

Abstract
We report a case of chronic pulmonary multi-drug-resistant tuberculosis. Despite 14 years of treatment, Mycobacterium tuberculosis was persistently isolated from sputum. Following treatment cessation the patient remained well, although M. tuberculosis was isolated from sputum for a further 8 years. Genome sequencing of eight serial M. tuberculosis isolates cultured between 1991 and 2011 revealed 17 mutations (0.8 mutations per genome year− 1). Eight of these were persisting mutations and only two mutations were detected in the 7 years following cessation of treatment in 2004. In four isolates there were mixed alleles, suggesting the likely presence of bacterial subpopulations. The initial 1991 isolate demonstrated genotypic resistance to isoniazid (katG W91R), rifampicin (rpoB S531L), ethambutol (embB M306V), streptomycin (gidB L16R), quinolones (gyrA S95T) and P-aminosalicylic acid (thyA T202A). Subsequent resistance mutations developed for pyrazinamide (pncA I31F) and ethionamide (ethA frameshift). Such information might have been instructive when developing a treatment regimen. In retrospect and with the benefit of high-resolution genomic hindsight we were able to determine that the patient received only one or two active anti-tuberculous agents for most of their treatment. Additionally, mutations in bacA and Rv2326c were detected, which may have contributed to the persistent but mild disease course. BacA is likely to be associated with maintenance of chronic infection and Rv2326c with a decreased bacterial metabolic state. These results expand our understanding of M. tuberculosis evolution during human infection and underline the link between antibiotic resistance and clinical persistence.
Keywords: antibiotic resistance, clinical persistence, evolution, Mycobacterium tuberculosis
Data Summary
Supplementary Table S1 is available through Fihare. http://dx.doi.org/10.6084/m9.figshare.1561411
Sequence read data for the study isolates have been submitted to the EMBL Sequence Read Archive (ENA). https://www.ebi.ac.uk/ena/data/view/ERP011175
Impact Statement
Tuberculosis (TB) is a major cause of human disease globally. The causative bacterium, Mycobacterium tuberculosis, has evolved with humans for many thousands of years. TB has wide-ranging disease manifestations. Factors related to the human host and the state of the immune system are well known to influence the disease progression of TB, while factors relating to the bacterium itself have been relatively elusive. Whole genome sequencing involves unravelling the entire sequence of bacterial DNA, and technology to do this has become increasingly available. Here we describe an unusual case of multi-drug-resistant TB, and present the results of whole genome sequencing of serial isolates collected over a 21-year period. This has enabled us to further our understanding of the evolution of the TB bacterium within the human host. This gives us insight into the development of drug resistance mutations and other mutations that may alter the disease course, and ultimately contributes to increased understanding of the biology of this important human disease.
Introduction
Infection with Mycobacterium tuberculosis is a major cause of human morbidity and mortality, and has wide-ranging clinical manifestations. While host factors, particularly T-cell-mediated immunity, are well known to influence disease presentation and outcome, bacterial factors are also important, but elusive to pinpoint. The majority of studies assessing strain-specific factors have looked at associations between lineage and disease manifestation; at present there is little known about the role of specific genes in determining disease phenotype (Coscolla & Gagneux, 2010). In recent years, technology allowing large-scale whole genome sequencing (WGS) has become increasingly available, which is facilitating a rapid increase in our understanding of the evolution of bacterial populations and in particular molecular mechanisms of drug resistance.
Here we have used genomics to investigate the evolution of M. tuberculosis during human pulmonary infection. We describe a case of chronic pulmonary multi-drug-resistant tuberculosis (TB), and report the results of WGS analysis of eight serial M. tuberculosis isolates collected from the same patient over a 21-year period. We describe the heterogeneity in sequencing results suggestive of dynamic bacterial subpopulations, and provide an estimate of the mutation rate of M. tuberculosis during active infection. We describe the baseline resistance-associated mutations and subsequent mutations that explain the changing pattern of drug resistance, and the potential influence of emergent mutations in the genes encoding putative exporter proteins, bacA, Rv1819c, Rv2326c and mshA, on the disease course.
Methods
Primary culture of M. tuberculosis from clinical specimens was performed using pyruvate-enriched Lowenstein–Jensen and Brown & Buckle media. Specimens received from 2007 onwards were also cultured within Mycobacteria Growth Indicator Tubes. Primary M. tuberculosis culture and susceptibility testing was also performed using the radiometric BACTEC 460TB system (Becton Dickinson) from 1991 to 2007, and from 2007 onwards using the BACTEC MGIT 960 system (Becton Dickinson). DNA was extracted from isolates cultured from sputum specimens as described by Ross et al. (1991). Isolate ID numbers and isolation dates are as follows: Mtb_41, 3 January 1991; Mtb_323, 14 May 1992; Mtb_682, 6 October 1993; Mtb_1087, 30 May 1995: Mtb_2573, 6 February 2002; Mtb_2855, 18 June 2003; Mtb_4891, 16 March 2010; Mtb_5288, 7 June 2011.
