

Abstract
Outbreaks of Salmonella Enteritidis have long been associated with contaminated poultry and eggs. In the summer of 2014 a large multi-national outbreak of Salmonella Enteritidis phage type 14b occurred with over 350 cases reported in the United Kingdom, Germany, Austria, France and Luxembourg. Egg supply network investigation and microbiological sampling identified the source to be a Bavarian egg producer. As part of the international investigation into the outbreak, over 400 isolates were sequenced including isolates from cases, implicated UK premises and eggs from the suspected source producer. We were able to show a clear statistical correlation between the topology of the UK egg distribution network and the phylogenetic network of outbreak isolates. This correlation can most plausibly be explained by different parts of the egg distribution network being supplied by eggs solely from independent premises of the Bavarian egg producer (Company X). Microbiological sampling from the source premises, traceback information and information on the interventions carried out at the egg production premises all supported this conclusion. The level of insight into the outbreak epidemiology provided by whole-genome sequencing (WGS) would not have been possible using traditional microbial typing methods.
Keywords: Salmonella, whole-genome sequencing, traceback investigation, foodborne outbreak
Data Summary
FASTQ sequences were deposited in the NCBI Short Read Archive under the BioProject PRJNA248792 (http://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA248792)
Supplementary material is available at the following git repository https://github.com/timdallman/sent_14b.git
Impact Statement
In this article we show how the phylogenetic relationships between isolates in a foodborne outbreak can be informative in revealing underlying epidemiological trends.
We were able to show a clear statistical correlation between the topology of the UK cases, the egg distribution network and the phylogenetic network of outbreak isolates. This indicated that the phylogeny clustered into distinct clades related to the source of eggs.
This study shows the benefit of whole-genome sequencing of pathogens in revealing the true epidemiology behind an outbreak allowing inferences to be made about source diversity and food-chain contamination.
Introduction
Salmonella Enteritidis outbreaks in humans are often linked to contaminated foodstuffs produced by the poultry industry (Harker et al., 2014; Lane et al., 2014). The incidence of Salmonella Enteritidis in the United Kingdom and in Europe has decreased significantly following the implementation of a vaccination program and other control measures in chicken flocks(Poirier et al., 2008), but outbreaks associated with contaminated eggs continue to occur (Hugas & Beloeil, 2014).
In 2014 a large multi-national outbreak of Salmonella Enteritidis phage type 14b was linked to consumption of eggs (Inns et al., 2015). Over 350 cases were reported in the United Kingdom, Germany, Austria, France and Luxembourg. Egg supply network investigation and microbiological sampling identified the source to be a Bavarian egg producer.
Whole-genome sequencing (WGS) is increasingly used for surveillance of food-borne pathogens (Dallman et al., 2015b; Quick et al., 2015; Jenkins et al., 2015) and prospective typing of Salmonella Enteritidis isolates can identify possible outbreaks in real-time (den Bakker et al., 2014; Deng et al., 2014). Salmonella Enteritidis outbreaks can be investigated using WGS as part of the case definition, as strains of a given outbreak are typically monophyletic (i.e. representing a single evolutionary pathway), with limited diversity between outbreak isolates (Deng et al., 2014; Wuyts et al., 2015; Taylor et al., 2015). WGS has however revealed significant polyclonal contamination within chicken production farms (Allard et al., 2013).
Phylogenetic methods that explore the relationships between microbial genomes have been used to study the emergence (Dallman et al., 2015a; Holt et al., 2012), geographical diffusion (Baker et al., 2015; He et al., 2013) and transmission of infections (Bryant et al., 2013; Eyre et al., 2013). Phylogenetic topologies can also be informative in terms of source attribution in outbreak investigations (Harris et al., 2013). In this study we explore the relationships between the observed phylogeny of 400 clinical, environmental and food isolates obtained in this outbreak and the distribution network of the implicated foodstuff, as ascertained by national and international traceback investigations.
Methods
Strains.
