

Abstract
We used genomics to study the evolution of meticillin-resistant Staphylococcus aureus (MRSA) during a complex, protracted clinical infection. Preparing closed MRSA genomes from days 0 and 115 allowed us to precisely reconstruct all genetic changes that occurred. Twenty-three MRSA blood cultures were also obtained during treatment, yielding 44 colony morphotypes that varied in size, haemolysis and antibiotic susceptibility. A subset of 15 isolates was sequenced and shown to harbour a total of 37 sequence polymorphisms. Eighty per cent of all mutations occurred from day 45 onwards, which coincided with the appearance of discrete chromosome expansions, and concluded in the day 115 isolate with a 98 kb tandem DNA duplication. In all heterogeneous vancomycin-intermediate Staphylococcus aureus isolates, the chromosomal amplification spanned at least a 20 kb region that notably included mprF, a gene involved in resistance to antimicrobial peptides, and parC, an essential DNA replication gene with an unusual V463 codon insertion. Restoration of the chromosome after serial passage under non-selective growth was accompanied by increased susceptibility to antimicrobial peptide killing and reduced vancomycin resistance, two signature phenotypes that help explain the clinical persistence of this strain. Elevated expression of the V463 parC was deleterious to the cell and reduced colony size, but did not alter ciprofloxacin susceptibility. In this study, we identified large DNA expansions as a clinically relevant mechanism of S. aureus resistance and persistence, demonstrating the extent to which bacterial chromosomes remodel in the face of antibiotic and host immune pressures.
Keywords: Staphylococcus aureus, antibiotic resistance, chromosome duplication, genomics, mutation, evolution
Data Summary
Supplementary Table S4 is available through FigShare, http://dx.doi.org/10.6084/m9.figshare.1394626 (2015) Table S4
Complete genome sequences available through FigShare pending GenBank release: http://dx.doi.org/10.6084/m9.figshare.1391325 (2015) JKD6229.gb http://dx.doi.org/10.6084/m9.figshare.1391322 (2015) JKD6210.gb http://dx.doi.org/10.6084/m9.figshare.1391323 (2015) JKD6210p.gb
Software used available through GitHub https://github.com/tseemann/prokka (2015) and http://www.vicbioinformatics.com/software.shtml
Sequence read data for the study isolates have been submitted to the EMBL Sequence Read Archive (ENA). http://www.ebi.ac.uk/ena/data/view/ERP010276
Impact Statement
Bacterial antibiotic resistance is an increasingly serious global problem. It is well known that resistance gene acquisition or small changes in specific bacterial genes called point mutations can lead to antibiotic resistance. In this study, we reveal yet another means by which the hospital superbug Staphylococcus aureus is evading our best antibiotic treatments. We have discovered that during human infection the bacterium makes multiple copies of additional segments of its DNA, increasing the number of genes in specific sections of its chromosome. We show that such gene expansion helps S. aureus to beat the last-line antibiotic vancomycin and evade human immune responses. This study reveals yet another tool in the arsenal of S. aureus that helps explain why it is such a formidable pathogen and often so difficult to treat.
Introduction
Nosocomial infections are a major contributor to human morbidity and mortality globally. Staphylococcus aureus, a human commensal and well-endowed opportunistic pathogen, is one of the major culprits, especially antibiotic-resistant strains such as meticillin-resistant S. aureus (MRSA) (Lowy, 1998). The success of S. aureus as a hospital-acquired pathogen is in part due to the capacity of the bacterium to continually evolve and adapt to the hospital environment, frequently leading to enhanced antimicrobial resistance through resistance gene acquisition or point mutations in specific genes (Chambers & Deleo, 2009) and other genetic changes promoting hospital adaptation (Paulander et al., 2013).
The application of new genomics tools to human pathogens has provided insight into the evolutionary dynamics and adaptive mechanisms during colonization or chronic infection that promote persistence and transition from colonizer to invading pathogen (McAdam et al., 2011; Smith et al., 2006; Young et al., 2012). To date, these studies have demonstrated that, contrary to traditional concepts, the bacterial population is heterogeneous during chronic colonization or infection with the accumulation of nucleotide polymorphisms potentially contributing to niche adaptation in the host. During human infection, antibiotic exposure provides a major selection pressure driving such in vivo adaptation (Howden et al., 2011; Mwangi et al., 2007), with some antibiotic resistance-associated mutations having been shown to promote resistance to host innate immunity, providing an important link between antibiotic resistance and bacterial persistence (Gao et al., 2013). Despite these recent studies, our understanding of how bacterial pathogens, particularly S. aureus, evolve and adapt during persistent infection remains incomplete.
Technological advances have provided the capacity to sequence large numbers of bacterial genomes in a cost-effective, time-efficient manner, providing the potential for new insights into bacterial adaptation during infection. Whilst a growing number of bacterial species are now represented by closed, fully annotated genomes, proportionally most bacterial genome sequences are incomplete (Chain et al., 2009). Comparative approaches based on read-mapping and single nucleotide polymorphism (SNP) or short insertion/deletion (indel) detection are routinely used to analyse and compare incomplete genome sequences (Gao et al., 2010; Howden et al., 2011; McAdam et al., 2011; Mwangi et al., 2007; Young et al., 2012). These approaches detect important changes occurring during infection and have demonstrated the heterogeneous nature of infecting bacterial populations, but they may fail to detect other significant genetic changes, such as the expansion of multicopy insertion sequences (McEvoy et al., 2013), chromosomal duplications (Sandegren & Andersson, 2009) or large genome inversions (Cui et al., 2012). Fully assembled, closed genome sequences and other tools such as optical mapping that generate high-resolution ordered bacterial genome maps provide the means to readily uncover additional chromosomal changes that occur in bacterial species during in vivo adaptation.
We have previously sequenced and partially assembled a pair of clinical S. aureus from a persistent bloodstream infection with the accumulation of resistance to multiple antimicrobials (Gao et al., 2010). We have shown that point mutations in rpoB, relA and rlmN each contributed to the evolution of antimicrobial resistance and immune escape. However, all the phenotypic changes observed in the final clinical isolate were not fully explained by these three mutations, such as small colony size and vancomycin-intermediate resistance. To better understand the evolutionary dynamics of S. aureus during this persistent infection we fully assembled, annotated and recompared the initial (JKD6210) and the final (JKD6229) isolate isolated after 115 days of infection. Additionally, we sequenced 15 intermediate isolates obtained throughout the course of treatment. To the best of our knowledge, this is the first study to show that during human infection, bacterial chromosome expansion in association with sequential acquisition of functionally significant point mutations caused substantial phenotypic changes in the organism, which together promoted antimicrobial resistance and host adaptation.
