

Abstract
Background
In recent years, novel hepadnaviruses, hepeviruses, hepatoviruses, and hepaciviruses have been discovered in various species of bat around the world, indicating that bats may act as natural reservoirs for these hepatitis viruses. In order to further assess the distribution of hepatitis viruses in bat populations in China, we tested the presence of these hepatitis viruses in our archived bat liver samples that originated from several bat species and various geographical regions in China.
Methods
A total of 78 bat liver samples (involving two families, five genera, and 17 species of bat) were examined using nested or heminested reverse transcription PCR (RT-PCR) with degenerate primers. Full-length genomic sequences of two virus strains were sequenced followed by phylogenetic analyses.
Results
Four samples were positive for hepadnavirus, only one was positive for hepevirus, and none of the samples were positive for hepatovirus or hepacivirus. The hepadnaviruses were discovered in the horseshoe bats, Rhinolophus sinicus and Rhinolophus affinis, and the hepevirus was found in the whiskered bat Myotis davidii. The full-length genomic sequences were determined for one of the two hepadnaviruses identified in R. sinicus (designated BtHBVRs3364) and the hepevirus (designated BtHEVMd2350). A sequence identity analysis indicated that BtHBVRs3364 had the highest degree of identity with a previously reported hepadnavirus from the roundleaf bat, Hipposideros pomona, from China, and BtHEVMd2350 had the highest degree of identity with a hepevirus found in the serotine bat, Eptesicus serotinus, from Germany, but it exhibited high levels of divergence at both the nucleotide and the amino acid levels.
Conclusions
This is the first study to report that the Chinese horseshoe bat and the Chinese whiskered bat have been found to carry novel hepadnaviruses and a novel hepevirus, respectively. The discovery of BtHBVRs3364 further supports the significance of host switches evolution while opposing the co-evolutionary theory associated with hepadnaviruses. According to the latest criterion of the International Committee on Taxonomy of Viruses (ICTV), we hypothesize that BtHEVMd2350 represents an independent genotype within the species Orthohepevirus D of the family Hepeviridae.
Keywords: Bat, Natural reservoir, Hepadnavirus, Hepevirus, Genome characterization
Background
Nearly 60% of emerging infectious diseases in humans are zoonotic, with up to 70% of them being found to originate from wildlife [1]. Bats have been identified as natural reservoirs of many viruses. Some of these viruses cause outbreaks of severe disease in humans [2], including the Ebola virus, the lyssavirus, the severe acute respiratory syndrome coronavirus, and henipaviruses [3]. Interestingly, these viruses rarely cause apparent clinical signs in bats [2]. Bats possess unique characteristics that may contribute to their ability to act as a major natural reservoir for viruses, including a high level of species diversity, a long lifespan, a high population density, and high levels of spatial mobility [4].
Previous studies mainly focused on bat-borne viruses that are transmitted via respiratory droplets [3]. However, in recent years, several hepatitis virus-related sequences, including those associated with hepadnaviruses, hepeviruses, hepatoviruses, and hepaciviruses, have been found in bats across the globe, indicating the importance of bats as the natural reservoirs of these viruses [5–9].
Hepatitis viruses include hepatitis viruses A, B, C, D, and E, which cause human hepatitis diseases. Hepatitis A virus (HAV) is classified as belonging to the genus Hepatovirus in the family Picornaviridae. Hepatitis B virus (HBV) is classified as belonging to the genus Orthohepadnavirus in the family Hepadnaviridae. Hepatitis C virus (HCV) is classified as belonging to the genus Hepacivirus in the family Flaviriridae. Hepatitis D virus (HDV) is considered to be a subviral satellite because it can only propagate in the presence of HBV. Hepatitis E virus (HEV) is classified as belonging to the genus Orthohepevirus in the family Hepeviridae. Hepatovirus-related sequences have been identified in 13 species of bat collected in North America, Europe, and Africa [5]. Hepadnavirus-related sequences have been discovered in five species of bat collected in Panama, Gabon, Myanmar, and China [6, 8–10]. Highly diverse hepacivirus-related sequences have been detected in 20 species of bat across the world [11]. Hepevirus-related sequences have been discovered in bats in Ghana, Panama, and Germany [7]. These results indicate that bats may be important reservoirs of these hepatitis viruses (Table 1).