Since April 2014 all presumptive isolates of Salmonella in England and Wales received by the Gastrointestinal Bacteria Reference Unit at Public Health England (PHE) have undergone WGS. As of 1st August 2015, 3844 sequences of Salmonella enterica serovar Enteritidis had been analysed for routine surveillance purposes. A set of 44 isolates from Germany, France, Austria and Luxembourg were sequenced as part of this investigation with the inclusion criteria as follows; clinical isolates belonging to phage type 14b, clinical isolates with matching multi-locus variable number tandem repeat analysis (MLVA) profile 2-12-7-3-2, and implicated food or environmental samples. Of all the isolates sequenced from the UK and mainland Europe, 401 strains of Salmonella Enteritidis matched the outbreak single-nucleotide polymorphism (SNP) address 1.2.3.38.38.38 (Ashton et al., 2015), these included all the isolates previously described by Inns et al. (2015). The strains are described in Table S1, available in the online version of this paper.
Food chain investigations.
Rapid Alert System for Food and Feed (RASFF) notifications were issued on 09 July 2014 (France), 31 July 2014 (Austria) and 1 August 2014 (France), which linked S. Enteritidis outbreaks in France and Austria to chicken eggs from Company X in Germany. Company X had four separate premises, three in Germany and one in the Czech Republic; all are operationally independent. All four sites used young chickens (pullets) from two locations: one in Germany and one in the Czech Republic. Food supply network investigations involved obtaining information on the supply of eggs from Company X to UK distributors and tracing onward supply to other UK companies. In addition, supply network investigations were conducted in England to trace supplies of chicken and chicken eggs consumed by cases to their source as described in Inns et al. (2015). In total, 198 of the 287 (69 %) confirmed UK cases could be plausibly linked to eggs supplied by one company, Company X, with no traceback information available for the other cases.
Sequencing.
Sequencing was performed by the PHE Genome Sequencing Unit using Nextera library preparation on a HiSeq 2500 (Illumina) run in fast mode according to the manufacturers’ instructions, which yielded 2×100 base pair paired-end reads. At the Robert Koch Institute, University of Birmingham and Institut Pasteur, libraries from genomic DNA were created using the Nextera library preparation kit and subsequently run on the Illumina MiSeq sequencer using Illumina’s v3 Reagent Kit to produce 2×300 base pair paired-end reads.
High-quality Illumina reads were mapped to the Salmonella enterica Enteritidis reference genome (GenBank:AM933172) using BWA-MEM (Li & Durbin, 2010). SNPs were then identified using GATK2 (McKenna et al., 2010) in unified genotyper mode. Core genome positions that had a high quality SNP (>90 % consensus, minimum depth 10×, GQ >=30) in at least one strain were extracted and RaxML v8.17 (Stamatakis, 2014) used to derive the maximum-likelihood phylogeny of the isolates under the GTRCAT model of evolution. Single-linkage SNP clustering was performed as previously described (Ashton et al., 2015). FASTQ reads from all sequences in this study can be found at the PHE Pathogens BioProject at the National Center for Biotechnology Information (Accession PRJNA248792).
Timed phylogenies.
Timed phylogenies were reconstructed using BEAST-MCMC v1.80 (Drummond et al., 2012) after first removing regions of the genome predicted to have undergone recombination using Gubbins v1.3 (Croucher et al., 2015). Alternative clock models and population priors were computed and assessed based on Bayes Factor (BF) tests using Tracer v1.6. The highest supported model was a relaxed lognormal clock rate under a constant population size. All models were run with a chain length of one billion. A maximum clade credibility tree was constructed using TreeAnnotator v1.75 (Drummond et al., 2012).
Network comparison.
Distances on the distribution network were measured as the number of intermediate nodes on the shortest path between a pair of cases. For instance, the distance between two cases infected in the same restaurant was one. The genetic distance between isolates of two different cases was measured as the patristic distances between the corresponding tips of the phylogeny. A Monte Carlo Mantel test (Mantel, 1967) was used to investigate the degree of correlation between the supply network and genetic distance matrices, using 167 cases that were both sequenced and documented on the distribution network, with 9999 random permutations of the data. Because the relationship may be driven by cases infected by the same source, we also tested correlations between distances based on pairs of cases with different sources of infection only.
