

Abstract
Background
DICER1 syndrome is a pediatric cancer predisposition condition causing a variety of tumor types in children and young adults. In this report we studied a family with two relatives presenting a variety of neoplastic conditions at childhood.
Methods
Germ-line mutation screening of the complete coding region of the DICER1 gene in genomic DNA from the proband was performed. The presence of somatic DICER1 mutation and further alterations in driver genes was investigated in genomic DNA obtained from available tumor samples.
Results
A nonsense germ-line mutation in DICER1 causing a truncated protein at the IIIb domain level was identified segregating within a family including two affected relatives who developed in one case cystic nephroma and pleuropulmonary blastoma, and rhabdomyosarcoma and multinodular goiter in the other. Additional in trans DICER1 missense somatic mutations in the IIIb DICER1 domain were found both in the cystic nephroma and in the rhabdomyosarcoma, suggesting that neoplasms in this family might arise from the unusual two-hit mechanism for DICER-derived tumorigenesis in which after the presence of a truncated constitutive protein, a neomorphic DICER1 activity is somatically adquired. Additional genetic alterations, such as TP53 mutations, were identified in the rhabdomyosarcoma.
Conclusions
Besides DICER1 loss of standard activity, oncogenic cooperation of other genes, as mutated TP53, may involve developing higher grade tumors within this syndrome. Given the broad clinical spectrum that may arise, genetic counseling and close surveillance must be offered to all family members at risk of DICER1 syndrome.
Keywords: DICER1 mutations, DICER1 syndrome
Background
Germ-line mutations in DICER1 have been described in the so-called DICER1 syndrome, a pleiotropic pediatric cancer predisposition condition causing a variety of tumor types in children and young adults, including pleuropulmonary blastoma (PPB), cystic nephroma (CN), rhabdomyosarcoma (RMS), multinodular goiter, ovarian Sertoli-Leydig cell tumor and other neoplastic conditions. DICER1 is a multidomain protein, containing two endoribonuclease III domains. In the majority of cases, germ-line mutations are nonsense, frameshift or splice-site mutations leading to premature truncation of the protein, resulting in loss of RNAseIII function [1–3]. RNA processing endoribonucleases are required for the biogenesis of microRNAs (miRNAs), cleaving precursor miRNAs into mature miRNAs which, in turn, post-transcriptionally regulate messenger RNA expression [2]. Disregulation of miRNAs is implicated in several human diseases, as they participate in many different biological processes. Thus, mutations in DICER1 have the potential to affect many biological functions and originate different phenotypes. In this communication, germ-line and somatic mutations in DICER1 are reported within a family with two relatives presenting a variety of neoplastic conditions at childhood.
Methods
Subjects
Our studied pedigree comprised 8 individuals. All subjects or their parents/legal guardians gave written informed consent for genetic research studies and peripheral blood samples were taken. Available frozen tumor tissue samples were obtained from HUCA Tumor Bank. Written informed consent for sample banking and research use was obtained at the time of the surgery.
DNA, RNA and cDNA samples
DNA from peripheral blood and tumor tissues was extracted using DNAzol (Molecular Research Center, USA), following manufacturer’s instructions. RNA was obtained from frozen CN and ERMS frozen tissue samples with Tri-reagent® (Ambion). cDNA was synthesized with RNA ImProm-II Reverse Transcriptase (Promega Corporation, Madison, WI, USA), following manufacturer’s instructions.
Mutational analyses
Individual coding exons of the DICER1 and TP53 gene including flanking intronic regions were amplified by PCR. Primer sequences are available on request. Purified sequence reactions were resolved on a capillary automated Sanger sequencing (ABIPrism310, Applied Biosystems, Thermo Fisher Scientific). GenBank reference sequence accession numbers were NM_177438, NG_016311, and NP_803187 for DICER1 and NG_017013.2, NM_000546.4 for TP53 gene.
