

Abstract
Behavioral, physiological, and anatomical evidence indicates that the dorsal and ventral zones of the hippocampus have distinct roles in cognition. How the unique functions of these zones might depend on differences in synaptic and neuronal function arising from the strikingly different gene expression profiles exhibited by dorsal and ventral CA1 pyramidal cells is unclear. To begin to address this question, we investigated the mechanisms underlying differences in synaptic transmission and plasticity at dorsal and ventral Schaffer collateral (SC) synapses in the mouse hippocampus. We find that, although basal synaptic transmission is similar, SC synapses in the dorsal and ventral hippocampus exhibit markedly different responses to θ frequency patterns of stimulation. In contrast to dorsal hippocampus, θ frequency stimulation fails to elicit postsynaptic complex-spike bursting and does not induce LTP at ventral SC synapses. Moreover, EPSP-spike coupling, a process that strongly influences information transfer at synapses, is weaker in ventral pyramidal cells. Our results indicate that all these differences in postsynaptic function are due to an enhanced activation of SK-type K+ channels that suppresses NMDAR-dependent EPSP amplification at ventral SC synapses. Consistent with this, mRNA levels for the SK3 subunit of SK channels are significantly higher in ventral CA1 pyramidal cells. Together, our findings indicate that a dorsal-ventral difference in SK channel regulation of NMDAR activation has a profound effect on the transmission, processing, and storage of information at SC synapses and thus likely contributes to the distinct roles of the dorsal and ventral hippocampus in different behaviors.
SIGNIFICANCE STATEMENT Differences in short- and long-term plasticity at Schaffer collateral (SC) synapses in the dorsal and ventral hippocampus likely contribute importantly to the distinct roles of these regions in cognition and behavior. Although dorsal and ventral CA1 pyramidal cells exhibit markedly different gene expression profiles, how these differences influence plasticity at SC synapses is unclear. Here we report that increased mRNA levels for the SK3 subunit of SK-type K+ channels in ventral pyramidal cells is associated with an enhanced activation of SK channels that strongly suppresses NMDAR activation at ventral SC synapses. This leads to striking differences in multiple aspects of synaptic transmission at dorsal and ventral SC synapses and underlies the reduced ability of ventral SC synapses to undergo LTP.
Keywords: complex-spike bursting, EPSP amplification, EPSP-spike coupling, hippocampal LTP, NMDA receptor, SK channel
Introduction
Although the hippocampus has been intensively studied because of its crucial role in spatial navigation as well as learning and memory, hippocampal lesions also have profound effects on innate fear and anxiety (Deacon et al., 2002; Kjelstrup et al., 2002; Bannerman et al., 2003; Pentkowski et al., 2006). Intriguingly, these different aspects of hippocampal function are segregated into distinct anatomical zones, with the dorsal hippocampus having a key role in memory formation, whereas the ventral hippocampus is more importantly involved in emotions and anxiety (Moser and Moser, 1998; Kjelstrup et al., 2002; Bannerman et al., 2003; Fanselow and Dong, 2010; Strange et al., 2014). Consistent with this functional segmentation along the dorsal-ventral axis of the hippocampus, both direct and indirect connections link the dorsal hippocampus to brain regions with roles in spatial information processing and memory, whereas the ventral hippocampus has prominent connections with brain regions involved in emotions and anxiety (Fanselow and Dong, 2010; Strange et al., 2014).
In contrast to the extensive differences in afferent and efferent connections of the dorsal and ventral regions of the hippocampus, the general structure of the internal circuitry of the hippocampus (i.e., the trisynaptic circuit) is preserved throughout the dorsal-ventral axis. This suggests that, along the dorsal-ventral axis, the hippocampus uses a stereotyped circuit to implement a common computational algorithm to process information. From this perspective, the distinct functions of the dorsal and ventral hippocampus are driven primarily by their unique patterns of connectivity with different brain regions. This notion, however, is complicated by dorsal-ventral differences in the cellular and synaptic physiology of hippocampal neurons that likely alter how information is processed by the trisynaptic circuit. For example, a remarkable level of heterogeneity in gene expression exists in both CA1 and CA3 pyramidal cells along the dorsal-ventral axis of the hippocampus (Leonardo et al., 2006; Thompson et al., 2008; Dong et al., 2009; Cembrowski et al., 2016a). Moreover, a variety of ion channels are differentially expressed in dorsal and ventral CA1 pyramidal cells (Marcelin et al., 2012; Dougherty et al., 2013; Hönigsperger et al., 2015). Any role these differences have in determining how information processing varies in the trisynaptic circuit of dorsal and ventral hippocampus has yet to be established, however. Interestingly, both short-term facilitation and LTP are reduced at Schaffer collateral (SC) synapses in the ventral hippocampus (Papatheodoropoulos et al., 2000; Maggio and Segal, 2007; Papatheodoropoulos, 2015). Although these differences could support important dorsal-ventral differences in information processing, the molecular mechanisms responsible for the divergent properties of synaptic plasticity in these regions are unclear. Furthermore, although θ oscillations in the ventral hippocampal CA1 region recorded in vivo are significantly less regular, lower in amplitude, and sometimes absent during behavioral states associated with robust θ oscillations in the dorsal CA1 region (Patel et al., 2012), it remains unclear whether there are important differences in how synapses, cells, and/or networks within the dorsal and ventral hippocampus process θ frequency patterns of activity.
Here, we investigated the mechanisms underlying dorsal-ventral differences in LTP induction using θ pulse stimulation (TPS) patterns of SC fiber stimulation (Thomas et al., 1998). We find that dorsal and ventral SC synapses exhibit markedly distinct responses to TPS and, in contrast to dorsal hippocampus, TPS fails to induce LTP at ventral synapses. EPSP amplification is also strongly reduced at ventral synapses and, as a result, both EPSP-spike (E-S) coupling and EPSP-evoked complex spike (CS) bursting are significantly weaker in ventral pyramidal cells. Our results indicate that all of these differences in postsynaptic function arise from a common mechanism: an enhanced activation of Ca2+-activated SK-type K+ channels that strongly inhibits NMDAR activation at ventral SC synapses. Together, our findings indicate that differences in the expression and/or activity of SK channels provide a potent mechanism for modulating multiple aspects of synaptic function involved in the transfer, processing, and storage of information.
Materials and Methods
Animals and slice preparation.
Male C57Bl/6N mice (Charles River Laboratories; 2–3 months old; housed in a 12/12 light/dark cycle) and Syt7 knock-out mice and wild-type littermates of both sexes (RRID: IMSR_JAX:004950, The Jackson Laboratory; 2–3 months old; housed in a 12/12 light/dark cycle) were deeply anesthetized with isoflurane; and following cervical dislocation, the brain was rapidly removed and submerged in ice-cold, oxygenated (95% O2/5% CO2) ACSF containing (in mm) as follows: 124 NaCl, 4 KCl, 25 NaHCO3, 1 NaH2PO4, 2 CaCl2, 1.2 MgSO4, and 10 glucose (Sigma-Aldrich). On a cold plate, the brain hemispheres were separated, blocked, and the hippocampi removed. The 400-μm-thick slices were then cut using a manual tissue chopper. Slices from the dorsal and ventral thirds of the hippocampus were used. Slices were maintained (at 30°C) in interface-type chambers that were continuously perfused (2–3 ml/min) with ACSF and allowed to recover for at least 2 h before recordings. All experimental techniques were approved by the Institutional Care and Use Committee at the University of California, Los Angeles.
Electrophysiological recordings.
A bipolar, nichrome wire stimulating electrode was placed in stratum radiatum of the CA1 region and used to activate Schaffer collateral/commissural fiber synapses. For extracellular recordings, evoked fEPSPs (basal stimulation rate = 0.02 Hz) were recorded in stratum radiatum using a glass microelectrode (A-M Systems) filled with ACSF (resistance ranged from 5 to 10 mΩ). Interface-type recording chambers were used for extracellular recordings while whole-cell current- and voltage-clamp recordings were done using slices maintained in submerged-slice type chambers. Whole-cell current-clamp recordings were used to examine pairing-induced LTP, intrinsic excitability, E-S coupling, and EPSP amplification. In these experiments, recording electrodes (4–8 mΩ) were filled with a K+-based electrode-filling solution containing (in mm) as follows: 122.5 K-gluconate, 17.5 KCl, 10 HEPES, 0.2 EGTA, 10 Na2-phosphocreatine, 4 Mg-ATP, 0.3 Na-GTP, pH 7.3 (290 mOsm). Whole-cell voltage-clamp recordings (evoked EPSCs and mEPSCs) were performed using a Cs+-based electrode-filling solution containing (in mm) as follows: 102 mm Cs-gluconate, 20 mm CsCl, 10 K-gluconate, 10 mm TEA-Cl, 5 mm QX314, 20 HEPES, 0.2 EGTA, 4 Mg-ATP, 0.3 Na-GTP, and 20 mm HEPES, pH 7.3 (290 mOsm). For experiments performed in picrotoxin (Sigma-Aldrich; 100 μm) the CA3 region was removed; and, unless noted otherwise, the slices were bathed in a modified ACSF containing 2.4 mm KCl, 4.0 mm CaCl2, and 2.4 mm MgSO4. Series resistance compensation was used in all voltage-clamp recordings except in experiments examining miniature postsynaptic currents. All cells were allowed to equilibrate for ≥3 min after break-in before starting experiments. Recordings where series resistance was ≥30 mΩ or unstable were discarded. Unless noted, reported membrane potentials are not adjusted for junction potentials.
LTP induction protocols.
The induction of LTP by TPS was examined using a 30-s-long train of 5 Hz stimulation (150 pulses). At the start of each experiment, the maximal fEPSP amplitude was determined and the intensity of presynaptic fiber stimulation was then adjusted to evoke fEPSPs with an amplitude ∼50% of the maximal amplitude. Unlike more conventional θ-burst stimulation protocols, where bursts of presynaptic stimulation are delivered at 5 Hz, only single pulses of stimulation are delivered during TPS. The average slope of fEPSPs evoked between 40 and 45 min after TPS (normalized to pre-TPS baseline) was used for statistical comparisons. In experiments using whole-cell current-clamp recordings, LTP was induced by pairing EPSPs evoked by SC fiber stimulation with tonic postsynaptic depolarization. Slices were bathed in modified ACSF containing picrotoxin and the strength of SC fiber stimulation was adjusted to evoke ∼15 mV EPSPs (Vm = −80 mV). Following a 10 min baseline (basal stimulation rate = 0.05 Hz), 100 pulses of presynaptic fiber stimulation delivered at 2 Hz were paired with postsynaptic depolarization to −10 mV. The average slope of EPSPs elicited 25–30 min after pairing (normalized to baseline) was used for statistical comparisons.