In addition, we accounted for the potential bias stemming from the existence of distinct genetic clades in the sampled isolates using a partial Mantel test (Legendre & Legendre, 2012). In this analysis, a linear regression is used to predict patristic distances as a function of a binary clade membership distance (0=same clade; 1=different clades) and distances on the food distribution network. Effects of each covariate and their interaction were tested using the classical ANOVA framework (Legendre & Legendre, 2012).
All analyses were carried out in the R software (Team, 2014), using the packages igraph (Csardi & Nepusz, 2006) for graph distances, adephylo (Jombart et al., 2010) for phylogenetic distances computations and ade4(Dray et al., 2007, p. 4) for the Mantel test.
Results
The phylogeny of 401 isolates implicated in the outbreak resolved into three clades all supported with bootstraps >95 (Fig. 1a). The isolates formed a single five-SNP single-linkage cluster with a maximum distance between any two genomes of 23 SNPs. Within England, the outbreak consisted of five point-source, geographically distinct incidents and 101 sporadic cases. Isolates from the point-source outbreaks resolved into distinct sub-clades in the outbreak phylogeny with a maximum SNP distance of 2 (mode of 0). Supply network information plausibly linked 198 cases in England to eggs supplied by one company, Company X (Fig. 1b). Multiple trace-back pathways to the implicated source of infection were identified (Inns et al., 2015).
Fig. 1.
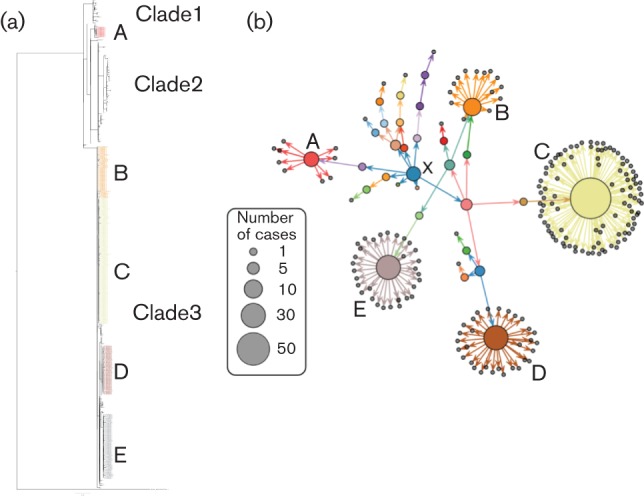
(a) Maximum-likelihood phylogeny based on whole-genome sequences of 401 isolates implicated in the outbreak rooted against an unrelated isolate of S. Enteritidis isolated from Luxembourg. (b) Distribution network for the 167 cases that were both sequenced and documented on the network, with arrows representing likely contaminations. Black circles represent cases, while internal nodes (sources) are represented as coloured disks, with a size proportional to the number of subsequent infections. The five point-source outbreaks associated with three Chinese restaurants (B, C, D), a hospital (E) and kebab grill (A) are coloured on the phylogeny and labelled on the trace-back network. Company X is the blue centroid sphere.
According to the Monte-Carlo Mantel test, genetic distances between isolates significantly increased with the distance (number of intermediate nodes) on the distribution network (r=0.62, P=0.0001). This relationship remained significant when considering only pairs of cases from different direct exposures (r=0.46; t-test: P=2.2×10−16) and was robust to non-linearities between distances (Spearman ρ=0.60, P=2.2×10−16). This relationship also remained significant when accounting for the existence of distinct genetic clades (ANOVA: F=18,023; P<2×10−16; Fig. 2). In fact, most of the variance in patristic distances could be explained by clade membership and by the accumulation of mutations during the outbreak (R2=0.94; P<2×10−16), with 0.18 % [CI95 %: (0.17 %; 0.18 %)] genome diversity accumulated on average between nodes of the food network within a given clade (Fig. 2).
Fig. 2.