Methods
Isolate details
The details of the clinical case have been reported previously (Gao et al., 2010). The first clinical isolate (JKD6210) was susceptible to vancomycin, rifampicin, ciprofloxacin and linezolid; however, after a complicated treatment course including all four of these antibiotics, a small colony variant (SCV JKD6229) was isolated in the spinal aspirate on day 115 after hospital admission. The SCV JKD6229 was a heterogeneous vancomycin-intermediate S. aureus (hVISA) and resistant to rifampicin and ciprofloxacin. It also displayed reduced susceptibility to linezolid. During the 115 days of infection a total of 24 specimens (23 blood cultures and one spinal aspirate) were culture positive for S. aureus. In total, 44 different colony morphologies were isolated from the positive cultures. Details of the isolates selected from these cultures are listed in Table S1 (available in the online Supplementary Material). All isolates were grown on horse blood agar (HBA) or in heart infusion broth at 37 °C unless otherwise stated. Colony size was measured on HBA after 20 h incubation.
δ-Haemolysin assay
The S. aureus accessory gene regulator (agr) activity was measured by δ-haemolysin production on Trypticase soy agar containing 5 % sheep blood using S. aureus strain RN4220, as described previously (Traber et al., 2008). Evidence of enhanced haemolysis between RN4220 and a test isolate was considered positive for δ-haemolysin production. Repression of agr was defined as an absence of δ-haemolysin production.
Antimicrobial susceptibility testing
Isolates were subcultured onto HBA for 18–24 h before performing any antimicrobial susceptibility testing. Vancomycin and teicoplanin macro E-tests were performed by applying 200 μl 2.0 McFarlane bacterial suspension onto brain heart infusion (BHI) agar. The plates were incubated for 48 h before readings were taken. An oxacillin E-test was performed using 0.5 McFarlane suspension onto 2 % NaCl BBL Mueller–Hinton II agar according to the manufacturer's instructions. Agar dilution was used for all other susceptibility testing, according to the Clinical Laboratory Standards Institute. Vancomycin population analysis profile testing was performed on all isolates to detect hVISA, as described previously (Howden et al., 2011).
Antimicrobial peptide susceptibility testing
Flow cytometry and propidium iodide-stained bacterial cells (excitation, 535 nm; emission, 620 nm; Sigma) were used to determine membrane permeabilization after treatment with antimicrobial peptides (hNP-1 or hBD-2, 20 μg ml− 1) as described previously (Gao et al., 2013). Heat-killed bacteria (95 °C for 15 min) and non-peptide-exposed bacteria were used as positive and negative controls, respectively. Flow cytometry was performed (LSRFortessa; Becton Dickinson) in 10 mM K+ minimal essential medium, pH 7.2. Fluorescence of a minimum of 5 × 103 cells was acquired for statistical analysis. The analysis was performed in biological triplicates for each peptide/strain combination and data were analysed using FACSDiva software (BD Biosciences).
Closed genome sequences
The complete genome assemblies for Sa_JKD6210 and Sa_JKD6229 were based on Ion Torrent mate-pair libraries. The libraries were prepared using a demonstration protocol (Ion Torrent p/n 4472004, release date 27 September 2011) with the 5500 SOLiD mate-pair library kit. Libraries were sequenced on an Ion Torrent Personal Genome Machine with a 316D chip and 200 bp chemistry, running TorrentSuite version 3.2.1 (Life Technologies). After processing, 1 629 524 reads for JKD6210 and 2 760 456 reads for JKD6229 contained the mate-pair linker sequence with a mean insert size of 5211 and 5152 bp, respectively. Primary de novo assembly was performed using Newbler (version 2.8), resulting in three scaffolds (2 762 210 bp) and two scaffolds (2 727 771 bp), respectively. Genome finishing was managed using Geneious Pro (version 7.2) with gaps closed by PCR, primer walking and Sanger sequencing. Final assemblies were validated by reference to high-resolution NcoI chromosome optical maps using MapSolver (version 3.2.0; OpGen). Ion Torrent sequencing errors were corrected by mapping Illumina MiSeq 250 bp paired-end reads from each genome back to the finished sequences (see below). Annotation was performed using Prokka version 1.10 (Seemann, 2014). Chromosome sequences were also compared by blastn analysis and visualized using act (Artemis Comparison Tool; Sanger). Sequences were screened for bacteriophage-associated sequences using phast (Zhou et al., 2011).
Whole-genome sequencing and analysis
Whole-genome sequencing was performed on the Illumina MiSeq platform, with Nextera XT library preparation and paired-end 250 bp sequencing chemistry. The raw reads were filtered using Trimmomatic (Bolger et al., 2014) to remove ambiguous base calls, Nextera adaptors, bases at Phred quality 10 and reads < 50 bp. Resulting FASTQ sequence read files were read-mapped to the completed JKD6210 genome using Bowtie version 2.1.0 (Langmead & Salzberg, 2012) with default parameters and consensus calling to identify SNPs (indels excluded) using Nesoni version 0.109 (https://github.com/Victorian-Bioinformatics-Consortium/nesoni). A list of the isolates sequenced and summaries of the output for each are shown in Table S1. An alignment file based on the variable nucleotide positions amongst all isolates was used to infer a whole-genome haplotype network using the median-joining algorithm as implemented in SplitsTree version 4.13.1 (Huson & Bryant, 2006).
Quantitative PCR (qPCR)
Real-time qPCR was used to assay the copy number of selected genes within the chromosomal repeats. The primers are listed in Table S2. Phire Hot Start II DNA polymerase (Thermo Scientific) was used in the qPCR and Evagreen dye (Biotium) was added into reactions in 1 : 20 ratio. The PCR was cycled at 98 °C for 15 s, 52 °C for 18 s and 72 °C for 15 s. The dye signals were sampled during the annealing steps. Standard curves were generated for each pair of primers, using 10-fold serial dilutions of S. aureus genomic DNA. The housekeeping gene gyrB was used as a reference gene to normalize the results.
Allelic exchange
By allelic exchange with pKOR1 (Bae & Schneewind, 2006), we introduced the alr1 15 bp deletion from BPH1123 into JKD6229. A 2 kb amplicon centred on the deletion was amplified from BPH1123 genomic DNA with primers listed in Table S2 and cloned into pKOR1. The plasmid was electroporated into JKD6229 after isolation from RN4220, with subsequent allelic exchange as described previously (Bae & Schneewind, 2006). Sanger sequencing of the region confirmed the authenticity of the mutation.
Inducible ParC expression
The full-length parC genes from JKD6210 (parC WT), SCV JKD6229 (parC V463) and BPH1116G20 (parC G296D, V463) were amplified by PCR (oligonucleotides used are listed in Table S2). The PCR products were digested with KpnI/EcoRI, ligated into plasmid pRAB11 (Helle et al., 2011) and transformed into Escherichia coli strain IM08B (Monk et al., 2015). The sequences of the parC alleles cloned into pRAB11 were confirmed by Sanger sequencing. The plasmids were directly transformed into JKD6210 with parC expression induced by addition of anhydrotetracycline. JKD6210 was also transformed with empty vector as a negative control. The resulting plasmids and strains are listed in Table S3. To assay the impact of inducible parC expression, the transformed isolates were grown on Mueller–Hinton agar containing chloramphenicol (10 μg ml− 1) with anhydrotetracycline (5 ng ml− 1) and incubated for 24 h.
