

Abstract
The study was designed to determine the role of long noncoding RNA (lncRNA), metastasis-associated lung adenocarcinoma transcript 1 (Malat1), in ischemic stroke outcome. Primary mouse brain microvascular endothelial cells (BMECs) were cultured and treated with Malat1 GapmeR before 16 h oxygen and glucose depravation (OGD). Cell death was assayed by LDH and MTT methods. Malat1 knock-out and wild-type mice were subjected to 1 h of middle cerebral artery occlusion (MCAO) and 24–72 h of reperfusion. To explore the underlying mechanism, apoptotic and inflammatory factors were measured by qPCR, ELISA, and Western blotting. The physical interaction between Malat1 and apoptotic or inflammatory factors was measured by RNA immunoprecipitation. Increased Malat1 levels were found in cultured mouse BMECs after OGD as well as in isolated cerebral microvessels in mice after MCAO. Silencing of Malat1 by Malat1 GapmeR significantly increased OGD-induced cell death and Caspase 3 activity in BMECs. Silencing of Malat1 also significantly aggravated OGD-induced expression of the proapoptotic factor Bim and proinflammatory cytokines MCP-1, IL-6, and E-selectin. Moreover, Malat1 KO mice presented larger brain infarct size, worsened neurological scores, and reduced sensorimotor functions. Consistent with in vitro findings, significantly increased expression of proapoptotic and proinflammatory factors was also found in the cerebral cortex of Malat1 KO mice after ischemic stroke compared with WT controls. Finally, we demonstrated that Malat1 binds to Bim and E-selectin both in vitro and in vivo. Our study suggests that Malat1 plays critical protective roles in ischemic stroke.
SIGNIFICANCE STATEMENT Accumulative studies have demonstrated the important regulatory roles of microRNAs in vascular and neural damage after ischemic stroke. However, the functional significance and mechanisms of other classes of noncoding RNAs in cerebrovascular pathophysiology after stroke are less studied. Here we demonstrate a novel role of Malat1, a long noncoding RNA that has been originally identified as a prognostic marker for non-small cell lung cancer, in cerebrovascular pathogenesis of ischemic stroke. Our experiments have provided the first evidence that Malat1 plays anti-apoptotic and anti-inflammatory roles in brain microvasculature to reduce ischemic cerebral vascular and parenchymal damages. Our studies also suggest that lncRNAs can be therapeutically targeted to minimize poststroke brain damage.
Keywords: apoptosis, cerebral ischemia, cerebral vascular endothelial cell, inflammation, Malat1, stroke
Introduction
Stroke is one of the leading causes of death worldwide and a major cause of long-term disability (Ouyang et al., 2013). Although many clinical trials for stroke therapy have been completed, the only effective treatment to date is thrombolysis. The development of effective therapies in the clinical setting is seriously limited by several reasons, including the rapid development of brain injury following ischemia, the complex interplays among signaling pathways, and the treatment window for specific targets (Yin et al., 2010a).
Noncoding RNAs (ncRNAs) are defined as untranslated regulatory RNA molecules. In humans, DNA sequences responsible for nonprotein coding regions comprise at least 98% of the total genome (Yin et al., 2014). Noncoding RNAs can be approximately divided into small (<200 nt) noncoding RNAs, which include microRNAs, and long noncoding RNAs (>200 nt, lncRNAs) (Bartel and Chen, 2004). They have within the past decade emerged as key mediators of posttranscriptional gene expression/function in pathological aspects of ischemic stroke. Accumulative studies have demonstrated the important regulatory roles of microRNAs in vascular and neural damage after ischemic stroke. However, the functional significance and mechanisms of other classes of noncoding RNAs in cerebrovascular pathophysiology after stroke are less studied. Recently, Mehta et al. (2015) reported that lncRNA FosDT promotes ischemic brain injury by interacting with REST-associated chromatin modifying proteins, suggesting that lncRNAs can be therapeutically targeted to minimize poststroke brain damage.
Malat1 is a highly abundant and evolutionary conserved lncRNA, which was first described to be associated with metastasis of lung tumors (Ji et al., 2003). A previous study reported that Malat1 promoted aggressive renal cell carcinoma through Ezh2 (Hirata et al., 2015). Malat1 silencing significantly suppresses the proliferation of esophageal squamous cell carcinoma at the G2/M phase (Wang et al., 2015). Furthermore, it was reported that Malat1 regulated endothelial cell function and vessel growth (Michalik et al., 2014). However, the function of Malat1 in the cerebral vasculature after stroke has not been studied.
Our previous study using RNA-sequencing (RNA-seq) technology has identified Malat1 as one of the most highly upregulated oxygen-glucose deprivation (OGD)-responsive endothelial lncRNAs (Zhang et al., 2016). Here we further studied the functional role of Malat1 both in cultured mouse brain microvascular endothelial cells (BMECs) and in a Malat1-deficient mouse model. We found increased Malat1 levels both in vitro and in vivo after ischemic insults. In cultured BMECs, downregulation of Malat1 by GapmeRs significantly increased OGD-induced cell death and Caspase 3 activity. Further, we showed that Malat1 played antiapoptotic and anti-inflammatory roles in cultured BMECs after OGD and in mice after ischemic stroke. Moreover, RNA immunoprecipitation studies indicate that Malat1 physically binds to Bim and E-selectin. Collectively, these results suggest that Malat1 plays critical protective roles in ischemic stroke.
Materials and Methods
Experimental mice.
Malat1 KO mice on a C57BL/6J (RRID: IMSR_JAX:000664) background were kindly provided by Dr. David L. Spector (Zhang et al., 2012). Generally, Malat1 KO mice are viable and fertile with normal appearance, behavior, growth, and litter size. The animal study protocol was approved by the University of Pittsburgh Animal Care and Use Committee.
Mouse model of transient focal cerebral ischemia.
Focal cerebral ischemia was induced in mice by intraluminal middle cerebral artery occlusion (MCAO) as described previously (Yin et al., 2010b). Briefly, male mice (8–10 weeks, 23–25 g) were anesthetized with 1.5%–3% isoflurane (Henry Schein Animal Health). A 2 cm length of a 7–0 rounded tip nylon suture was gently advanced from the external carotid artery up to the internal carotid artery until regional cerebral blood flow was reduced to <16% of baseline. After 60 min of MCA occlusion, blood flow was restored by removing the suture, and the mice were allowed to recover for 1–7 d. Changes in cerebral blood flow, arterial blood gases, mean arterial pressure, and heart rate were monitored in animals 30 min before, during, and 30 min after MCAO. The rectal temperature was controlled at 37.0 ± 0.5°C during surgery.
