

Abstract
Triggering Receptor Expressed on Myeloid cells 2 (TREM2), which is expressed on myeloid cells including microglia in the CNS, has recently been identified as a risk factor for Alzheimer's disease (AD). TREM2 transmits intracellular signals through its transmembrane binding partner DNAX-activating protein 12 (DAP12). Homozygous mutations inactivating TREM2 or DAP12 lead to Nasu–Hakola disease; however, how AD risk-conferring variants increase AD risk is not clear. To elucidate the signaling pathways underlying reduced TREM2 expression or loss of function in microglia, we respectively knocked down and knocked out the expression of TREM2 in in vitro and in vivo models. We found that TREM2 deficiency reduced the viability and proliferation of primary microglia, reduced microgliosis in Trem2−/− mouse brains, induced cell cycle arrest at the G1/S checkpoint, and decreased the stability of β-catenin, a key component of the canonical Wnt signaling pathway responsible for maintaining many biological processes, including cell survival. TREM2 stabilized β-catenin by inhibiting its degradation via the Akt/GSK3β signaling pathway. More importantly, treatment with Wnt3a, LiCl, or TDZD-8, which activates the β-catenin-mediated Wnt signaling pathway, rescued microglia survival and microgliosis in Trem2−/− microglia and/or in Trem2−/− mouse brain. Together, our studies demonstrate a critical role of TREM2-mediated Wnt/β-catenin pathway in microglial viability and suggest that modulating this pathway therapeutically may help to combat the impaired microglial survival and microgliosis associated with AD.
SIGNIFICANCE STATEMENT Mutations in the TREM2 (Triggering Receptor Expressed on Myeloid cells 2) gene are associated with increased risk for Alzheimer's disease (AD) with effective sizes comparable to that of the apolipoprotein E (APOE) ε4 allele, making it imperative to understand the molecular pathway(s) underlying TREM2 function in microglia. Our findings shed new light on the relationship between TREM2/DNAX-activating protein 12 (DAP12) signaling and Wnt/β-catenin signaling and provide clues as to how reduced TREM2 function might impair microglial survival in AD pathogenesis. We demonstrate that TREM2 promotes microglial survival by activating the Wnt/β-catenin signaling pathway and that it is possible to restore Wnt/β-catenin signaling when TREM2 activity is disrupted or reduced. Therefore, we demonstrate the potential for manipulating the TREM2/β-catenin signaling pathway for the treatment of AD.
Keywords: Akt/GSK3β signaling pathway, Alzheimer's disease, cell survival, microglia, TREM2, Wnt/β-catenin signaling pathway
Introduction
Several rare mutations in the Triggering Receptor Expressed on Myeloid cells 2 (TREM2) gene are associated with increased risk for Alzheimer's disease (AD), with effective sizes comparable to that of the apolipoprotein E (APOE) ε4 allele (Guerreiro et al., 2013; Jonsson and Stefansson, 2013; Jonsson et al., 2013). TREM2 is an immunoreceptor expressed on myeloid cells, including immature dendritic cells, osteoclasts, macrophages, and microglia, and transmits intracellular signals through its transmembrane binding partner DNAX-activating protein 12 (DAP12). In the brain, TREM2 is one of the most highly expressed receptors in microglia and has been found to modulate microglia-mediated phagocytic clearance of apoptotic neurons and inflammatory responses (Hickman and El Khoury, 2014). Individuals with genetic mutations inactivating TREM2 or DAP12 develop Nasu–Hakola disease with cystic-like lesions of the bone and brain demyelination, respectively, leading to fractures and presenile dementia (Paloneva et al., 2002; Klünemann et al., 2005), implying a critical role of TREM2 in maintaining the homeostasis of the CNS.
Microglia, which account for 5–10% of the total cell population in mammalian brains, play important roles in the brain innate immune system (Lawson et al., 1990; Block et al., 2007; Polazzi and Monti, 2010; Aguzzi et al., 2013; Malik et al., 2015). Microglia rapidly extend processes to the sites of injury, migrate to lesion sites, recognize pathogens, and ramify and mount immune responses, including the release of cytokines and phagocytosis of damaged debris (Ransohoff and Perry, 2009). In the brains of both AD patients and mouse models, microglia are found to be closely associated with amyloid plaques and exhibit an “activated” proinflammatory phenotype (Perlmutter et al., 1990; Frautschy et al., 1998; Lee and Landreth, 2010). Microglia maintain their numbers through self-repopulation and alter their appearance and numbers during AD pathogenesis (Ajami et al., 2007; Wes et al., 2016). TREM2 is specifically expressed in microglia in the healthy brain (Hickman et al., 2013), whereas TREM2 deficiency has been shown to reduce microgliosis in 5×FAD mice with amyloid plaques not fully enclosed by microglia (Wang et al., 2015; Wang et al., 2016; Yuan et al., 2016). Although it has been suggested that microglial TREM2 modulates immune responses and affects amyloid pathology in AD (Painter et al., 2015; Colonna and Wang, 2016; Ulrich and Holtzman, 2016), the molecular pathways underlying TREM2-regulated microglial survival remain unclear.
Here, we demonstrate that TREM2 promotes microglial survival by activating the Wnt/β-catenin signaling pathway through posttranslational regulation of β-catenin. In addition, treatment with Wnt3a, LiCl, or TDZD-8, which activates Wnt/β-catenin signaling, promotes microglial survival and microgliosis in Trem2−/− microglia and in the brains of Trem2−/− mice. Our studies suggest that therapeutic strategies targeting the TREM2-mediated Wnt/β-catenin signaling pathway may restore microglial functions that hold promise for the treatment of AD.
Materials and Methods
Reagents.
Cyclin D1 (RRID: AB_2259616), c-Myc (RRID: AB_2151827), Bcl-2 (RRID: AB_1903907) antibodies, phospho-Akt (Ser473) antibody (RRID: AB_329825), Akt antibody (RRID: AB_915783), phospho-p38 MAPK (Thr180/Tyr182) antibody (RRID: AB_331641), p38 MAPK antibody (RRID: AB_330713), p44/42 MAPK (Erk1/2) antibody (RRID: AB_330744), phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody (RRID: AB_331646), cleaved Caspase-3 (Asp175) antibody (RRID: AB_2341188), phospho-GSK-3β (Ser9) antibody (RRID: AB_331405) were all from Cell Signaling Technology. Anti-β-actin antibody (RRID: AB_306371), goat anti-mouse IgG, and the bromodeoxyuridine (BrdU) incorporation assay kit were from Abcam. Recombinant mouse granulocyte-macrophage colony stimulating factor (GM-CSF) was from R&D Systems. LiCl, β-catenin (RRID: AB_476831) antibody, TDZD-8, kainic acid (KA), and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) were from Sigma-Aldrich. The In Situ Cell Death Detection Kit was from Roche. ReverTra Ace qPCR RT Master Mix was from Toyobo. The Basic Glial Cells Nucleofector Kit was from Lonza.
Animals and treatment procedures.