Hot spot mutations in KRAS, NRAS, EGFR, PIK3CA and BRAF were sought by real-time quantitative allele-specific PCR amplification using commercial kits (RAS Mutation Screening Panel, Entrogen, USA; therascreen EGFR RGQ PCR Kit V2, Qiagen, UK; cobas PIK3CA Mutation Test, Roche, USA; cobas 4800 BRAF V600 Mutation Test, Roche, USA), following manufacturer’s instructions. Deletions or amplifications in PDGRFA, TP53, CDKN2A, CDK4, RB1, EGFR, PTEN, and MMDM2 genes and in the chromosomal regions 1p and 19q were studied by MLPA (Multiplex Ligation-dependent Probe Amplification) using commercial kits (P0471, P088, and P105 probemixes, MRC-Holland, The Netherlands), following manufacturer’s instructions.
Results
Case report
Index case was a 2-year-old girl presenting a cystic nephroma at 11 months of age that required surgical resection of her left kidney. A computed tomography of the chest revealed a cystic mass in the left lower lobe adjacent to the diaphragm. At 14 months of age, a left lower lobectomy was performed and a pathological diagnosis of type I PPB was established (Fig. 1a and b).
Fig. 1.
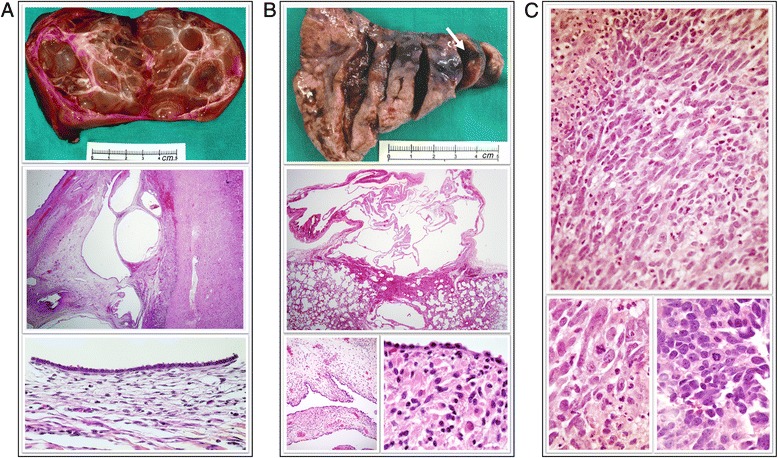
Morphologic study of the reported tumors. a. Left nephrectomy from the proband. Top: Gross morphology image of the resected kidney presenting a thin-wall multicystic parenchyma. Middle: Medium power view of a haematoxylin and eosin staining (HE) showing multiple cysts of different size. Scarce renal tissue is preserved. Bottom: Higher power view of the cysts surface showing a single layer of flattened/cuboidal cells with well oriented nuclei, acidophilus cytoplasm, and without atypia. b. Left lower lobectomy from the proband. Top: Gross morphology image of the resected lobe. An arrow is pointing to the cystic lesion of 12 mm largest size. Middle: At histological level, a unique benign cystic lesion was identified (HE). Bottom left: A single cubic-cell layer lining the epithelium was observed in most of the cystic lesion. Bottom right: Only very few and isolated clumps of primitive mesenchymal cells and small clusters of cells with rhabdomyomatous differentiation were found along the whole tissue sample. c. Histological images of the rhabdomyosarcoma from the proband’s cousin. Top: HE staining showing dense neoplastic cell proliferation in a solid-storiform pattern. Bottom: Intense pleomorphism is observed, with fusiform-oval cells and common mitotic figures, with focal anaplastic cells
Germ-line mutational analysis
Considering patient’s tumors nature, genetic mutation screening of the complete coding region of the DICER1 gene in genomic DNA from proband’s blood was performed. It led to identify a nonsense truncating mutation affecting Q1783 residue, codified in exon 24 (c.5387C > T; p.Q1783*). This mutation was found in heterozygosity and predicted to truncate the protein by the RNaseIIIb domain of the enzyme. We studied segregation of this germ-line mutation with diverse pathologies in 7 available relatives and identified the proband’s mother and grandmother as carriers of the mutation (Fig. 2). In addition, a 21-year-old female cousin of the proband who was diagnosed of an embryonal RMS (Fig. 1c) at age 14 and multinodular goiter at age 20, was also germ-line carrier of the DICER1 mutation. Interestingly, thyroid affection was also reported in most of the family members, being multinodular goiter with calcifications the only remarkable pathological phenotype present in the proband’s mother and grandmother. Of those participating in the study, 4 out of 5 affected of thyroid alterations carried the p.Q1783* mutation.