Whole genome DNA sequencing was performed on an Illumina MiSeq using 250 bp paired-end reads, with library preparation using Nextera XT (Illumina). Prior to mapping, reads were trimmed to remove adaptor sequences and low-quality bases (less than Q10). Reads less than 50 nt in length were removed. Resulting sequence fastq files were aligned to the M. tuberculosis H37Rv reference chromosome (NC_000962.3) using Snippy v2.6 (https://github.com/tseemann/snippy) and Nesoni v0.130 (https://github.com/Victorian-Bioinformatics-Consortium/nesoni) to identify single nucleotide polymorphisms (SNPs). Mapping was constrained to non-repetitive sequences, representing 98.4 % coverage of the H37Rv reference. Average depth of coverage per genome was as follows: Mtb_41, 215 × ; Mtb_323, 151 × ; Mtb_682, 38 × ; Mtb_1087, 46 × ; Mtb_2573, 109 × ; Mtb_2855, 34 × ; Mtb_4891, 128 × ; Mtb_5288, 12 × . The low coverage of Mtb_5288 was below our minimum target coverage of 30 × but additional DNA from this isolate was unavailable so was included here with the caveat of potential SNP-calling unreliability. Bi-allelic mutations were called when read-mapping indicated the presence of an alternative nucleotide in greater than 10 % of reads across a given position in the reference. The Variant Call Format (VCF) files describing the SNPs for each isolate were uploaded to P (http://pathogenseq.lshtm.ac.uk/phytblive/index.php) to determine the closest of the seven M. tuberculosis complex lineages and as an initial screen for mutations linked to antibiotic resistance. Resistance mutations were also identified with reference to the TB Drug Resistance Mutation Database (http://www.tbdreamdb.com/) (Sandgren et al., 2009). IS6110 insertion site mapping was performed with IS_mapper (https://github.com/jhawkey/IS_mapper), using NC_000962.3 as a reference (Hawkey et al., 2015).
Results and Discussion
Case history
In January 1991, a 35-year-old East Timorese female presented with haemoptysis, sweats and 5 kg weight loss over 6 months. She had arrived in Melbourne, Australia, in October 1990, and had been treated for pulmonary TB with an unknown treatment regimen during the year leading up to her arrival in Australia. The patient's admission chest radiograph demonstrated left upper lobe consolidation, and sputum was smear-positive for acid-fast bacilli (AFB). She commenced treatment with isoniazid, rifampicin, pyrazinamide and ethambutol for a presumptive diagnosis of TB. M. tuberculosis was subsequently isolated from sputum. Multiple changes were made to the treatment regimen as summarized in Fig. 1. Susceptibility testing demonstrated resistance to isoniazid and rifampicin but susceptibility to ethambutol and pyrazinamide, confirming a diagnosis of multidrug-resistant TB (MDR-TB). Susceptibility testing results are summarized in Fig. 2. The patient remained symptomatic with persistent cough and haemoptysis, and remained smear-positive for AFB and culture-positive for M. tuberculosis. In April 1992, the patient underwent left upper lobectomy, and excision of the apical segment of the left lower lobe. Following this, the patient's cough resolved. She remained well from that time, although she was able to produce sputum that was intermittently smear-positive for AFB, and persistently culture positive for M. tuberculosis. In July 2004, the patient remained well and all medications were stopped, although the preceding sputum specimen had been culture-positive for M. tuberculosis. The patient was reviewed regularly between then and 2010 and remained well. She was still well in 2010 when a repeat sputum specimen was smear-negative but culture-positive for M. tuberculosis. Treatment was not reinitiated. Subsequent sputum specimens in 2011 and 2012 were culture-positive for M. tuberculosis but two sputum specimens in 2014 were culture-negative.
Fig. 1.

MDR-TB therapy administered, 1991–2004. Depicted in red on the left are the antibiotics used with the date that each treatment was initiated and the duration indicated by the blue horizontal bars. Red triangles indicate the date sputum samples were collected from which M. tuberculosis was isolated in culture. The isolate ID numbers are shown above the sample collection date.
Fig. 2.