All animal procedures were in strict accordance with the National Institutes of Health's Guidelines for the Care and Use of Laboratory Animals and were approved by the Animal Ethics Committee of Xiamen University. TREM2 knock-out mice (Trem2−/−; C57BL/6N) and the corresponding control C57BL/6N mice were obtained from the UC Davis Knockout Mouse Project (KOMP) repository as described previously (Jay et al., 2015) and were bred in the Animal Centre of Xiamen University. Trem2−/− or WT male mice at 8 weeks of age were intraperitoneally injected with 25 mg/kg KA and the mouse brains were dissected 3 d later. For LiCl treatment, Trem2−/− or WT male mice at 8 weeks of age were intraperitoneally injected with 200 mg/kg LiCl for 3 consecutive days and the mouse brains were dissected. The coronal sections from Trem2−/− or WT mouse brains were subjected to Iba-1 immunofluorescence staining for microglia. The brain lysates from Trem2−/− or WT mouse brains were subjected to Western blotting.
Primary microglial culture and treatment.
Primary microglial cells were prepared as described previously (Liu et al., 1994; Zhu et al., 2010; Zheng et al., 2016) with modification. Briefly, mixed glial cells from newborn (postnatal 1–3 d old) Trem2−/− or WT C57BL/6N pups were cultured in Dulbecco modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin/streptomycin in poly-l-lysine (Sigma-Aldrich)-coated cell culture flasks (Corning). The medium was replaced the next day with fresh DMEM plus 10% FBS and 25 ng/ml GM-CSF. Microglia cells were harvested by shaking gently after 8–10 d in culture as described previously (Zheng et al., 2016). The isolated WT microglia were subjected to TREM2 knock-down by electroporation. Isolated Trem2−/− or WT microglia were subjected to cell cycle analysis or plated for MTT assay, BrdU incorporation assay, or terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate nick-end labeling (TUNEL) staining. For rescue experiments, Trem2−/− or WT microglia were treated with conditioned medium from parental L cell (LCM)- or Wnt3a-expressing cultures (Wnt3aCM) or with 5 or 10 mm LiCl or with 5 μm TDZD-8 for 24 h and used for qRT-PCR, Western blotting, MTT assay, BrdU incorporation assay, or TUNEL staining.
Trem2 knock-down by siRNA.
Electroporation in primary microglia was performed as described previously (Zheng et al., 2016). Briefly, knock-down of TREM2 with Trem2-specific siRNA (Invitrogen) was achieved by electroporation using an Amaxa Nucleofector and a glial-specific Nucleofector kit (Lonza) according to the manufacturer's instructions. Each electroporation reaction contained 4 × 106 cells and 200 nm siRNA. Transfected cells were plated and used for qRT-PCR, Western blotting, MTT assay, or BrdU incorporation assay.
Immunofluorescence staining.
The coronal cortex and hippocampus of the brain slices were fixed with 4% PFA in PBS for 30 min at room temperature (RT). Before staining, the slices were washed thoroughly with PBS. The slices were then permeabilized with 0.1% Triton X-100 for 15 min at RT. The nonspecific binding sites were blocked with 5% normal goat serum in PBS for 30 min at RT and the slices were incubated with the mouse anti-Iba1 primary antibody (1:100; Wako Chemicals) overnight at 4°C. The slices were then washed three times with the blocking solution and incubated with the Alexa Fluor 488-conjugated goat anti-mouse secondary antibody (1:500; Life Technology) for 2 h at RT. The slices were washed three times with PBS and mounted on object glasses with Vectashield mounting medium with DAPI (Vector Laboratories). The images were captured with an Olympus FV10-ASW confocal microscope. Five different fields per high-power field (HPF) in hippocampi and cortices of at least five sections per mouse from five mice were selected for quantifying the number of microglial cells. The number, processes, and cell body area of microglia double stained with DAPI and Iba1 were quantified per HPF in hippocampi and cortices with Olympus FV10-ASW 4.0 Viewer software.
TUNEL staining.
The numbers of apoptotic and necrotic cells were determined by TUNEL. Briefly, cells were washed with PBS, fixed in 4% polyformaldehyde (Sigma-Aldrich) for 15 min, washed again with PBS, and then stained with TUNEL according to the manufacturer's instructions (In Situ Cell Death Detection Kit, TMR red; Roche Diagnostics). DAPI was used to stain cell nuclei. Seven different fields per HPF from three independent experiments were randomly selected for quantifying the number of TUNEL-positive cells. The TUNEL-positive cell nuclei were counted manually and compared with the total microglia number counted manually as DAPI and given as a percentage.
Cell cycle analysis.
Trem2−/− or WT microglia were collected for cell cycle analysis. Cells were washed twice with PBS and resuspended in 1 ml of DAPI stain solution (25 μg/ml) and incubated for 15 min at 37°C. DNA was quantified 1 h after staining. The analysis was performed by flow cytometry (Guava easyCyte 8 HT; Millipore) and cell population frequencies (G0/G1, S, and G2/M) were determined using FlowJo software.
Reverse transcription and quantitative real-time PCR.
Total RNA was isolated from tissues or cells using TRIzol reagent (Thermo Fisher Scientific). Reverse transcription was performed using ReverTra Ace qPCR RT Master Mix (Toyobo) and the resulting cDNA was used for qRT-PCR. The set of β-actin primers was used as an internal control for each specific gene amplification. The relative levels of expression were quantified and analyzed by using Bio-Rad iCycleriQ software. The real-time value for each sample was averaged and compared using the CT method, where the amount of target RNA (2−ΔΔCT) was normalized to the endogenous β-actin reference (ΔCT) and then normalized against control levels. The primers sequences used to amplify target genes Trem2, β-catenin, Cyclin D1, c-Myc, and β-actin were as follows: Trem2-forward: 5′-TCATAGGGGCAAGACACCT-3′; Trem2-reverse: 5′-GCTGCTCATCTTACTCTTTGTC-3′; β-catenin-forward: 5′-ATGGAGCCGGACAGAAAAGC-3′; β-catenin-reverse: 5′-CTTGCCACTCAGGGAAGGA-3′; Cyclin D1-forward: 5′-GCGTACCCTGACACCAATCTC-3′; Cyclin D1-reverse: 5′-CTCCTCTTCGCACTTCTGCTC-3′; c-Myc-forward: 5′-ATGCCCCTCAACGTGAACTTC-3′; c-Myc-reverse: 5′-CGCAACATAGGATGGAGAGCA-3′; β-actin-forward: 5′-AGTGTGACGTTGACATCCGTA-3′; and β-actin-reverse: 5′-GCCAGAGCAGTAA TCTCCTTC-3′.
BrdU incorporation assay.
BrdU incorporation assay was performed using BrdU cell proliferation ELISA kit (Abcam) according to the manufacturer's protocol. Briefly, microglia were seeded at a density of 2 × 104 cells/well and BrdU was added to the cells for 18 h for incorporation. Cells were then fixed, permeabilized, and denatured to enable the detection of incorporated BrdU through anti-BrdU antibody. After the incubation of horseradish peroxidase-conjugated secondary antibody, the colored reaction product was quantified using a spectrophotometer (Varioan Flash; Thermo Fisher Scientific) at a dual wavelength of 450/550 nm. BrdU-positive ELISA signals were normalized against total cell numbers to eliminate the possibility that the results were affected by potential changes of viable cells.
MTT assay.