Basal properties of excitatory and inhibitory synaptic transmission.
Basal synaptic strength at SC synapses was determined by comparing presynaptic fiber volleys and fEPSP slopes elicited by different intensities of presynaptic fiber stimulation. Quantal size and release probability were determined from the amplitude and frequency (Prange and Murphy, 1999), respectively, of miniature EPSCs (mEPSCs) recorded at −80 mV in pyramidal cells bathed in standard ACSF containing TTX (1 μm; Alomone Labs) and picrotoxin. A template-based event detection routine in pClamp 10 (Molecular Devices) was used to identify mEPSCs. Detected events <6 pA were excluded from the analysis. The use-dependent inhibition of NMDAR-mediated EPSCs by MK-801 (Huettner and Bean, 1988) was also used to examine basal release probability (Hessler et al., 1993; Rosenmund et al., 1993). In these experiments, slices were bathed in CNQX (Alomone Labs; 20 μm) and picrotoxin and pyramidal cells were voltage-clamped at 40 mV. Evoked currents were set at ∼300 pA, and a baseline was established. Synaptic stimulation was stopped for 10 min during bath application of MK-801 (Abcam; 40 μm), and then 100 pulses of synaptic stimulation were delivered at 0.1 Hz. For each cell, the activity-dependent, MK-801 inhibition of NMDAR-mediated EPSCs was measured using a weighted decay time constant (τw) calculated from double exponential fits to the time course of the inhibition using the following equation:
where τf and τs are the time constants of the fast and slow components, and αf and αs are the amplitudes of fast and slow components.
The ratio of NMDAR to AMPAR-mediated EPSCs was measured by adjusting the strength of presynaptic fiber stimulation to elicit EPSCs with a peak amplitude of ∼−200 pA at Vm = −80 mV and then recording EPSCs at Vm = 40 mV. The AMPAR- and NMDAR-mediated components of the synaptic currents were estimated by measuring the EPSC amplitude at 5 and 50 ms after EPSC onset, respectively, from the average 40 mV waveform for each cell. To determine the NMDA current decay time constant, the decay of the average EPSC waveform at 40 mV for each cell was fit with a double exponential and a weighted decay time constant was calculated for comparison.
E-S coupling was measured using whole-cell current-clamp techniques to record postsynaptic responses elicited by different intensities of SC fiber stimulation (membrane potential was set to −70 mV using current injection). Five responses were elicited at each stimulation intensity, and the size of EPSPs was measured using the average EPSP slope. To examine EPSP amplification, current through the recording electrode was used to set Vm = −80 mV and the strength of SC fiber stimulation was adjusted to evoke ∼5 mV EPSPs. Steady-state current injection was then used to examine the effect of membrane depolarization (up to −40 mV) on EPSP amplitude and duration. For each cell, the average of 5 EPSPs recorded at each membrane potential tested was used for analysis. d-APV and apamin were acquired from Alomone Labs.
Inhibitory synaptic transmission was assessed in the following ways. The ratio of excitation to inhibition was measured by voltage clamping and holding cells at the reversal potential for GABA (−42 mV) and for glutamate (15 mV) receptors, which had been measured in this preparation and was not different between dorsal and ventral pyramidal cells. Charge transfer at each reversal potential was computed and the ratio taken for comparison. The recording electrode solution used in these experiments contained the following (in mm): 115 Cs-gluconate, 10 K-gluconate, 13 TEA-Cl, 0.2 EGTA, 20 HEPES, 5 QX314, 5 Na-posphocreatine, 0.3 Na-GTP, and 4 Mg-ATP, pH 7.3 (290 mOsm). Spontaneous and miniature IPSCs (mIPSCs) were recorded in cells voltage-clamped at −70 mV using recording electrodes filled with a solution containing the following (in mm): 140 CsCl, 4 NaCl, 1 MgCl2, 0.1 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 5 QX314, and 10 HEPES, pH 7.3. The external solution contained kynurenate (Abcam; 3 mm) to block AMPAR- and NMDAR-mediated currents and TTX was also present during mIPSC recordings. The very high frequency of spontaneous and mIPSCs made it difficult to resolve single events; thus, total charge transfer during 10-s-long recording intervals was measured. The TPS-induced depression of evoked IPSCs (Vm = −70) was also measured using CsCl-based electrode solutions and kynurenate to block excitatory synaptic transmission. Bath application of picrotoxin (100 μm) or bicuculline (Abcam; 40 μm) completely blocked currents recorded in the presence of kynurenate, confirming that the responses are GABAA receptor-mediated.
Intrinsic excitability.
Resting membrane potential was the steady-state membrane voltage with no current injection measured ≥3 min after break-in but before any kind of current injection to the neuron. All other intrinsic properties were measured with steady-state current injected to hold cells at −57 mV (∼−70 mV after correction for calculated junction potential), which was the approximate resting membrane potential for pyramidal cells (Table 1). All protocols were repeated three times per cell and averaged. For input resistance, 500 ms current steps of 0 to −200 pA were injected in −20 pA increments. Steady-state responses were measured as the average change in voltage in the last 100 ms of the pulse. The slope of a regression line fitted to the voltage versus current data was used to calculate input resistance. Current sag was also measured during these experiments and computed as the initial voltage trough during the −100 and −200 pA steps minus the steady-state voltage change. Rheobase was determined by injecting 1 ms square pulses in 100 pA steps and recording the strength of the first pulse to elicit an action potential. Action potential threshold, height, after-hyperpolarization, and after-depolarization were computed from these waveforms as described by Jensen et al. (1996), except for threshold, which was the average of the thresholds computed using Methods II, VI, and VII from Sekerli et al. (2004). Firing frequency versus injected current was measured as the number of spikes per 500 ms step in 25 pA increments from 0 to 150 pA.
Table 1.
Intrinsic excitability in dorsal (D) and ventral (V) CA1 pyramidal cells
| Parameter | Mean ± SEM (range); n | p | Test |
|---|---|---|---|
| Input resistance (mΩ) | D: 83.6 ± 3.4 (56.5 to 99.3); n = 13 | 0.892 | Mann–Whitney |
| V: 87.5 ± 7.6 (51.7 to 128.9); n = 12 | |||
| Resting membrane potential (mV)a | D: −75.2 ± 1.1 (−80 to −66); n = 13 | 0.052 | Mann–Whitney |
| V: −72.9 ± 0.8 (−79 to −69); n = 12 | |||
| Sag with −100 pA injection (mV) | D: −2.7 ± 0.2 (−0.9 to −4.0); n = 13 | 0.724 | Mann–Whitney |
| V: −3.2 ± 0.4 (−0.9 to −5.8); n = 12 | |||
| Sag with −200 pA injection (mV) | D: −5.2 ± 0.4 (−7.2 to −2.0); n = 13 | 0.724 | Mann–Whitney |
| V: −6.1 ± 0.8 (−9.9 to −1.9); n = 12 | |||
| Rheobase (pA) | D: 892 ± 136 (533 to 2433); n = 13 | 0.956 | Mann–Whitney |
| V: 1072 ± 188 (300 to 2200); n = 12 | |||
| AP threshold (mV)a | D: −49.0 ± 1.0 (−52.6 to −38.3); n = 13 | 0.072 | Mann–Whitney |
| V: −46.4 ± 1.4 (−52.7 to −36.7); n = 11 | |||
| AP height (mV) | D: 95.5 ± 1.5 (81.0 to 105.0); n = 13 | 0.093 | Mann–Whitney |
| V: 90.1 ± 2.4 (72.9 to 98.2); n = 11 | |||
| AP after-depolarization peak (mV) | D: −44.7 ± 0.7 (−48.4 to −38.7); n = 13 | 0.224 | Mann–Whitney |
| V: −42.8 ± 1.4 (−48.9 to −31.6); n = 11 | |||
| AP after-hyperpolarization peak (mV) | D: −45.8 ± 0.9 (−51.8 to −39.0); n = 13 | 0.214 | Student's t |
| V: −44.1 ± 1.0 (−39.3 to −50.5); n = 11 |
aAdjusted for calculated junction potential of 13.4 mV.
qRT-PCR.
Isolated CA1 regions were microdissected from dorsal and ventral hippocampal slices, snap frozen on dry ice, and then stored at −80°C until use. RNA was then purified from samples (each containing 8 CA1 regions) using a combined Trizol (Invitrogen) and column (QIAGEN) purification protocol. The 500 μl of Trizol was added to each sample, followed by the standard Trizol protocol until the phase separation step. The top aqueous layer was then removed, mixed 1:1 with 100% ethanol, and added to the column, followed by the manufacturer protocol for column purification.
Equal amounts of dorsal and ventral purified RNA (100 ng per RT reaction) were added into the reverse transcription reaction using the SuperScript III First Strand Synthesis System with random hexamer priming. qPCR was then performed on the cDNA using SYBR Green-based PCR (Applied Biosystems) using primers specific for Kcnn2, Kcnn3, and Hprt1 in technical triplicates per reaction. Normalization was done using ΔCt by first normalizing Kcnn2/3 expression levels to Hprt of that sample, then by comparing the normalized Kcnn2/3 ventral Ct values to dorsal Ct values.
Exon-spanning primers for Kcnn2 were 5′-GTCGCTGTATTCTTTAGCTCTG, 3′-ACGCTCATAAGTCATGGC; and Kcnn3 were 5′-GCTCTGATTTTTGGGATGTTTG, 3′-CGATGATCAAACCAAGCAGGATGA (both from Bond et al. (2004)). Hprt1 primers were 5′-TGTTGTTGGATATGCCCTTG, 3′-GGCCACAGGACTAGAACACC.
Statistical analysis.
Values are reported as mean ± SEM. After evaluating data for normality and homoscedasticity, Student's t tests or, where appropriate, Mann–Whitney U tests were used to determine statistical significance between two groups. All other comparisons used either one-way or two-way ANOVAs. Student-Newman-Keuls post hoc tests were used in cases where multiple comparisons were required. Each test and sample sizes are documented in the figure legends.