Table 1.
Hepatitis virus-like sequences detected in bats
| Virus family | Order: Chiroptera | Sampling site (s) (year) | Isolation source | Genomic sequence | Reference | ||
|---|---|---|---|---|---|---|---|
| Family | Genera | Species | |||||
| Hepatoviridae | Emballonuridae | Coleura | C. afra | Ghana (2011), Gabon (2009) | Feces, blood | Full | [5] |
| Pteropodidae | Eidolon | E. helvum | Ghana (2010/2011) | Blood | Full | ||
| Natalidae | Natalus | N. lanatus | Costa Rica (2010) | Feces | Full | ||
| Rhinolophidae | Rhinolophus | R. landeri | Ghana (2011) | Feces | Full | ||
| R. ferrumequinum | Romania (2008/2009), Bulgaria (2008/2009), Luxemburg (2011) | Feces | Full | ||||
| Rhinolophus | R. hipposideros | Bulgaria (2008/2009), Spain (2010) | Feces | Full | |||
| Vespertilionidae | Glauconycteris | G. spec. | Côte d’Ivoire (2013) | Intestines | Full | ||
| Miniopterus | M. cf. manavi | Madagascar (2014) | Liver | Full | |||
| M. schreibersii | Romania (2008) | Feces | Full | ||||
| Myotis | M. dasycneme | Germany (2008) | Feces | Full | |||
| M. myotis | Romania (2008), Germany (2008/2010) | Feces | Full | ||||
| Nyctalus | N. noctula | Romania (2008/2009), Germany (2009) | Feces | Full | |||
| Pipistrellus | P. kuhlii | Ukraine (2010/2011) | Feces | Full | |||
| Hepadnaviridae | Vespertilionidae | Miniopterus | Miniopterus spp. | Myanmar (2010) | Liver | Full | [8] |
| Hipposideridae | Hipposideros | H. pomona | China (2011) | Liver | Full | [9] | |
| H. cf. ruber | Gabon (2009) | Blood | Full | [6] | |||
| Phyllostomidae | Uroderma | U. bilobatum | Panama (2010/2011) | Blood | Full | ||
| Rhinolophidae | Rhinolophus | R. alcyone | Gabon (2009) | Blood | Full | ||
| R. sinicus | China (2012) | Liver | Full | This study | |||
| R. affinis | China (2013) | Liver | Partial | ||||
| Hepaciviridae | Emballonuridae | Taphozous | Not identified | Cameroon (2010) | Blood | Partial | [11] |
| Hipposideridae | Hipposideros | H. vittatus | Kenya (2010) | Blood | Full | ||
| H. gigas | Nigeria (2008/2010) | Blood | Partial | ||||
| Molossidae | Chaerephon | Not identified | Cameroon (2010), Kenya (2010) | Blood | Partial | ||
| Mops | M. condylurus | DRC (2011) | Blood | Full | |||
| Otomops | O. martiensseni | Kenya (2010) | Blood | Full | |||
| Nyctinomops | N. macrotis | Mexico (2011) | Blood | Partial | |||
| Phyllostomidae | Artibeus | A. watsoni | Mexico (2011) | Blood | Partial | ||
| Carollia | C. perspicillata | Guatemala (2010), Mexico (2011) | Blood | Full | |||
| Desmodus | D. rotundus | Guatemala (2010), | Blood | Partial | |||
| Glossophaga | G. commissarisi | Mexico (2011) | Blood | Partial | |||
| Sturnira | S. lilium | Guatemala (2010) | Blood | Full | |||
| S. ludovici | Mexico (2011) | Blood | Partial | ||||
| Trachops | T. cirrhosus | Mexico (2011) | Blood | Partial | |||
| Pteropodidae | Eidolon | E. helvum | Cameroon (2010), DRC (2011) | Blood | Full | ||
| Epomophorus | E. labiatus | Kenya (2010) | Blood | Partial | |||
| Megaloglossus | M. woermanni | DRC (2011) | Blood | Full | |||
| Rousettus | R. aegyptiacus | Kenya (2010), Nigeria (2008/2010) | Blood | Full | |||
| Vespertilionidae | Scotoecus | Not identified | Kenya (2010) | Blood | Partial | ||
| Scotophilus | S. dingani | Kenya (2010) | Blood | Full | |||
| S. nigrita | Nigeria (2008/2010) | Blood | Partial | ||||
| Unknown | Nigeria (2008/2010), Mexico (2010/2011) | Blood | Partial | ||||
| Hepeviridae | Hipposideridae | Hipposideros | H. abae | Ghana (2009) | Feces | Partial | [7] |
| Phyllostomidae | Vampyrodes | V. caraccioli | Panama (2011) | Blood | Partial | ||
| Vespertilionidae | Eptesicus | E. serotinus | Germany (2008) | Liver | Full | ||
| Myotis Myotis |
M. bechsteinii | Germany (2008) | Feces | Partial | |||
| M. daubentonii | Germany (2008) | Feces | Partial | ||||
| M. davidii | China (2011) | Liver | Full | This study | |||
DRC Democratic Republic of the Congo
There are around 120 species of bat in China; however, only limited information has been reported regarding the hepatitis viruses, a novel Orthohepadnavirus in pomona roundleaf bats from Yunnan province was identified in 2015 [9]. In this study, we report the discovery of four novel hepadnaviruses and a hepevirus in our archived bat liver samples that had been collected from several bat species and various geographical regions in China.