Cell viability was analyzed using the MTT assay. Briefly, microglia were cultured in 96-well plates at a concentration of 3 × 104/well, incubated for 24 h, and treated with or without LCM, Wnt3aCM, or LiCl. After 24 h, MTT solution was added to each well at a final concentration of 0.5 mg/ml and the cells were further incubated for 4 h. At the end of incubation, formazan crystals resulting from MTT reduction were dissolved by addition of 100 μl of dimethyl sulfoxide per well. The absorbance was measured at 570 nm using an automated ELISA plate reader.
Western blotting analysis.
All cells were lysed in RIPA buffer (Boster) and total protein concentrations were determined with a BCA Protein Assay Kit (Thermo Fisher Scientific). Total protein (20 μg) was loaded for each sample into 8%, 10%, or 12% SDS-PAGE. Western blotting was performed as described previously (Liu et al., 2014). Briefly, gels were transferred onto PVDF membranes (Millipore). Primary antibodies were incubated overnight at 4°C. Immunoreactive bands were detected by ECL Western blotting detection reagents (Millipore) and quantified using ImageJ Software.
Statistical analysis.
All quantified data represent mean ± SEM. Statistical significance was determined by ANOVA and Tukey's post hoc test (GraphPad Prism 5.0) when more than two groups were compared and Student's t test when one group was compared with the control group. p < 0.05 was considered significant.
Results
Decreased microglial survival and microgliosis in Trem2−/− microglia and in the brains of Trem2−/− mice
To address the potential roles of TREM2 in regulating microglia viability, Trem2 was knocked down in primary microglia by two independent siRNAs targeting distinct regions of TREM2 and the knock-down efficiency was assessed by qRT-PCR (Fig. 1A). Cell survival was suppressed significantly in Trem2-knock-down microglia (Fig. 1B) and in microglia derived from Trem2 knock-out (Trem2−/−) mice (Fig. 1D) as examined by MTT assay. To determine whether the decreased viability of Trem2-deficient microglia resulted from a decrease in microglia proliferation, an increase in apoptosis, or both, we first assessed microglia proliferation using a BrdU incorporation assay. We found that knock-down of Trem2 in primary microglia suppressed microglia proliferation (Fig. 1C). Cell proliferation was also reduced in microglia derived from Trem2−/− mice compared with those from WT mice (Fig. 1E). To quantify the apoptotic status of primary microglia from WT control or Trem2−/− mice by TUNEL staining, we assessed the number of TUNEL-positive cells (green) compared with DAPI-positive total cells (blue) in the microglia culture (Fig. 1F). The number of TUNEL-positive apoptotic cells was increased in Trem2−/− microglia compared with WT microglia (Fig. 1F). To further confirm this effect, cell lysates from WT or Trem2−/− microglia were analyzed by Western blotting for cleaved Caspase-3 (c-Casp3), the activation of which is required for the induction of apoptosis; pro-Caspase-3 (pro-Casp3), the inactive precursor to c-Casp3; and Bcl-2, an anti-apoptotic protein. We found that the amount of c-Casp3 was increased, whereas the amounts of pro-Casp3 and the Bcl-2 were decreased in Trem2−/− microglia compared with WT microglia (Fig. 1G), suggesting that Trem2 depletion both suppresses microglial proliferation and accelerates microglial apoptosis.
Figure 1.

TREM2 deficiency in microglia leads to decreased cell viability and increased cell death. A, Knock-down of Trem2 with two independent siRNAs in mouse primary microglia was confirmed by qRT-PCR. B, Knock-down of Trem2 suppressed microglial viability compared with the control nontarget (NT) group as examined by MTT assay. C, Trem2 knock-down suppressed microglial proliferation as assessed by BrdU incorporation assay. D, Decreased cell viability in Trem2−/− microglia compared with WT cells. E, Proliferation was suppressed in Trem2−/− microglia compared with WT cells. F, Trem2−/− microglia were cultured for the indicated times and apoptosis and necrosis were assessed by TUNEL staining. TUNEL-positive, apoptotic cells (green) were quantified as a percentage of total DAPI-positive cells (blue). The number of TUNEL-positive cells increased in Trem2−/− microglia compared with WT cells. G, Cell lysates from WT or Trem2−/− microglia were analyzed for pro-Caspase3 (pro-Casp3), cleaved Caspase-3 (c-Casp3), and Bcl-2 levels by Western blotting. Protein levels were quantified by densitometry ratio to α-tubulin for comparison. The level of c-Casp3 was increased, whereas pro-Casp3 and Bcl-2 were suppressed in Trem2−/− microglia compared with WT cells. Data are plotted as mean ± SEM (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001.
To evaluate the role of TREM2 in regulating microglial activation in response to injury in vivo, Trem2−/− or WT mice were intraperitoneally injected with 25 mg/kg KA, a potent analog of glutamate commonly used to induce excitotoxicity and subsequent neuronal death associated with various neurodegenerative disorders (Zheng et al., 2011; Luo et al., 2013). By analyzing cell number, cell processes, and cell bodies of microglia, we found that KA treatment resulted in marked microglia morphological change (increased cell body and processes) and a dramatic increase in the number of activated microglia in WT mouse brain; however, this response was significantly reduced in TREM2-deficient mice, in particular in the hippocampus (Fig. 2A–I). These results indicate that TREM2 expression and function are essential in the regulation of microglial survival and activation upon neuronal injury.
Figure 2.

TREM2 deficiency leads to reduced microgliosis in response to KA treatment in mouse brain. Trem2−/− or WT mice were intraperitoneally injected with 25 mg/kg KA at 8 weeks of age and mouse brains were dissected 3 d later. A, Coronal sections from Trem2−/− or WT mouse brains were stained with Iba-1 (green) for microglia. Representative images are shown. B, D, Number of microglia double stained with DAPI (blue) and Iba1 (green) was quantified per HPF in Trem2−/− or WT hippocampi and cortices. C, E, Microglia number response to KA was assessed by quantifying the cell number ratio of KA group to control group in WT or Trem2−/− brains. F, G, Quantification of microglial cell body (area, in square micrometers) per HPF in the hippocampi and cortices of WT or Trem2−/− brains with or without KA treatment. H, I, Quantification of microglial process per HPF in the hippocampi and cortices of WT or Trem2−/− brains with or without KA treatment. Scale bar, 100 μm. Data are plotted as mean ± SEM (n = 5). *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant.
TREM2 depletion in microglia leads to cell cycle arrest at the G1/S checkpoint
Given that TREM2 mediates microglial proliferation and cell death, which are tightly coupled with cell-cycle regulators and apoptotic stimuli, we investigated how TREM2 modulates cell cycle progression by FACS analysis. The number of cells at different cell cycle phases (G1/G0, S, or G2/M) was quantified as a percentage of the whole for comparison (Fig. 3A). We found that TREM2 deficiency in microglia resulted in an increased of G1/G0 phase cell population and a decreased percentage of cells in the S phase (Fig. 3B). Given that Cyclin D1 functions as a regulatory subunit of cyclin-dependent kinase 4 (CDK4) or CDK6, the activity of which is required for the cell cycle G1/S transition (Agami and Bernards, 2000) and that c-Myc is implicated in cell proliferation and growth, we also examined the expression of Cyclin D1 and c-Myc in WT and Trem2−/− microglia. Trem2−/− microglia, compared with WT cells, exhibited reduced Cyclin D1 and c-Myc mRNA when examined by qRT-PCR (Fig. 3C). Similarly, Cyclin D1 and c-Myc protein levels were also suppressed in Trem2−/− microglia compared with WT cells (Fig. 3D). Together, our results demonstrate that TREM2 deficiency affects critical cell cycle regulators, leading to a marked reduction of microglia proliferation and viability, with an accumulation of cells in the G0/G1 phase of the cell cycle.