Results
CS bursting and LTP deficits in ventral CA1 following θ frequency stimulation
TPS protocols induce LTP at SC synapses by activating a rich set of circuit and synaptic temporal dynamics that lead to CS bursting in CA1 pyramidal cells (Thomas et al., 1998; Fink and O'Dell, 2009; Wójtowicz and Mozrzymas, 2015), a pattern of action potential (AP) firing exhibited by pyramidal cells in vivo (Kandel and Spencer, 1961; Rank, 1973). Thus, understanding how these dynamics vary in the dorsal and ventral hippocampal CA1 region can provide insights into how dorsal-ventral differences in gene expression impact synaptic plasticity, neuronal excitability, and circuit function. Therefore, in our initial experiments, we examined both the short- and long-term changes in synaptic transmission induced by a 150 pulse train of TPS. As expected from previous work (Thomas et al., 1998), TPS elicited complex, time-varying changes in both synaptic transmission and postsynaptic firing in dorsal slices (Fig. 1). Following an initial facilitation of synaptic transmission, EPSPs began to elicit bursts of multiple population spikes (Fig. 1A,B), which are due to CS bursts of postsynaptic APs (Thomas et al., 1998). Although the facilitation gradually faded and synaptic transmission at later time points was depressed, EPSPs continued to elicit CS bursts for the remainder of the stimulation train (Fig. 1B). In stark contrast, the dynamics of synaptic transmission during TPS were less dramatic at SC synapses in the ventral hippocampus and, surprisingly, EPSPs failed to elicit postsynaptic CS bursts (Fig. 1A,B). Notably, CS bursting provides the postsynaptic depolarization needed for NMDAR activation and LTP induction by TPS (Thomas et al., 1998). Thus, this difference in EPSP-evoked CS bursting suggested that SC synapses in the dorsal and ventral hippocampus would exhibit an equally dramatic difference in their ability to undergo TPS-induced LTP. Indeed, TPS induced robust LTP in dorsal hippocampal slices but had no lasting effect on synaptic strength in ventral slices (Fig. 1C). In contrast, similar amounts of potentiation were induced in dorsal and ventral pyramidal cells when low-frequency SC fiber stimulation was paired with strong postsynaptic depolarization during whole-cell current-clamp recordings (Fig. 1D). This indicates that the failure of TPS to induce LTP at SC synapses in the ventral hippocampus is not simply due to a reduced capacity for LTP. Instead, it suggests that dorsal-ventral differences in TPS-induced LTP are due to differences in presynaptic and/or postsynaptic properties that regulate the ability of SC EPSPs to evoke postsynaptic CS bursts.
Figure 1.

θ frequency modulation of transmission at SC synapses in the dorsal and ventral hippocampal CA1 regions. A, Examples of fEPSPs evoked by TPS in dorsal (top) and ventral hippocampal slices (bottom). Note the prominent, multiple population spikes elicited during TPS in dorsal hippocampal slices. Calibration: 2 mV, 5 ms. B, fEPSP slopes (left) and number of population spikes (right) elicited during TPS (n = 6 dorsal and n = 7 ventral slices). Shading represents SEM. C, TPS fails to induce LTP at SC fiber synapses in ventral hippocampus. At 45 min after TPS (delivered at time = 0), fEPSPs were potentiated to 160 ± 8% of baseline in dorsal slices (n = 6) and were 109 ± 6% of baseline in ventral slices (n = 7, p = 1.8 × 10−4, Student's t test: t(11) = 5.516). Traces at right represent superimposed fEPSPs recorded during baseline and 45 min after TPS. Calibration: 2 mV, 5 ms. D, Pairing presynaptic fiber stimulation (100 pulses at 2 Hz) with postsynatpic depolarization (at time = 0) induces similar amounts of LTP in dorsal and ventral pyramidal cells. At 30 min after pairing, EPSPs were 193 ± 4.6% of baseline in dorsal cells (n = 7) and 180 ± 10% of baseline in ventral cells (n = 6, p = 0.22, Student's t test: t(11) = 1.291). Traces represent superimposed EPSPs recorded during baseline and 30 min after LTP induction. Calibration: 5 mV, 20 ms.
Properties of basal synaptic transmission at dorsal and ventral SC fiber synapses
In addition to the weaker facilitation observed during TPS (Fig. 1B), ventral SC synapses also exhibited significantly reduced paired-pulse facilitation compared with dorsal synapses (Fig. 2A). In contrast, basal synaptic strength, determined by comparing the amplitudes of presynaptic fiber volleys and fEPSP slopes for responses elicited by different intensities of SC fiber stimulation, was the same (Fig. 2B). Moreover, there was no difference in the amplitude of mEPSCs in dorsal and ventral CA1 pyramidal cells (Fig. 2C), indicating that quantal size is similar at excitatory synapses in the dorsal and ventral hippocampus. The frequency of mEPSCs was also similar (Fig. 2D), suggesting that the probability of transmitter release is the same at dorsal and ventral synapses (Prange and Murphy, 1999). Consistent with this notion, the activity-dependent inhibition of NMDAR-mediated EPSCs by the channel blocker MK-801 (Huettner and Bean, 1988), which can be used to measure the probability of evoked release at SC synapses (Hessler et al., 1993; Rosenmund et al., 1993), was not significantly different in dorsal and ventral CA1 pyramidal cells (Fig. 2F,G). Thus, although SC synapses in the dorsal and ventral hippocampus exhibit striking differences in activity-dependent forms of plasticity, basal transmission appears to be essentially identical at these synapses.
Figure 2.

Paired-pulse facilitation, basal synaptic strength, and transmitter release probability at excitatory synapses in CA1 region of dorsal and ventral hippocampus. A, Paired-pulse facilitation is significantly lower at ventral SC fiber synapses. **p < 0.001 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). p = 1.6 × 10−14, F(1,57) = 104.7 (n = 12 dorsal and n = 9 ventral slices). Right, Traces represent fEPSPs evoked by stimulation pulses delivered with a 50 ms interpulse interval. Calibration: 1 mV, 10 ms. B, Comparison of presynaptic fiber volley amplitudes and postsynaptic fEPSP slopes for responses elicited by different intensities of SC fiber stimulation in dorsal (n = 17) and ventral (n = 18) hippocampal slices (p = 0.982, Student's t test: t(33) = 0.022). C, D, Amplitude (C) and frequency (D) of mEPSCs in dorsal and ventral CA1 pyramidal cells (n = 1843 events from 7 dorsal cells and 1815 events from 7 ventral cells). There was no significant difference in either the mean amplitude (dorsal: 23.0 ± 1.6 pA; ventral: 23.7 ± 1.0 pA, p = 0.71, Student's t test: t(11) = 0.378) or frequency of mEPSCs (dorsal: 1.7 ± 0.1 Hz; ventral: 1.7 ± 0.1 Hz, p = 0.94, Student's t test: t(11) = 0.076). E, Examples of mEPSCs recorded from dorsal and ventral pyramidal cells. Calibration: 20 pA, 100 ms. F, Example of the use-dependent block of NMDAR-mediated EPSCs in the presence of 40 μm MK-801 in a dorsal CA1 pyramidal cell. Synaptic stimulation was omitted during the first 10 min of MK-801 application. Inset, NMDAR-mediated currents recorded before, immediately after resuming synaptic stimulation in presence of MK-801, and after 20 stimulation pulses in presence of MK-801. Calibration: 100 pA, 50 ms. G, The activity-dependent inhibition of NMDAR-mediated EPSCs by MK-801 is not significantly different in dorsal (n = 6) and ventral (n = 7) pyramidal cells. Weighted decay time constants for the MK-801 inhibition of NMDAR EPSCs were 209 ± 10 s and 214 ± 33 s in dorsal and ventral cells, respectively (p = 0.88, Student's t test: t(11) = 0.155).
Assessing the importance of activity-dependent presynaptic facilitation in the induction of θ frequency CS bursting and LTP
The robust initial facilitation of transmission at SC fiber synapses during TPS in the dorsal hippocampus and its near absence at ventral synapses (Fig. 1B) suggested that dorsal-ventral differences in short-term, presynaptic plasticity may have a central role in regulating CS bursting and LTP induced by θ frequency stimulation. To examine this, we used mice lacking the Syt7 gene that encodes synaptotagmin VII, a presynaptic Ca2+ sensor recently shown to have a crucial role in paired-pulse and frequency facilitation at SC fiber synapses (Jackman et al., 2016). Consistent with previous findings (Jackman et al., 2016), both paired-pulse facilitation (Fig. 3A) and the facilitation of synaptic transmission during the initial phase of TPS were almost completely abolished in slices from Syt7−/− mice (Fig. 3B). The ability of EPSPs to elicit CS bursts was also altered in Syt7−/− mutants, with the onset of EPSP-evoked bursting during TPS being significantly delayed (Fig. 3C,D). Moreover, TPS induced significantly less LTP in slices from Syt7−/− mice (Fig. 3E), most likely due to the reduction in EPSP-evoked bursting during the stimulation train (Thomas et al., 1998). Together, these findings indicate that, although not essential, frequency facilitation of EPSPs during TPS does contribute to LTP induction by significantly advancing the onset of CS bursting.
Figure 3.

Facilitation, CS bursting, and LTP induction are reduced in dorsal hippocampal slices from Syt7−/− mutant mice. A, Paired-pulse facilitation is significantly reduced in dorsal hippocampal slices from Syt7−/− mice. **p < 0.001 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). p = 3.7 × 10−24, F(1,36) = 609.2 (n = 18 slices from 6 wild-type and n = 15 slices from 5 Syt7−/− mutant mice). B, C, fEPSP slopes (B) and number of population spikes (C) elicited during TPS in slices from wild-type (n = 18 slices from 6 mice) and Syt7−/− mutants (n = 15 slices from 5 mice). Shading represents SEM. The latency to first burst (first stimulation pulse to elicit 2 or more population spikes) was 28 ± 3.1 pulses in slices from wild-type mice and 55 ± 6.7 pluses in slices from Syt7−/− mice (p = 4.1 × 10−4, Student's t test: t(9) = 5.445). D, Examples of fEPSPs elicited during TPS in slices from wild-type (top) and Syt7−/− mutant mice. Calibration: 2 mV, 5 ms. E, TPS stimulation-induced LTP is reduced in Syt7−/− mutant mice (p = 0.014, Student's t test: t(9) = 3.045). At 45 min after TPS (delivered at time = 0), fEPSPs were potentiated to 148 ± 4% of baseline in slices from wild-type littermates (p = 8.38 × 10−7, paired t test: t(17) = 7.521) and were 121 ± 8% of baseline in slices from Syt7−/− mutants (p = 1.86 × 10−3, paired t test: t(14) = 3.824). Results from the same experiments shown in B, C.