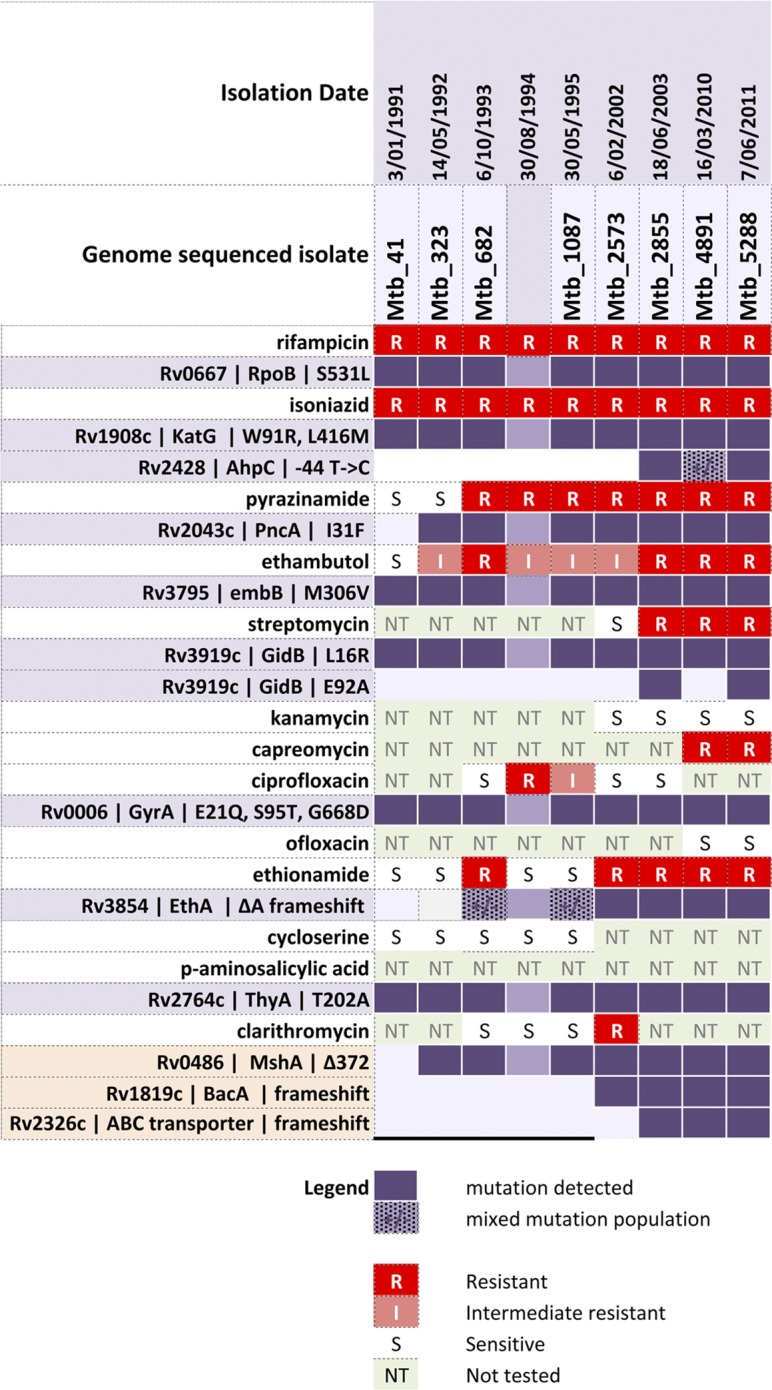
Summary of M. tuberculosis mutations occurring over 20 years. The left-hand column shows antibiotic (white rows with red blocks for resistance) and resistance gene/locus implicated (mauve rows with purple blocks indicate presence of the resistance-conferring mutation). Note that a DNA sample was not available for sequencing the isolate from 30 August 1994 although resistance testing was performed.
Initial genotypic multi-drug resistance
Taking advantage of DNA preparations stored by the reference laboratory from initial genotyping analysis, eight M. tuberculosis genomes were sequenced spanning the 20 years of the 21-year treatment and observation period. Genome comparisons of the initial 1991 isolate (isolate 41) against a database of 1601 M. tuberculosis-complex genome sequences showed that isolate 41 was a lineage 4 strain (lineage 4.3.4.1), and confirmed that it contained mutations in rpoB and katG, which corresponded with phenotypic resistance to rifampicin and isoniazid (Fig. 2). Isolate 41 also had a mutation in embB, which is associated with ethambutol resistance (Sreevatsan et al., 1997b). Initial susceptibility testing in 1991 suggested the isolate was sensitive to this agent. Subsequent isolates demonstrated reduced ethambutol susceptibility.