Figure 3.

TREM2 deletion in microglia leads to cell cycle arrest at the G1/S checkpoint. A, Trem2−/ −or WT microglia were fixed, stained with DAPI, and analyzed by flow cytometry. B, Ratio of stage-specific (G1/G0, S, or G2/M) cell numbers to total cell numbers was quantified for comparison. The number of G1/G0 phase cells increased, whereas S-phase cells decreased in Trem2−/− microglia compared with WT microglia. C, Cyclin D1 and c-Myc mRNA levels were decreased in Trem2−/− microglia compared with WT cells as quantified by qRT-PCR. D, Cell lysates from Trem2−/− microglia were analyzed for Cyclin D1 and c-Myc by Western blotting. Protein levels were quantified by densitometry normalized to α-tubulin. Note that Cyclin D1 and c-Myc were downregulated in Trem2−/− microglia compared with WT cells. Data are plotted as mean ± SEM (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001.
β-catenin is downregulated in Trem2-deficient microglia
Because depletion of TREM2 in microglia reduced cell viability dramatically, we investigated whether the ERK1/2, p38 mitogen-activated protein kinase (MAPK), and β-catenin signaling pathways, which are involved in the activation and proliferation of myeloid cells including macrophages (Otero et al., 2009; Otero et al., 2012; Xing et al., 2015), are also involved in regulating microglial cell survival. We found that β-catenin was suppressed, but the expression of neither phosphorylated p38MAPK (p-P38) nor ERK1/2 (p-ERK1/2) was changed, in Trem2 knock-down microglia compared with the nontarget control cells (Fig. 4A,B). The decrease in β-catenin and lack of significant changes in p-P38 or p-ERK1/2 levels were further confirmed in microglia from Trem2−/− mice (Fig. 4C,D). Therefore, the β-catenin signaling pathway may play a critical role in TREM2-regulated microglial survival.
Figure 4.

β-catenin was downregulated in Trem2-depleted microglia. A, Trem2 was knocked down by two independent siRNAs. Protein levels of β-catenin, phosphorylated p38 (p-P38), phosphorylated ERK1/2 (p-ERK1/2), total p38, total ERK1/2 from nontarget (NT), or Trem2 knock-down microglia were analyzed by Western blotting. B, Protein levels were quantified by densitometry and presented as ratios to α-tubulin. β-catenin was suppressed in Trem2 knock-down microglia compared with NT microglia. The amount of p-P38 or p-ERK1/2 was not altered in Trem2 knock-down microglia compared with NT microglia. C, D, Cell lysates of WT or Trem2−/− microglia were analyzed by Western blotting. The amount of β-catenin was reduced in Trem2−/− microglia. Protein levels were quantified by densitometry and are presented as ratios to α-tubulin. p-P38 or p-ERK1/2 levels were not altered in Trem2−/− microglia compared with WT microglia. Data are plotted as mean ± SEM (n = 3). **p < 0.01; ns, not significant.
TREM2 stabilizes β-catenin by inhibiting the degradation of β-catenin via Akt/GSK3β signaling pathway
Given that β-catenin is downregulated in Trem2−/− microglia, we first investigated whether TREM2 mediates the transcription of β-catenin by qPCR analysis. We found that there was no difference in β-catenin mRNA levels between Trem2−/− and WT microglia (Fig. 5A), indicating that TREM2 did not affect the transcription of β-catenin in microglia. To investigate whether TREM2 deficiency affects the stability of β-catenin, Trem2−/− or WT microglia were treated with cycloheximide, which inhibits protein synthesis, for various periods of time. We found that β-catenin was degraded more rapidly in the Trem2−/− microglia than in WT cells (Fig. 5B,C). Because β-catenin is ubiquitinated and degraded through the proteasomal pathway (Aberle et al., 1997; Orford et al., 1997), we sought to determine whether a proteasomal inhibitor can suppress β-catenin degradation in Trem2−/− microglia. After incubation with the proteasome inhibitor MG132, β-catenin levels were increased in both Trem2−/− and WT microglia (Fig. 5B,D). Together, these results demonstrate that TREM2 stabilizes β-catenin by suppressing its proteasomal degradation.
Figure 5.

Accelerated degradation of β-catenin in Trem2-depleted microglia. A, β-catenin mRNA levels were not altered in Trem2−/− microglia compared with WT cells as quantified by qRT-PCR. B, Trem2−/− or WT microglia were treated with cycloheximide (CHX; 100 μg/ml) or MG132 (10 μm) for the indicated times and cell lysates were analyzed by Western blotting. C, Protein levels were quantified by densitometry and normalized to GAPDH. Upon CHX treatment, β-catenin protein levels increased significantly in WT microglia at 4 and 5 h compared with Trem2−/− cells. D, When cells were treated with MG132, β-catenin protein level increased in both Trem2−/− and WT microglia in a time-dependent manner. Data are plotted as mean ± SEM (n = 3). *p < 0.05; **p < 0.01.
To elucidate the specific pathway through which TREM2 suppresses proteasomal degradation of β-catenin, we examined the potential role of the PI3K/Akt signaling pathway, which has been shown to be downstream of TREM2-mediated immune modulation (Sun et al., 2013; Zhu et al., 2014) and to regulate β-catenin (Cross et al., 1995; Seira and Del Río, 2014). We thus examined the phosphorylation status of Akt and GSK-3β in WT and Trem2−/− microglia, given that phosphorylation of GSK3β at the serine 9 residue (GSK3βS9) leads to GSK3β inactivation (Cross et al., 1995). As shown in Figure 6, A–C, phosphorylation of AktS473 and GSK-3βS9 was decreased significantly in Trem2 knock-down microglia and in Trem2−/− microglia (Fig. 6D–F). Therefore, these results indicate that TREM2 stabilizes β-catenin by inhibiting the proteosomal degradation of β-catenin via the Akt/GSK3β signaling pathway.
Figure 6.