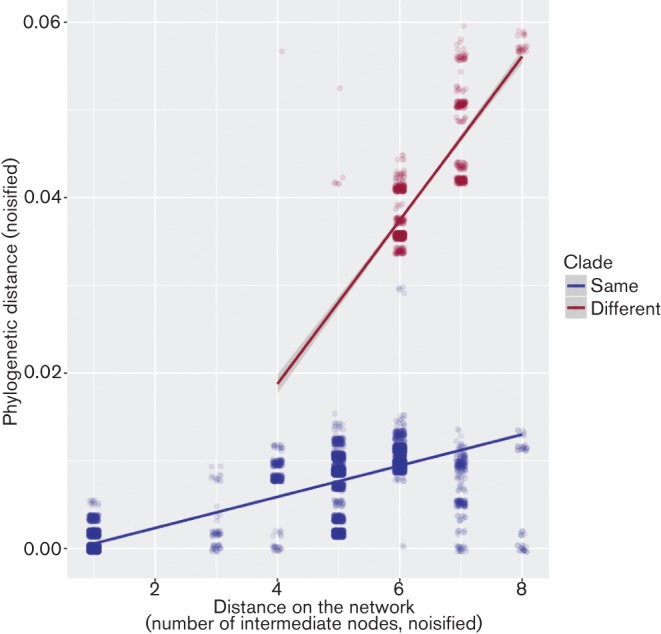
Scatterplot showing the relationship between the phylogenetic distance and the distance between cases on the traceback network, after accounting for the existence of two distinct genetic clades in sampled isolates. Each dot represents a pairwise comparison between two cases. Data have been slightly noisified to better visualise overlapping points. Dots are colored according to clade memberships, with pairs of isolates from the same clade in blue, and from different clades in red. Lines indicate predictions of a linear model using different slopes for each group.
The outbreak occurred over a period of 17 weeks in England and Wales. To test the hypothesis that this length of time was insufficient for the observed level of nucleotide diversity to occur during the outbreak period two timed phylogenies were constructed. Firstly a mutation rate of 3.4 [95 % highest posterior density (HPD) 2.6–5.1] mutations per genome per year was estimated based on 142 diverse (50 SNP cluster) representatives of the Salmonella Enteritidis PHE collection. This mutation rate is three times faster than that predicted by Deng et al. (2014), although the predicted time to most recent common ancestor of the lineages is consistent, suggesting that the differences lie in the SNP-calling algorithms. Secondly a mutation rate of 8.35 (95 % HPD 5.8–11.0) mutations per genome per year was estimated based on the outbreak isolates only. The faster short-term mutation rate between Salmonella outbreak samples has previously been described (Hawkey et al., 2013) and maybe due to intense sampling of minority variants pre-fixation. Regardless of these discrepancies no analysis predicts a mutation rate that provides sufficient time for accumulation of the diversity observed during this international outbreak. The time to the most recent common ancestor for the three clades was estimated to be 2.9 years (95 % HPD 2.5–3.2 years) (Fig. S1). The correlation between phylogenetics and the egg distribution network could however be explained by either a single sampling event from a single diverse source into different parts of the food network or by multiple sampling from compartmentalised diversity in the source population into different parts of the food network.
A set of 44 isolates from countries other than England were sequenced, including isolates from clinical cases, implicated foodstuffs and both environmental sampling and eggs from the implicated egg producer (Table 1). Twelve sequences from isolates directly sampled from premises A, or from eggs that could be directly linked by batch number to premises A of company X clustered unilaterally into clade 1 of the phylogeny (Fig. 1a). Clade 1 included three clinical cases from Germany and three cases from England. Two further clinical isolates from France in clade 1 were from cases where trace-back information linked them to eggs from premises A of company X. Two egg isolates from France could be traced to premises A due to their egg-mark. Environmental sampling from premises A yielded six isolates of S. Enteritidis from eggs, three from poultry and one from the production environment; all clustered into clade 1. After the link with clinical cases in France was identified, premises A was deep cleaned following culling of the flock (Fig. 3).
Table 1. Strain list of samples from outside the United Kingdom .