In addition to the mutations associated with resistance to first-line agents, isolate 41 also harboured mutations associated with resistance to streptomycin, quinolones and p-aminosalicylic acid (PAS). This included a mutation in gidB linked to streptomycin resistance (Okamoto et al., 2007). Streptomycin susceptibility testing was not done until 1998, at which point the isolate tested susceptible to streptomycin, although after 5 years of treatment with streptomycin phenotypic resistance subsequently developed in 2003. There were no mutations in aminoglycoside-resistance-conferring genes including rrs, rpsL or eis (Zaunbrecher et al., 2009). Isolate 41 had three gyrA mutations, one of which (S95T) has been previously reported in association with reduced susceptibility to quinolones (Kapur et al., 1995). Quinolone susceptibility testing was not done until 1993, at which point the isolate tested susceptible to ciprofloxacin. However, subsequent decreased susceptibility to ciprofloxacin was observed in four of 15 isolates during the 10 years of treatment with this agent from 1994 to 2003. Isolate 41 also possessed a characteristic mutation in thyA that is associated with resistance to PAS (Rengarajan et al., 2004) (Fig. 2). However, phenotypic PAS susceptibility testing was never undertaken and it is uncertain whether the patient had previously received PAS as part of TB treatment in East Timor.
At the outset of treatment in 1991, the patient was therefore infected with an M. tuberculosis strain harbouring genotypic resistance to four first-line drugs (rifampicin, isoniazid, ethambutol and streptomycin) and two second-line drugs (quinolones and PAS). If a genomic approach had been available in 1991, knowledge of this resistance profile at the commencement of treatment may have prevented exposure to unnecessary toxic drugs, and allowed composition of an effective regimen that included enough active drugs to prevent emergence of further resistance. Phenotypic susceptibility testing is known to be unreliable for ethambutol, pyrazinamide and some second-line agents, which may account for the discordant phenotype/genotype results (Hillemann et al., 2013). While the increased availability of WGS has led to a rapid increase in available sequence data, there is significant remaining uncertainty surrounding the phenotypic implications of putative drug resistance mutations. It is also likely that some antibiotic resistance-associated genes are yet to be identified (Witney et al., 2015). A standardized method for large-scale identification of drug resistance mutations has recently been proposed (Walker et al., 2015).
Additional drug resistance mutations arising during 14 years of treatment
Treatment with pyrazinamide began in January 1991, and pyrazinamide resistance was first detected in June 1993. Genome analysis uncovered a resistance-conferring pncA mutation in May 1992, 13 months prior to the first detection of phenotypic resistance (Fig. 2). Prothionamide treatment began in June 1991, with ethionamide resistance observed in October 1993 and the appearance of an ethionamide resistance mutation in ethA at that time (Fig. 2). Heterogeneity was initially present; 26 % of sequence reads for the October 1993 isolate demonstrated the resistance-conferring et mutation and the remainder were consistent with wild-type (Table S1, available in the online Supplementary Material). This population dynamic subsequently shifted such that the ΔethA mutant was dominant and the wild-type sequence was no longer detected (Fig. 2, Table 1). An additional mutation linked to isoniazid resistance, an intergenic nucleotide substitution 44 bp upstream of ahpC, was identified in the June 2003 isolate (Sreevatsan et al., 1997a). Also in June 2003, a second gidB mutation (E92A) was identified at the same time that streptomycin resistance was detected. To our knowledge, this mutation has not been previously identified as being associated with streptomycin resistance. We hypothesize that the combination of the existing L16R with the subsequent E92A mutation may have had an additive effect and led to emergence of phenotypic resistance to streptomycin. Of note, there was re-emergence of the wild-type allele in March 2010 but reversion to E92A by June 2011.
Table 1. Mutations detected among the seven M. tuberculosis isolates over 20 years compared with initial isolate 41–, Deletion; bold type and bold border indicate persisting mutation; ‘X/Y’ indicates bi-allelic base call.

The lag between genotypic and phenotypic resistance and the heterogeneity of sequence reads is likely to be due to the presence of subpopulations of M. tuberculosis with different resistance profiles. Heterogeneity in susceptibility results for different M. tuberculosis colonies cultured from the same sputum specimen has previously been described (Mariam et al., 2011; Sun et al., 2012). There is evidence to suggest that such heterogeneity is associated with increased likelihood of a poor clinical outcome (Zetola et al., 2014). With this constellation of drug resistance-associated mutations, the patient received a regimen with only one or two active agents for the majority of her 14 years of treatment.