Therefore, we next tested whether enhancing stimulation strength during θ frequency stimulation would induce CS bursting and LTP in ventral slices from wild-type mice. In these experiments, the intensity of presynaptic fiber stimulation during the first 50 pulses of stimulation (the time period where CS bursting develops in dorsal slices) was increased to mimic (or exceed) the initial facilitation of transmission that occurs during TPS in dorsal slices (Fig. 4A). Despite more robust synaptic activation during TPS, CS bursting (Fig. 4B) and LTP (Fig. 4C) failed to occur, suggesting that a lack of facilitation at ventral SC synapses is not primarily responsible for the absence of TPS-induced CS bursting and LTP at these synapses.
Figure 4.

Increasing the strength of presynaptic fiber stimulation does not enable EPSP-evoked CS bursting and LTP induction in ventral hippocampus. A, B, fEPSP slopes (A) and number of population spikes (B) elicited during TPS in ventral slices in experiments where the strength of presynaptic fiber stimulation during the first 50 pulses of TPS was increased to mimic (1.1× basal stimulation pulse duration, n = 6) or exceed (1.2× basal stimulation pulse duration, n = 6) the facilitation that occurs during the first 50 pulses of TPS in dorsal hippocampal slices. C, Increasing the intensity of presynaptic fiber stimulation during TPS (delivered at time = 0) fails to enable LTP induction in ventral hippocampal slices. At 45 min after TPS, fEPSPs were 112.7 ± 5.9% of baseline in experiments where stimulation strength was increased 1.1-fold at start of TPS and were 110 ± 2.5% of baseline in experiments where the stimulation strength was increased to 1.2-fold (p = 0.928, one-way ANOVA: F(2,15) = 0.075). D, E, fEPSP slopes (D) and number of population spikes (E) elicited by TPS in interleaved control experiments where the strength of presynaptic fiber stimulation was held constant throughout the experiment (n = 8 dorsal and n = 6 ventral slices). F, TPS-induced LTP in interleaved control experiments. At 45 min after TPS, fEPSPs were potentiated to 161 ± 5.7% of baseline in dorsal slices and were 111 ± 6% of baseline in ventral slices (p = 6.6 × 10−5, Student's t test: t(12) = 5.962).
Previous studies have shown that another type of presynaptic plasticity, the short-term depression of feedforward inhibition that occurs during θ frequency SC fiber stimulation, can have a crucial role in the induction of LTP (Davies et al., 1991; Mott and Lewis, 1991). Thus, we also examined whether there might be dorsal-ventral differences in both short-term plasticity and the basal properties of inhibitory synaptic transmission. We found, however, that the TPS-induced depression of IPSCs was essentially identical in dorsal and ventral pyramidal cells (Fig. 5A). Moreover, spontaneous IPSCs (Fig. 5B,C) and IPSC/EPSC ratios for synaptic currents evoked by SC fiber stimulation (Fig. 5D,E) were not different in dorsal and ventral pyramidal cells. Thus, there do not appear to be significant differences in inhibitory transmission that could contribute the distinct effects of TPS on LTP induction and CS bursting in dorsal and ventral CA1 regions.
Figure 5.
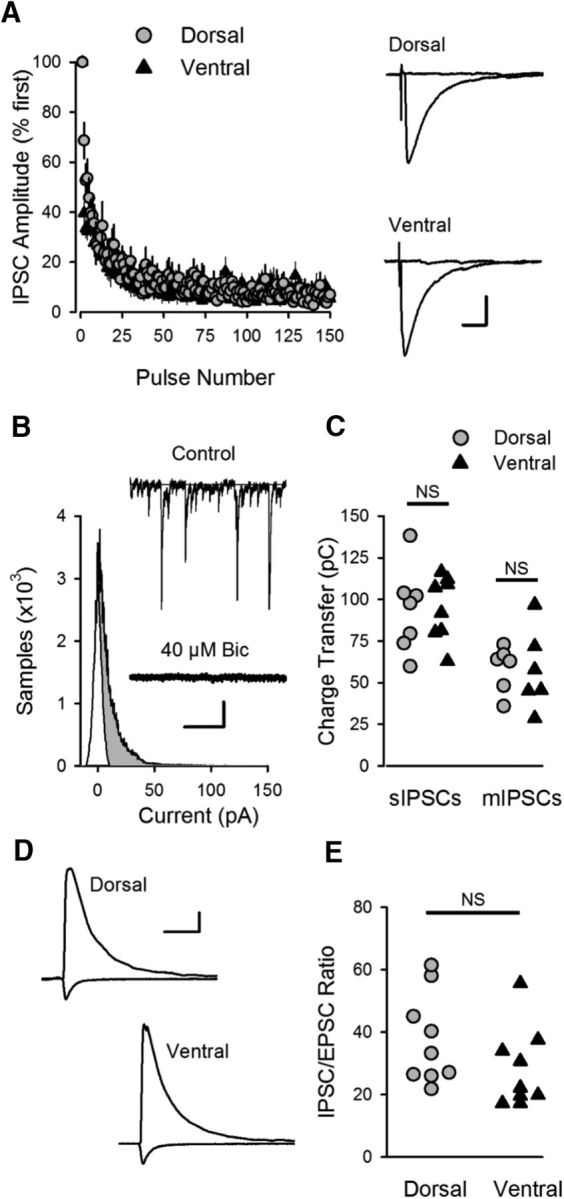
Inhibitory synaptic transmission in CA1 region of dorsal and ventral hippocampus. A, Activity-dependent depression of IPSCs during TPS is similar in dorsal (n = 8) and ventral (n = 9) CA1 pyramidal cells. Traces represent superimposed IPSCs evoked by the first and last pulse of TPS in dorsal and ventral CA1 pyramidal cells. Calibration: 15 ms, 100 pA. B, All points histogram for spontaneous IPSCs in the presence and absence of the GABAA receptor blocker bicuculline (40 μm). Shaded region represents IPSC charge transfer. Traces represent spontaneous IPSCs (sIPSCs) recorded from a dorsal CA1 pyramidal cell before (control) and after application of bicuculline. Calibration: 0.5 s, 40 pA. C, Charge transfer for sIPSCs and mIPSCs (mIPSCs were recorded in presence of 1 μm TTX). Charge transfer for sIPSCs was 94.3 ± 8.4 pC in dorsal cells (n = 8) and 95.2 ± 6.8 pC in ventral cells (n = 8 ventral cells, p = 0.937, Student's t test: t(14) = 0.08). Charge transfer for mIPSCs was 58.5 ± 5.6 pC in dorsal cells (n = 6) and 57.6 ± 9.8 pC in ventral cells (n = 6, p = 0.939, Student's t test: t(10) = 0.078). D, Examples of evoked IPSCs (Vm = 15 mV) and EPSCs (Vm = −42 mv) in dorsal and ventral pyramidal cells. Calibration: 40 ms, 250 pA. E, IPSC/EPSC ratios in dorsal (37.7 ± 4.8, n = 9) and ventral pyramidal cells (27.6 ± 3.8, n = 10) were not significantly different (p = 0.117, Student's t test: t(17) = 1.651).
Weaker E-S coupling and EPSP amplification in ventral CA1
We next examined whether differences in postsynaptic excitability might contribute to dorsal-ventral differences in TPS-induced CS bursting and LTP. Although previous studies have found differences in intrinsic excitability in dorsal and ventral CA1 pyramidal cells (Dougherty et al., 2012, 2013; Marcelin et al., 2012; Hönigsperger et al., 2015; Malik et al., 2016), under our experimental conditions rheobase and threshold for APs elicited by somatic current injection were not significantly different (Table 1). There was also no significant difference in the number of APs elicited by somatic injection of pulses of depolarizing current (Fig. 6A,B). Importantly, somatic current injection might fail to detect differences in ionic conductances present in dendritic spines that regulate depolarization mediated by excitatory synaptic inputs (Ngo-Anh et al., 2005; Bloodgood and Sabatini, 2007; Faber et al., 2008; Gu et al., 2008; Wang et al., 2014). Thus, we also examined intrinsic excitability by measuring the ability of different sizes of EPSPs, evoked by different intensities of presynaptic fiber stimulation, to elicit APs, a phenomenon known as E-S coupling. Although AP firing in response to somatic current injections was the same in dorsal and ventral pyramidal cells (Fig. 6A,B), E-S coupling was significantly weaker in ventral pyramidal cells (Fig. 6C,D).
Figure 6.

Weaker E-S coupling in ventral CA1 pyramidal cells. A, Depolarization induced by somatic current injection elicits similar numbers of APs in dorsal and ventral pyramidal cells (n = 13 dorsal and n = 12 ventral cells, p = 0.694, two-way ANOVA: F(1,138) = 0.155). B, Traces represent examples of APs elicited by 50 and 100 pA current injections in dorsal and ventral cells. Calibration: 20 mV, 100 ms. C, The ability of EPSPs to elicit postsynaptic APs is significantly reduced in ventral pyramidal cells. *p < 0.01 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). **p < 0.001 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). p = 2.3 × 10−7, F(1,62) = 33.794 (n = 10 dorsal and n = 7 ventral cells). D, Traces represent examples of postsynaptic responses evoked by small (top, EPSP slope ≈ 5 mV/ms) and larger EPSPs (bottom, EPSP slope ≈ 8 mV/ms). Calibration: 10 mV, 20 ms.
To gain further insight into the mechanisms responsible for the dorsal-ventral difference in E-S coupling, we examined the voltage-dependent amplification of EPSPs that occurs with membrane depolarization in pyramidal cells (Stuart and Sakmann, 1995; Andreasen and Lambert, 1999; Fricker and Miles, 2000; Carter et al., 2012). In these experiments, inhibitory synaptic transmission was blocked with picrotoxin (100 μm) to pharmacologically isolate EPSPs, and the strength of SC fiber stimulation was adjusted to evoke small, subthreshold EPSPs (∼5 mV at Vm = −80 mV). Synaptic responses were then recorded at postsynaptic membrane potentials between −80 and −40 mV (Fig. 7A). Although robust EPSP amplification was observed in dorsal pyramidal cells, there was little, if any, depolarization-induced amplification of EPSPs in ventral pyramidal cells (Fig. 7A,B).
Figure 7.