Traceback informtion relates to premises A or B from company X. ?, No traceback information available.
| Strain | Country | Clade | Traceback | Source |
|---|---|---|---|---|
| H143980751 | Germany | 1 | A | Egg |
| H143980752 | Germany | 1 | A | Egg |
| H143980753 | Germany | 1 | A | Egg |
| H143980754 | Germany | 1 | A | Egg |
| H143980755 | Germany | 1 | A | Egg |
| H143980756 | Germany | 1 | A | Egg |
| H143360569 | France | 1 | A | Human |
| H143360570 | France | 1 | A | Egg |
| H143360571 | France | 1 | A | Egg |
| 201405122 | France | 1 | A | Human |
| 14-06145 | Germany | 1 | ? | Human |
| 14-05226 | Germany | 1 | A | Environmental |
| 14-05225 | Germany | 1 | A | Poultry |
| 14-05224 | Germany | 1 | A | Poultry |
| 14-05227 | Germany | 1 | A | Poultry |
| 14-06012 | Germany | 1 | ? | Human |
| 14-06175 | Germany | 1 | ? | Human |
| H143720773 | Luxembourg | 2 | ? | Human |
| H143980750 | Germany | 2 | B | Egg |
| H143980757 | Germany | 2 | B | Egg |
| H143380471 | Austria | 2 | ? | Human |
| H143380472 | Austria | 2 | ? | Human |
| H143380473 | Austria | 2 | ? | Human |
| H143380474 | Austria | 2 | ? | Human |
| H143360568 | France | 2 | ? | Human |
| 201405861 | France | 2 | ? | Human |
| 201405760 | France | 2 | B | Human |
| 201405756 | France | 2 | B | Human |
| 201405757 | France | 2 | B | Human |
| 14-04296 | Germany | 2 | ? | Human |
| 14-04310 | Germany | 2 | ? | Human |
| 14-04552 | Germany | 2 | ? | Human |
| 14-04639 | Germany | 2 | ? | Human |
| 14-04870 | Germany | 2 | ? | Human |
| 14-05946 | Germany | 2 | ? | Human |
| 14-06388 | Germany | 2 | ? | Human |
| H143380470 | Austria | 3 | ? | Human |
| H143380475 | Austria | 3 | ? | Human |
| 14-05567 | Germany | 3 | ? | Human |
| 14-05569 | Germany | 3 | ? | Human |
| 14-05795 | Germany | 3 | ? | Human |
| 14-05568 | Germany | 3 | ? | Human |
| H143360566 | France | Non-outbreak | ? | Human |
| H143360567 | France | Non-outbreak | ? | Human |
| 14-06148 | Germany | Non-outbreak | ? | Human |
Fig. 3.

The epidemic curve of English cases over nine months with the interventions at sites of company X indicated by vertical lines. Cases are coloured by phylogenetic clade.
Two isolates from eggs sampled at premises B of company X clustered in clade 2. Clade 2 included five clinical cases from France, of which three were linked to eggs from premises B of company X, as well as seven clinical cases from Germany, four from Austria, one from Luxembourg and 32 from England. Nine of the English cases were linked to a point source outbreak at a kebab restaurant.
Clade 3 included the majority of isolates from England, including isolates from four separate point source outbreaks. The clade contains four isolates from clinical cases in Germany and two isolates from clinical cases in Austria. Although cases were linked through exposure traceback to company X, no isolates from clade 3 were detected at premises A or B.
Fig. 3 shows the distribution of cases over time coloured by clade with interventions involving flock destruction and deep cleaning at premises A and B also depicted. The intervention at premises A preceded the majority of cases. Cases in Clade 3 continued after the interventions at premises A and B. The timeline and epidemiological data are congruent with the phylogenetic analysis. Isolates from clade 1 were not detected after interventions at premises A of company X (removal of the flock and cessation of egg delivery). Clade 2 strains were rarely detected from 26 days (estimated shelf life for eggs) after interventions at premises B of company X (cessation of egg delivery to the UK). Clade 3 strains, which were detected neither at premises A nor at premises B of company X, continued to be detected from May to November 2014 after interventions at premises A and B, but detections largely ceased from 26 days after the cessation of egg deliveries from the Czech premises of company X.