Very low M. tuberculosis mutation rate during active infection
The mutation rate for M. tuberculosis compared with other pathogens is low and estimated at 0.3-0.5 mutations per genome year− 1 (Ford et al., 2011), and is thought to be similar in latent, active and recently reactivated disease (Ford et al., 2013). Across the eight genomes collected over a 21-year period of active infection, 17 mutations were detected, representing a mutation rate of 0.8 mutations per genome year− 1 if all mutations are considered (Tables 1 and S1). The majority of mutations occurred during antibiotic treatment. There were only two new mutations detected between cessation of treatment in 2004 and the final isolate in 2011. We attempted to examine the background mutation rate by counting only synonymous mutations. However, all within-CDS (coding DNA sequence) nucleotide mutations that became fixed in the population over 20 years were also non-synonymous, i.e. they changed the amino acid sequence. Furthermore, the two intergenic SNPs that became fixed (positions 2726149 and 3726078) were probably promoter-associated mutations and so had a functional consequence (Table 1). These observations indicate a mutation rate much lower than previous reports, with no background mutations occurring over 20 years of active infection.
The genome of each isolate harboured eight copies of IS6110 with no insertion site variation between genomes (data not shown). The isolates all harboured an IS6110 copy in the or region between dnaA and dnaN, as has been described previously by Turcios et al. (2009).
M. tuberculosis population heterogeneity
In four isolates there were mixed alleles for some loci, where read-mapping indicated two potential sequence variants at a particular chromosome position (Tables 1 and S1). This observation highlights the interconnected and shifting nature of the bacterial population within the patient, and is consistent with the presence of subpopulations, with dynamic changes occurring as a result of selection pressures such as antibiotic exposure and host immune responses.
Clinical persistence-associated mutations
In February 2002, a loss-of-function frameshift mutation was identified in bacA. This gene encodes a putative membrane protein (BacA) and its orthologues have been implicated in the maintenance of chronic infection and/or symbiosis in Brucella abortus (Lier et al., 2000) and Sinorhizobium melliloti (Glazebrook et al., 1993) and their respective hosts. In M. tuberculosis, bacA is thought to encode an ABC transporter. A laboratory-derived M. tuberculosis bacA deletion mutant exhibited reduced susceptibility to aminoglycosides and prolonged survival in a murine lung infection model (221 days) compared with wild-type (156 days), indicating a role for BacA in chronicity of infection (Domenech et al., 2009). Interestingly, bacA has been reported to be upregulated in the context of rifampicin monoresistance, providing another example of a link between this gene and antibiotic exposure (Li et al., 2015).
Between February 2002 and June 2003, a nucleotide insertion occurred causing a frameshift mutation in Rv2623c, a putative ABC transporter ATP-binding protein (Yousuf et al., 2015). Expression of the transcriptional regulator Rv0494 is increased in M. tuberculosis during conditions of starvation such as during dormancy (Yousuf et al., 2015). Furthermore, Rv0494 has been shown to downregulate expression of Rv2623c (Yousuf et al., 2015). This suggests that latency is associated with decreased expression of Rv2623c. We propose that the Rv2623c loss-of-function mutation may reduce M. tuberculosis metabolic activity, and that this may have contributed to the patient's relatively indolent disease progression.
An in-frame deletion of codon 372 of Rv0486 mshA first occurred in May 1992. MshA is an enzyme required for production of a mycothiol precursor, 1-O-(2-acetamido-2-deoxy-α-d-glucopyranosyl)-d-myo-inositol (Newton et al., 2006). Mycothiol is a glutathione-like thiol produced by M. tuberculosis and other Actinobacteria. Mycothiol has an important role in defence against alkylating agents and reactive oxygen and nitrogen species, and is essential for mycobacterial growth (Newton et al., 2008). MshA mutagenesis studies indicate that this enzyme is required for mycobacterial growth and survival (Buchmeier & Fahey, 2006). MshA also has been implicated in drug resistance, arising during patient treatment with ethionamide (Vilchèze et al., 2008). The mshA codon-372 deletion may alter bacterial fitness but the impact of this change is uncertain.
An intergenic substitution mutation − 9 bp upstream of Rv1819c was observed in the 2011 isolate. This mutation appears to have altered the ribosome binding site (RBS) for this CDS, changing the motif ‘GGAG’ to ‘GGAA’. Rv1819c endodes a PE/PPE family protein PE15. PE15 has been associated with altered inflammatory immune responses and intracellular survival and we speculate that altered expression of PE15 may also help to explain the persistence of this infection (Tiwari et al., 2012).