Dorsal-ventral difference in NMDAR-mediated EPSP amplification at SC synapses. A, Average EPSPs recorded from 10 dorsal and 12 ventral cells at the indicated membrane potentials. Shading represents SEM. Right, Traces represent superimposed EPSPs recorded at −80 and −40 mV. Calibration: 2 mV, 20 ms. B, Increase in EPSP integrals with depolarization (EPSP amplification) is significantly smaller in ventral pyramidal cells. *p < 0.05 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). **p < 0.001 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). p = 6.5 × 10−8, F(1,120) = 33.23. C, EPSP amplification in the presence of the NMDAR blocker d-APV (50 μm) (n = 9 dorsal and n = 10 ventral cells). D, Amplification determined from the ratio of EPSP integrals at −40 and −80 mV for all cells for results shown in B, C. In control recordings, the amplification in dorsal cells (2.7 ± 0.23) was significantly greater than that seen in ventral pyramidal cells (1.3 ± 0.12). Blocking NMDARs significantly reduced amplification in dorsal cells (1.3 ± 0.16) but had no effect on amplification in ventral pyramidal cells (1.3 ± 0.17). **p < 0.001 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). p = 4.9 × 10−7, F(3,38) = 16.544.
Consistent with previous findings indicating that NMDAR activation significantly contributes to EPSP amplification in CA1 pyramidal cells (Fricker and Miles, 2000), bath application of the NMDAR antagonist d-APV (50 μm) strongly inhibited EPSP amplification in dorsal CA1 pyramidal cells (Fig. 7C,D). Blocking NMDARs had no effect, however, on EPSPs in ventral pyramidal cells (Fig. 7C,D). These findings indicate that NMDAR activation is strongly downregulated at ventral SC synapses. Moreover, because NMDAR activation has a crucial role in CS bursting in dorsal CA1 pyramidal cells in vivo (Grienberger et al., 2014), we hypothesized that the absence of robust NMDAR-mediated EPSP amplification in ventral pyramidal cells might account for the absence of CS bursting during TPS in the ventral hippocampus.
SK channel suppression of NMDAR-dependent EPSP amplification and CS bursting
Given the prominent role of NMDAR activation in EPSP amplification in dorsal CA1 pyramidal cells, one simple explanation for the absence of EPSP amplification in ventral CA1 pyramidal cells is that NMDAR expression is lower at ventral SC synapses. Indeed, the NMDAR-mediated component of fEPSPs measured using extracellular recording techniques is smaller at SC synapses in the ventral hippocampus (Maggio et al., 2015). Previous studies have found, however, that the presence of Ca2+-activated, SK-type K+ channels in dendritic spines can potently inhibit activation of NMDARs by subthreshold EPSPs (Faber et al., 2005; Ngo-Anh et al., 2005; Bloodgood and Sabatini, 2007). Thus, another possibility was that differential expression of SK channels might underlie the differences in EPSP amplification between dorsal and ventral pyramidal cells. Consistent with this second possibility, the NMDAR-mediated component of EPSCs elicited by SC fiber stimulation was not significantly different in dorsal and ventral pyramidal cells when postsynaptic SK channels (and other K+ channels) were blocked by intracellular perfusion of Cs+ and TEA (Fig. 8A). This indicates that the absence of NMDAR-mediated EPSP amplification in ventral CA1 pyramidal cells is not due to a dorsal-ventral difference in postsynaptic NMDAR content. In contrast, using qRT-PCR, we found that, although there was no dorsal-ventral difference in mRNA levels for the SK2 subunit of the SK channel, mRNA levels for SK3 subunits were ∼1.6-fold higher in isolated CA1 regions obtained from ventral hippocampal slices (Fig. 9A). Although this suggests that SK3 subunit mRNA is more highly expressed in ventral pyramidal cells, measurements of mRNA levels in samples prepared from hippocampal slices reflect levels of SK channel subunit mRNA expression in multiple cell types. Thus, we also analyzed data generated from a recent RNA-Seq study specifically examining differences in gene expression in dorsal and ventral CA1 pyramidal cells (Cembrowski et al., 2016a). This analysis, performed using Hipposeq (Cembrowski et al., 2016b), revealed that SK3 subunit mRNA levels are twofold to ninefold higher in ventral CA1 pyramidal cells (Fig. 9B). Based on these findings, we directly tested the potential impact of dorsal-ventral differences in SK channel expression and/or function on NMDAR-dependent EPSP amplification by examining the effects of the selective SK channel antagonist apamin. Blocking SK channels with apamin (100 nm) significantly enhanced EPSP amplification in ventral pyramidal cells, abolishing the dorsal-ventral difference in EPSP amplification observed previously (Fig. 8B,C). In addition, blocking NMDARs with APV prevented the apamin-induced enhancement of EPSP amplification in ventral pyramidal cells (Fig. 8B,C), indicating that SK channel activation suppresses EPSP amplification in ventral pyramidal cells by inhibiting NMDAR activation. Indeed, although APV had no effect on EPSPs evoked at −40 mV in ventral pyramidal cells under control conditions, an APV-sensitive component of EPSPs was present when SK channels were blocked with apamin (Fig. 8D).
Figure 8.
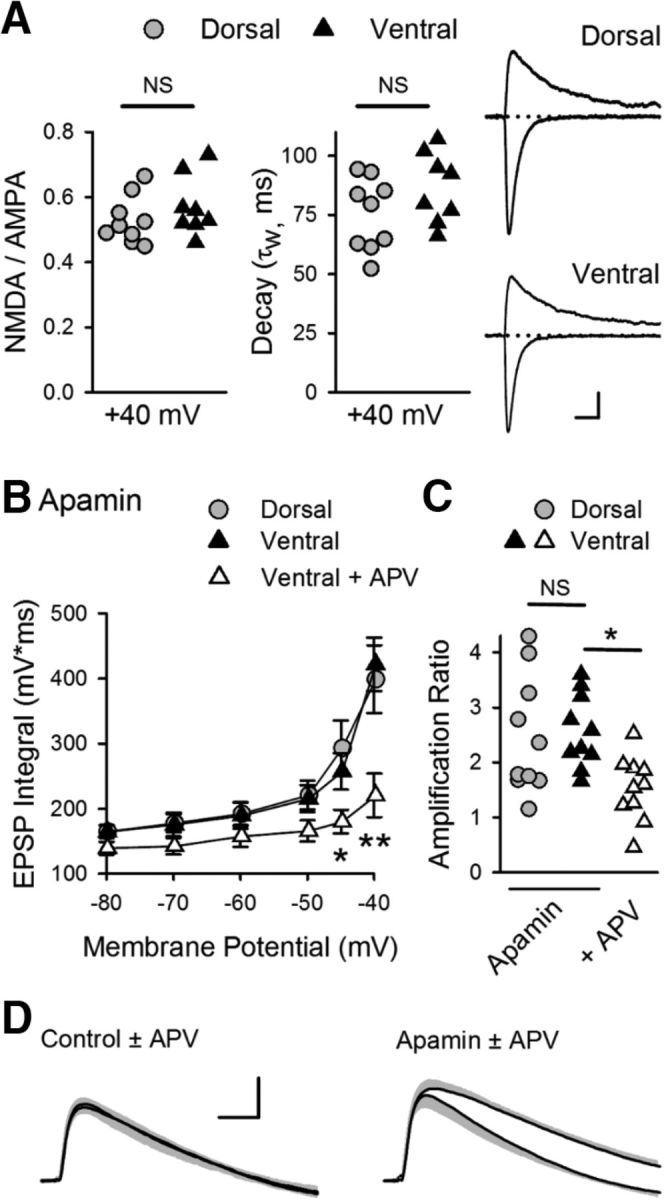
SK channel suppression of NMDAR activation and EPSP amplification at SC synapses in ventral hippocampus. A, Ratio of NMDAR- and AMPAR-mediated components (left) and decay time constants (middle) for EPSCs recorded at Vm = 40 mV in dorsal and ventral pyramidal cells (n = 9 dorsal and n = 8 ventral cells). Ratios were 0.52 ± 0.02 and 0.57 ± 0.03 in dorsal and ventral cells, respectively (p = 0.312, Student's t test: t(15) = 1.046). Decay time constants were 75 ± 5.1 ms in dorsal cells and 86 ± 5.2 ms in ventral cells (p = 0.15, Student's t test: t(15) = 1.517). Right, Traces represent example EPSCs recorded at −80 and 40 mV. Calibration: 75 pA, 20 ms. B, Blocking SK channels with 100 nm apamin enables NMDAR-mediated EPSP amplification in ventral pryamidal cells. *p < 0.05 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). **p < 0.001 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). p = 4.1 × 10−7, F(2,162) = 16.12 (n = 10 dorsal and n = 10 ventral cells in apamin, n = 10 ventral cells in apamin plus d-APV). C, Amplification for all cells in the presence of apamin or apamin plus d-APV. Amplification in the presence of apamin was 2.5 ± 0.34 and 2.6 ± 0.21 for dorsal and ventral pyramidal cells, respectively, and 1.7 ± 0.17 for ventral cells in the presence of apamin + d-APV. *p < 0.05 (one-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). p = 0.013, F(2,27) = 5.154. D, Traces represent superimposed average EPSPs in ventral pyramidal cells elicited at Vm = −40 mV in the absence and presence of APV in control cells (left, from experiments shown in Fig. 7B, C) and in the presence of apamin (right). Shading represents SEM. Calibration: 2 mV, 20 ms.
Figure 9.

SK channel subunit expression in dorsal and ventral hippocampus. A, Levels of Kcnn2 (SK2) and Kcnn3 (SK3) mRNA expression in isolated CA1 regions microdissected from dorsal and ventral hippocampal slices. Lines indicate values obtained from tissue samples from the same hippocampus. B, Expression levels for SK channel subunit mRNA in dorsal and ventral CA1 pyramidal cells obtained using Hipposeq (Cembrowski et al., 2016b) analysis of hippocampal RNA-seq database generated by Cembrowski et al. (2016a).