Assessment of infarct volume, neurological deficit, and sensorimotor function.
Mice were killed on day 1, and infarct area was measured from the brain slices using 2% TTC staining as described previously (Yin et al., 2013). Neurobehavioral deficits were determined by the adhesive removal test, foot fault test, and cylinder test at 1, 3, 5, and 7 d of reperfusion (Wang et al., 2014). Following cerebral ischemia, mice were also tested for neurological deficits and scored on a 5 point scale: 0, no observable neurological deficits (normal); 1, failure to extend right forepaw (mild); 2, circling to the contralateral side (moderate); 3, falling to the right (severe); 4, mice could not walk spontaneously; 5, depressed level of consciousness (very severe).
Isolation of cerebral vessels.
The mouse brains were removed, and cerebral microvessels were isolated using a previously described method (Yin et al., 2006). The final vessel pellet was stored at −80°C until various biochemical assays were performed.
Cell cultures and OGD.
Mouse primary BMECs were purchased from Cell Biologics (catalog #C57-6023). Mouse N2A neuroblastoma (N2A) cells were purchased from ATCC (catalog #CCL-131, RRID: CVCL_0470). Mouse BMECs and N2A cells (2–10 passages) were grown to 85%–95% confluency before use. To mimic ischemia-like conditions in vitro, mouse BMEC cultures were exposed to OGD for 16 h (Yin et al., 2002) and mouse N2A cultures (2–4 passages) were exposed to OGD for 4 h (Yin et al., 2010a). In some experiments, BMECs were treated with 50 nm Malat1 GapmeR or Malat1 Control (Ctrl) for 48 h before OGD exposure.
Assessment of cell death and caspase-3 activity.
The extent of mouse BMEC and N2A cell death was assessed by MTT and LDH assays. Caspase-3 activity in isolated cerebral microvessels was detected by a commercial Caspase-3 Colorimetric Activity Assay Kit (Millipore) according to manufacturer's instructions.
Quantitative real-time PCR.
Total RNA was isolated from BMEC cultures or cerebral cortex by using an RNeasy Mini Kit (QIAGEN) or Trizol (Invitrogen). Quantitative real-time RT-PCR was performed with a Bio-Rad CFX Connect thermocycler, iScript cDNA synthesis kit, and iTaq Universal SYBR Green supermix (Bio-Rad). Specific primers used for the reaction are as follows: Malat1 forward, 5′-ggcggaattgctggtagttt-3′; Malat1 reverse, 5′-agcatagcagtacacgcctt-3′; Bim forward, 5′-cggatcggagacgagttca-3′; Bim reverse, 5′-ttcagcctcgcggtaatca-3′; Bax forward, 5′-caggatgcgtccaccaaga-3′; Bax reverse, 5′-ccatattgctgtccagttcatctc-3′; MCP-1 forward, 5′-gcaccagcaccagccaactctcact-3′; MCP-1 reverse, 5′-cattccttcttggggtcagcacag-3′; IL-6 forward, 5′-agttgccttcttgggactga-3′; IL-6 reverse, 5′-tccacgatttcccagagaac-3′; E-selectin forward, 5′-agctacccatggaacacgac-3′; E-selectin reverse, 5′-acgcaagttctccagctgtt-3′; cyclophilin forward, 5′-actcctcatttagatgggcatca-3′; cyclophilin reverse, 5′-gagtatccgtacctccgcaaa-3′. The relative mRNA expression was normalized to cyclophilin RNA levels (Yin et al., 2011). PCR experiments were repeated 3 times, each using separate sets of cultures.
Western blot.
Samples from the mouse cerebral cortex or BMEC cultures were homogenized in lysis buffers, and total protein was isolated as described previously (Zhu et al., 2009). Various primary antibodies were used, including Bim (1:500; Cell Signaling Technology catalog #2933 RRID: AB_1030947), Bax (1:1000; Cell Signaling Technology catalog #2772 RRID: AB_329921), E-selectin (4 μg/ml; Abcam catalog #ab18981 RRID: AB_470289), MCP-1 (1:500; Cell Signaling Technology catalog #2029 RRID: AB_1264199), IL-6 (1:500; Cell Signaling Technology catalog #12912), and β-actin (1:4000; Sigma-Aldrich catalog #A5441 RRID: AB_476744).
In vitro cytokine analysis.
Mouse BMEC cultures were treated with 50 nm Malat1 GapmeR or Malat1 Ctrl for 48 h before OGD exposure for 16 h. MCP-1 and IL-6 in supernatants were measured by ELISA (R&D Biosystems).
RNA immunoprecipitation.
RNA-immunoprecipitation (RIP) was performed using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore) according to the manufacturer's instructions. Anti-Bim (Thermo Fisher Scientific catalog #MA5-14848 RRID: AB_10981505) or anti-E-selectin (Abcam catalog #ab6630 RRID: AB_305609) antibodies were used for RIP. Coprecipitated RNAs from total extracts of OGD-treated BMEC cultures or mouse ischemic brains were analyzed by qPCR using primers specific for Malat1. Total RNA (input control) and the isotype control were assayed simultaneously to show the binding specificity between Malat1 and Bim/E-selectin.
RNA sequencing and bioinformatics analysis.
RNA-seq was performed using a previously described method (Zhang et al., 2016).
RNA FISH.
To detect Malat1 in situ expression, BMEC cells were fixed in 4% formaldehyde for 15 min at room temperature and then permeabilized with 0.5% Triton X-100 on ice for 10 min. Cells were washed in PBS 3× for 10 min and rinsed once in 2× SSC before hybridization. Hybridization was performed using Malat1 FITC-labeled locked nucleic acid (LNA) probes (Exiqon) in a moist chamber at 37°C for 8 h.
Statistical analysis.
Quantitative data are expressed as mean ± SEM. Differences among three or more groups were statistically analyzed by one-way ANOVA followed by Bonferroni's post hoc test. Comparisons between two experimental groups were based on a two-tailed t test. A p value <0.05 was considered significant.