The patient initially had severe symptoms of TB and evidence of advanced and progressive pulmonary infection. Her disease burden was reduced significantly with a lobectomy. This procedure was not curative, however, and M. tuberculosis persisted thereafter for many years. Despite culture positivity at the time of treatment cessation, the disease did not progress in this patient; she appeared to remain in homeostasis with the organism for many years subsequently. We hypothesize that the mutations in bacA, Rv2326c and potentially mshA contributed to the persistent but mild disease course in this patient. However, we also recognize that each of these genes has been linked previously in various ways to resistance, and thus the driving pressures for changes in these loci may have been the antibiotics, not host immune pressures per se. Note, however, that in other pathogens resistance mutations can confer persister phenotypes. Studies of clinical Staphylococcus aureus show that rifampicin-resistance-conferring mutations in rpoB lead to increased resistance to innate immune killing mechanisms and increased in vivo persistence (Gao et al., 2013).
It is also likely that the resistance-associated mutations observed in this study have altered bacterial fitness. Resistance mutations have varying effects on the fitness of M. tuberculosis. These effects are often moderated by compensatory mutations at other sites (Müller et al., 2013). For example, mutations in rpoB are frequently associated with decreased fitness, although the effect can be decreased by compensatory mutations in rpoB or rpoB (Gagneux et al., 2006). Strains with mutations leading to low or no fitness cost account for the majority of drug-resistant isolates from clinical specimens; there is evidence that these strains are positively selected for over time when heteroresistance is present, and are more easily transmitted than resistant strains with high fitness cost (Müller et al., 2013).
Conclusion
We describe an unusual case of MDR-TB characterized by treatment failure but an indolent course. Initial phenotypic susceptibility testing revealed resistance to isoniazid and rifampicin, although at treatment outset there was unknown genotypic resistance to these drugs as well as to ethambutol, quinolones, streptomycin and PAS. Subsequent resistance-associated mutations for pyrazinamide and ethionamide emerged. If this genotypic resistance profile is considered reliable, the patient was therefore treated with just one or two active drugs for the majority of the 14-year treatment duration. The lag between genotypic and phenotypic resistance and the heterogeneity of sequence reads are suggestive of bacterial subpopulations with different resistance profiles, although some discordance between genotypic and phenotypic results may be due to unreliable phenotypic susceptibility testing. The majority of mutations identified occurred during the period of multi-drug treatment and were associated with drug resistance. The additional emergence of mutations in bacA, RV2326c, Rv1819 and mshA may have contributed to the indolent but persistent disease course. Our study follows a previous report that used genomics similarly to describe the evolution of XDR-TB in a single patient over a 4-year period (Eldholm et al., 2014). This report also uncovered considerable within-sample genomic heterogeneity, as well as identifying mutations and expression changes in M. tuberculosis export-associated genes that might be linked with increased resistance (Eldholm et al., 2014). WGS of serial M. tuberculosis isolates from single patient series are providing important real-world insights into within-host evolution of M. tuberculosis during periods with and without treatment. Findings from these studies have implications for treatment but also for aiding interpretation of genome sequence variation in the context of M. tuberculosis transmission.
Acknowledgements
We acknowledge funding support from the Victorian Infectious Diseases Service and the NHMRC Centre for Clinical Excellence in Tuberculosis Control. We are grateful to the patient for her consent to be involved in this study.