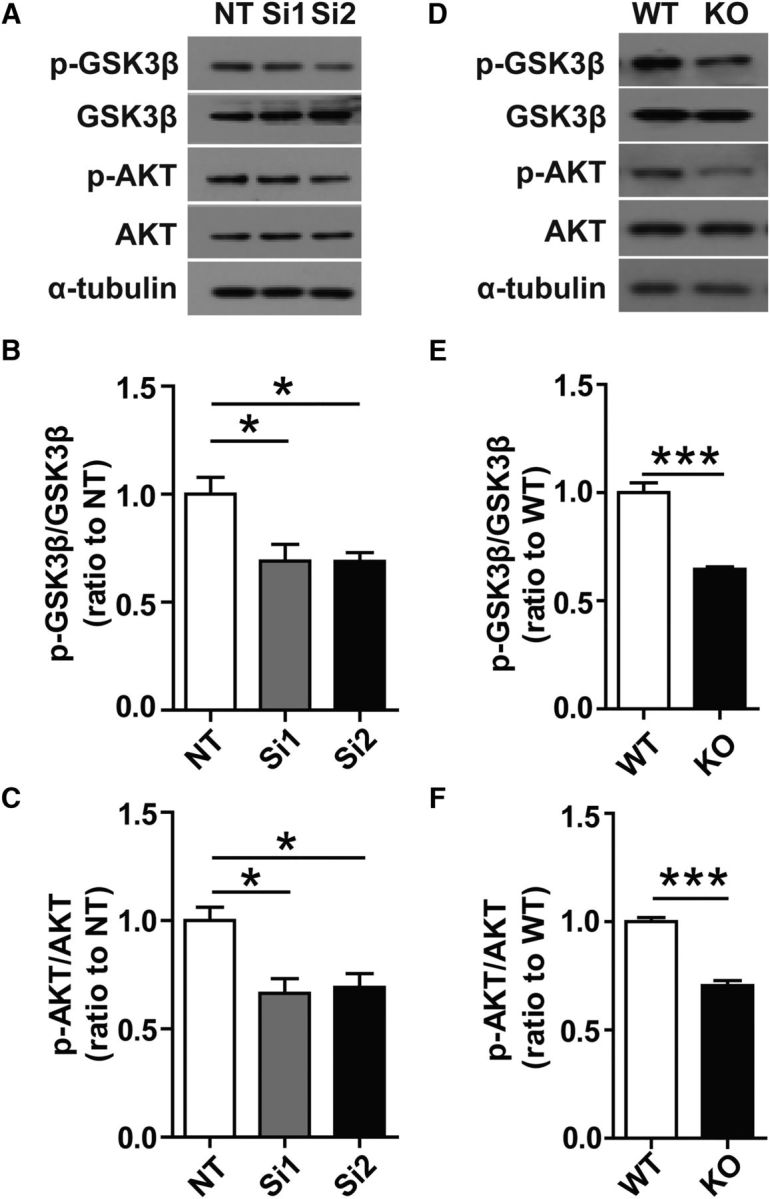
Decreased phosphorylation of GSK3β and AKT in Trem2-depleted microglia. A, Protein levels in cell lysates from Trem2 knock-down microglia were analyzed by Western blotting. Representative Western blots of phosphorylated GSK3βS9, phosphorylated AktS473, total GSK3β, and total Akt are shown. B, C, Protein levels were quantified by densitometry and normalized to total GSK3β/Akt and α-tubulin. The levels of phosphorylated GSK3βS9 and phosphorylated AktS473 were decreased in Trem2 knock-down microglia compared with nontarget (NT) cells. D, Cell lysates from Trem2−/− or WT microglia were analyzed to assess the levels of the indicated proteins by Western blotting. Representative Western blots of phosphorylated GSK3βS9, phosphorylated AktS473, total GSK3β, and total Akt are shown. E, F, Protein levels were quantified by densitometry and normalized to total GSK3β/Akt and α-tubulin. The levels of phosphorylated GSK3βS9 and phosphorylated AktS473 were decreased in Trem2−/− microglia compared with WT cells. Data are plotted as mean ± SEM (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001.
Wnt3a restores β-catenin level and cell growth in Trem2−/− microglia
β-catenin is a key mediator in the canonical Wnt signaling pathway (Clevers, 2006). After activation by Wnt ligand-receptor binding, β-catenin escapes proteosomal degradation and translocates into the nucleus, where it binds to target genes that modulate various biological processes, including cell proliferation, differentiation, and survival (Mosimann et al., 2009). To determine whether activating Wnt/β-catenin signaling can restore cell survival in Trem2−/− microglia, Trem2−/− or WT microglia were treated with Wnt3aCM or control L cell CM for 24 h. Wnt3a CM increased β-catenin in WT and Trem2−/− microglia compared with L cell CM (Fig. 7A,B). Importantly, Wnt3aCM promoted the cell viability and proliferation suppressed in Trem2−/− microglia as examined by MTT or BrdU incorporation assay (Fig. 7C, D). These results demonstrate that activation of the Wnt/β-catenin signaling pathway partially restores the proliferation and survival of Trem2−/− microglia.
Figure 7.

Wnt3a restores β-catenin level and cell growth in Trem2−/− microglia. A, Trem2−/− or WT microglia were treated with LCM or Wnt3aCM for 24 h. The β-catenin level was analyzed by Western blotting. B, Amount of β-catenin was increased in Trem2−/− microglia treated with Wnt3aCM compared with the cells treated with LCM. Protein levels were quantified by densitometry and presented as ratios to GAPDH. C, Wnt3a promoted the cell viability of Trem2−/− microglia. D, Wnt3a partially rescued the cell proliferation of Trem2−/− microglia as examined by BrdU incorporation assay. Data are plotted as mean ± SEM (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001.
LiCl, a GSK-3β inhibitor, rescues β-catenin signaling and the survival of Trem2−/− microglia in vivo and in vitro
Given that Wnt3a enhanced β-catenin level and cell survival in Trem2−/− microglia, we next evaluated how LiCl, a GSK-3β inhibitor that can activate the Wnt signaling pathway by stabilizing β-catenin (Kramer et al., 2012), affects Trem2−/− microglial survival. When cells were treated with LiCl for 24 h, β-catenin increased significantly in Trem2−/− microglia (Fig. 8A,B). Moreover, both cell viability and proliferation increased in Trem2−/− microglia upon LiCl treatment, although it did not completely restore the effect to the levels in WT microglia (Fig. 8C,D). These results were further confirmed by treatment with TDZD-8, a specific GSK-3β inhibitor, for 24 h in WT or Trem2−/− microglia, which enhanced β-catenin level in Trem2−/− microglia (Fig. 8E,F).
Figure 8.

LiCl and TDZD-8 rescue β-catenin signaling and cell survival in Trem2−/− microglia. A, Trem2−/− or WT microglia were treated with or without LiCl for 24 h. β-catenin level was analyzed by Western blotting. B, LiCl increased β-catenin in Trem2−/− microglia compared with control treated cells. C, LiCl enhanced the viability of Trem2−/− microglia. D, LiCl restored Trem2−/− microglial proliferation as examined by BrdU incorporation assay. E, Trem2−/− or WT microglia were treated with or without 5 μm TDZD-8 for 24 h. β-catenin level was analyzed by Western blotting. F, TDZD-8 increased β-catenin in Trem2−/− microglia compared with vehicle treated cells. Data are plotted as mean ± SEM (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001.
In addition, we examined the ability of LiCl to modulate the survival of microglia in Trem2−/− mice in vivo. LiCl intraperitoneal treatment significantly restored the number of microglia in Trem2−/− mouse brain (Fig. 9A,B), suggesting that LiCl can rescue suppressed microglial proliferation in Trem2−/− mice. We confirmed that the levels of β-catenin, GSK-3β, c-Myc, and Cyclin D1 were increased significantly in the hippocampus of Trem2−/− mouse brain in response to LiCl treatment, although their levels were not completely restored to those in WT mice (Fig. 9C). Together, our findings demonstrate that treatment with LiCl, which activates Wnt/β-catenin signaling, rescues microglial cell death and microgliosis resulting from TREM2 deficiency.