Methods
Samples
A total of 78 liver tissue samples were collected from dead bats caused by accident during sampling, which comprised two families, five genera, and 17 species, and used for virus screening (Table 2). Different tissues (heart, liver, spleen, lung, kidney, brain and intestine) were collected separately and used for analyzing virus tissue tropism. The animals were firstly identified based on their morphology and then the species that they belonged to were further confirmed using DNA sequencing of the mitochondrial cytochrome b (CytB) gene following previously described methods [12].
Table 2.
Detection of hepadnavirus and hepevirus in bats in China between 2008 and 2013
| Family | Genus | Species | No. of samples | No. of hepadnavirus positive samples | No. of hepevirus positive samples | Sampling site (s) (year) |
|---|---|---|---|---|---|---|
| Vespertilionidae | Myotis | M. adversus | 4 | Yunnan (2008) | ||
| M. pilosus | 7 | Yunnan (2008) | ||||
| M. chinensis | 1 | Hubei (2008) | ||||
| M. davidii | 12 | 1 | Hubei (2011), Yunnan (2011) | |||
| M. ikonnikovi | 3 | Yunan (2009), Hubei (2011) | ||||
| M. formosus | 1 | Yunnan (2011) | ||||
| Miniopterus | M. fuliginosus | 1 | Henan (2010) | |||
| Aselliscus | A. stoliczkanus | 1 | Yunnan (2012) | |||
| Rhinolophidae | Rhinolophus | R. sinicus | 19 | 2 | Hubei (2008/2011), Sichuan (2011), Yunnan (2009/2012/2013) |
|
| R. monoceros | 3 | Hubei (2011), Chongqing (2011) | ||||
| R. affinis | 7 | 2 | Henan (2010), Hubei (2011) Yunnan (2012/2013), | |||
| R. pusillus | 4 | Yunnan (2012/2013) | ||||
| R. pearsonii | 1 | Chongqing (2011) | ||||
| Hipposideros | H. pratti | 2 | Hubei (2010) | |||
| H. armiger | 4 | Sichuan (2011) | ||||
| H. pomona | 7 | Yunnan (2013) | ||||
| H. larvatus | 1 | Yunnan (2011) | ||||
| Total | 5 genera | 17 species | 78 | 4 | 1 |
RNA extraction and PCR
RNA was extracted from tissue using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) following manufacturer’s instructions, and cDNA was synthesized using Moloney Murine Leukemia Virus (M-MLV) Reverse Transcriptase (Promega, Madison, WI, USA). The extracted RNA from liver was tested by nested or heminested reverse transcription PCR (RT-PCR) using degenerate primers based on the conserved domain of the RNA-dependent RNA polymerase (RdRp) gene of viruses in the genus Hepatovirus, the polymerase gene of viruses in the family Hepadnaviridae, the RdRp gene of viruses in the genus Hepacivirus [11], and the RdRp gene of viruses in the family Hepeviridae [7] (Table 3). Standard precautions were taken to avoid contamination of the PCR procedure, and no false-positives were observed in the negative controls. The PCR products underwent gel purification with MinElute Gel Extraction Kit (Qiagen, Germany) and they were sequenced with both forward and reverse primers using the 3100 Sequencer (ABI, Waltham, MA, USA).