Conclusion
Whole-genome sequencing allows the identification of linked cases of infection with unprecedented resolution. Due to the sequential nature of mutational drift, phylogenetic methods can be used to study variation in genomes and reveal ancestral relationships. The topology of such a phylogeny in the case of foodborne disease may reveal information about source diversity and how that diversity was sampled. In this study, sequences from a large outbreak of S. Enteritidis linked to the consumption of eggs originating from a Bavarian producer revealed a phylogeny with three clades forming a monophyletic cluster within the S. Enteritidis population. Isolates from five point-source outbreaks clustered themselves into distinct monophyletic clusters with minimal variation.
Traceback information in France led to sampling of two premises of Company X in Bavaria. Eggs, environmental samples and isolates from cases linked to premises A clustered phylogenetically into Clade 1 whereas eggs, environmental samples and isolates from cases linked to premises B clustered phylogenetically into Clade 2. Although the majority of cases in Clade 3 were linked via the supply network to Company X, clade 3 strains were not identified in samples from premises A or B. Interventions including flock destruction and disinfection at premises A and premises B coincided with a cessation of cases from Clades 1 and 2. Cases in clade 3 continued sporadically for several months after these interventions, however the frequency decreased following the import suspension of eggs from the Czech premises of company X. We conclude that the origin of Clade 3 is most likely to be another location within company X or its suppliers. The three clades’ common ancestor existed approximately three years previously.
The supply network investigation for English cases revealed a complex egg distribution network consisting of several distinct distribution chains. We were able to show a clear statistical correlation between the topology of the UK egg distribution network and the phylogenetic network of outbreak isolates. This correlation can most plausibly be explained by different parts the egg distribution network being supplied by eggs solely from independent premises of Company X. This resulted in a phylogeny that clusters into distinct clades related to the source of eggs. When accounting for these clades, almost all of the genetic variation could be explained by the process of diffusion of isolates over the food distribution network (R2=0.94; P<2×10−16).
In this paper, we used a simple approach to quantify the extent of the association between the phylogeny of sampled isolates and the food distribution network, which is only partially known. Our approach captures the topology of the food distribution network using distances between cases, computed as the shortest path between the corresponding nodes. While crude, this measure should be robust to the addition of nodes and edges, as long as the closest food providers linking the cases have been reported. Therefore, our results should remain identical when considering the full food distribution network.
This is the first time, to our knowledge, that phylogenetic data have been combined with food supply network data in the context of an infectious disease outbreak investigation. Our results suggest that combining whole-genome sequencing with information on the food distribution network permits a more detailed exploration of possible sources of infection in outbreak situations and to inform interventions. We recommend that further work be undertaken to develop and standardise the methods used to compare phylogenetic and food supply network information, to enable use of these techniques in future outbreaks to help identify sources and guide the implementation of public health control measures to prevent further illness.
The level of insight into the outbreak epidemiology provided by WGS would not have been possible using traditional microbial typing methods routinely employed for Salmonella outbreak investigation such as MLVA, PFGE and phage typing. WGS provided the high-resolution typing needed that allowed the effectiveness of interventions at premises A and B to be observed. Similarly the robust, high discrimination of WGS provided the evidence for driving traceback in specific directions, which is of particular importance in complex foodborne source identification investigations. Finally the digital nature of WGS data allowed data to be readily exchanged and analysed between four institutions in different countries.
Acknowledgements
We would like to acknowledge Catherine Ragimbeau who performed MLVA on the isolate from Luxembourg, Nathalie Jourdan-da silva of the French Institute for Public Health Surveillance for her epidemiological input, Renaud Lailler at the French Agency for Food, Environmental and Occupational Health & Safety (ANSES) who isolated the French egg isolates and Claire Jenkins at PHE for her critical input on the manuscript.
‘The research was funded by the National Institute for Health Research Health Protection Research Unit (NIHR HPRU) in Gastrointestinal Infections at University of Liverpool in partnership with Public Health England (PHE). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, the Department of Health or Public Health England.