Fig. 2.
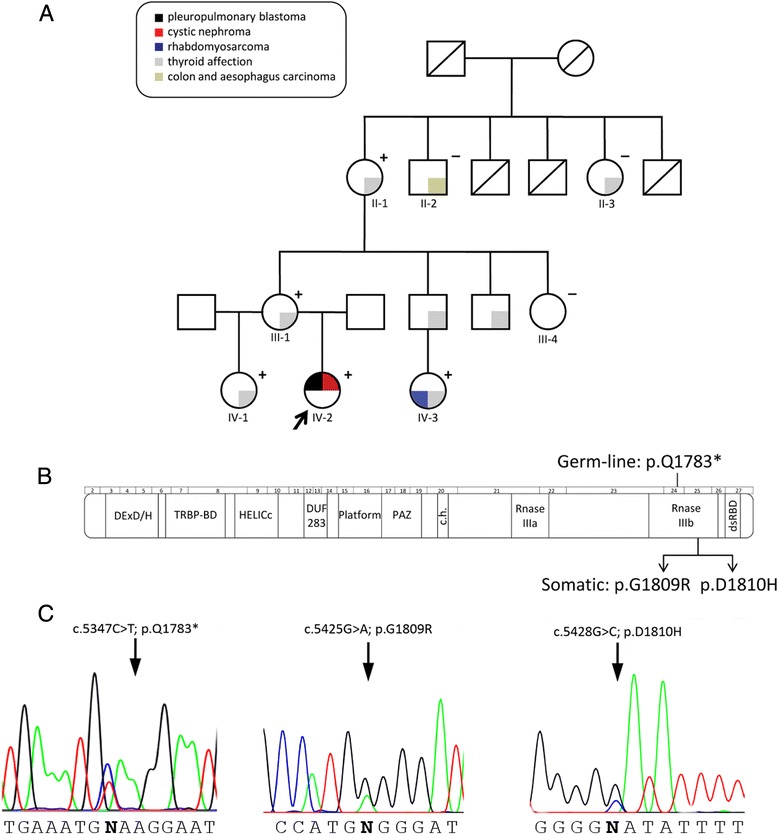
a. Pedigree of the studied family. Clinical phenotypes are represented with colors. Individuals screened for DICER1 mutations are indicated with + or — if the germ-line mutation was present or absent, respectively. The index patient is marked with an arrow. b. Schematic representation of DICER1 protein structure and corresponding gene exons. Positions of the here identified mutations are indicated. c. Electropherograms corresponding to the germ-line c.5347C > T (p.Q1783*) and somatic c.5425G > A (p.G1809R); c.5428G > C (p.D1810H) mutations
Somatic mutation analysis
The presence of an additional somatic mutation in the remaining DICER1 allele was investigated in genomic DNA obtained from CN and ERMS available tumor samples. Tumor tissue from the PPB was not accessible for molecular studies since the number of tumor cells was very low (Fig. 1b) and the block tissues were exhausted for histological diagnosis. We found a missense heterozygous c.5425G > A (p.G1809R) mutation in DNA obtained from CN, whereas ERMS harbored a c.5428G > C (p.D1810H) substitution, also in heterozygosis (Fig. 2b and c).
Somatic mutational analyses in additional putative oncogenic driver genes with clinical relevance were studied. No alterations in KRAS, NRAS, EGFR, PIK3CA, BRAF, PDGRFA, TP53, CDKN2A, CDK4, RB1, EGFR, PTEN, and MMDM2 genes were detected in the CN. In contrast, in genomic DNA from ERMS we found TP53 to carry the c.404G > A (p.C135Y) mutation in heterozygosity. ERMS tumor also harbored additional gene alterations: low amplification of EGFR, PDGFRA, and CDK4 genes, and loss of heterozygosity of CDKN2A and 19q12-19q13.43 chromosomal regions.
Mutation allelic configuration
We aimed to investigate if the somatic and germ-line DICER1 mutations were separate events on different alleles. For this purpose, we analyzed cDNA synthesized from ERMS tumor RNA. Due to the fact that germ-line c.5387C > T (p.Q1783*) mutation creates an HpyCH4V restriction site, we designed a PCR amplification and HpyCH4V digestion assay in which the somatic mutations would be included in the restriction fragment generated by the presence of germ-line mutation. Sequencing analysis of germ-line mutated restriction fragments revealed that germ-line and somatic mutations lied in trans.