Figure 9.

LiCl rescues β-catenin signaling and cell survival in Trem2−/− mouse brain. A, Trem2−/− or WT mice at 8 weeks of age were intraperitoneally injected with LiCl (200 mg/kg) for 3 consecutive days and mouse brains were harvested and dissected. Brain sections from Trem2−/− or WT mice were double stained with Iba-1 (green) and DAPI (blue) for microglia. Representative images are shown. B, Number of microglia double stained with DAPI (blue) and Iba1 (green) was quantified per HPF in hippocampi and cortices. C, Protein levels in brain lysates were analyzed by Western blotting. Protein levels were quantified by densitometry and normalized to total GSK3β and/or α-tubulin. The levels of β-catenin, Cyclin D1, c-Myc, and phosphorylated GSK3βS9 were increased in Trem2−/− mice treated with LiCl compared with WT mice. Scale bar, 100 μm. Data are plotted as mean ± SEM (n = 5). *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
Our findings shed new light on the relationship between Wnt/β-catenin signaling and the TREM2/DAP12 pathway and offer a pathological mechanism through which reduced TREM2, or TREM2 loss-of-function, might impair microglial survival in AD pathogenesis. We provide evidence that TREM2 promotes microglial survival by activating the Wnt/β-catenin signaling pathway, which plays essential roles in many biological processes during embryonic development and disease pathogenesis, including cell proliferation, survival, migration, and polarity, fate decisions, and self-renewal (Logan and Nusse, 2004; Clevers, 2006; Clevers and Nusse, 2012; Chen et al., 2013). We also show that knock-down or depletion of TREM2, a receptor specifically expressed in microglia (Colonna, 2003; Hickman and El Khoury, 2014), results in increased apoptosis and decreased proliferation, which can be partially restored by Wnt3a or LiCl or TDZD-8 treatment. Together, our data suggest the potential promise of manipulating the TREM2/β-catenin signaling pathway for the treatment of AD.
Such findings come on the heels of new evidence suggesting that TREM2-mediated signaling may play a role in microglial proliferation/survival in response to injury and aging, although controversy surrounding TREM2 has focused on the differences of the phenotypes observed in murine models. For example, Poliani et al. (2015) found that there was no significant difference in the abundance of microglia between WT and Trem2−/− mice until 2 years of age and most prominently in the corpus callosum. However, Cantoni et al. (2015) showed that microglia proliferation and abundance were diminished in Trem2−/− mice in the context of demyelination, suggesting a critical role of TREM2 in the survival and proliferation of microglia. Recent studies also demonstrate that a lack of TREM2 in osteoclast precursors impairs proliferation and β-catenin activation in response to macrophage colony-stimulating factor (M-CSF) (Otero et al., 2012), whereas deficiencies in DAP12, a required adaptor for TREM2 signaling, resulted in a profound reduction of microglia in aged mice (Otero et al., 2009). Whether Wnt/β-catenin signaling intersects with CSF-1R/M-CSF pathway and whether these pathways are modulated by TREM2/DAP12 signaling in microglia need to be further addressed in future studies.
TREM2 activation leads to DAP12 phosphorylation via Src family kinases, initiating the downstream signaling cascades including PI3K, PKC, and ERK(Ulrich and Holtzman, 2016). In examining the effects of TREM2 depletion, we did not observe a change in the level or activation of ERK1/2 or p38MAPK signaling pathways despite their role in mediating a variety of cellular processes, including proliferation, cell survival, and cell death (Clevers and Nusse, 2012; Cossa et al., 2013). Rather, TREM2 deficiency led to a dramatic downregulation of Wnt/β-catenin signaling in microglia, decreased microglial survival, and enhanced cell death. Therefore, our findings provide strong evidence that TREM2 regulates the proliferation and survival of microglia by activating the Wnt/β-catenin signaling pathway. Treatment with Wnt3a, LiCl, or TDZD-8 did not completely rescue the relevant phenotypes in Trem2−/− microglia and increased β-catenin levels both in WT and in Trem2−/− microglia, indicating that the TREM2-regulated Wnt/β-catenin signaling pathway might be a critical, but not necessarily a sole, pathway in regulating microglial proliferation and survival. Upregulation of Wnt/β-catenin signaling may promote microglial survival and function, which may represent an alternative therapeutic strategy for neurodegenerative diseases associated with TREM2-mediated microglial dysfunction. Furthermore, because Wnt/β-catenin signaling is essential for normal CNS development (Ciani and Salinas, 2005) and embryonic microglia have been shown to regulate neural precursor pool size and the outgrowth of dopaminergic axons in the developing brain (Cunningham et al., 2013; Squarzoni et al., 2014), understanding whether TREM2 mediates microglial function through modulating Wnt/β-catenin pathway during neurodevelopment is of great interest.
Our results further indicate that, through the cross talk between TREM2/β-catenin pathways, TREM2 may play a critical role in regulating microglial proliferation and cell viability by modulating the proteins involved in cell cycle control. Cell cycle progression through the G1 phase and the G1/S transition are events highly regulated by a complex mechanism involving the activation of cell cycle proteins, including Cyclin D1 and CDKs, which regulate proliferation and cell cycle progression and were suppressed in Trem2−/− microglia, supported our observation of G1/S checkpoint arrest and increased microglia apoptosis under the conditions of TREM2 depletion. Uncovering how TREM2 regulates the expression or the kinase activity of cell cycle proteins warrants further investigation.
As indicated by our results, TREM2 may interact with DAP12 to activate PI3K/Akt signaling, thereby inactivating GSK3β and stabilizing β-catenin (Fig. 10; Papkoff and Aikawa, 1998; Sun et al., 2013). Akt, a PI3-kinase activated protein kinase, plays an important role in the cell death/survival pathway during neuronal development and injury repair (Franke et al., 1997; Seira and Del Río, 2014) and activation of Akt has been linked to the promotion of microglial cell proliferation (Kim et al., 2004; Ito et al., 2005; Suh et al., 2005). The PI3K/Akt pathway represents a downstream signaling pathway in TREM2-mediated immune modulation (Sun et al., 2013; Zhu et al., 2014). In brief, PI3K/Akt phosphorylates GSK3β on Ser9 to render it inactive, which in turn promotes β-catenin stabilization and induction of responsive genes that enhance cell survival (Papkoff and Aikawa, 1998; Chen et al., 2001; You et al., 2004; Li et al., 2006). In this study, we found that Trem2−/− microglia exhibit reduced phosphorylation of AktS473 and GSK-3βS9 and accelerated degradation of β-catenin. These findings suggest that, under normal conditions, TREM2 promotes microglial proliferation and survival after injury through the activation of Akt/β-catenin pathways. We also provide strong evidence that upregulation of Wnt/β-catenin signaling via Wnt3a ligand or LiCl or TDZD-8 treatment, which suppresses GSK-3β, restores β-catenin signaling significantly and promotes Trem2−/− microglial survival in vitro and in vivo. Suppressing GSK-3β may enable the accumulated β-catenin to translocate into the nucleus, where it engages TCF transcription factors, YAP1/TBX5, or the TAZ complex, driving the expression of antiapoptotic/survival genes such as Cyclin D1, c-Myc, and Bcl-2 (Clevers and Nusse, 2012; Rosenbluh et al., 2014).