Supplementary Data
Supplementary File 1
Abbreviations:
- WGS
whole-genome sequencing
- MLVA
multi-locus variable number tandem repeat analysis
References
- Allard M. W., Luo Y., Strain E., Pettengill J., Timme R., Wang C., Li C., Keys C. E., Zheng J., et al. (2013). On the evolutionary history population genetics and diversity among isolates of Salmonella Enteritidis PFGE pattern JEGX01.0004. PLoS One 8e55254. 10.1371/journal.pone.0055254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton P., Nair S., Peters T., Tewolde R., Day M., Doumith M., Green J., Jenkins C., Underwood A., et al. (2015). Revolutionising public health reference microbiology using whole genome sequencing: Salmonella as an exemplar. bioRxiv 33225. [Google Scholar]
- Baker K. S., Dallman T. J., Ashton P. M., Day M., Hughes G., Crook P. D., Gilbart V. L., Zittermann S., Allen V. G., et al. (2015). Intercontinental dissemination of azithromycin-resistant shigellosis through sexual transmission: a cross-sectional study. Lancet Infect Dis 15913–921. 10.1016/S1473-3099(15)00002-X [DOI] [PubMed] [Google Scholar]
- Bryant J. M., Grogono D. M., Greaves D., Foweraker J., Roddick I., Inns T., Reacher M., Haworth C. S., Curran M. D., et al. (2013). Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. The Lancet 3811551–1560. 10.1016/S0140-6736(13)60632-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher N. J., Page A. J., Connor T. R., Delaney A. J., Keane J. A., Bentley S. D., Parkhill J., Harris S. R.(2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res 43e15. 10.1093/nar/gku1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csardi G., Nepusz T.(2006). The igraph software package for complex network research. InterJournal Complex Syst 16951–9. [Google Scholar]
- Dallman T. J., Ashton P. M., Byrne L., Perry N. T., Petrovska L., Ellis R. J., Allison L., Hanson M., Holmes A., et al. (2015b). Whole genome sequencing for national surveillance of Shiga toxin producing Escherichia coli O157. Clin Infect Dis 3305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallman T. J., Ashton P. M., Byrne L., Perry N. T., Petrovska L., Ellis R., Allison L., Hanson M., Holmes A., other authors(2015a). Applying phylogenomics to understand the emergence of Shiga-toxin-producing Escherichia coli O157:H7 strains causing severe human disease in the UK. Microbial Genomics 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Bakker H. C., Allard M. W., Bopp D., Brown E. W., Fontana J., Iqbal Z., Kinney A., Limberger R., Musser K. A., et al. (2014). Rapid whole-genome sequencing for surveillance of Salmonella enterica serovar enteritidis. Emerg Infect Dis 201306–1314. 10.3201/eid2008.131399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X., Desai P. T., den Bakker H. C., Mikoleit M., Tolar B., Trees E., Hendriksen R. S., Frye J. G., Porwollik S., et al. (2014). Genomic epidemiology of Salmonella enterica serotype Enteritidis based on population structure of prevalent lineages. Emerg Infect Dis 201481–1489. 10.3201/eid2009.131095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray S., Dufour A.-B., et al. (2007). The ade4 package: implementing the duality diagram for ecologists. Journal of Statistical Software 221–20. 10.18637/jss.v022.i04 [DOI] [Google Scholar]
- Drummond A. J., Suchard M. A., Xie D., Rambaut A.(2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution 291969–1973. 10.1093/molbev/mss075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre D. W., Cule M. L., Wilson D. J., Griffiths D., Vaughan A., O'Connor L., Ip C. L., Golubchik T., Batty E. M., et al. (2013). Diverse sources of C. difficile infection identified on whole-genome sequencing. N Engl J Med 3691195–1205. 10.1056/NEJMoa1216064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harker K. S., Lane C., Gormley F. J., Adak G. K.(2014). National outbreaks of Salmonella infection in the UK, 2000-2011. Epidemiol Infect 142601–607. 10.1017/S0950268813001210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris S. R., Cartwright E. J., Török M. E., Holden M. T., Brown N. M., Ogilvy-Stuart A. L., Ellington M. J., Quail M. A., Bentley S. D., et al. (2013). Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis 13130–136. 10.1016/S1473-3099(12)70268-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkey J., Edwards D. J., Dimovski K., Hiley L., Billman-Jacobe H., Hogg G., Holt K. E.