Discussion
In this report we identify germ-line and somatic DICER1 gene mutations in a Caucasian family where two young females were diagnosed, one with cystic nephroma and pleuropulmonary blastoma, and the other with embryonal rhabdomyosarcoma and multinodular goiter. This pedigree demonstrates that genetic alteration in the DICER1 gene can cause a wide clinical spectrum of carcinomas, in accordance with recent studies [1]. Both affected patients shared the nonsense c.5347C > T germ-line DICER1 mutation, predicted to truncate the protein, and thus likely impairing activity of the DICER1 enzyme [3].
Interestingly, thyroid affection was also reported in most of the family members. However, we were not able to establish definitive association of the DICER1 mutation with any specific thyroid disease due to lack of access to detailed clinical histories or affected tissue samples from these patients. As goiter is an endemic disease in Asturias (North of Spain) [4], from where the family is native, it cannot be discarded the existence of further risk factors for thyroid affection within this pedigree.
We identified additional somatic missense DICER1 mutations in CN and ERMS tumors (p.G1809R and p.D1810H, respectively), affecting metal ion-binding regions of the protein and thus presumably affecting catalytic activity of RNAseIIIb (but not RNAaseIIIa) domain [3]. In contrast with the initially proposed theory of haploinsufficiency of this tumor suppressor gene as the cause underlying DICER1 syndrome [5], our results support the hypothesis that patients harboring a loss-of-function germ-line DICER1 mutation acquire a second somatic hit during tumorigenesis, resulting in a modified enzyme activity, at the RNAseIIIb level [6]. Loss of RNAseIIIb activity of DICER1 protein has been shown to selectively reduce the generation of 5p miRNAs, with no influence on 3p miRNAs processing, and thus generating a deregulation of control in gene expression [7, 8]. As we have established in our samples, since the second somatic mutation lies in trans, both alleles are compromised. This need of a compound disruption of DICER1 may explain the low penetrance of germ-line DICER1 mutations alone.
Given the wide clinical phenotypes observed, additional somatic gene mutations might be involved in the carcinogenic mechanisms of the syndrome. The malignant nature of ERMS tumor is underlined by the presence of additional genomic alterations, especially TP53 mutations. The TP53 gene c.404G > A mutation (p.C135Y) identified in ERMS results in the loss of one of the two p53 intramolecular disulfide bonds. Consequently, the protein conformation and subcellular location are altered [9, 10]. Moreover, it has been proposed that mutant p53 can down-regulate DICER1 expression, enhancing the metastasic potential of tumor cells [11]. Therefore, we suggest that, in addition to DICER1 loss of full activity, oncogenic cooperation of other genes, such as mutated TP53, may involve developing of higher grade tumors. ERMS tumor also harbored genomic alterations in several frequent driver genes.
Conclusions
Given the variety of tumors developed within this family, and the risk of ovarian cancer (Sertoli-Leydig type) [1, 3, 6] and thyroid affection in adults, a close surveillance might be offered to all at risk family members of DICER1 syndrome pedigrees.
Acknowledgments
We show our gratitude to Dr. M.J. Antuña (Unidad de Oncología Pediátrica, HUCA) for helpful discussion and advice. We thank Fundación Caja Rural de Asturias for financial collaborative support to Laboratorio de Oncología Molecular (HUCA). IUOPA is supported by Fundación Bancaria Caja de Ahorros de Asturias.
Funding
This work is supported by a research grant from Caja Rural Asturias to Laboratorio de Oncología Molecular (HUCA).
Availability of data and materials
Data and materials are available upon request.
Authors’ contributions
LF-M contributed sequencing mutational analysis, and co-wrote the manuscript; JAV collaborated with clinical data and study design; ISR, ASP and MGA contributed with MLPA and quantitative PCR mutational analysis; HET and SF provided pathological analysis and samples; AP and PB supplied clinical familial samples and data; MB designed the study, directed and supervised research and co-wrote the manuscript. All authors approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent for publication of clinical details was obtained from all the studied members of the family or their parents. The documents have been incorporated to the clinical history registry of each patient.