Results
Induction of Malat1 levels in cultured BMECs after OGD and mouse cerebral microvessels after focal cerebral ischemia
To uncover the transcriptomic effects of ischemic stimuli on cerebral vascular endothelium, mouse BMECs were subjected to OGD for 16 h. We then performed RNA-seq by using RNA isolated from control and OGD-treated cultures from four biological replicates (Zhang et al., 2016). Malat1 was identified among the three most highly upregulated lncRNA transcripts and showed a 6- to 7-fold increased expression in BMEC cultures after 16 h OGD (Fig. 1A) as well as in isolated cerebral microvessels from mice that were subjected to transient 1 h MCAO and 24 h reperfusion (Fig. 1B) by quantitative real-time PCR (qPCR) analysis. Of note, Malat1 was not pathologically increased in whole mouse brains in response to the same ischemic insult (Fig. 1B). The expression and nuclear localization of Malat1 were also shown in BMECs by in situ hybridization using Malat1 FITC-labeled LNA probes (Exiqon) but not by the control probe (Fig. 1C). These results suggest that upregulated Malat1 may play an important role in cerebral endothelial pathogenesis after ischemic stroke.
Figure 1.
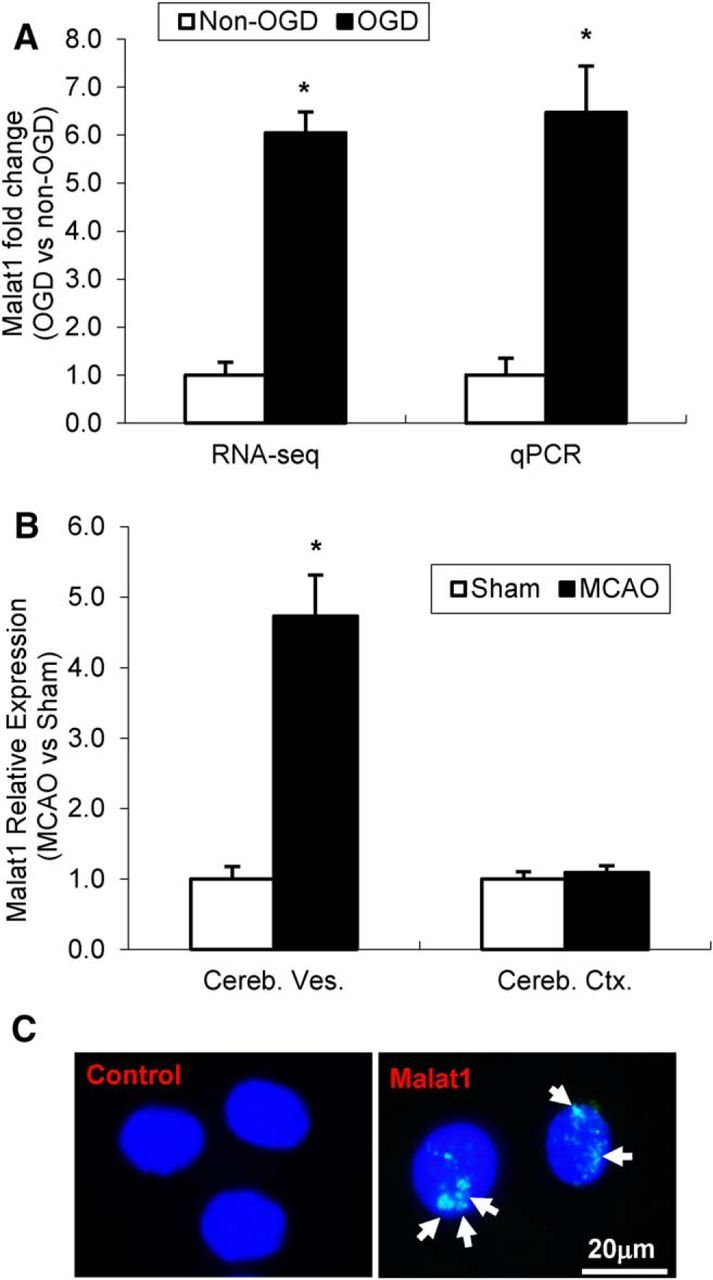
Malat1 expression in mouse BMEC cultures and mouse cerebral microvessels following focal cerebral ischemia. A, RNA-seq and qPCR data showing the expression of Malat1 in cultured BMECs. Malat1 was significantly induced after OGD treatment. B, Malat1 level was also examined in isolated cerebral microvessels (Cereb. Ves.) and cerebral cortex (Cereb. Ctx.) in mice subjected to 1 h MCAO and 24 h reperfusion (n = 3). Significantly increased expression of Malat1 was found in isolated cerebral microvessels after MCAO. C, In situ hybridization detection of Malat1 nuclear distribution (arrows) in BMECs. Data are mean ± SEM. *p < 0.05 versus Non-OGD or Sham group. Scale bar, 20 μm.
Silencing of Malat1 augments ischemia-induced endothelial cell death in vitro
OGD treatment for 16 h induced dramatic cell death in mouse BEMC cultures in our previous study. Because Malat1 was highly expressed and most profoundly increased by OGD, we further explored the function of Malat1 in mouse endothelial cells by manipulating Malat1 expression. We used fluorescein-labeled Malat1 GapmeR as a genetic approach to achieve Malat1 silencing in mouse BMEC cultures. GapmeRs are single-stranded oligonucleotides that consist of a DNA stretch flanked by LNA nucleotides (Dullea et al., 2014). Base-pairing with the targeted lncRNA in the nucleus induced degradation by an RNase H-dependent mechanism (Lanford et al., 2010). Successful delivery of LNA-Malat1 GapmeR into mouse BMECs was confirmed by fluorescence microscopy (Fig. 2A), and the basal and OGD-induced Malat1 levels (Fig. 2B) were significantly decreased by transfection of BMECs with LNA-Malat1 GapmeR for 48 h versus transfection with the Malat1 control. Treatment with Malat1 GapmeR significantly increased OGD-induced cerebral vascular endothelial cell death compared with the Malat1 ctrl group, as evidenced by LDH quantitation (Fig. 2C) and MTT assays (Fig. 2D). Malat1 GapmeR also significantly augmented OGD-activated caspase 3 activity (Fig. 2E) in mouse BMECs. We also examined Malat1 expression in neuroblastoma cell line N2A by qPCR analysis. Malat1 was expressed at relatively low level in N2A cells yet still can be induced by OGD treatment (Fig. 2F). LDH (Fig. 2G) and MTT (Fig. 2H) assays demonstrated that exposure to OGD for 4 h induced dramatic N2A cell death, and silencing of Malat1 by GapmeR did not affect cell death further.
Figure 2.