Repeated subculture of intermediate isolate BPH1116
Isolate BPH1116 was subcultured in BHI broth and passaged daily for 20 days. This involved transfer of 5 μl overnight culture (OD600 >2.0) into 10 ml fresh BHI broth. Subculture 20 was named BPH1116-G20. This isolate underwent whole-genome sequencing and qPCR was carried out to assess loss of chromosomal duplications.
Results
Isolate characterization during persistent infection
In a previous study, we used genomics to investigate the molecular basis for a S. aureus SCV phenotype with reduced linezolid susceptibility, isolated from a single patient over the course of a protracted infection (Gao et al., 2010). Draft genome sequences were obtained for the initial clinical isolate that demonstrated normal colony morphology (JKD6210), and an SCV isolate that had acquired resistance to multiple antimicrobials and was isolated after 115 days of persistent and recurrent infection (JDK6229). Only four mutations (two single nucleotide substitutions, two codon insertions) and the loss of a 25 kb plasmid were found in the SCV. Allelic exchange experiments partially restored the growth defects, but did not fully explain the antimicrobial resistance of the SCV. To better understand the genetic changes that led to the full SCV phenotype, we conducted a more detailed comparative and functional genomic investigation that began by closing and annotating JKD6210 (initial isolate) and JKD6229 (final isolate). We then sequenced 15 of the 44 S. aureus isolates that were cultured from 24 clinical samples (23 blood cultures) that had been submitted to the laboratory during the patient's treatment. The S. aureus isolates from these samples exhibited extensive phenotypic heterogeneity, especially after day 14 of infection, initially observed as a range of colony morphotypes. For each blood culture plate, a representative of each distinct colony morphotype was stored at − 80 °C for later analysis.
To determine the phenotypic heterogeneity of the different colony types that emerged during the infection, each morphotype was assayed for growth characteristics, haemolysis patterns and antibiotic susceptibility profiles (Fig. 1). This testing included 44 isolates from the 24 clinical samples. The colony diameter sizes were highly variable over the course of the infection (0.6–3.9 mm) and whilst the initial clinical isolate was homogeneous with normal colony morphology, the subsequent cultures demonstrated mixed colony morphotypes (Fig. 1a). Colonies of reduced size were first isolated from the day 5 blood culture. Most blood cultures after day 14 produced mixed populations with varying colony sizes ranging from 0.6 to 2.5 mm (Fig. 1a).
Fig. 1.

Phenotypic diversity of isolates during the persistent infection. (a) The colony size, antibiogram, vancomycin population analysis profile area under curve (VAN PAP/AUC) result and δ toxin activity of all 44 isolates are summarized. Increasing vancomycin resistance is indicated by colour (green is susceptible and red is resistant). Likewise, reduced colony size is indicated by red, whilst normal colony size is indicated by green. The sequential increase in antibiotic resistance is highlighted in red, with R indicating no growth and S indicating growth on the relevant antibiotic concentration. CIP, ciprofloxacin; RIF, rifampicin; VAN, vancomycin; TP, teicoplanin; FOX, cefoxitin; OX, oxicillin. (b) Analysis of δ-haemolysin production in 15, sequenced intermediate isolates demonstrated significant heterogeneity. Clinical isolate BPH1118 had no δ-haemolysin activity, whilst BPH1141 and BPH1141 had weak activity.
Antibiogram and vancomycin population analysis profile testing demonstrated the sequential acquisition of low-level vancomycin resistance in the bacterial population, although some vancomycin-susceptible populations (BPH1123, BPH1143, BPH1146 and BPH1148) were still present after day 14 where the hVISA strains were dominant in the infection (Fig. 1a). The δ-haemolysin assay is a surrogate marker for activity of the accessory gene regulator (agr) locus in S. aureus, the major quorum-sensing regulator of virulence. This assay suggested that strains BPH1118 (day 47), BPH1141 (day 61) and BPH1146 (day 113) had defective agr function, whilst other strains had intact agr function (Fig. 1a).
These phenotypic comparisons clearly indicated that a heterogeneous population of S. aureus infected the patient. To better understand the evolution of the bacterial population during this persistent infection, additional genome sequencing was performed. This included the full assembly and closing the genomes for the first and last clinical isolates, and generating draft genome sequences for the 15 intermediary isolates (Table S1).
Comparison of fully assembled chromosomes reveals a large chromosomal duplication
To enable a thorough analysis of the 15 additional draft genomes sequences, we fully assembled the chromosome for the initial and the final clinical isolates, JKD6210 (single circular chromosome 2 748 898 bp, circular plasmid 25 003 bp) and JKD6229 (single circular chromosome 2 847 260 bp). We utilized a combination of Ion Torrent mate-pair and Illumina paired-end libraries to produce high-quality closed genomes. High-resolution NcoI optical maps were additionally generated for both isolates to guide and validate in silico scaffolding. Unexpectedly, the optical maps suggested an ∼100 kbp repeated region in the SCV JKD6229 chromosome (Fig. 2a). The in silico assembly of JKD6229 confirmed the optical mapping and demonstrated a 98 231 bp sequence in a tandem duplication from recA nucleotide position 1 356 561 to trpB position 1 454 925 (JKD6210 numbering). The duplication created a hybrid trpB/recA gene (Fig. 2). The impact of the duplication on phenotype is assessed in a later section.
Fig. 2.

Complete and correct assembly of JKD6210 and JKD6229. (a) Comparison of high-resolution NcoI optical maps with in silicoNcoI predictions confirming correct assembly of the chromosome for each isolate. Depicted also is the 98 kb duplicated region. (b) Expanded view of the 98 kb duplication showing the flanking protein-coding DNA sequence impacted by the mutation, and the location of parC and mprF within a 20 kb internal segment. Shown also is 50 bp of sequence either side of the trpB/recA junction.
Analysis of the two closed chromosomes also uncovered previously unrecognized mutations in SCV JKD6229, including three small codon deletions in rplC, spoU and entS, and one in-frame insertion in rocA (Fig. 3, Table S4).
Fig. 3.

Population genotype and phenotype summary of all sequenced isolates. (a) Median-joining network graph showing the genetic relationship between the 17 clinical S. aureus isolates obtained over the duration of treatment, inferred from whole-genome sequence alignments against JKD6210.Numbers on graph edges indicate SNPs. (b) Mutation summary, culminating in SCV JKD6229 as shown by purple blocks. Lower row contains a heatmap summary of changing vancomycin susceptibility. (c) Subset of the phenotype data for each isolate presented in Fig. 1 for ease of comparison with the corresponding genotype, including heatmap of mean colony size (mm) fluctuations, haemolysis and antibiogram results generated by agar dilution. Red blocks denote resistance.