Enabling CS bursting and LTP in ventral CA1 by SK channel inhibition
To determine how the lack of EPSP amplification in ventral pyramidal cells influences E-S coupling and CS bursting, we next examined its effect on EPSP-evoked AP firing in dorsal and ventral pyramidal cells. First, the intensity of SC stimulation was adjusted to evoke large EPSPs (∼15 mV) that were below threshold for action potential generation at Vm = −70 mV, and the ability of EPSPs to elicit APs was then tested at membrane potentials between −70 and −50 mV. In addition, inhibitory synaptic transmission was suppressed with picrotoxin to mimic the activity-dependent depression of IPSCs that occurs during TPS (Fig. 5A). Under these conditions, EPSPs reliably elicited robust, CS bursts in dorsal pyramidal cells at membrane potentials depolarized to ≥−60 mV, with little or no bursting observed in ventral pyramidal cells at all membrane potentials tested (Fig. 10A). Consistent with the notion that NMDAR activation facilitates CS bursting, bursts of postsynaptic APs were rarely seen in cells where NMDARs were blocked by intracellular application of MK-801 (40 μm) (Fig. 10B). To determine whether SK channel inhibition of the NMDAR-mediated component of EPSPs is responsible for the lack of EPSP-evoked bursting in ventral pyramidal cells, we next repeated these experiments in the presence of apamin. Although apamin had little effect on bursting in dorsal cells, it strongly facilitated EPSP-evoked CS bursting in ventral pyramidal cells (Fig. 10C). Importantly, apamin did not facilitate EPSP-evoked CS bursting in ventral pyramidal cells where intracellular perfusion of MK-801 was used to block NMDARs, demonstrating both the crucial role for NMDAR activation in EPSP-evoked CS bursting and the direct role of SK channels in suppressing NMDAR activation. Together, these findings indicate that NMDAR-mediated EPSP amplification has an important role in the ability of excitatory synaptic inputs to trigger CS-like bursting in CA1 pyramidal cells and that activation of SK channels prevents EPSP-evoked bursting in ventral CA1 pyramidal cells by suppressing NMDAR activation and EPSP amplification.
Figure 10.
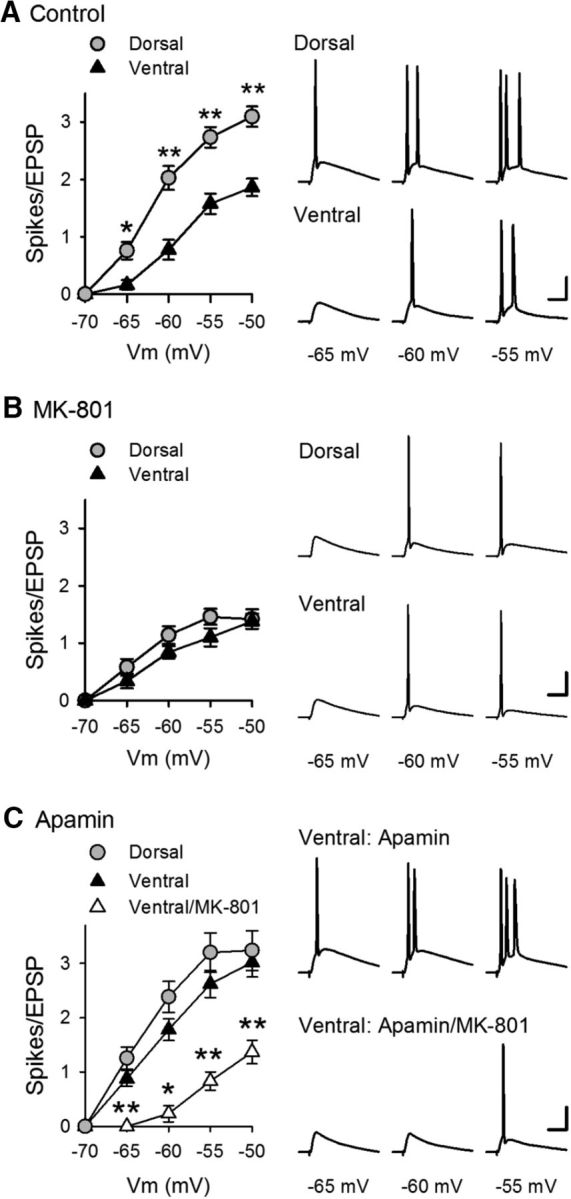
Enhancement of EPSP-evoked CS bursting by blockade of SK channels in ventral CA1 pyramidal cells. A, Effect of membrane depolarization on EPSP-evoked AP firing in dorsal (n = 15) and ventral (n = 16) CA1 pyramidal cells. *p < 0.01 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). **p < 0.001 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). p = 1.8 × 10−15, F(1,120) = 83.715. Traces represent postsynaptic responses elicited at the indicated membrane potentials. B, EPSPs fail to elicit bursting when NMDARs are inhibited with intracellular application of 40 μm MK-801 (n = 10 dorsal and n = 10 ventral cells). C, Inhibiting SK channels with 100 nm apamin enables NMDAR-dependent EPSP-evoked bursting in ventral CA1 pyramidal cells (n = 11 dorsal cells and n = 10 ventral cells in the presence of apamin and 6 ventral cells in the presence of apamin with intracellular application of MK-801). *p < 0.05 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). **p < 0.001 (two-way ANOVA with Student-Newman-Keuls post hoc multiple-comparisons test). p = 5.5 × 10−14, F(2,84) = 44.902. Traces represent postsynaptic responses elicited in ventral pyramidal cells in the presence of apamin with and without intracellular MK-801. Calibration: 20 mV, 20 ms.
Finally, we examined whether enhanced SK suppression of NMDAR-dependent amplification and resultant CS bursting at ventral SC synapses is responsible for the robust dorsal-ventral difference in TPS-induced LTP by examining the effects of apamin on TPS-induced CS bursting and LTP. As we observed in our initial experiments (Fig. 1), EPSPs failed to elicit CS bursting and LTP during TPS in ventral hippocampal slices under control conditions. However, inhibiting SK channels with apamin enabled robust CS bursting (Fig. 11A,B) and strongly facilitated the induction of LTP in ventral hippocampal slices (Fig. 11C) while having no effect on LTP induction in dorsal hippocampal slices (Fig. 11D).
Figure 11.

Inhibiting SK channels enables TPS-induced CS bursting and LTP in ventral hippocampal slices. A, Traces represent examples of fEPSPs elicited during TPS in ventral hippocampal slices during control experiments and in the presence of apamin. Calibration: 1 mV, 10 ms. B, Summary of EPSP-evoked bursting during TPS in ventral hippocampal slices in the absence (control, n = 8) and presence of apamin (n = 7). Results are from the same experiments shown in C. C, Apamin enables LTP induction by TPS (delivered at time = 0) in ventral hippocampal slices. At 45 min after TPS, fEPSPs were potentiated to 149 ± 6.2% of baseline in the presence of apamin compared with 105 ± 2.9% of baseline in control experiments (p = 1.6 × 10−5, Student's t test: t(13) = 6.65). Traces represent superimposed fEPSPs recorded during baseline and 45 min after TPS in control experiments (top) and in experiments with apamin (bottom). Calibration: 1 mV, 5 ms. D, Apamin has no effect on TPS-induced LTP in dorsal hippocampal slices. At 45 min after TPS, fEPSPs were potentiated to 145 ± 3.8% of baseline in control experiments (n = 7) and 144 ± 4.2% of baseline in the presence of apamin (n = 7, p = 0.85, Student's t test: t(12) = 0.199).
Discussion
Results from numerous anatomical and behavioral studies provide strong support for the idea that the dorsal and ventral zones of the hippocampus have fundamentally distinct roles in spatial learning, anxiety, and emotions (Moser and Moser, 1998; Fanselow and Dong, 2010; Strange et al., 2014). It is unclear, however, whether the different functional roles of the dorsal and ventral hippocampus simply reflect the distinct connections of the dorsal and ventral hippocampus with other brain regions or whether the dorsal and ventral hippocampus also process information in different ways. The fact that the overall anatomical organization of the trisynaptic circuit is preserved along the entire dorsal-ventral axis suggests that a common, core algorithm is implemented throughout the hippocampus. Our results, however, challenge this view and suggest that, at least within the CA1 component of this circuit, the distinct functional roles of the dorsal and ventral hippocampus are supported by pronounced differences in both presynaptic and postsynaptic function that can fundamentally influence the processing and storage of information.
Consistent with previous work (Papatheodoropoulos and Kostopoulos, 2000; Papatheodoropoulos, 2015), both paired-pulse facilitation (Fig. 2A) and the initial facilitation of transmission that occurs at the start of TPS (Fig. 1B) were significantly lower at ventral SC synapses. Surprisingly, although differences in short-term plasticity are often associated with differences in the basal probability of neurotransmitter release from presynaptic terminals, basal release probability at dorsal and ventral SC synapses was the same. This dissociation indicates that a difference in the efficacy of transmission at SC synapses in the dorsal and ventral hippocampus will only emerge during patterns of presynaptic activity that occur at frequencies high enough to engage the mechanisms underlying short-term plasticity. Thus, the distinct properties of short-term plasticity at dorsal and ventral SC fiber synapses likely give rise to profound dorsal-ventral differences in basic forms of synaptic computation, such as pattern detection and frequency filtering that are thought to be mediated by short-term plasticity (Abbott and Regehr, 2004).
Our results indicate that differences in long-term plasticity can also support the segregation of hippocampal function along its dorsal-ventral axis. Specifically, we find that, although θ frequency patterns of synaptic stimulation induce robust LTP at SC synapses in the dorsal CA1 region, activation of SK-type K+ channels potently suppresses NMDAR activation and prevents TPS-induced LTP at SC synapses in the ventral hippocampus. Consistent with our findings, both SK2 and SK3 subunits of SK channels are present in dendritic spines (Lin et al., 2008; Ballesteros-Merino et al., 2014) and activation of SK channels by increases in spine Ca2+ acts in a feedback fashion to limit membrane depolarization and inhibit NMDAR activation (Faber et al., 2005; Ngo-Anh et al., 2005; Bloodgood and Sabatini, 2007). Moreover, inhibiting SK channels with the selective blocker apamin facilitates LTP induction (Behnisch and Reymann, 1998; Stackman et al., 2002; Faber et al., 2005; Buchanan et al., 2010). Our findings confirm and extend these earlier observations by demonstrating that regional differences in SK channel activity provide a mechanism for generating functionally distinct types of synapses where θ frequency patterns of synaptic activity can have profoundly different effects on both synaptic transmission and long-term synaptic plasticity. Importantly, our findings also indicate that, in addition to their canonical role as a detector of coincident presynaptic and postsynaptic activity, NMDARs also have a crucial role in EPSP-evoked CS bursting. Thus, differences in SK channel regulation of NMDAR activation at SC-CA1 pyramidal cell synapses also underlie the profound dorsal-ventral disparity in EPSP amplification and E-S coupling, processes that fundamentally impact input-output transformations at synapses.
