasnik M. A.; Braisted A. C.; Wells J. A. (1997) Structural mimicry of a native protein by a minimized binding domain. Proc. Natl. Acad. Sci. U. S. A. 94, 10080–10085. 10.1073/pnas.94.19.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik E.; Michalak E. M.; Villunger A.; Adams J. M.; Strasser A. (2007) Ultraviolet radiation triggers apoptosis of fibroblasts and skin keratinocytes mainly via the BH3-only protein Noxa. J. Cell Biol. 176, 415–424. 10.1083/jcb.200608070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales A. A.; Olsson A.; Celsing F.; Osterborg A.; Jondal M.; Osorio L. M. (2005) High expression of bfl-1 contributes to the apoptosis resistant phenotype in B-cell chronic lymphocytic leukemia. Int. J. Cancer 113, 730–737. 10.1002/ijc.20614. [DOI] [PubMed] [Google Scholar]
- Vogler M. (2012) BCL2A1: the underdog in the BCL2 family. Cell Death Differ. 19, 67–74. 10.1038/cdd.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth M.; Pettitt A.; Varadarajan S.; Cohen G. M. (2016) BH3 profiling and a toolkit of BH3-mimetic drugs predict anti-apoptotic dependence of cancer cells. Br. J. Cancer 114, 638–641. 10.1038/bjc.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman M. D.; Nyman T.; Welin M.; Lehtio L.; Flodin S.; Tresaugues L.; Kotenyova T.; Flores A.; Nordlund P. (2008) Completing the family portrait of the anti-apoptotic Bcl-2 proteins: crystal structure of human Bfl-1 in complex with Bim. FEBS Lett. 582, 3590–3594. 10.1016/j.febslet.2008.09.028. [DOI] [PubMed] [Google Scholar]
- Stebbins J. L.; Santelli E.; Feng Y.; De S. K.; Purves A.; Motamedchaboki K.; Wu B.; Ronai Z. A.; Liddington R. C.; Pellecchia M. (2013) Structure-based design of covalent Siah inhibitors. Chem. Biol. 20, 973–982. 10.1016/j.chembiol.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird G. H.; Bernal F.; Pitter K.; Walensky L. D. (2008) Synthesis and biophysical characterization of stabilized alpha-helices of BCL-2 domains. Methods Enzymol. 446, 369–386. 10.1016/S0076-6879(08)01622-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird G. H.; Gavathiotis E.; LaBelle J. L.; Katz S. G.; Walensky L. D. (2014) Distinct BimBH3 (BimSAHB) stapled peptides for structural and cellular studies. ACS Chem. Biol. 9, 831–837. 10.1021/cb4003305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBelle J. L.; Katz S. G.; Bird G. H.; Gavathiotis E.; Stewart M. L.; Lawrence C.; Fisher J. K.; Godes M.; Pitter K.; Kung A. L.; Walensky L. D. (2012) A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J. Clin. Invest. 122, 2018–2031. 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky L. D.; Bird G. H. (2014) Hydrocarbon-stapled peptides: principles, practice, and progress. J. Med. Chem. 57, 6275–6288. 10.1021/jm4011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hind C. K.; Carter M. J.; Harris C. L.; Chan H. T.; James S.; Cragg M. S. (2015) Role of the pro-survival molecule Bfl-1 in melanoma. Int. J. Biochem. Cell Biol. 59, 94–102. 10.1016/j.biocel.2014.11.015. [DOI] [PubMed] [Google Scholar]
- Rega M. F.; Wu B.; Wei J.; Zhang Z.; Cellitti J. F.; Pellecchia M. (2011) SAR by interligand nuclear overhauser effects (ILOEs) based discovery of acylsulfonamide compounds active against Bcl-x(L) and Mcl-1. J. Med. Chem. 54, 6000–6013. 10.1021/jm200826s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matutes E.; Owusu-Ankomah K.; Morilla R.; Garcia Marco J.; Houlihan A.; Que T. H.; Catovsky D. (1994) The immunological profile of B-cell disorders and proposal of a scoring system for the diagnosis of CLL. Leukemia 8, 1640–1645. [PubMed] [Google Scholar]
- Kashyap M. K.; Kumar D.; Villa R.; La Clair J. J.; Benner C.; Sasik R.; Jones H.; Ghia E. M.; Rassenti L. Z.; Kipps T. J.; Burkart M. D.; Castro J. E. (2015) Targeting the spliceosome in chronic lymphocytic leukemia with the macrolides FD-895 and pladienolide-B. Haematologica 100, 945–954. 10.3324/haematol.2014.122069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.