Genome sequence comparisons of the intermediate isolates confirm a heterogeneous population
Fifteen intermediate isolates were selected for whole-genome sequencing. Sequence reads were mapped to the JKD6210 completed chromosome and a full list of variants was produced (Figs 3 and S1, Table S4). A haplotype network was inferred using the median-joining algorithm from alignment of the resulting variable nucleotides (indels excluded). This network demonstrated the heterogeneous structure of the S. aureus population within the patient, as it evolved from the single progenitor strain (Fig. 3a). The majority of mutations arose after the exposure to rifampicin, ciprofloxacin and vancomycin from day 14 onwards, and this was also reflected in dramatically diverse colony morphotypes. There were eight persisting mutations that were absent in JKD6210 and accumulated in one or more intermediate strains to persist through to JKD6229 (Fig. 3). The critical time period for the emergence of the persistent SCV was between days 14 and 45, when the mutations in rpoB, relA, parC and rplC first appeared in BPH1116. This was the first hVISA strain isolated during the infection (Fig. 3a). All four of these mutations were sustained in isolates recovered during the infection except for the absence of the rplC mutation in two vancomycin-sensitive S. aureus (VSSA) strains (Fig. 3a, b, Table S3). The mutations in rlmN and rocA were detected at day 57 after linezolid was first employed, with the rlmN mutation shown to inactivate RlmN methylase activity and directly associate with linezolid resistance (Gao et al., 2010; LaMarre et al., 2011). Mutations in spoU and entS appeared in isolates from the day 113 blood culture.
An analysis of the mutations that appeared in one or more of the intermediate isolates but did not persist through to JKD6229 was also undertaken (Fig. S1, Table S4). Some of these changes helped explain the varying phenotypes of the intermediate isolates. For example, BPH1118 had five additional changes that included an agrA loss-of-function mutation that explained the absence of δ-haemolysis in this isolate (Fig. 1a). A 15 bp deletion in alr1 was found in the VSSA isolate BPH1123,. This gene encodes an alanine racemase. Transfer of the alr1 allele from BPH1123 into the hVISA JKD6229 increased the susceptibility of the strain to vancomycin, which fits with the sensitivity of BPH1123 to vancomycin and oxacillin (Fig. S2).
Large chromosomal duplications contribute to antibiotic and antimicrobial peptide resistance
Given the discovery of the chromosomal duplication in JKD6229, we examined changing the read-coverage density across the JKD6210 chromosome to investigate possible chromosomal duplications amongst the intervening isolates (Fig. 4). Two distinct high-coverage regions suggestive of chromosomal repeats were noted, first detected in BPH1116 at day 45 (Fig. 4a). Region 1 was variable in length, up to ∼98 kb and spanned the genes SA1128–SA1204 (S. aureus N315 locus_tag nomenclature). This was the same region duplicated in JKD6229 (Fig. 2). Region 1 also appeared to be duplicated in five other isolates (Fig. 4). We used the unique sequence in JKD6229 across the trpB/recA junction (Fig. 2b) to screen for the same duplication site within the sequence reads of the intermediary isolates. Only JKD6229 contained this sequence, indicating that this region was independently duplicated amongst the intermediary isolates.
Fig. 4.

Detection of chromosomal expansions in intermediate isolates. (a) Heatmap sequence coverage summary of the 15 isolates against the chromosome of JKD6210, showing a region of likely chromosomal amplification of variable length from 10 to 98 kb (region 1) present in six isolates and a ∼35 kb region (region 2), corresponding to phiSA3. (b) Three coding sequences from region 1 and 2 were selected as qPCR targets for verifying the copy number of the amplified regions shown in (a). The copy numbers of these coding sequences were aligned with coverage summary and confirm the expected copy number changes. Isolates that demonstrated reduced vancomycin susceptibility are highlighted in red, whilst green indicates vancomycin susceptibility. All isolates containing chromosomal expansions had reduced vancomycin susceptibility. P1–P3, region 1; P4–P6, region 2 (phiSA3).
Region 2 was ∼35 kbp and spanned the staphylococcal bacteriophage phiSA3. Higher read-coverage across this region was suggestive of mixed cell populations with integrated and excised circular phage forms, as the strains BPH1117 and BPH1136 were still producing α-haemolysin, which is inhibited if β-haemolysin is made (Fig. 1a). The β-haemolysin gene is disrupted on phiSA3 integration. We therefore conducted an in silico screen of the reads from all isolates for the presence of the sequence across the junction of the circular form of phiSA3. Reads from S. aureus isolate BPH1116 onwards indicated the presence of excised, circular forms of phiSA3. This result explains the higher read-coverage across region 2. We further confirmed the read-coverage results by qPCR targeting three protein-coding DNA sequences within each of region 1 and region 2 (containing phiSA3) (Fig. 4a). Read-coverage and mapping locations across region 1 indicated there were duplications of variable length (20–98 kb) and copy number (Fig. 4a).
Intriguingly, the read-coverage and confirmatory qPCR showed that only isolates with reduced vancomycin susceptibility contained chromosome expansions, suggesting a possible contribution of this DNA amplification to antibiotic resistance. Isolate BPH1122 contained the shortest example of the region 1 chromosome duplication, covering 11 coding sequences from SA1183 to SA1193 (Fig. 4). These coding sequences included genes encoding proteins involved in DNA replication, in particular chromosome segregation (parC, parE), and mprF, encoding multiple peptide resistance factor (MprF). In S. aureus, mutations leading to increased activity of MprF are associated with enhanced resistance to host defence peptides and reduced susceptibility to daptomycin (Bayer et al., 2014). Only one mutation was detected across this region compared with JKD6210 and this was the V463 codon insertion that was present in all copies of parC, first observed in the day 45 isolate BPH1116 (Figs 3a and 4). Mutations in parC are associated with fluoroquinolone resistance, but usually occur at or around position 80 (Schmitz et al., 1998).
To investigate the functional impact of the region 1 chromosomal duplication in S. aureus, isolate BPH1116 (3 × 60 kb tandem repeated sections) was serially subcultured for 20 days, generating BPH1116G20 which was shown by qPCR and genome sequencing to have lost the chromosomal expansion (Fig. S3). When BPH1116 (day 45) was compared with JKD6210 (day 0), it exhibited a reduced colony size and susceptibility to vancomycin (Fig. 1a), with 3 × 60 kb copies of the region 1 chromosomal duplication present (Fig. 4). BPH1116G20 was tested for the above phenotypes and additionally screened for the resistance to host defence peptides, due to the presence of mprF in the amplified region. BPH1116G20 displayed increased colony size, susceptibility to vancomycin and host defence peptides compared with BPH1116 (Fig. 5).
Fig. 5.