(2013). Evidence of microevolution of Salmonella Typhimurium during a series of egg-associated outbreaks linked to a single chicken farm. BMC Genomics 14800. 10.1186/1471-2164-14-800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M., Miyajima F., Roberts P., Ellison L., Pickard D. J., Martin M. J., Connor T. R., Harris S. R., Fairley D., et al. (2013). Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet 45109–113. 10.1038/ng.2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt K. E., Baker S., Weill F. X., Holmes E. C., Kitchen A., Yu J., Sangal V., Brown D. J., Coia J. E., et al. (2012). Shigella sonnei genome sequencing and phylogenetic analysis indicate recent global dissemination from Europe. Nat Genet 441056–1059. 10.1038/ng.2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugas M., Beloeil P.(2014). Controlling Salmonella along the food chain in the European Union - progress over the last ten years. Eurosurveillance 1920804. 10.2807/1560-7917.ES2014.19.19.20804 [DOI] [PubMed] [Google Scholar]
- Inns T., Lane C., Peters T., Dallman T., Chatt C., McFarland N., Crook P., Bishop T., Edge J., et al. (2015). A multi-country Salmonella Enteritidis phage type 14b outbreak associated with eggs from a German producer: 'near real-time' application of whole genome sequencing and food chain investigations, United Kingdom, May to September 2014. Euro Surveill 2021098. 10.2807/1560-7917.ES2015.20.16.21098 [DOI] [PubMed] [Google Scholar]
- Jenkins C., Dallman T. J., Launders N., Willis C., Byrne L., Jorgensen F., Eppinger M., Adak G. K., Aird H., et al. (2015). Public health investigation of two outbreaks of Shiga toxin-producing Escherichia coli O157 associated with consumption of watercress. Appl 813946–3952. 10.1128/AEM.04188-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart T., Balloux F., Dray S.(2010). adephylo: new tools for investigating the phylogenetic signal in biological traits. Bioinformatics 261907–1909. 10.1093/bioinformatics/btq292 [DOI] [PubMed] [Google Scholar]
- Lane C. R., LeBaigue S., Esan O. B., Awofisyo A. A., Adams N. L., Fisher I. S., Grant K. A., Peters T. M., Larkin L., et al. (2014). Salmonella enterica serovar Enteritidis, England and Wales, 1945-2011. Emerg Infect Dis 201097–1104. 10.3201/eid2007.121850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre P., Legendre L. F.(2012). Numerical ecology Elsevier.
- Li H., Durbin R.(2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel N.(1967). The detection of disease clustering and a generalized regression approach. Cancer Res 27209–220. [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 201297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier E., Watier L., Espie E., Weill F. X., De Valk H., Desenclos J. C.(2008). Evaluation of the impact on human salmonellosis of control measures targeted to Salmonella Enteritidis and Typhimurium in poultry breeding using time-series analysis and intervention models in France. Epidemiol Infect 1361217–1224. 10.1017/S0950268807009788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J., Ashton P., Calus S., Chatt C., Gossain S., Hawker J., Nair S., Neal K., Nye K., et al. (2015). Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol 16114. 10.1186/s13059-015-0677-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A.(2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 301312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A. J., Lappi V., Wolfgang W. J., Lapierre P., Palumbo M. J., Medus C., Boxrud D.(2015). Characterization of foodborne outbreaks of Salmonella enterica serovar Enteritidis with whole-genome sequencing single nucleotide polymorphism-based analysis for surveillance and outbreak detection. J Clin Microbiol 533334–3340. 10.1128/JCM.01280-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team R. C.(2014). R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria 2012 ISBN 3-900051-07-0. [Google Scholar]
- Wuyts V., Denayer S., Roosens N. H., Mattheus W., Bertrand S., Marchal K., Dierick K., De Keersmaecker S. C.(2015). Whole genome sequence analysis of Salmonella Enteritidis PT4 outbreaks from a national reference laboratory's viewpoint. PLoS Curr 7. 10.1371/currents.outbreaks.aa5372d90826e6cb0136ff66bb7a62fc [DOI] [PMC free article] [PubMed] [Google Scholar]
Data Bibliography
- Dallman, T. J., Ashton, P. A., Jenkins, C. & Grant, K. NCBI Short Read Archive: PRJNA248792 (2015).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File 1