Figure 10.

Schematic model of TREM2 in the regulation of β-catenin signaling and microglial survival. TREM2 interacts with DAP12 to activate PI3K/Akt signaling. Akt phosphorylates the serine 9 residue of GSK3β (GSK3βS9), leading to GSK3β inactivation and stabilization of β-catenin. β-catenin accumulates in the cytoplasm and then enters the nucleus, where it regulates the expression of target genes such as Cyclin D1, c-Myc, and Bcl-2.
Our studies demonstrate that TREM2 is a crucial determinant of microglial survival and microgliosis and that TREM2 loss of function may lead to compromised microglial survival and function in neurodegenerative diseases. Although it remains to be determined whether TREM2 mutations or other abnormalities lead to impaired microglial survival, our results demonstrate that it is possible to restore the TREM2-regulated β-catenin pathway under conditions in which TREM2 activity is disrupted or reduced. In particular, targeting the Wnt/β-catenin pathway downstream of TREM2 may represent a novel therapeutic approach to combat the microglial dysfunction associated with chronic inflammation-related neurodegenerative diseases such as AD.
Footnotes
This work was supported by the Natural Science Foundation of Fujian Province of China (Grant 2016J01411 to H.Z.); the Educational Department of Fujian Province of China (Grant JZ160403 to H.Z.); the National Institutes of Health (Grants R01AG027924, R01AG035355, and R01AG046205 to G.B.); the National Natural Science Foundation of China (Grant 81100842 to H.Z., Grant U1505227 to G.B., and Grants 81370459 and 31400914 to X.-F.C.); Tanz Family Funds; and the Alzheimer's Association (Grant C4C-15–369446 to H.X.).
The authors declare no competing financial interests.
References
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997) beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J 16:3797–3804. 10.1093/emboj/16.13.3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agami R, Bernards R (2000) Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell 102:55–66. 10.1016/S0092-8674(00)00010-6 [DOI] [PubMed] [Google Scholar]
- Aguzzi A, Barres BA, Bennett ML (2013) Microglia: scapegoat, saboteur, or something else? Science 339:156–161. 10.1126/science.1227901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10:1538–1543. 10.1038/nn2014 [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69. 10.1038/nrn2038 [DOI] [PubMed] [Google Scholar]
- Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R, Yuede CM, Galimberti D, Olivecrona G, Klein RS, Cross AH, Otero K, Piccio L (2015) TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol 129:429–447. 10.1007/s00401-015-1388-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HL, Chew LJ, Packer RJ, Gallo V (2013) Modulation of the Wnt/beta-catenin pathway in human oligodendroglioma cells by Sox17 regulates proliferation and differentiation. Cancer Lett 335:361–371. 10.1016/j.canlet.2013.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J, Wang CY (2001) Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J Cell Biol 152:87–96. 10.1083/jcb.152.1.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani L, Salinas PC (2005) WNTs in the vertebrate nervoussystem: from patterning to neuronal connectivity. Nat Rev Neurosci 6:351–362. [DOI] [PubMed] [Google Scholar]
- Clevers H. (2006) Wnt/beta-catenin signaling in development and disease. Cell 127:469–480. 10.1016/j.cell.2006.10.018 [DOI] [PubMed] [Google Scholar]
- Clevers H, Nusse R (2012) Wnt/beta-catenin signaling and disease. Cell 149:1192–1205. 10.1016/j.cell.2012.05.012 [DOI] [PubMed] [Google Scholar]
- Colonna M. (2003) TREMs in the immune system and beyond. Nat Rev Immunol 3:445–453. 10.1038/nri1106 [DOI] [PubMed] [Google Scholar]
- Colonna M, Wang Y (2016) TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci 17:201–207. 10.1038/nrn.2016.7 [DOI] [PubMed] [Google Scholar]
- Cossa G, Gatti L, Cassinelli G, Lanzi C, Zaffaroni N, Perego P (2013) Modulation of sensitivity to antitumor agents by targeting the MAPK survival pathway. Current Pharmaceutical Design 19:883–894. 10.2174/138161213804547187 [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789. 10.1038/378785a0 [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Martínez-Cerdeño V, Noctor SC (2013) Microglia regulatethe number of neural precursor cells in the developing cerebral cortex. J Neurosci 33:4216–4233. 10.1523/JNEUROSCI.3441-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC (1997) PI3K: downstream AKTion blocks apoptosis. Cell 88:435–437. 10.1016/S0092-8674(00)81883-8 [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152:307–317. [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis Group (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368:117–127. 10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, El Khoury J (2014) TREM2 and the neuroimmunology of Alzheimer's disease. Biochem Pharmacol 88:495–498. 10.1016/j.bcp.2013.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J (2013) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16:1896–1905. 10.1038/nn.3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Sawada M, Haneda M, Fujii S, Oh-Hashi K, Kiuchi K, Takahashi M, Isobe K (2005) Amyloid-beta peptides induce cell proliferation and macrophage colony-stimulating factor expression via the PI3-kinase/Akt pathway in cultured Ra2 microglial cells. FEBS Lett 579:1995–2000. 10.1016/j.febslet.2005.02.048 [DOI] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, Cotleur AC, Butovsky O, Bekris L, Staugaitis SM, Leverenz JB, Pimplikar SW, Landreth GE, Howell GR, Ransohoff RM, Lamb BT (2015) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J Exp Med 212:287–295. 10.1084/jem.20142322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368:107–116. 10.1056/NEJMoa1211103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson K (2013) TREM2 and neurodegenerative disease. N Engl J Med 369:1568–1569. 10.1056/NEJMc1306509 [DOI] [PubMed] [Google Scholar]
- Kim WK, Hwang SY, Oh ES, Piao HZ, Kim KW, Han IO (2004) TGF-beta1 represses activation and resultant death of microglia via inhibition of phosphatidylinositol 3-kinase activity. J Immunol 172:7015–7023. 10.4049/jimmunol.172.11.7015 [DOI] [PubMed] [Google Scholar]
- Klünemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, De Bleecker JL, Rossor MN, Marienhagen J, Klein HE, Peltonen L, Paloneva J (2005) The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology 64:1502–1507. 10.1212/01.WNL.0000160304.00003.CA [DOI] [PubMed] [Google Scholar]
- Kramer T, Schmidt B, Lo Monte F (2012) Small-molecule inhibitors of GSK-3: structural insights and their application to Alzheimer's disease models. Int J Alzheimers Dis 2012:381029. 10.1155/2012/381029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S (1990) Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39:151–170. 10.1016/0306-4522(90)90229-W [DOI] [PubMed] [Google Scholar]