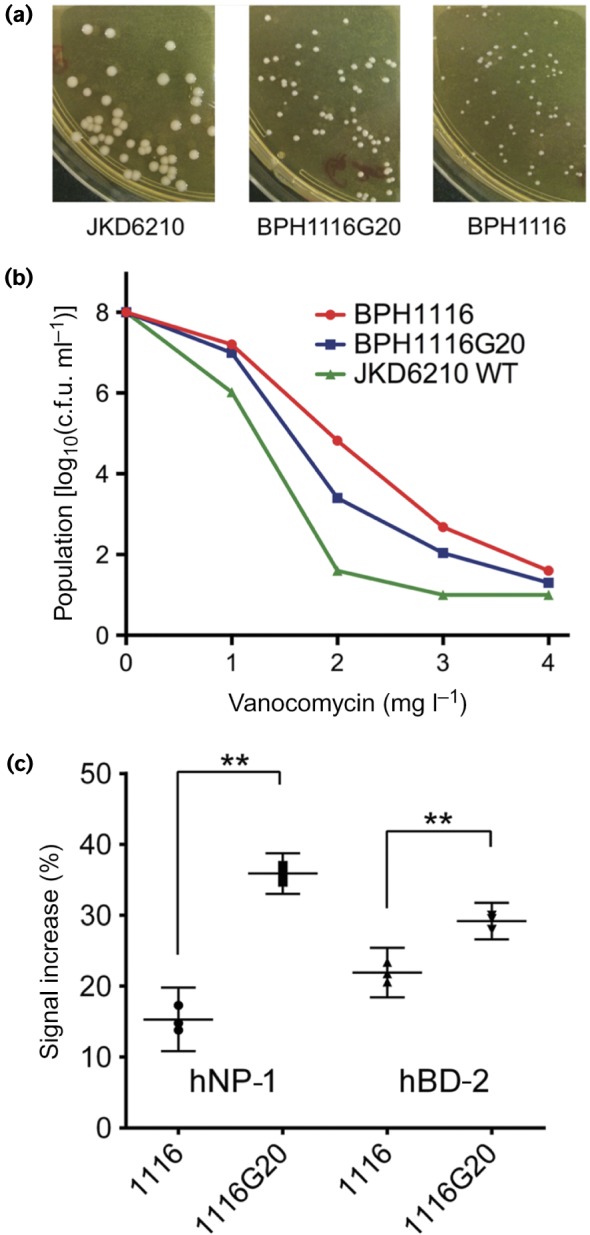
Impact of chromosomal expansion on key phenotypes. (a) Comparisons of colony morphology, showing partial restoration of colony size after loss of the chromosomal amplification spanning parC and mprF in isolate BPH1116G20.(b) Increased susceptibility to vancomycin upon loss of the chromosomal amplification as measured by population analysis profile. (c) Increased sensitivity to antimicrobial peptide killing upon loss of the chromosomal amplification as indicated by enhanced propidium iodide staining, as measured by flow cytometry. The y-axis shows the increase in propidium iodide-mediated fluorescence for antimicrobial peptide-exposed versus non-exposed S. aureus. Results indicate means and 95 % confidence intervals for biological triplicates. **Significantly different, P < 0.001, unpaired t-test.
In addition to the loss of the chromosomal duplication, genome sequencing of BPH1116G20 identified two additional non-synonymous point mutations, one each in the genes parC and abgT (AbgTG326R). The gene abgT encodes a putative p-aminobenzoyl-glutamate transport protein, which may be involved in uptake of folate degradation products (Green et al., 2010). The additional mutation in parC that yielded ParCG296D/V463 (compared with JKD6210) was particularly interesting given the essential nature of parC and that three copies of ParCV463 were present in the chromosomal duplication within BPH1116 (Fig. 4). Despite numerous attempts, we were unable to introduce the V463 allele into the WT parC of JKD6210.
Deleterious effect of overexpression of parC V463 in S. aureus
We therefore took an alternate approach and placed the three different parC alleles [WT, V463 and the double mutation (V463, G296D)] under a Tet-inducible promoter in the plasmid pRAB11 and expressed them in the JKD6210 background. Using only a very low level of inducer (concentrations of >80 ng anhydrotetracycline ml− 1 were toxic for all alleles, including WT), the growth characteristics of the two parC mutants were compared with controls (Fig. 6). Expression of parC V463 had a profound impact on growth; a phenotype that was rescued in the presence of the G296D mutation suggesting that this mutation is compensatory (Fig. 6). We also assessed the susceptibility of each strain to ciprofloxacin by E-test ( < 5 ng anhydrotetracycline ml− 1), but observed no change in resistance for either of the mutants compared with WT (data not shown). We also wondered if the ParCV463 allele induced a mutator phenotype in S. aureus. To test this, we compared the frequency of occurrence of spontaneous rifampicin resistance in each of the strains < 5 ng anhydrotetracycline ml− 1 induction, but we observed no difference in mutation frequency between strains harbouring mutants and WT parC alleles (observed range 2.1 × 10− 8 to 3.6 × 10− 8).
Fig. 6.
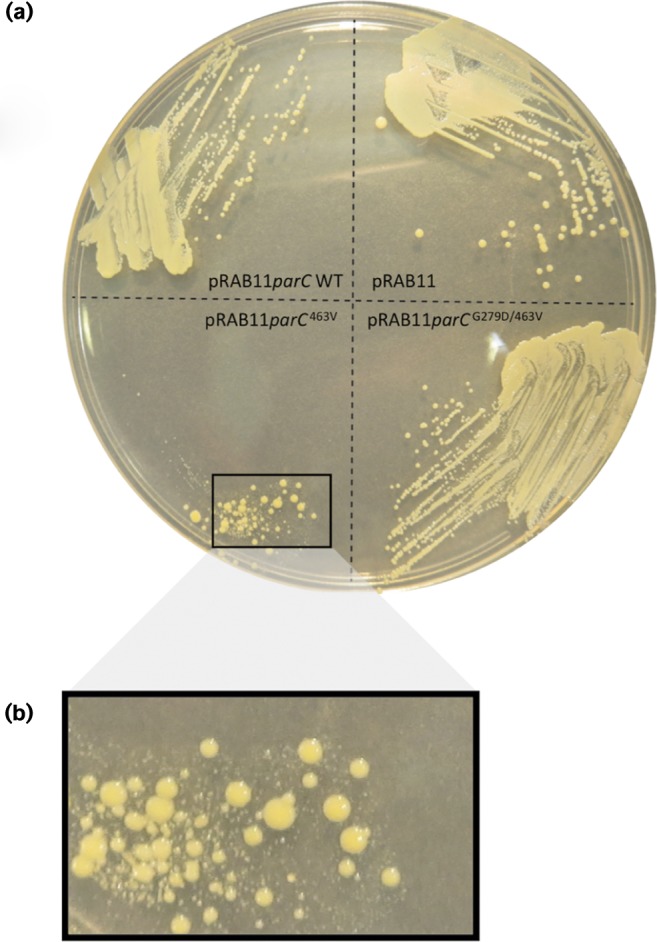
Impact of ParC mutations on growth of S. aureus JKD6210.(a) Expression of three different parC alleles in JKD6210. The alleles were cloned into the Tet-inducible expression vector pRAB11 and transformed into JKD6210. Shown is growth of each strain on Mueller–Hinton agar in the presence of 5 ng anhydrotetracycline ml− 1.(b) Impact of overexpression of parC with the V463 mutation on growth and colony size of JKD6210.