Supplementary Data
Supplementary Data
Abbreviations:
- AFB
acid-fast bacilli
- CDS
coding DNA sequence
- MDR-TB
multidrug-resistant TB
- PAS
p-aminosalicylic acid
- SNP
single nucleotide polymorphism
- TB
tuberculosis
- WGS
whole genome sequencing
References
- Buchmeier N., Fahey R. C. (2006). The mshA gene encoding the glycosyltransferase of mycothiol biosynthesis is essential in Mycobacterium tuberculosis Erdman FEMS Microbiol Lett 26474–79 10.1111/j.1574-6968.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- Coscolla M., Gagneux S. (2010). Does M. tuberculosis genomic diversity explain disease diversity? Drug Discov Today Dis Mech 7e43–e59 10.1016/j.ddmec.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenech P., Kobayashi H., Lier K., Walker G. C., Barry C. E., III (2009). BacA, an ABC transporter involved in maintenance of chronic murine infections with Mycobacterium tuberculosis J Bacteriol 191477–485 10.1128/JB.01132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldholm V., Norheim G., von der Lippe B., Kinander W., Dahle U. R., Caugant D. A., Mannsåker T., Mengshoel A. T., Dyrhol-Riise A. M., Balloux F. (2014). Evolution of extensively drug-resistant Mycobacterium tuberculosis from a susceptible ancestor in a single patient Genome Biol 15490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford C. B., Lin P. L., Chase M. R., Shah R. R., Iartchouk O., Galagan J., Mohaideen N., Ioerger T. R., Sacchettini J. C., other authors (2011). Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection Nat Genet 43482–486 10.1038/ng.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford C. B., Shah R. R., Maeda M. K., Gagneux S., Murray M. B., Cohen T., Johnston J. C., Gardy J., Lipsitch M., Fortune S. M. (2013). Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis Nat Genet 45784–790 10.1038/ng.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux S., Long C. D., Small P. M., Van T., Schoolnik G. K., Bohannan B. J. (2006). The competitive cost of antibiotic resistance in Mycobacterium tuberculosis Science 3121944–1946 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- Gao W., Cameron D. R., Davies J. K., Kostoulias X., Stepnell J., Tuck K. L., Yeaman M. R., Peleg A. Y., Stinear T. P., Howden B. P. (2013). The RpoB H481Y rifampicin resistance mutation and an active stringent response reduce virulence and increase resistance to innate immune responses in Staphylococcus aureus J Infect Dis 207929–939 10.1093/infdis/jis772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazebrook J., Ichige A., Walker G. C. (1993). Rhizobium meliloti homolog of the Escherichia coli peptide-antibiotic transport protein SbmA is essential for bacteroid development Genes Dev 71485–1497 10.1101/gad.7.8.1485. [DOI] [PubMed] [Google Scholar]
- Hawkey J., Hamidian M., Wick R. R., Edwards D. J., Billman-Jacobe H., Hall R. M., Holt K. E. (2015). ISMapper: identifying transposase insertion sites in bacterial genomes from short read sequence data BMC Genomics 16667. 10.1186/s12864-015-1860-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillemann D., Hoffner S., Cirillo D., Drobniewski F., Richter E., Rüsch-Gerdes S., Baltic-Nordic TB-Laboratory Network, TB PAN-NET & ECDC ERLN-TB Networks (2013). First evaluation after implementation of a quality control system for the second line drug susceptibility testing of Mycobacterium tuberculosis joint efforts in low and high incidence countries PL One 8e76765. 10.1371/journal.pone.0076765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur V., Li L. L., Hamrick M. R., Plikaytis B. B., Shinnick T. M., Telenti A., Jacobs W.R., Jr, Banerjee A., Cole S., other authors (1995). Rapid Mycobacterium species assignment and unambiguous identification of mutations associated with antimicrobial resistance in Mycobacterium tuberculosis by automated DNA sequencing Arch Pathol Lab Med 119131–138. [PubMed] [Google Scholar]
- Lier K., Phillips R. W., Grippe V. K., Roop R.M., II, Walker G. C. (2000). Similar requirements of a plant symbiont and a mammalian pathogen for prolonged intracellular survival Science 2872492–2493 10.1126/science.287.5462.2492. [DOI] [PubMed] [Google Scholar]
- Li G., Zhang J., Guo Q., Wei J., Jiang Y., Zhao X., Zhao L. L., Liu Z., Lu J., Wan K. (2015). Study of efflux pump gene expression in rifampicin-monoresistant Mycobacterium tuberculosis clinical isolates J Antibiot (Tokyo) 68431–435 10.1038/ja.2015.9. [DOI] [PubMed] [Google Scholar]
- Mariam S. H., Werngren J., Aronsson J., Hoffner S., Andersson D. I. (2011). Dynamics of antibiotic resistant Mycobacterium tuberculosis during long-term infection and antibiotic treatment PLoS One 6e21147. 10.1371/journal.pone.0021147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller B., Borrell S., Rose G., Gagneux S. (2013). The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis Trends Genet 29160–169 10.1016/j.tig.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton G. L., Ta P., Bzymek K. P., Fahey R. C. (2006). Biochemistry of the initial steps of mycothiol biosynthesis J Biol Chem 28133910–33920 10.1074/jbc.M604724200. [DOI] [PubMed] [Google Scholar]