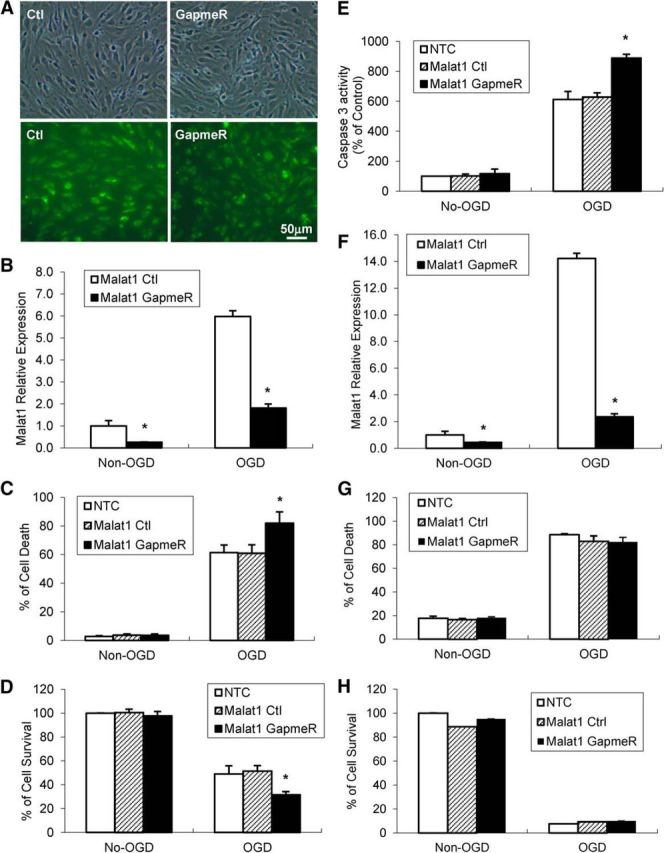
Silencing of Malat1 by LNA-GapmeRs increases OGD-induced mouse BMEC cell death. GapmeRs are DNA oligonucleotides with LNA residues at the 3′ and 5′ ends that induce RNase H-mediated degradation of nuclear RNA. GapmeRs targeting Malat1 or scrambled control GapmeRs were transfected at 50 nm. A, Images were taken 48 h after transfection. GFP+ cells indicated successfully transfected cells. B, qPCR showed significantly reduced Malat1 levels in BMECs after transfection in the absence or presence of 16 h OGD exposure. C, D, Exposure to OGD results in obvious BMEC cell death detected by LDH assay (C) and MTT assay (D). Increased OGD-induced cell death in Malat1 GapmeR-transfected BMECs compared with Malat1 Ctrl-transfected cells. E, Exposure to OGD increases caspase 3 activation in BMEC cells transfected with Malat1 GapmeR. F, qPCR showed significantly reduced Malat1 levels in N2A cells after transfection in the absence or presence of 4 h OGD exposure. G, H, Exposure to OGD resulted in remarkable N2A cell death detected by LDH (G) and MTT assay (H). However, silencing of Malat1 by GapmeR had no effects on OGD-induced N2A cell death. Data are mean ± SEM (n = 3). *p < 0.05 versus Malat1 control + Non-OGD or OGD group. Scale bar, 50 μm.
Genetic deficiency of Malat1 potentiates ischemic brain injury in mice
Given that Malat1 plays endothelial protective roles in vitro, we next determined whether Malat1 has any impact on ischemic brain injury in vivo. We used a silencing strategy using Malat1 knock-out (KO) mice on a C57BL/6J background (Zhang et al., 2012). Malat1 KO and littermate control (WT) mice were subjected to transient MCAO for 1 h followed by 24 h reperfusion. In comparison with the Malat1 WT mice, Malat1 KO mice showed a larger cerebral infarct volume (Fig. 3A,B) and a significantly more severe neurological deficit (Fig. 3C) in response to ischemic insults. These results suggest that Malat1 silencing exacerbates ischemic brain damage.
Figure 3.

Effects of Malat1 genetic deficiency on ischemic infarction and neurological outcomes. Malat1 KO and littermate control (Malat1 WT) mice were subjected to 1 h MCAO and 24 h reperfusion. A, The 2% TTC-stained coronal sections are shown at different brain levels posterior to the frontal pole. B, C, Quantitative analysis was performed on infarct volume (B) and neurological deficits (C) in mice after stroke. In comparison with Malat1 WT mice, Malat1 KO mice showed larger ischemia-induced brain infarction (n = 11) and worsened neurological outcomes (n = 11). Data are mean ± SD. *p < 0.05 versus the WT group.
Inhibitory recovery of sensorimotor functions in Malat1 KO mice after focal cerebral ischemia
Stroke in humans often causes extensive sensorimotor dysfunction. We therefore used a series of neurobehavioral tests to examine functional outcomes of Malat1 deficiency in stroke mice. Malat1 KO and littermate WT mice were subjected to 1 h MCAO followed by 1–7 d reperfusion, and assessed by adhesive tape removal, foot fault, and cylinder tests. Malat1 KO mice showed significantly increased touch time (Fig. 4A), removal time (Fig. 4B), and fault steps (Fig. 4C) compared with Malat1 WT controls after MCAO. In addition, Malat1 KO mice exhibited less balanced use of both limbs compared with Malat1 WT mice (Fig. 4D). Together, these results suggest that Malat1 plays a critical role in prompting neurological functional recovery from ischemic brain insults.
Figure 4.

Genetic deletion of Malat1 gene aggravates sensorimotor functions of mice after MCAO. A–D, Focal cerebral ischemia was induced in Malat1 WT and Malat1 KO mice by 1 h MCAO followed by 1–7 d reperfusion. Ischemic mice (n = 8) were subjected to adhesive tape removal (A, B), foot fault (C), and cylinder (D) tests for examination of sensorimotor functions. In comparison with Malat1 WT mice, Malat1 KO mice showed increased touch time, removal time, fault steps, and less balanced use of both limbs after MCAO. Data are mean ± SD. *p < 0.05 versus the Malat1 WT group.