Discussion
Recent advances in genomics have provided the tools to precisely unravel the genomic population structure during bacterial infection. However, utilizing these technologies to understand pathogen populations and evolution during clinical infections requires carefully collected and curated clinical isolates. Most studies to date investigating the evolution of S. aureus during clinical infections, in particular in relation to evolving antibiotic resistance, have focused on horizontal gene transfer events (Berglund & Söderquist, 2008; Bloemendaal et al., 2010; Weigel et al., 2003), or the sequential acquisition of SNPs or small indels (Cameron et al., 2012; Friedman et al., 2006; Howden et al., 2008, 2011; Mwangi et al., 2007; Weigel et al., 2003), usually by comparing consensus sequences from a pair of clinical isolates with divergent susceptibility profiles. In this study, by carefully characterizing phenotypic diversity within the infecting S. aureus population recovered from a serious and persistent S. aureus infection, we identified a continuously evolving bacterial population within the patient that facilitated adaptation to higher levels of antibiotic resistance, reduced susceptibility to host innate immune factors and altered virulence. The phenotypic and genomic diversity revealed within a single patient infection highlights the population complexity of high-burden bacterial infections, and also demonstrates that selecting single colonies for sequencing from such cases will not reveal this genomic diversity.
Here, we also showed that high-quality closed genome sequences are important for gaining a deeper understanding of how S. aureus is evolving during infection. The large chromosomal duplications we observed were readily detected by comparisons of completed genomes, but were easily missed by the more common automated de novo assembly or read-mapping approaches.
The acquisition of mutations during the infection was rapid in an evolutionary sense, driven by the highly selective nature of the environment, including host defence mechanisms and multiple antimicrobial exposures. Whilst eight mutations were sequentially acquired and maintained in the final clinical isolate (Fig. 3), a number of which have been demonstrated to confer survival advantages through activation of the stringent response (RelA fig128Y), reduced host innate immune susceptibility (RelA fig128Y, RpoB H481Y) or enhanced antimicrobial resistance (RpoB H481Y, RlmN 345E insertion) (Gao et al., 2010, 2013), 39 SNPs or indels appeared in an apparently transient manner and were not selectively maintained. Investigation of one transient 12 bp deletion in alr1, encoding alanine racemase 1 (which converts l-alanine to d-alanine, a cell wall precursor), found in isolate BPH1123, restored vancomycin susceptibility in the day 50 isolate (Fig. S2). The mutation resulted in increased antibiotic susceptibility, possibly explaining its lack of persistence. The additional mutations maintained in strain JKD6229 are also predicted to contribute to the altered growth and reduced antibiotic susceptibility of this isolate. The 5 bp frame-shift mutation in rocA is predicted to result in a dysfunctional oxidoreductase and impaired l-proline degradation in the arginine deaminase pathway. This may result in a lower efficiency of the arginine deaminase pathway to provide ATP. The downregulation of rocA was previously reported in a hemB-mutated SCV (Seggewiss et al., 2006), suggesting that this insertion may contribute to the SCV phenotype. The 12 bp deletion in rplC results in a 4 aa deletion in ribosomal protein L3, a mutation that may contribute to the slow growth of the isolate (Cui et al., 2010) and the reduced linezolid susceptibility (Locke et al., 2009). The impacts of the frame-shift mutations in spoU, encoding the TrmH family RNA methyltransferase, and entS, encoding a putative major facilitator transporter, are unclear (Table S4).
Aside from SNPs and indels, distinct chromosomal rearrangements and duplications have received increasing attention as mediators of bacterial evolution and adaptation (Sandegren & Andersson, 2009; Sun et al., 2012), especially as mediators of enhanced antimicrobial resistance (Sandegren & Andersson, 2009). These gene duplications and amplifications generate extensive and reversible genetic variation that facilitates bacterial adaptation (Andersson & Hughes, 2009). The most striking feature of the genomic analysis of isolates from this infection was the variable chromosomal duplications that were uncovered only in isolates demonstrating an hVISA phenotype. Gene duplications and amplifications have been studied predominantly in vitro, reported rarely clinically and, to the best of our knowledge, have never been reported during human infection with S. aureus. Interestingly, the size, copy number and precise break-points of the duplicated regions were variable amongst the intermediate isolates, with the smallest detected region containing only 11 coding sequences, whilst the largest duplication was ∼98 kb. These data indicate that chromosome duplications were occurring independently in this region over the course of the infection. Several mechanisms have been proposed to explain the occurrence of gene duplications and amplifications (Sandegren & Andersson, 2009). Directly oriented repeated sequences such as insertion sequence elements or rRNA operons can mediate a RecA-dependent non-equal homologous recombination. However, the absence of repeated sequences flanking the duplicated region here suggests a RecA-independent mechanism, which may include phenomena such as DNA break-mediated rolling circle replication (Sandegren & Andersson, 2009). Once a region has been duplicated then RecA-dependent mechanisms may be involved in producing additional copies, using the tandem duplication as substrate for homologous recombination. Such a process could explain the presence of 3 × 60 kb copies of the region observed in BPH1116 (Fig. 4).
Chromosomal duplications confer a fitness cost on bacteria (Adler et al., 2014), hence they tend to be unstable. Serial passage of isolate BPH1116 resulted in loss of the chromosomal duplication, and comparison of this isolate (BPH1116G20) with the isolate containing the duplication revealed important phenotypic consequences of the duplication that included enhanced antibiotic resistance and reduced susceptibility to human antimicrobial peptides. The smallest duplicated region (11 coding sequences) included mprF, which has been linked previously to changes in vancomycin susceptibility (Ruzin et al., 2003), and host antimicrobial peptide and daptomycin resistance by altering the surface charge of the cell surface (Ernst & Peschel, 2011; Mishra et al., 2009). Resistance to two human antimicrobial peptides, hNP-1 and hBD-2, was therefore analysed in the intermediate isolate BPH1116 containing multiple copies of mprF and the lab-derived BPH1116G20, which only has one copy. As predicted, BPH1116 was more resistant to killing by antimicrobial peptides than BPH1116G20.
We were particularly interested in the role of the unusual V463 codon insertion mutation in the duplicated copies of parC (also called grlA). Quinolone resistance in S. aureus is most frequently caused by substitution mutations at codon 80, within the quinolone resistance-determining region (Schmitz et al., 1998). Mutations at this position confer high-level quinolone resistance (typically >32 μg ml− 1). The V463 mutation appears uncommon and, to the best of our knowledge, has not been, reported previously. We expected this mutation might explain the shift observed in susceptibility to ciprofloxacin at day 45 from 0.5 to 1.0 mg l− 1 (Fig. 1). Inducible expression of the V463 parC allele in JKD6210 saw a substantial impact on colony size and growth, but there was no change in antibiotic susceptibility (Fig. 6). In other bacteria, quinolone resistance mutations can dramatically alter the minimal selective concentration (MSC), sometimes at antibiotic concentrations 230-fold less than the MIC (Gullberg et al., 2011). The MSC is the concentration of antibiotic where the fitness cost of the mutation is balanced by the selective effect of the drug (Sandegren & Andersson, 2009) and we speculate that the two copies of the V463 mutation might be lowering the MSC, giving the strain a competitive advantage in the patient, in circumstances where the concentration of antibiotic (in this case ciprofloxacin) was below the MIC. Experiments to test this hypothesis remain to be performed. Quinolones have also been reported to mediate disturbances in DNA fork progression and replication repair, and we speculate that the ciprofloxacin exposure might have had such an effect (Didier et al., 2011).