Although our results suggest that dorsal-ventral differences in SK3 subunit expression contribute to increased SK channel activity at SC synapses in the ventral hippocampus, they do not rule out the possibility that a dorsal-ventral difference in the coupling between SK channels and sources of Ca2+ influx, such as NMDARs and voltage-activated Ca2+ channels, might also be involved. Moreover, the spine localization of SK channels is highly activity-dependent (Lin et al., 2008, 2010), suggesting that factors in addition to overall levels of expression may contribute. The specific increase in SK3 is of interest, though, as elevated SK3 subunit expression contributes to age-related deficits in both LTP and memory (Blank et al., 2003).
An important implication of our results is that differences in the susceptibility of dorsal and ventral SC synapses to LTP induction during θ frequency stimulation are achieved by placing differing inhibitory constraints on LTP induction. In dorsal CA1 pyramidal cells, even large EPSPs fail to induce AP bursting when inhibitory synaptic transmission is intact (Fig. 4C,D) but readily elicit CS bursts when inhibitory synaptic transmission is reduced (Fig. 7A). Thus, GABAergic inhibition acts as the sole inhibitory constraint that counteracts EPSP amplification and the activity-dependent suppression of IPSCs during TPS is sufficient to enable the EPSP-evoked CS bursting necessary for LTP induction. Although the suppression of IPSCs during TPS is similar in ventral pyramidal cells (Fig. 5A), ventral synapses have the additional potent inhibitory constraint of elevated SK channel activity that prevents θ frequency patterns of presynaptic activity from engaging the cellular and molecular machinery necessary for LTP induction. Moreover, although we find that presynaptic facilitation does not appear to be either necessary or sufficient for TPS-induced LTP, the disruption of facilitation in Syt7−/− mutants was associated with a reduction in both EPSP-evoked CS bursting and LTP induction during TPS at dorsal SC synapses. Thus, it seems likely that the dorsal-ventral difference in facilitation also has an important role in regulating the ability of SC synapses in the dorsal and ventral hippocampus to undergo LTP in response to θ frequency patterns of presynaptic activity. In contrast, the induction machinery for LTP, itself, appears to be unchanged between the dorsal and ventral CA1 regions (Figs. 1D, 11C). Thus, levels of postsynaptic phasic inhibition, presynaptic facilitation, and SK channel activity likely work together to confer differential pattern selectivity for LTP induction at dorsal and ventral SC synapses. This implies that LTP is used to store information at both dorsal and ventral SC synapses but is recruited by very different patterns of presynaptic activity.
Although the optimal activity patterns for inducing LTP at ventral SC synapses remain unclear, SK channels are inhibited by adrenergic receptor activation (Maingret et al., 2008) and SK channel activity in dendritic spines is down-regulated by metabotropic glutamate receptor activation (Tigaret et al., 2016) as well as β-adrenergic (Faber et al., 2008) and muscarinic receptor activation (Buchanan et al., 2010; Giessel and Sabatini, 2010). Thus, in addition to coincident presynaptic and postsynaptic activity, the induction of LTP at SC synapses in the ventral hippocampus may be highly state-dependent and require the release of modulatory neurotransmitters, such as acetylcholine or norepinephrine, to overcome the SK channel inhibition of NMDAR activation. Interestingly, a variety of neuromodulatory projections, including noradrengeric inputs from the locus ceruleus, are denser in ventral hippocampus (Haring and Davis, 1985; Strange et al., 2014). Neurons in the locus ceruleus exhibit elevated levels of tonic activity during heightened states of arousal and fire high-frequency bursts of APs in response to stressful, noxious, and/or novel stimuli (Aston-Jones and Cohen, 2005; Sara and Bouret, 2012). Thus, norepinephrine, as well as other modulatory neurotransmitters that regulate SK channel activity, may serve as an essential trigger for plasticity in the ventral hippocampus during behaviors that contain greater emotional valence.
Our results also indicate a previously unappreciated strong interrelationship among several fundamental properties of synaptic transmission that have been studied for decades. These are NMDA receptor activation, EPSP amplification, and E-S coupling. As integrate-and-fire neurons, pyramidal cells are not only sensitive to the rising slope and peak of the EPSP but also the duration and magnitude of the EPSP envelope (Shadlen and Newsome, 1994; Sakai et al., 1999; Rauch et al., 2003). Because of this, the amplification of EPSP amplitude and duration due to NMDAR activation at depolarized membrane potentials strongly facilitates the ability of synaptic inputs to elicit postsynaptic APs (Fig. 10). This indicates that, rather than acting as passive detectors of coincident presynaptic and postsynaptic activity, NMDARs also have an active role in regulating postsynaptic firing. Thus, low levels of NMDAR activation can act in a positive feedback manner by promoting postsynaptic AP firing and bursting, which, by helping to relieve the voltage-dependent Mg2+ block of NMDAR ion channels, leads to even stronger NMDAR activation. This phenomenon, which our results indicate allows even low-frequency patterns of synaptic activity to produce the strong levels of NMDAR activation needed for LTP induction, is likely to be a common feature of NMDAR-mediated synaptic transmission at excitatory synapses throughout the brain. Moreover, by providing an activity-dependent, negative feedback mechanism to limit NMDAR activation, SK channel activation interacts with these phenomena to regulate multiple aspects of synaptic transmission and plasticity.
Footnotes
This work was supported by National Institute of Mental Health Grant R01MH060919-15 to T.J.O. W.E.B. was supported by National Institute of Neurological Disorders and Stroke Grant T32NS007101-34. S.A.J. was supported by National Institute of General Medical Sciences Grant T32GM065823-11. R.G. was supported by an ARCS Foundation Scholar Award. We thank Dr. Seth Grant for helpful comments on the manuscript.
The authors declare no competing financial interests.
References
- Abbott LF, Regehr WG (2004) Synaptic computation. Nature 431:796–803. 10.1038/nature03010 [DOI] [PubMed] [Google Scholar]
- Andreasen M, Lambert JD (1999) Somatic amplification of distally generated subthreshold EPSPs in rat hippocampal pyramidal neurones. J Physiol 519:85–100. 10.1111/j.1469-7793.1999.0085o.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Cohen JD (2005) An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci 28:403–450. 10.1146/annurev.neuro.28.061604.135709 [DOI] [PubMed] [Google Scholar]
- Ballesteros-Merino C, Watanabe M, Shigemoto R, Fukazawa Y, Adelman JP, Luján R (2014) Differential subcellular localization of SK3-containing channels in the hippocampus. Eur J Neurosci 39:883–892. 10.1111/ejn.12474 [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Grubb M, Deacon RM, Yee BK, Feldon J, Rawlins JN (2003) Ventral hippocampal lesions affect anxiety but not spatial learning. Behav Brain Res 139:197–213. 10.1016/S0166-4328(02)00268-1 [DOI] [PubMed] [Google Scholar]
- Behnisch T, Reymann KG (1998) Inhibition of apamin-sensitive calcium dependent potassium channels facilitate the induction of long-term potentiation in the CA1 region of rat hippocampus in vitro. Neurosci Lett 253:91–94. 10.1016/S0304-3940(98)00612-0 [DOI] [PubMed] [Google Scholar]
- Blank T, Nijholt I, Kye MJ, Radulovic J, Spiess J (2003) Small-conductance, Ca2+-activated K+ channel SK3 generates age-related memory and LTP deficits. Nat Neurosci 6:911–912. 10.1038/nn1101 [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL (2007) Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron 53:249–260. 10.1016/j.neuron.2006.12.017 [DOI] [PubMed] [Google Scholar]
- Bond CT, Herson PS, Strassmaier T, Hammond R, Stackman R, Maylie J, Adelman JP (2004) Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J Neurosci 24:5301–5306. 10.1523/JNEUROSCI.0182-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan KA, Petrovic MM, Chamberlain SE, Marrion NV, Mellor JR (2010) Facilitation of long-term potentiation by muscarinic M(1) receptors is mediated by inhibition of SK channels. Neuron 68:948–963. 10.1016/j.neuron.2010.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BC, Giessel AJ, Sabatini BL, Bean BP (2012) Transient sodium current at subthreshold voltages: activation by EPSP waveforms. Neuron 75:1081–1093. 10.1016/j.neuron.2012.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cembrowski MS, Bachman JL, Wang L, Sugino K, Shields BC, Spruston N (2016a) Spatial gene-expression gradients underlie prominent heterogeneity of CA1 pyramidal neurons. Neuron 89:351–368. 10.1016/j.neuron.2015.12.013 [DOI] [PubMed] [Google Scholar]
- Cembrowski MS, Wang L, Sugino K, Shields BC, Spruston N (2016b) Hipposeq: a comprehensive RNA-seq database of gene expression in hippocampal principal neurons. eLife 5 10.7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CH, Starkey SJ, Pozza MF, Collingridge GL (1991) GABA autoreceptors regulate the induction of LTP. Nature 349:609–611. 10.1038/349609a0 [DOI] [PubMed] [Google Scholar]
- Deacon RM, Bannerman DM, Rawlins JN (2002) Anxiolytic effects of cytotoxic hippocampal lesions in rats. Behav Neurosci 116:494–497. 10.1037/0735-7044.116.3.494 [DOI] [PubMed] [Google Scholar]
- Dong HW, Swanson LW, Chen L, Fanselow MS, Toga AW (2009) Genomic-anatomic evidence for distinct functional domains in hippocampal field CA1. Proc Natl Acad Sci U S A 106:11794–11799. 10.1073/pnas.0812608106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty KA, Islam T, Johnston D (2012) Intrinsic excitability of CA1 pyramidal neurones from the rat dorsal and ventral hippocampus. J Physiol 590:5707–5722. 10.1113/jphysiol.2012.242693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty KA, Nicholson DA, Diaz L, Buss EW, Neuman KM, Chetkovich DM, Johnston D (2013) Differential expression of HCN subunits alters voltage-dependent gating of h-channels in CA1 pyramidal neurons from dorsal and ventral hippocampus. J Neurophysiol 109:1940–1953. 10.1152/jn.00010.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Delaney AJ, Sah P (2005) SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat Neurosci 8:635–641. 10.1038/nn1450 [DOI] [PubMed] [Google Scholar]