Silencing of Malat1 increases proapoptotic Bim expression after ischemic insults
There is evidence suggesting that Malat1 is involved in genome stability (Tripathi et al., 2010) and cell cycle checkpoint regulation (Yang et al., 2013), all contributing to uncontrolled cell growth. To explore the underlying mechanisms contributing to the protective role of Malat1, we first investigated the mRNA expressional profiles of a number of Bcl-2 family of apoptosis regulators, including Bim, Bid, Bad, Bax, Bmf, and DP5 in mouse BMEC cultures with the presence or absence of Malat1 GapmeR. We found that silencing of Malat1 significantly aggravated OGD-induced mRNA levels of Bim (Fig. 5A), but not other proapoptotic genes tested (data not shown) by qPCR. The increased Bim expression in Malat1 GapmeR-treated BMECs at the protein level was also confirmed by Western blotting (Fig. 5B). Consistent with in vitro observations, increased Bim mRNA (Fig. 5C) and protein (Fig. 5D) levels were also found in the cerebral cortex of Malat1 KO mice subjected to 1 h MCAO followed by 24 h (Fig. 5) or 72 h (data not shown) reperfusion in comparison with Malat1 WT. These results suggested that Malat1 appeared to have antiapoptotic effects in cerebral endothelium by regulating the key apoptotic cell death regulator Bim.
Figure 5.

The effect of Malat1 silencing on proapoptotic factors in mouse BMECs after OGD and in mouse brain after focal cerebral ischemia. A, Cultured BMECs were transfected by LNA-Malat1 GapmeR and scramble control for 48 h, and then subjected to 16 h OGD. The expression levels of Bim and Bax were determined by qPCR and normalized to cyclophilin (n = 3). B, The protein levels of Bim and Bax were determined by Western blotting, with β-actin as the loading control. C, Stroke was induced in Malat1 WT and Malat1 KO mice by 1 h MCAO followed by 24 h reperfusion. Expression levels of Bim and Bax were determined by qPCR and normalized to cyclophilin (n = 6/group). D, The protein levels of Bim and Bax were determined, with β-actin as the loading control. Silencing of Malat1 increased the expression of the proapoptotic factor Bim in cultured BMECs and in the mouse ischemic brain regions after MCAO. Experiments were repeated three times, and representative blots are displayed. Data are mean ± SEM. *p < 0.05 versus Malat1 Ctrl + ODG or Malat1 WT + MCAO group. #p < 0.05 versus Malat1 Ctrl + Non-OGD or Malat1 WT + sham group.
Silencing of Malat1 increases proinflammatory cytokine levels in BMEC cultures
It is widely accepted that endothelial inflammation and subsequent impairment of endothelial function contribute to ischemic brain injury (Sandoval and Witt, 2008). We therefore determined whether altered expression of Malat1 could regulate the inflammatory response in BMEC cultures. By qPCR, we measured mRNA abundance of three well-known proinflammatory cytokines, E-selectin (Fig. 6A), MCP-1 (Fig. 6B), and IL-6 (Fig. 6C). The 16 h of OGD treatment significantly increased the mRNA production of all three proinflammatory factors. Silencing of Malat1 by GapmeR further aggravated OGD-induced E-selectin, MCP-1, and IL-6 mRNA levels. We also observed increased E-selectin expression in Malat1 GapmeR-treated BMECs at the protein level by Western blotting (Fig. 6D) and upregulated MCP-1 (Fig. 6E) and IL-6 (Fig. 6F) levels in Malat1 GapmeR-treated BMEC culture medium by ELISA. These data suggested that Malat1 has anti-inflammatory roles in cerebral endothelium in an in vitro ischemic stroke model.
Figure 6.

Effects of Malat1 silencing on major proinflammatory cytokines in mouse BMECs after OGD. A–C, Cultured BMECs were transfected with LNA-Malat1 GapmeR and scramble control for 48 h, and then subjected to 16 h OGD. The expression levels of E-selectin (A), MCP-1 (B), and IL-6 (C) were determined by qPCR and normalized to cyclophilin (n = 3). D, The protein level of E-selectin was determined by Western blotting, with β-actin as the loading control. Experiments were repeated three times, and representative blots are displayed. E, F, Protein levels of MCP-1 (E) and IL-6 (F) in Malat1 Ctrl and Malat1 GapmeR-treated BMEC culture medium were analyzed by ELISA (n = 3). Silencing of Malat1 by GapmeR treatment increased the expression of proinflammatory cytokines MCP-1, IL-6, and E-selectin in cultured BMECs after 16 h OGD. Data are mean ± SD. *p < 0.05 versus Malat1 Ctrl + OGD group. #p < 0.05 versus Malat1 Ctrl + Non-OGD group.
Genetic deficiency of Malat1 augments inflammation in mouse brain after MCAO
To confirm the anti-inflammatory roles of Malat1 in vivo, Malat1 WT and Malat1 KO mice were subjected to 1 h MCAO followed by 24 h (Fig. 7) or 72 h (data not shown) reperfusion. Consistent with in vitro results, significantly increased expression of E-selectin (Fig. 7A), MCP-1 (Fig. 7B), and IL-6 (Fig. 7C) at mRNA and protein (Fig. 7D) levels were found in Malat1 KO mouse cortex after ischemic stroke in comparison with Malat1 WT controls. Collectively, it appears that Malat1 plays cerebrovascular protective roles by inhibiting inflammatory responses induced by ischemic insults.
Figure 7.

Effects of Malat1 genetic deficiency on proinflammatory cytokines in mouse brain after focal cerebral ischemia. A–C, Stroke was induced in Malat1 WT and KO mice by 1 h MCAO followed by 24 h reperfusion. Total RNA was extracted from Malat1 WT and Malat1 KO mouse cortex 24 h after MCAO, and expression levels of E-selectin (A), MCP-1 (B), and IL-6 (C) were determined by qPCR and normalized to cyclophilin (n = 6/group). D, Total protein was extracted and subjected to gel electrophoresis. The protein levels of E-selectin, MCP-1, and IL-6 were determined, with β-actin as the loading control. Increased expression levels of the proinflammatory cytokines MCP-1, IL-6, and E-selectin were induced in Malat1 KO mice 24 h after MCAO compared with Malat1 WT mice. Experiments were repeated three times, and representative blots are displayed. Data are mean ± SEM. *p < 0.05 versus Malat1 WT + MCAO group. #p < 0.05 versus Malat1 WT + Sham group.