This clinical case represents a remarkably resistant and persistent clinical infection. Our genomic deconstruction of the infection provides a window into the complex bacterial dynamics of deep-seated staphylococcal infections, demonstrating for the first time that multi-copy chromosome duplications and amplifications, in addition to the sequential acquisition of diverse mutations, lead to drug-resistant, clinically persistent S. aureus isolates.
Acknowledgements
This research was supported by the National Health and Medical Research Council (NHMRC) of Australia (1008973). B. P. H. and T. P. S. are recipients of NHMRC fellowships (1023526 and 1008549, respectively). S. G. and T. S. were supported in part by the VLSCI Life Sciences Computation Centre, a collaboration between Melbourne, Monash and La Trobe Universities, and an initiative of the Victorian Government, Australia.
Supplementary Data
Supplementary Data
Abbreviations:
- hVISA
heterogeneous vancomycin-intermediate Staphylococcus aureus
- MRSA
meticillin-resistant Staphylococcus aureus
- MSC
minimal selective concentration
- qPCR
quantitative PCR
- SCV
small colony variant
- SNP
single nucleotide polymorphism
- VSSA
vancomycin-sensitive Staphylococcus aureus
References
- Adler M., Anjum M., Berg O.G., Andersson D.I., Sandegren L. (2014). High fitness costs and instability of gene duplications reduce rates of evolution of new genes by duplication-divergence mechanisms Mol Biol Evol 311526–1535 10.1093/molbev/msu111. [DOI] [PubMed] [Google Scholar]
- Andersson D.I., Hughes D. (2009). Gene amplification and adaptive evolution in bacteria Annu Rev Genet 43167–195 10.1146/annurev-genet-102108-134805. [DOI] [PubMed] [Google Scholar]
- Bae T., Schneewind O. (2006). Allelic replacement in Staphylococcus aureus with inducible counter-selection Plasmid 5558–63 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Bayer A.S., Mishra N.N., Sakoulas G., Nonejuie P., Nast C.C., Pogliano J., Chen K.T., Ellison S.N., Yeaman M.R., Yang S.J. (2014). Heterogeneity of mprF sequences in methicillin-resistant Staphylococcus aureus clinical isolates: role in cross-resistance between daptomycin and host defense antimicrobial peptides Antimicrob Agents Chemother 587462–7467 10.1128/AAC.03422-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund C., Söderquist B. (2008). The origin of a methicillin-resistant Staphylococcus aureus isolate at a neonatal ward in Sweden-possible horizontal transfer of a staphylococcal cassette chromosome mec between methicillin-resistant Staphylococcus haemolyticusStaphylococcus aureus Clin Microbiol Infect 141048–1056 10.1111/j.1469-0691.2008.02090.x. [DOI] [PubMed] [Google Scholar]
- Bloemendaal A.L., Brouwer E.C., Fluit A.C. (2010). Methicillin resistance transfer from Staphylocccus epidermidis to methicillin-susceptible Staphylococcus aureus in a patient during antibiotic therapy PLoS One 5e11841. 10.1371/journal.pone.0011841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data Bioinformatics 302114–2120 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron D.R., Ward D.V., Kostoulias X., Howden B.P., Moellering R.C., Jr, Eliopoulos G.M., Peleg A.Y. (2012). Serine/threonine phosphatase Stp1 contributes to reduced susceptibility to vancomycin and virulence in Staphylococcus aureus J Infect Dis 2051677–1687 10.1093/infdis/jis252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain P.S., Grafham D.V., Fulton R.S., Fitzgerald M.G., Hostetler J., Muzny D., Ali J., Birren B., Bruce D.C., other authors (2009). Genomics. Genome project standards in a new era of sequencing Science 326236–237 10.1126/science.1180614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers H.F., Deleo F.R. (2009). Waves of resistance: Staphylococcus aureus in the antibiotic era Nat Rev Microbiol 7629–641 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L., Isii T., Fukuda M., Ochiai T., Neoh H.M., Camargo I.L., Watanabe Y., Shoji M., Hishinuma T., Hiramatsu K. (2010). An RpoB mutation confers dual heteroresistance to daptomycin and vancomycin in Staphylococcus aureus Antimicrob Agents Chemother 545222–5233 10.1128/AAC.00437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L., Neoh H.M., Iwamoto A., Hiramatsu K. (2012). Coordinated phenotype switching with large-scale chromosome flip-flop inversion observed in bacteria Proc Natl Acad Sci U S A 109E1647–E1656 10.1073/pnas.1204307109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didier J.P., Villet R., Huggler E., Lew D.P., Hooper D.C., Kelley W.L., Vaudaux P. (2011). Impact of ciprofloxacin exposure on Staphylococcus aureus genomic alterations linked with emergence of rifampin resistance Antimicrob Agents Chemother 551946–1952 10.1128/AAC.01407-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst C.M., Peschel A. (2011). Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids Mol Microbiol 80290–299 10.1111/j.1365-2958.2011.07576.x. [DOI] [PubMed] [Google Scholar]
- Friedman L., Alder J.D., Silverman J.A. (2006). Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus Antimicrob Agents Chemother 502137–2145 10.1128/AAC.00039-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W., Chua K., Davies J.K., Newton H.J., Seemann T., Harrison P.F., Holmes N.E., Rhee H.W., Hong J.I., other authors (2010). Two novel point mutations in clinical Staphylococcus aureus reduce linezolid susceptibility and switch on the stringent response to promote persistent infection PLoS Pathog 6e1000944. 10.1371/journal.ppat.1000944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W., Cameron D.R., Davies J.K., Kostoulias X., Stepnell J., Tuck K.L., Yeaman M.R., Peleg A.Y., Stinear T.P., Howden B.P. (2013). The RpoB H481Y rifampicin resistance mutation and an active stringent response reduce virulence and increase resistance to innate immune responses in Staphylococcus aureus J Infect Dis 207929–939 10.1093/infdis/jis772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green J.M., Hollandsworth R., Pitstick L., Carter E.L. (2010). Purification and characterization of the folate catabolic enzyme p-aminobenzoyl-glutamate hydrolase from Escherichia coli J Bacteriol 1922407–2413 10.1128/JB.01362-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullberg E., Cao S., Berg O.G., Ilbäck C., Sandegren L., Hughes D., Andersson D.I. (2011). Selection of resistant bacteria at very low antibiotic concentrations PLoS Pathog 7e1002158. 10.1371/journal.ppat.1002158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helle L., Kull M., Mayer S., Marincola G., Zelder M.E., Goerke C., Wolz C., Bertram R. (2011). Vectors for improved Tet repressor-dependent gradual gene induction or silencing in Staphylococcus aureus Microbiology 1573314–3323 10.1099/mic.0.052548-0. [DOI] [PubMed] [Google Scholar]