- Faber ES, Delaney AJ, Power JM, Sedlak PL, Crane JW, Sah P (2008) Modulation of SK channel trafficking by beta adrenoceptors enhances excitatory synaptic transmission and plasticity in the amygdala. J Neurosci 28:10803–10813. 10.1523/JNEUROSCI.1796-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, Dong HW (2010) Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65:7–19. 10.1016/j.neuron.2009.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink AE, O'Dell TJ (2009) Short trains of θ frequency stimulation enhance CA1 pyramidal neuron excitability in the absence of synaptic potentiation. J Neurosci 29:11203–11214. 10.1523/JNEUROSCI.1450-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker D, Miles R (2000) EPSP amplification and the precision of spike timing in hippocampal neurons. Neuron 28:559–569. 10.1016/S0896-6273(00)00133-1 [DOI] [PubMed] [Google Scholar]
- Giessel AJ, Sabatini BL (2010) M1 muscarinic receptors boost synaptic potentials and calcium influx in dendritic spines by inhibiting postsynaptic SK channels. Neuron 68:936–947. 10.1016/j.neuron.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grienberger C, Chen X, Konnerth A (2014) NMDA receptor-dependent multidendrite Ca2+ spikes required for hippocampal burst firing in vivo. Neuron 81:1274–1281. 10.1016/j.neuron.2014.01.014 [DOI] [PubMed] [Google Scholar]
- Gu N, Hu H, Vervaeke K, Storm JF (2008) SK (KCa2) channels do not control somatic excitability in CA1 pyramidal neurons but can be activated by dendritic excitatory synapses and regulate their impact. J Neurophysiol 100:2589–2604. 10.1152/jn.90433.2008 [DOI] [PubMed] [Google Scholar]
- Haring JH, Davis JN (1985) Differential distribution of locus coeruleus projections to the hippocampal formation: anatomical and biochemical evidence. Brain Res 325:366–369. 10.1016/0006-8993(85)90342-7 [DOI] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R (1993) The probability of transmitter release at a mammalian central synapse. Nature 366:569–572. 10.1038/366569a0 [DOI] [PubMed] [Google Scholar]
- Hönigsperger C, Marosi M, Murphy R, Storm JF (2015) Dorsoventral differences in Kv7/M-current and its impact on resonance, temporal summation and excitability in rat hippocampal pyramidal cells. J Physiol 593:1551–1580. 10.1113/jphysiol.2014.280826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE, Bean BP (1988) Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc Natl Acad Sci U S A 85:1307–1311. 10.1073/pnas.85.4.1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman SL, Turecek J, Belinsky JE, Regehr WG (2016) The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature 528:88–91. 10.1038/nature16507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MS, Azouz R, Yaari Y (1996) Spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol 492:199–210. 10.1113/jphysiol.1996.sp021301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER, Spencer WA (1961) Electrophysiology of hippocampal neurons: II. After potentials and repetitive firing. J Neurophysiol 25:243–259. [DOI] [PubMed] [Google Scholar]
- Kjelstrup KG, Tuvnes FA, Steffenach HA, Murison R, Moser EI, Moser MB (2002) Reduced fear expression after lesions of the ventral hippocampus. Proc Natl Acad Sci U S A 99:10825–10830. 10.1073/pnas.152112399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardo ED, Richardson-Jones JW, Sibille E, Kottman A, Hen R (2006) Molecular heterogeneity along the dorsal-ventral axis of the murine hippocampal CA1 field: a microarray analysis of gene expression. Neuroscience 137:177–186. 10.1016/j.neuroscience.2005.08.082 [DOI] [PubMed] [Google Scholar]
- Lin MT, Luján R, Watanabe M, Adelman JP, Maylie J (2008) SK2 channel plasticity contributes to LTP at Schaffer collateral-CA1 synapses. Nat Neurosci 11:170–177. 10.1038/nn2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Luján R, Watanabe M, Frerking M, Maylie J, Adelman JP (2010) Coupled activity-dependent trafficking of synaptic SK2 channels and AMPA receptors. J Neurosci 30:11726–11734. 10.1523/JNEUROSCI.1411-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio N, Segal M (2007) Unique regulation of long term potentiation in the rat ventral hippocampus. Hippocampus 17:10–25. 10.1002/hipo.20237 [DOI] [PubMed] [Google Scholar]
- Maggio N, Shavit Stein E, Segal M (2015) Ischemic LTP: NMDA-dependency and dorso/ventral distribution within the hippocampus. Hippocampus 25:1465–1471. 10.1002/hipo.22467 [DOI] [PubMed] [Google Scholar]
- Maingret F, Coste B, Hao J, Giamarchi A, Allen D, Crest M, Litchfield DW, Adelman JP, Delmas P (2008) Neurotransmitter modulation of small-conductance Ca2+-activated K+ channels by regulation of Ca2+ gating. Neuron 59:439–449. 10.1016/j.neuron.2008.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Dougherty KA, Parikh K, Byrne C, Johnston D (2016) Mapping the electrophysiological and morphological properties of CA1 pyramidal neurons along the longitudinal hippocampal axis. Hippocampus 26:341–361. 10.1002/hipo.22526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelin B, Lugo JN, Brewster AL, Liu Z, Lewis AS, McClelland S, Chetkovich DM, Baram TZ, Anderson AE, Becker A, Esclapez M, Bernard C (2012) Differential dorso-ventral distributions of Kv4.2 and HCN proteins confer distinct integrative properties to hippocampal CA1 pyramidal cell distal dendrites. J Biol Chem 287:17656–17661. 10.1074/jbc.C112.367110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser MB, Moser EI (1998) Functional differentiation in the hippocampus. Hippocampus 8:608–619. 10.1002/(SICI)1098-1063(1998)8:6%3C608::AID-HIPO3%3E3.0.CO;2-7 [DOI] [PubMed] [Google Scholar]
- Mott DD, Lewis DV (1991) Facilitation of the induction of long-term potentiation by GABAB receptors. Science 252:1718–1720. 10.1126/science.1675489 [DOI] [PubMed] [Google Scholar]
- Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP (2005) SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci 8:642–649. 10.1038/nn1449 [DOI] [PubMed] [Google Scholar]
- Papatheodoropoulos C. (2015) Striking differences in synaptic facilitation along the dorsoventral axis of the hippocampus. Neuroscience 301:454–470. 10.1016/j.neuroscience.2015.06.029 [DOI] [PubMed] [Google Scholar]
- Papatheodoropoulos C, Kostopoulos G (2000) Decreased ability of rat temporal hippocampal CA1 region to produce long-term potentiation. Neurosci Lett 279:177–180. 10.1016/S0304-3940(99)01002-2 [DOI] [PubMed] [Google Scholar]
- Patel J, Fujisawa S, Berényi A, Royer S, Buzsáki G (2012) Traveling θ waves along the entire septotemporal axis of the hippocampus. Neuron 75:410–417. 10.1016/j.neuron.2012.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentkowski NS, Blanchard DC, Lever C, Litvin Y, Blanchard RJ (2006) Effects of lesions to the dorsal and ventral hippocampus on defensive behaviors in rats. Eur J Neurosci 23:2185–2196. 10.1111/j.1460-9568.2006.04754.x [DOI] [PubMed] [Google Scholar]
- Prange O, Murphy TH (1999) Correlation of miniature synaptic activity and evoked release probability in cultures of cortical neurons. J Neurosci 19:6427–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank JB., Jr (1973) Studies of single neurons in dorsal hippocampal formation and septum in unrestrained rats: I. Behavioral correlates and firing properties. Exp Neurol 41:461–531. [DOI] [PubMed] [Google Scholar]
- Rauch A, La Camera G, Luscher HR, Senn W, Fusi S (2003) Neocortical pyramidal cells respond as integrate-and-fire neurons to in vivo-like input currents. J Neurophysiol 90:1598–1612. 10.1152/jn.00293.2003 [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL (1993) Nonuniform probability of glutamate release at a hippocampal synapse. Science 262:754–757. 10.1126/science.7901909 [DOI] [PubMed] [Google Scholar]
- Sakai Y, Funahashi S, Shinomoto S (1999) Temporally correlated inputs to leaky integrate-and-fire models can reproduce spiking statistics of cortical neurons. Neural Netw 12:1181–1190. 10.1016/S0893-6080(99)00053-2 [DOI] [PubMed] [Google Scholar]
- Sara SJ, Bouret S (2012) Orienting and reorienting: the locus coeruleus mediates cognition through arousal. Neuron 76:130–141. 10.1016/j.neuron.2012.09.011 [DOI] [PubMed] [Google Scholar]
- Sekerli M, Del Negro CA, Lee RH, Butera RJ (2004) Estimating action potential thresholds from neuronal time-series: new metrics and evaluation of methodologies. IEEE Trans Biomed Eng 51:1665–1672. 10.1109/TBME.2004.827531 [DOI] [PubMed] [Google Scholar]
- Shadlen MN, Newsome WT (1994) Noise, neural codes and cortical organization. Curr Opin Neurobiol 4:569–579. 10.1016/0959-4388(94)90059-0 [DOI] [PubMed] [Google Scholar]
- Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP, Tzounopoulos T (2002) Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J Neurosci 22:10163–10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange BA, Witter MP, Lein ES, Moser EI (2014) Functional organization of the hippocampal longitudinal axis. Nat Rev Neurosci 15:655–669. 10.1038/nrn3785 [DOI] [PubMed] [Google Scholar]
- Stuart G, Sakmann B (1995) Amplification of EPSPs by axo-somatic sodium channels in neocortical pyramidal neurons. Neuron 15:1065–1076. 10.1016/0896-6273(95)90095-0 [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Watabe AM, Moody TD, Makhinson M, O'Dell TJ (1998) Postsynaptic complex spike bursting enables the induction of LTP by θ frequency synaptic stimulation. J Neurosci 18:7118–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CL, Pathak SD, Jeromin A, Ng LL, MacPherson CR, Mortrud MT, Cusick A, Riley ZL, Sunkin SM, Bernard A, Puchalski RB, Gage FH, Jones AR, Bajic VB, Hawrylycz MJ, Lein ES (2008) Genomic anatomy of the hippocampus. Neuron 60:1010–1021. 10.1016/j.neuron.2008.12.008 [DOI] [PubMed] [Google Scholar]
- Tigaret CM, Olivo V, Sadowski JH, Ashby MC, Mellor JR (2016) Coordinated activation of distinct Ca2+ sources and metabotropic glutamate receptors encodes Hebbian synaptic plasticity. Nat Commun 7:10289. 10.1038/ncomms10289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Lin MT, Adelman JP, Maylie J (2014) Distinct Ca2+ sources in dendritic spines of hippocampal CA1 neurons couple to SK and Kv4 channels. Neuron 81:379–387. 10.1016/j.neuron.2013.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wójtowicz T, Mozrzymas JW (2015) Diverse impact of neuronal activity at θ frequency on hippocampal long-term plasticity. J Neurosci Res 93:1330–1344. 10.1002/jnr.23581 [DOI] [PubMed] [Google Scholar]