Malat1 physically binds to Bim and E-selectin
lncRNAs are known to associate with chromatin and serve regulatory functions through sequence-specific hybridization and/or through structural and spatial mechanisms (Zhao et al., 2010). One of the most straightforward explanations for Malat1's inhibitory effects on proapoptotic factors and proinflammatory cytokines would be that Malat1 physically associates with them. To test this possibility, we first performed RNA immunoprecipitation with the Bim or E-selectin antibody from total extracts of OGD-treated BMEC cultures. We observed ∼16-fold enrichment of Malat1 in the anti-Bim immunoprecipitate and ∼31-fold enrichment of Malat1 in the anti-E-selectin immunoprecipitate compared with IgG controls (Fig. 8A). To further confirm the physical interaction of Malat1 with Bim/E-selectin in vivo, we performed RIP with the Bim or E-selectin antibody from fresh brain extracts of C57BL/6J mice that underwent 1 h MCAO followed by 24 h reperfusion. Consistently, we found ∼32-fold enrichment of Malat1 in the anti-Bim brain immunoprecipitate and ∼34-fold enrichment of Malat1 in the anti-E-selectin brain immunoprecipitate compared with IgG controls (Fig. 8B). Together, the results indicated that Malat1 physically associates with Bim and E-selectin in vitro and in vivo.
Figure 8.

Malat1 physically associates with Bim and E-selectin in mouse BMECs and mouse brains. A, B, Interaction between Malat1 and Bim or E-selectin revealed by RIP experiments. Total extracts of cultured BMECs after 16 h OGD (A) and mouse brains after 24 h MCAO (B) were immunoprecipitated with control IgG, anti-Bim, or anti-E-selectin antibody, and the complexes were analyzed for the presence of Malat1 by qPCR. Fold enrichment over the normal rabbit IgG was calculated by the ΔΔCt method (n = 3). Data are mean ± SEM of two independent experiments with triplicate wells. *p < 0.05 versus IgG group.
Discussion
There is growing evidence that lncRNAs are important regulators of physiological and pathological responses; however, their potential importance in cerebrovascular pathophysiology is only now emerging (Mehta et al., 2015). It was reported by Mehta et al. (2015) that the expression of lncRNA FosDT was induced in adult rats following transient MCAO. FosDT knockdown significantly ameliorated the postischemic motor deficits and reduced the infarct volume (Mehta et al., 2015). Malat1 was first described to be associated with metastasis of lung tumors (Ji et al., 2003). Ji et al. (2003) reported, for the first time, that expression of Malat1 was significantly associated with metastasis in non-small cell lung cancer patients. Also Malat1's association with metastasis was stage- and histology-specific (Ji et al., 2003). Other studies showed that aberrant expression of Malat1 correlates with tumor development (Xiao et al., 2015), progression (Lu et al., 2015), metastasis (Cai et al., 2016), and survival (Wei and Niu, 2015) in many cancer types. In addition to its oncogenic role in different cancers, Malat1 is also involved in other diseases, such as diabetes (Liu et al., 2014), myocardial infarction, and heart failure (Uchida and Dimmeler, 2015) and even in normal biologic processes, such as vessel growth (Thum and Fiedler, 2014), synaptogenesis (Liu et al., 2014), and myogenesis (Vausort et al., 2014). However, the functional significance of Malat1 in cerebrovascular physiology and pathophysiological disorders has never been studied.
In this study, we demonstrate a novel function of Malat1, which has been originally identified as a prognostic marker for non-small cell lung cancer, in modulating apoptotic and inflammatory responses in the mouse cerebral endothelium after in vitro and in vivo cerebral ischemic insults. This novel finding is strongly confirmed by our experimental evidence demonstrating that Malat1 expression was induced in mouse BMECs after OGD and in isolated cerebral microvessels in mice after MCAO and reperfusion. We also demonstrated that genetic deletion of Malat1 gene presented larger brain infarct size, worse neurological scores, and more severe sensorimotor dysfunction in mice after MCAO than WT controls. Moreover, we found that silencing of Malat1 augmented ischemia-induced endothelial loss, apoptosis, and inflammation in vitro and in vivo. Furthermore, we proved that Malat1 physically interacts with proapoptotic Bim and proinflammatory E-selectin. To our knowledge, our experiments have provided the first evidence that Malat1 plays anti-apoptotic and anti-inflammatory roles in brain microvasculature to reduce ischemic cerebral vascular and parenchymal damages.
It is well known that Malat1 is a nuclear-retained noncoding RNA (Zhao et al., 2016); however, several recent publications report the presence of Malat1 in the cytoplasm to exert its function (Dodd et al., 2013; Han et al., 2015), suggesting that localization of Malat1 may be cell type-specific. Michalik et al. (2014) determined the localization of several lncRNAs, including Malat1 in endothelial cells derived from various human vascular beds and found that Malat1 was highly enriched in the nuclear fraction (Michalik et al., 2014). The subcellular distribution of Malat1 in mouse BMEC cultures was determined in our study by in situ hybridization. It appears that Malat1 predominantly localized in the nucleus, which is consistent with most other published reports in HeLa cells (Tripathi et al., 2010), lung cancer cells (Tano et al., 2010), and THP-1 cells (Zhao et al., 2016).
There is evidence suggesting that Malat1 may be involved in cell cycle regulation. Its role in G2/M regulation has been specifically emphasized in several studies (Tripathi et al., 2010; Yang et al., 2013). Tripathi et al. (2010) reported that Malat1 depletion leads to an accumulated G2/M population, indicating a critical involvement of Malat1 in cell cycle checkpoints. Michalik et al. (2014) managed to silence Malat1 expression in human umbilical vein endothelial cells by either siRNA or LNA-GapmeR treatment and found reduced proliferative ability and reduced progression through the cell cycle in both treatments (Michalik et al., 2014). In our study, Malat1 silencing by GapmeR treatment in mouse BMEC cells augments OGD-induced cell death and caspase-3 activity (Fig. 2). We are among the first to examine the effects of Malat1 on cerebral vascular endothelial damage after ischemic insults. Ischemic endothelial cell death undergoes both necrosis and apoptosis. In rodent experimental stroke models, activation of both caspase-dependent and caspase-independent apoptotic pathways have been shown to be responsible for endothelial cell apoptosis. In response to various ischemic stimuli, many proapoptotic factors, such as BH3-only proteins, have been identified in cerebral endothelium (Yin et al., 2014). Bim belongs to the BH3-only subclass of the Bcl-2 family of apoptosis regulators. These proteins contain only one of the Bcl-2 homology regions and are essential initiators of apoptotic cell death (Kelekar and Thompson, 1998). We examined the expression profile of major BH3-only proapoptotic factors and found that Bim was significantly upregulated in BMEC cultures treated with Malat1 GapmeR after OGD compared with Malat1 controls and in Malat1 KO mice after MCAO in comparison with Malat1 WT littermates (Fig. 5), suggesting that Malat1 plays an anti-apoptotic role through inhibitory effects on Bim.