Ethics approval and consent to participate
The study was approved by the Comité de Ética de la Investigación del Principado de Asturias (study number 69/16). Written informed consent for genetic studies and publication of clinical details was obtained from all the studied members of the family or their parents, in the course of an individualized genetic counselling interview at the Familial Cancer Unit performed by two of the authors (PB, AP). Written informed consents have been incorporated to the clinical history registry of each patient.
Abbreviations
- CN
Cystic nephroma
- ERMS
Embryonal rhabdomyosarcoma
- HUCA
Hospital Universitario Central de Asturias
- IUOPA
Instituto Universitario de Oncología del Principado de Asturias
- MLPA
Multiplex ligation-dependent probe amplification
- PPB
Pleuropulmonary blastoma
- RMS
Rhabdomyosarcoma
Contributor Information
Lorena Fernández-Martínez, Email: Lorena.fdez@gmail.com.
José Antonio Villegas, Email: javillegas10@hotmail.com.
Íñigo Santamaría, Email: isr@hca.es.
Ana S. Pitiot, Email: anapitiot@uniovi.es
Marta G. Alvarado, Email: gonzalezamaria@uniovi.es
Soledad Fernández, Email: msoledad.fernandez@sespa.es.
Héctor Torres, Email: Ress_44@yahoo.com.
Ángeles Paredes, Email: marpafe@gmail.com.
Pilar Blay, Email: pilar.blay@sespa.es.
Milagros Balbín, Email: mbalbin@hca.es.
References
- 1.Slade I, Bacchelli C, Davies H, Murray A, Abbaszadeh F, Hanks S, et al. DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet. 2011;48:273–8. doi: 10.1136/jmg.2010.083790. [DOI] [PubMed] [Google Scholar]
- 2.Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965. doi: 10.1126/science.1174334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer. 2014;14:662–72. doi: 10.1038/nrc3802. [DOI] [PubMed] [Google Scholar]
- 4.Gómez de la Torre R, Enguix Armada A, García L, Otero J. Thyroid nodule disease in a previously endemic goiter area. An Med Interna. 1993;10:487–9. [PubMed] [Google Scholar]
- 5.Lambertz I, Nittner D, Mestdagh P, Denecker G, Vandesompele J, Dyer MA, Marine JC. Monoallelic but not biallelic loss of Dicer1 promotes tumorigenesis in vivo. Cell Death Differ. 2010;17:633–41. doi: 10.1038/cdd.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heravi-Moussavi A, Anglesio MS, Cheng SW, Senz J, Yang W, Prentice L, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med. 2012;366:234–42. doi: 10.1056/NEJMoa1102903. [DOI] [PubMed] [Google Scholar]
- 7.Anglesio MS, Wang Y, Yang W, Senz J, Wan A, Heravi-Moussavi A, et al. Cancer-associated somatic DICER1 hotspot mutations cause defective miRNA processing and reverse-strand expression bias to predominantly mature 3p strands through loss of 5p strand cleavage. J Pathol. 2013;229:400–9. doi: 10.1002/path.4135. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Chen J, Yang W, Mo F, Senz J, Yap D, et al. The oncogenic roles of DICER1 RNase IIIb domain mutations in ovarian Sertoli-Leydig cell tumors. Neoplasia. 2015;17:650–60. doi: 10.1016/j.neo.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nie L, Sasaki M, Maki CG. Regulation of p53 nuclear export through sequential changes in conformation and ubiquitination. J Biol Chem. 2007;282:14616–25. doi: 10.1074/jbc.M610515200. [DOI] [PubMed] [Google Scholar]
- 10.Zhuang J, Wang PY, Huang X, Chen X, Kang JG, Hwang PN. Mithochondrial disulfide relay mediates translocation of p53 and partitions its subcellular activity. Proc Natl Acad Sci U S A. 2013;110:17356–61. doi: 10.1073/pnas.1310908110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller PA, Trinidad AG, Caswell PT, Norman JC, Vousden KH. Mutant p53 regulates Dicer through p63-dependent and —independent mechanisms to promote an invasive phenotype. J Biol Chem. 2014;289:122–32. doi: 10.1074/jbc.M113.502138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data and materials are available upon request.