- Newton G. L., Buchmeier N., Fahey R. C. (2008). Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria Microbiol Mol Biol Rev 72471–494 10.1128/MMBR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S., Tamaru A., Nakajima C., Nishimura K., Tanaka Y., Tokuyama S., Suzuki Y., Ochi K. (2007). Loss of a conserved 7-methylguanosine modification in 16S rRNA confers low-level streptomycin resistance in bacteria Mol Microbiol 631096–1106 10.1111/j.1365-2958.2006.05585.x. [DOI] [PubMed] [Google Scholar]
- Rengarajan J., Sassetti C. M., Naroditskaya V., Sloutsky A., Bloom B. R., Rubin E. J. (2004). The folate pathway is a target for resistance to the drug para-aminosalicylic acid (PAS) in mycobacteria Mol Microbiol 53275–282 10.1111/j.1365-2958.2004.04120.x. [DOI] [PubMed] [Google Scholar]
- Ross B. C., Raios K., Jackson K., Sievers A., Dwyer B. (1991). Differentiation of Mycobacterium tuberculosis strains by use of a nonradioactive Southern blot hybridization method J Infect Dis 163904–907 10.1093/infdis/163.4.904. [DOI] [PubMed] [Google Scholar]
- Sandgren A., Strong M., Muthukrishnan P., Weiner B. K., Church G. M., Murray M. B. (2009). Tuberculosis Drug Resistance Mutation Database PL Med 6e1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreevatsan S., Pan X., Zhang Y., Deretic V., Musser J. M. (1997a). Analysis of the oxyR-ahpC region in isoniazid-resistant and-susceptible Mycobacterium tuberculosis complex organisms recovered from diseased humans and animals in diverse localities Antimicrob Agents Chemother 41600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreevatsan S., Stockbauer K. E., Pan X., Kreiswirth B. N., Moghazeh S. L., Jacobs W. R., Jr, Telenti A., Musser J. M. (1997b). Ethambutol resistance in Mycobacterium tuberculosis: critical role of embB mutations Antimicrob Agents Chemother 411677–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G., Luo T., Yang C., Dong X., Li J., Zhu Y., Zheng H., Tian W., Wang S., other authors (2012). Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients J Infect Dis 2061724–1733 10.1093/infdis/jis601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari B. M., Kannan N., Vemu L., Raghunand T. R. (2012). The Mycobacterium tuberculosis PE proteins Rv0285 and Rv1386 modulate innate immunity and mediate bacillary survival in macrophages PL One 7e51686. 10.1371/journal.pone.0051686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcios L., Casart Y., Florez I., de Waard J., Salazar L. (2009). Characterization of IS6110 insertions in the dnaA-dnaN intergenic region of Mycobacterium tuberculosis clinical isolates Clin Microbiol Infect 15200–203 10.1111/j.1469-0691.2008.02107.x. [DOI] [PubMed] [Google Scholar]
- Vilchèze C., Av-Gay Y., Attarian R., Liu Z., Hazbón M. H., Colangeli R., Chen B., Liu W., Alland D., other authors (2008). Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis Mol Microbiol 691316–1329 10.1111/j.1365-2958.2008.06365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker T. M., Kohl T. A., Omar S. V., Hedge J., Del Ojo Elias C., Bradley P., Iqbal Z., Feuerriegel S., Niehaus K. E., other authors (2015). Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study Lancet Infect Dis 151193–1202 10.1016/S1473-3099(15)00062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witney A. A., Gould K. A., Arnold A., Coleman D., Delgado R., Dhillon J., Pond M. J., Pope C. F., Planche T. D., other authors (2015). Clinical application of whole-genome sequencing to inform treatment for multidrug-resistant tuberculosis cases J Clin Microbiol 531473–1483 10.1128/JCM.02993-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousuf S., Angara R., Vindal V., Ranjan A. (2015). Rv0494 is a starvation-inducible, auto-regulatory Fa-like regulator from Mycobacterium tuberculosis Microbiology 161463–476 10.1099/mic.0.000017. [DOI] [PubMed] [Google Scholar]
- Zaunbrecher M. A., Sikes R. D., Jr, Metchock B., Shinnick T. M., Posey J. E. (2009). Overexpression of the chromosomally encoded aminoglycoside acetyltransferase eis confers kanamycin resistance in Mycobacterium tuberculosis Proc Natl Acad Sci U S A 10620004–20009 10.1073/pnas.0907925106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetola N. M., Modongo C., Moonan P. K., Ncube R., Matlhagela K., Sepako E., Collman R. G., Bisson G. P. (2014). Clinical outcomes among persons with pulmonary tuberculosis caused by Mycobacterium tuberculosis isolates with phenotypic heterogeneity in results of drug-susceptibility tests J Infect Dis 2091754–1763 10.1093/infdis/jiu040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Data Bibliography
- 1. Stinear, T. Fihare http://dx.doi.org/10.6084/m9.figshare.1561411 (2015).
- 2. Stinear, T. European Nucleotide Archive (ENA) http://www.ebi.ac.uk/ena/data/view/ERP011175 (2015).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data