Inflammatory mediators generated/released from various neural cells are involved in molecular cascades determining stroke outcome (Huang et al., 2006). During cerebral ischemia, cerebral endothelial cells are stimulated by inflammatory mediators in ischemic brains and undergo proinflammatory activation. Activated cerebral endothelium have the capacity to produce and/or secrete many inflammatory mediators, most important of which are the selectins (P-selectin, E-selectin), proinflammatory cytokines, and integrins, thus becoming a source of inflammation themselves in the cerebral vasculature (Ruetzler et al., 2001; Huang et al., 2006). In our study, silencing of Malat1 aggregated ischemia-triggered proinflammatory cytokines IL-6, MCP-1, and E-selectin levels in BMEC cultures (Fig. 6) and in mouse brains. It appears that Malat1 plays vaso/neuroprotective roles by inhibiting inflammatory responses induced by ischemic stroke. More systematic approaches, such as RNA-sequencing or microarray analysis, could be used in the future study to uncover all target genes that are responsible for Malat1-mediated protection after ischemic stroke.
The most straightforward explanation for Malat1 regulation of Bim or E-selectin would be through direct binding. Recently, Zhao et al. (2016) performed RIP with the p65 antibody from subcellular extracts of LPS-stimulated THP-1 macrophages, and observed ∼7-fold enrichment of nuclear Malat1 in the anti-p65 immunoprecipitate compared with the IgG control. We also used RIP experiments in our study, and we found ∼34-fold enrichment of Malat1 in the anti-E-selectin brain immunoprecipitate and ∼32-fold enrichment of Malat1 in the anti-Bim brain immunoprecipitate compared with IgG controls. This is the first study to document a direct physical association between Malat1 and Bim or E-selectin in vitro and in vivo.
In conclusion, we reported novel findings that Malat1 protects the cerebral microvasculature and parenchyma from cerebral ischemic insults by inhibiting endothelial cell death and inflammation. There is growing evidence showing that ischemia-induced cerebral endothelial cell injury, endothelial cell inflammation, and subsequent impairment of endothelial cell function increase cerebrovascular permeability and blood–brain barrier leakage, contributing to ischemic brain injury (Ruetzler et al., 2001; Ouyang et al., 2013). Therefore, the findings of the present study may have therapeutic implications. Future endeavors are needed to investigate whether pharmacological manipulation of the Malat1 gene affects stroke outcomes and neurological function.
Footnotes
This work was supported by National Institutes of Health Grants NS094930, NS091175, and NS086820 to K.-J.Y.
The authors declare no competing financial interests.
References
- Bartel DP, Chen CZ (2004) Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet 5:396–400. 10.1038/nrg1328 [DOI] [PubMed] [Google Scholar]
- Cai X, Liu Y, Yang W, Xia Y, Yang C, Yang S, Liu X (2016) Long noncoding RNA MALAT1 as a potential therapeutic target in osteosarcoma. J Orthop Res 34:932–941. 10.1002/jor.23105 [DOI] [PubMed] [Google Scholar]
- Dodd DW, Gagnon KT, Corey DR (2013) Digital quantitation of potential therapeutic target RNAs. Nucleic Acid Ther 23:188–194. 10.1089/nat.2013.0427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dullea R, Salatto C, Sciabola S, Chen T, Dimattia D, Gandhok H, Kreeger J, Weng Y, Clark T, Vage C, Stanton R (2014) Study of CRTC2 pharmacology using antisense oligonuceotides. Nucleic Acid Ther 24:127–138. 10.1089/nat.2013.0456 [DOI] [PubMed] [Google Scholar]
- Han X, Yang F, Cao H, Liang Z (2015) Malat1 regulates serum response factor through miR-133 as a competing endogenous RNA in myogenesis. FASEB J 29:3054–3064. 10.1096/fj.14-259952 [DOI] [PubMed] [Google Scholar]
- Hirata H, Hinoda Y, Shahryari V, Deng G, Nakajima K, Tabatabai ZL, Ishii N, Dahiya R (2015) Long noncoding RNA MALAT1 promotes aggressive renal cell carcinoma through Ezh2 and interacts with miR-205. Cancer Res 75:1322–1331. 10.1158/0008-5472.CAN-14-2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Upadhyay UM, Tamargo RJ (2006) Inflammation in stroke and focal cerebral ischemia. Surg Neurol 66:232–245. 10.1016/j.surneu.2005.12.028 [DOI] [PubMed] [Google Scholar]
- Ji P, Diederichs S, Wang W, Böing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, Thomas M, Berdel WE, Serve H, Müller-Tidow C (2003) MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 22:8031–8041. 10.1038/sj.onc.1206928 [DOI] [PubMed] [Google Scholar]
- Kelekar A, Thompson CB (1998) Bcl-2-family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol 8:324–330. 10.1016/S0962-8924(98)01321-X [DOI] [PubMed] [Google Scholar]
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Ørum H (2010) Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327:198–201. 10.1126/science.1178178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JY, Yao J, Li XM, Song YC, Wang XQ, Li YJ, Yan B, Jiang Q (2014) Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis 5:e1506. 10.1038/cddis.2014.466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Luo F, Liu Y, Liu X, Shi L, Lu X, Liu Q (2015) Posttranscriptional silencing of the lncRNA MALAT1 by miR-217 inhibits the epithelial-mesenchymal transition via enhancer of zeste homolog 2 in the malignant transformation of HBE cells induced by cigarette smoke extract. Toxicol Appl Pharmacol 289:276–285. 10.1016/j.taap.2015.09.016 [DOI] [PubMed] [Google Scholar]
- Mehta SL, Kim T, Vemuganti R (2015) Long noncoding RNA FosDT promotes ischemic brain injury by interacting with REST-associated chromatin-modifying proteins. J Neurosci 35:16443–16449. 10.1523/JNEUROSCI.2943-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalik KM, You X, Manavski Y, Doddaballapur A, Zörnig M, Braun T, John D, Ponomareva Y, Chen W, Uchida S, Boon RA, Dimmeler S (2014) Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res 114:1389–1397. 10.1161/CIRCRESAHA.114.303265 [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Stary CM, Yang GY, Giffard R (2013) microRNAs: innovative targets for cerebral ischemia and stroke. Curr Drug Targets 14:90–101. 10.2174/138945013804806424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruetzler CA, Furuya K, Takeda H, Hallenbeck JM (2001) Brain vessels normally undergo cyclic activation and inactivation: evidence from tumor necrosis factor-alpha, heme oxygenase-1, and manganese superoxide dismutase immunostaining of vessels and perivascular brain cells. J Cereb Blood Flow Metab 21:244–252. 10.1097/00004647-200103000-00008 [DOI] [PubMed] [Google Scholar]