- Howden B.P., Stinear T.P., Allen D.L., Johnson P.D., Ward P.B., Davies J.K. (2008). Genomic analysis reveals a point mutation in the two-component sensor gene graS that leads to intermediate vancomycin resistance in clinical Staphylococcus aureus Antimicrob Agents Chemother 523755–3762 10.1128/AAC.01613-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden B.P., McEvoy C.R., Allen D.L., Chua K., Gao W., Harrison P.F., Bell J., Coombs G., Bennett-Wood V., other authors (2011). Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR PLoS Pathog 7e1002359. 10.1371/journal.ppat.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D.H., Bryant D. (2006). Application of phylogenetic networks in evolutionary studies Mol Biol Evol 23254–267 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- LaMarre J.M., Howden B.P., Mankin A.S. (2011). Inactivation of the indigenous methyltransferase RlmN in Staphylococcus aureus increases linezolid resistance Antimicrob Agents Chemother 552989–2991 10.1128/AAC.00183-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. (2012). Fast gapped-read alignment with Bowtie 2 Nat Methods 9357–359 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke J.B., Hilgers M., Shaw K.J. (2009). Novel ribosomal mutations in Staphylococcus aureus strains identified through selection with the oxazolidinones linezolid and torezolid (TR-700) Antimicrob Agents Chemother 535265–5274 10.1128/AAC.00871-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowy F.D. (1998). Staphylococcus aureus infections N Engl J Med 339520–532 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- McAdam P.R., Holmes A., Templeton K.E., Fitzgerald J.R. (2011). Adaptive evolution of Staphylococcus aureus during chronic endobronchial infection of a cystic fibrosis patient PLoS One 6e24301. 10.1371/journal.pone.0024301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy C.R., Tsuji B., Gao W., Seemann T., Porter J.L., Doig K., Ngo D., Howden B.P., Stinear T.P. (2013). Decreased vancomycin susceptibility in Staphylococcus aureus caused by IS256 tempering of WalKR expression Antimicrob Agents Chemother 573240–3249 10.1128/AAC.00279-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra N.N., Yang S.J., Sawa A., Rubio A., Nast C.C., Yeaman M.R., Bayer A.S. (2009). Analysis of cell membrane characteristics of in vitro-selected daptomycin-resistant strains of methicillin-resistant Staphylococcus aureus Antimicrob Agents Chemother 532312–2318 10.1128/AAC.01682-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk I.R., Tree J.J., Howden B.P., Stinear T.P., Foster T.J. (2015). Complete bypass of restriction systems for major Staphylococcus aureus lineages MBio 6e00308–e00315 10.1128/mBio.00308-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mwangi M.M., Wu S.W., Zhou Y., Sieradzki K., de Lencastre H., Richardson P., Bruce D., Rubin E., Myers E., other authors (2007). Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing Proc Natl Acad Sci U S A 1049451–9456 10.1073/pnas.0609839104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulander W., Nissen Varming A., Baek K.T., Haaber J., Frees D., Ingmer H. (2013). Antibiotic-mediated selection of quorum-sensing-negative Staphylococcus aureus MBio 3e00459–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzin A., Severin A., Moghazeh S.L., Etienne J., Bradford P.A., Projan S.J., Shlaes D.M. (2003). Inactivation of mprF affects vancomycin susceptibility in Staphylococcus aureus Biochim Biophys Acta 1621117–121 10.1016/S0304-4165(03)00028-X. [DOI] [PubMed] [Google Scholar]
- Sandegren L., Andersson D.I. (2009). Bacterial gene amplification: implications for the evolution of antibiotic resistance Nat Rev Microbiol 7578–588 10.1038/nrmicro2174. [DOI] [PubMed] [Google Scholar]
- Schmitz F.J., Jones M.E., Hofmann B., Hansen B., Scheuring S., Lückefahr M., Fluit A., Verhoef J., Hadding U., other authors (1998). Characterization of grlAgrlBgyrA, and gyrB mutations in 116 unrelated isolates of Staphylococcus aureus and effects of mutations on ciprofloxacin MIC Antimicrob Agents Chemother 421249–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. (2014). Prokka: rapid prokaryotic genome annotation Bioinformatics 302068–2069 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- Seggewiss J., Becker K., Kotte O., Eisenacher M., Yazdi M.R., Fischer A., McNamara P., Al Laham N., Proctor R., other authors (2006). Reporter metabolite analysis of transcriptional profiles of a Staphylococcus aureus strain with normal phenotype and its isogenic hemB mutant displaying the small-colony-variant phenotype J Bacteriol 1887765–7777 10.1128/JB.00774-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.E., Buckley D.G., Wu Z., Saenphimmachak C., Hoffman L.R., D'Argenio D.A., Miller S.I., Ramsey B.W., Speert D.P., other authors (2006). Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients Proc Natl Acad Sci U S A 1038487–8492 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Ke R., Hughes D., Nilsson M., Andersson D.I. (2012). Genome-wide detection of spontaneous chromosomal rearrangements in bacteria PLoS One 7e42639. 10.1371/journal.pone.0042639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traber K.E., Lee E., Benson S., Corrigan R., Cantera M., Shopsin B., Novick R.P. (2008). agr function in clinical Staphylococcus aureus isolates Microbiology 1542265–2274 10.1099/mic.0.2007/011874-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel L.M., Clewell D.B., Gill S.R., Clark N.C., McDougal L.K., Flannagan S.E., Kolonay J.F., Shetty J., Killgore G.E., Tenover F.C. (2003). Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus Science 3021569–1571 10.1126/science.1090956. [DOI] [PubMed] [Google Scholar]
- Young B.C., Golubchik T., Batty E.M., Fung R., Larner-Svensson H., Votintseva A.A., Miller R.R., Godwin H., Knox K., other authors (2012). Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease Proc Natl Acad Sci U S A 1094550–4555 10.1073/pnas.1113219109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Liang Y., Lynch K.H., Dennis J.J., Wishart D.S. (2011). phast: a fast phage search tool Nucleic Acids Res 39 (Suppl), W347–W352 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Data Bibliography
- 1.Stinear, T. FigShare http://dx.doi.org/10.6084/m9.figshare.1394626 (2015) Table S4.
- 2.Stinear, T. FigShare http://dx.doi.org/10.6084/m9.figshare.1391325 (2015) JKD6229.gb.
- 3.Stinear, T. FigShare http://dx.doi.org/10.6084/m9.figshare.1391322 (2015) JKD6210.gb.
- 4.Stinear, T. FigShare http://dx.doi.org/10.6084/m9.figshare.1391323 (2015) JKD6210p.gb.
- 5.Seeman, T. GitHub https://github.com/tseemann/prokka (2015).
- 6.Stinear, T. European Nucleotide Archive http://www.ebi.ac.uk/ena/data/view/ERP010276 (2015).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data