- Lee CY, Landreth GE (2010) The role of microglia in amyloid clearance from the AD brain. J Neural Transm 117:949–960. 10.1007/s00702-010-0433-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K (2006) Winding through the WNT pathway during cellular development and demise. Histol Histopathol 21:103–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Tsai CW, Deak F, Rogers J, Penuliar M, Sung YM, Maher JN, Fu Y, Li X, Xu H, Estus S, Hoe HS, Fryer JD, Kanekiyo T, Bu G (2014) Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer's disease. Neuron 84:63–77. 10.1016/j.neuron.2014.08.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Brosnan CF, Dickson DW, Lee SC (1994) Macrophage colony-stimulating factor mediates astrocyte-induced microglial ramification in human fetal central nervous system culture. Am J Pathol 145:48–53. [PMC free article] [PubMed] [Google Scholar]
- Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20:781–810. 10.1146/annurev.cellbio.20.010403.113126 [DOI] [PubMed] [Google Scholar]
- Luo J, Elwood F, Britschgi M, Villeda S, Zhang H, Ding Z, Zhu L, Alabsi H, Getachew R, Narasimhan R, Wabl R, Fainberg N, James ML, Wong G, Relton J, Gambhir SS, Pollard JW, Wyss-Coray T (2013) Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J Exp Med 210:157–172. 10.1084/jem.20120412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M, Parikh I, Vasquez JB, Smith C, Tai L, Bu G, LaDu MJ, Fardo DW, Rebeck GW, Estus S (2015) Genetics ignite focus on microglial inflammation in Alzheimer's disease. Mol Neurodegener 10:52. 10.1186/s13024-015-0048-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann C, Hausmann G, Basler K (2009) Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol 10:276–286. 10.1038/nrm2654 [DOI] [PubMed] [Google Scholar]
- Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW (1997) Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. J Biol Chem 272:24735–24738. 10.1074/jbc.272.40.24735 [DOI] [PubMed] [Google Scholar]
- Otero K, Turnbull IR, Poliani PL, Vermi W, Cerutti E, Aoshi T, Tassi I, Takai T, Stanley SL, Miller M, Shaw AS, Colonna M (2009) Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol 10:734–743. 10.1038/ni.1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero K, Shinohara M, Zhao H, Cella M, Gilfillan S, Colucci A, Faccio R, Ross FP, Teitelbaum SL, Takayanagi H, Colonna M (2012) TREM2 and beta-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J Immunol 188:2612–2621. 10.4049/jimmunol.1102836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter MM, Atagi Y, Liu CC, Rademakers R, Xu H, Fryer JD, Bu G (2015) TREM2 in CNS homeostasis and neurodegenerative disease. Mol Neurodegener 10:43. 10.1186/s13024-015-0040-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, Bianchin M, Bird T, Miranda R, Salmaggi A, Tranebjaerg L, Konttinen Y, Peltonen L (2002) Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet 71:656–662. 10.1086/342259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papkoff J, Aikawa M (1998) WNT-1 and HGF regulate GSK3 beta activity and beta-catenin signaling in mammary epithelial cells. Biochem Biophys Res Commun 247:851–858. 10.1006/bbrc.1998.8888 [DOI] [PubMed] [Google Scholar]
- Perlmutter LS, Barron E, Chui HC (1990) Morphologic association between microglia and senile plaque amyloid in Alzheimer's disease. Neurosci Lett 119:32–36. 10.1016/0304-3940(90)90748-X [DOI] [PubMed] [Google Scholar]
- Polazzi E, Monti B (2010) Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog Neurobiol 92:293–315. 10.1016/j.pneurobio.2010.06.009 [DOI] [PubMed] [Google Scholar]
- Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, Colonna M (2015) TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest 125:2161–2170. 10.1172/JCI77983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH (2009) Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 27:119–145. 10.1146/annurev.immunol.021908.132528 [DOI] [PubMed] [Google Scholar]
- Rosenbluh J, Wang X, Hahn WC (2014) Genomic insights into WNT/beta-catenin signaling. Trends Pharmacol Sci 35:103–109. 10.1016/j.tips.2013.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seira O, Del Río JA (2014) Glycogen synthase kinase 3 beta (GSK3beta) at the tip of neuronal development and regeneration. Mol Neurobiol 49:931–944. 10.1007/s12035-013-8571-y [DOI] [PubMed] [Google Scholar]
- Squarzoni P, Oller G, Hoeffel G, Pont-Lezica L, Rostaing P, Low D, Bessis A, Ginhoux F, Garel S (2014) Microglia modulate wiring of theembryonic forebrain. Cell Rep 8:1271–1279. 10.1016/j.celrep.2014.07.042 [DOI] [PubMed] [Google Scholar]
- Suh HS, Kim MO, Lee SC (2005) Inhibition of granulocyte-macrophage colony-stimulating factor signaling and microglial proliferation by anti-CD45RO: role of Hck tyrosine kinase and phosphatidylinositol 3-kinase/Akt. J Immunol 174:2712–2719. 10.4049/jimmunol.174.5.2712 [DOI] [PubMed] [Google Scholar]
- Sun M, Zhu M, Chen K, Nie X, Deng Q, Hazlett LD, Wu Y, Li M, Wu M, Huang X (2013) TREM-2 promotes host resistance against Pseudomonas aeruginosa infection by suppressing corneal inflammation via a PI3K/Akt signaling pathway. Invest Ophthalmol Vis Sci 54:3451–3462. 10.1167/iovs.12-10938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Holtzman DM (2016) TREM2 function in Alzheimer's disease and neurodegeneration. ACS Chem Neurosci 7:420–427. 10.1021/acschemneuro.5b00313 [DOI] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160:1061–1071. 10.1016/j.cell.2015.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM, Colonna M (2016) TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med 213:667–675. 10.1084/jem.20151948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wes PD, Sayed FA, Bard F, Gan L (2016) Targeting microglia for the treatment of Alzheimer's disease. Glia 64:1710–1732. 10.1002/glia.22988 [DOI] [PubMed] [Google Scholar]
- Xing J, Titus AR, Humphrey MB (2015) The TREM2-DAP12 signaling pathway in Nasu-Hakola disease: a molecular genetics perspective. Res Rep Biochem 5:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You L, He B, Uematsu K, Xu Z, Mazieres J, Lee A, McCormick F, Jablons DM (2004) Inhibition of Wnt-1 signaling induces apoptosis in beta-catenin-deficient mesothelioma cells. Cancer Res 64:3474–3478. 10.1158/0008-5472.CAN-04-0115 [DOI] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J (2016) TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90:724–739. 10.1016/j.neuron.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Liu CC, Atagi Y, Chen XF, Jia L, Yang L, He W, Zhang X, Kang SS, Rosenberry TL, Fryer JD, Zhang YW, Xu H, Bu G (2016) Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiol Aging 42:132–141. 10.1016/j.neurobiolaging.2016.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XY, Zhang HL, Luo Q, Zhu J (2011) Kainic acid-induced neurodegenerative model: potentials and limitations. J Biomed Biotechnol 2011:457079. 10.1155/2011/457079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Li D, Wu Y, Huang X, Wu M (2014) TREM-2 promotes macrophage-mediated eradication of Pseudomonas aeruginosa via a PI3K/Akt pathway. Scand J Immunol 79:187–196. 10.1111/sji.12148 [DOI] [PubMed] [Google Scholar]
- Zhu W, Zheng H, Shao X, Wang W, Yao Q, Li Z (2010) Excitotoxicity of TNFalpha derived from KA activated microglia on hippocampal neurons in vitro and in vivo. J Neurochem 114:386–396. 10.1111/j.1471-4159.2010.06763.x [DOI] [PubMed] [Google Scholar]