- Sandoval KE, Witt KA (2008) Blood–brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 32:200–219. 10.1016/j.nbd.2008.08.005 [DOI] [PubMed] [Google Scholar]
- Tano K, Mizuno R, Okada T, Rakwal R, Shibato J, Masuo Y, Ijiri K, Akimitsu N (2010) MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett 584:4575–4580. 10.1016/j.febslet.2010.10.008 [DOI] [PubMed] [Google Scholar]
- Thum T, Fiedler J (2014) LINCing MALAT1 and angiogenesis. Circ Res 114:1366–1368. 10.1161/CIRCRESAHA.114.303896 [DOI] [PubMed] [Google Scholar]
- Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, Blencowe BJ, Prasanth SG, Prasanth KV (2010) The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 39:925–938. 10.1016/j.molcel.2010.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S, Dimmeler S (2015) Long noncoding RNAs in cardiovascular diseases. Circ Res 116:737–750. 10.1161/CIRCRESAHA.116.302521 [DOI] [PubMed] [Google Scholar]
- Vausort M, Wagner DR, Devaux Y (2014) Long noncoding RNAs in patients with acute myocardial infarction. Circ Res 115:668–677. 10.1161/CIRCRESAHA.115.303836 [DOI] [PubMed] [Google Scholar]
- Wang J, Shi Y, Zhang L, Zhang F, Hu X, Zhang W, Leak RK, Gao Y, Chen L, Chen J (2014) Omega-3 polyunsaturated fatty acids enhance cerebral angiogenesis and provide long-term protection after stroke. Neurobiol Dis 68:91–103. 10.1016/j.nbd.2014.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li M, Wang Z, Han S, Tang X, Ge Y, Zhou L, Zhou C, Yuan Q, Yang M (2015) Silencing of long noncoding RNA MALAT1 by miR-101 and miR-217 inhibits proliferation, migration, and invasion of esophageal squamous cell carcinoma cells. J Biol Chem 290:3925–3935. 10.1074/jbc.M114.596866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Niu B (2015) Role of MALAT1 as a prognostic factor for survival in various cancers: a systematic review of the literature with meta-analysis. Dis Markers 2015:164635. 10.1155/2015/164635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Tang K, Liu P, Chen K, Hu J, Zeng J, Xiao W, Yu G, Yao W, Zhou H, Li H, Pan Y, Li A, Ye Z, Wang J, Xu H, Huang Q (2015) lncRNA MALAT1 functions as a competing endogenous RNA to regulate ZEB2 expression by sponging miR-200s in clear cell kidney carcinoma. Oncotarget 6:38005–38015. 10.18632/oncotarget.5357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Yi F, Han X, Du Q, Liang Z (2013) MALAT-1 interacts with hnRNP C in cell cycle regulation. FEBS Lett 587:3175–3181. 10.1016/j.febslet.2013.07.048 [DOI] [PubMed] [Google Scholar]
- Yin KJ, Chen SD, Lee JM, Xu J, Hsu CY (2002) ATM gene regulates oxygen-glucose deprivation-induced nuclear factor-kappaB DNA-binding activity and downstream apoptotic cascade in mouse cerebrovascular endothelial cells. Stroke 33:2471–2477. 10.1161/01.STR.0000030316.79601.03 [DOI] [PubMed] [Google Scholar]
- Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, Bateman R, Song H, Hsu FF, Turk J, Xu J, Hsu CY, Mills JC, Holtzman DM, Lee JM (2006) Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J Neurosci 26:10939–10948. 10.1523/JNEUROSCI.2085-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Deng Z, Huang H, Hamblin M, Xie C, Zhang J, Chen YE (2010a) miR-497 regulates neuronal death in mouse brain after transient focal cerebral ischemia. Neurobiol Dis 38:17–26. 10.1016/j.nbd.2009.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Deng Z, Hamblin M, Xiang Y, Huang H, Zhang J, Jiang X, Wang Y, Chen YE (2010b) Peroxisome proliferator-activated receptor δ regulation of miR-15a in ischemia-induced cerebral vascular endothelial injury. J Neurosci 30:6398–6408. 10.1523/JNEUROSCI.0780-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Deng Z, Hamblin M, Zhang J, Chen YE (2011) Vascular PPARδ protects against stroke-induced brain injury. Arterioscler Thromb Vasc Biol 31:574–581. 10.1161/ATVBAHA.110.221267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Fan Y, Hamblin M, Zhang J, Zhu T, Li S, Hawse JR, Subramaniam M, Song CZ, Urrutia R, Lin JD, Chen YE (2013) KLF11 mediates PPARgamma cerebrovascular protection in ischaemic stroke. Brain 136:1274–1287. 10.1093/brain/awt002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Hamblin M, Chen YE (2014) Non-coding RNAs in cerebral endothelial pathophysiology: emerging roles in stroke. Neurochem Int 77:9–16. 10.1016/j.neuint.2014.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Arun G, Mao YS, Lazar Z, Hung G, Bhattacharjee G, Xiao X, Booth CJ, Wu J, Zhang C, Spector DL (2012) The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep 2:111–123. 10.1016/j.celrep.2012.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yuan L, Zhang X, Hamblin MH, Zhu T, Meng F, Li Y, Chen YE, Yin KJ (2016) Altered long non-coding RNA transcriptomic profiles in brain microvascular endothelium after cerebral ischemia. Exp Neurol 277:162–170. 10.1016/j.expneurol.2015.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Su Z, Song D, Mao Y, Mao X (2016) The long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced inflammatory response through its interaction with NF-kappaB. FEBS Lett 590:2884–2895. 10.1002/1873-3468.12315 [DOI] [PubMed] [Google Scholar]
- Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT (2010) Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell 40:939–953. 10.1016/j.molcel.2010.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WB, Zhu HL, Wu JJ, Wang BH, Wang QR (2009) [Cultivation and purification of high proliferative potential endothelial progenitor cells from murine bone marrow]. Fen Zi Xi Bao Sheng Wu Xue Bao 42:237–243. [PubMed] [Google Scholar]