Table 3.
Primers used for virus RT-PCR screening and virus quantification
| Primer | Sequence (5′-3′)a | Polarity | Targeted virus | Reference |
|---|---|---|---|---|
| HAV-3D-F1 | CYTATHTRAARGATGAGCTKAGA | + | Hepatovirus | This study |
| HAV-3D-F2 | ACRTCATCICCRTARCAIAGRA | + | ||
| HAV-3D-R1 | RTCIAARACWAGRGCNATYG | - | ||
| HAV-3D-R2 | TACCWAATCATRAATGGACT | - | ||
| HBV-pol-F1 | TAGACTSGTGGTGGACTTCTC | + | Hepadnavirus | This study |
| HBV-pol-F2 | AGTRAAYTGAGCCAGGAGAAAC | + | ||
| HBV-pol-R1 | TGCCATCTTCTTGTTGGTTC | - | ||
| HBV-pol-R1 | CATATAASTRAAAGCCAYACAG | - | ||
| BHV-1-F1 | GTAGCGGAGAAGATGTATCTGGG | + | Hepacivirus | [11] |
| BHV-1-R1 | GCCTTAGCCTTGAGAAAGCAGGTGAT | + | ||
| BHV-1-F2 | GAGAAGATGTATCTGGGGGACGT | + | ||
| BHV-1-R2 | AGAAAGCAGGTGATGGTATTGCC | + | ||
| BHV-2-F1 | CCAAARGTWGTBAAGGCTGTGCT | - | ||
| BHV-2-R1 | ACTTTGAKCCASGCAGTKARACAGTT | - | ||
| BHV-2-F2 | GCTGTGCTSAAGGAMGAGTACGGCT | + | ||
| BHV-2-R2 | CCASGCAGTKARACAGTTACTRGAG | - | ||
| DE-F4228 | ACYTTYTGTGCYYTITTTGGTCCITGGTT | + | Hepevirus | [7] |
| DE-R4598 | GCCATGTTCCAGAYGGTGTTCCA | - | ||
| DE-R4565 | CCGGGTTCRCCIGAGTGTTTCTTCCA | - | ||
| BtHEV-qF | ATGTCCGTGTTCAGGTTCC | + | Bat hepevirus | This study |
| BtHEV-qR | GCCAACCCTCATTTGCAAC | - | ||
| BtHBV-qF | TGTTGGTTCTCCTGGATTGGAG | + | Bat hepadnaviruses | This study |
| BtHBV-qR | TGAAGGAATGGGCCAGCAGGTG | - |
R: G/A; Y: C/T; S: G/C; W: A/T; M: A/C; K: G/T; H: A/C/T; N: A/T/C/G; I: inosine
Genomic sequencing
The complete genomic sequences of one hepadnavirus strain and one hepevirus strain were amplified using PCR with degenerate primers (the primers are available upon request). The genome ends were amplified using a 5′-Full RACE Kit (TaKaRa, Japan). The PCR products underwent gel purification with MinElute Gel Extraction Kit (Qiagen, Germany) and they were sequenced with both forward and reverse primers using the 3100 Sequencer. The sequencing chromatograms were inspected for overlapping multicolor peaks, which are an indicator of sequence heterogeneity in the amplicons. The PCR products were cloned using the pGEM-T Easy Vector System (Promega, Germany) and at least three clones for each PCR fragment were sequenced to obtain a consensus sequence.
Sequence analysis
The preliminary sequence management and analysis were carried out using Geneious version 9.1.3 (Biomatters Ltd., Auckland, New Zealand) and the sequence alignment and editing were performed using MAFFT [13]. The phylogenetic analysis of hepadnavirus used the neighbor-joining (NJ) method with Hasegawa-Kishino-Yano substitution model and complete deletion option and hepevirus used the maximum-likelihood (ML) method with the nucleotide percentage distance substitution matrix and the complete deletion option in MEGA version 7 [14]. The sequences and GenBank accession numbers of the representative viruses in the families Hepadnaviridae and Hepeviridae used in the phylogenetic analyses are presented in Figs. 1 and 2.
Fig. 1.
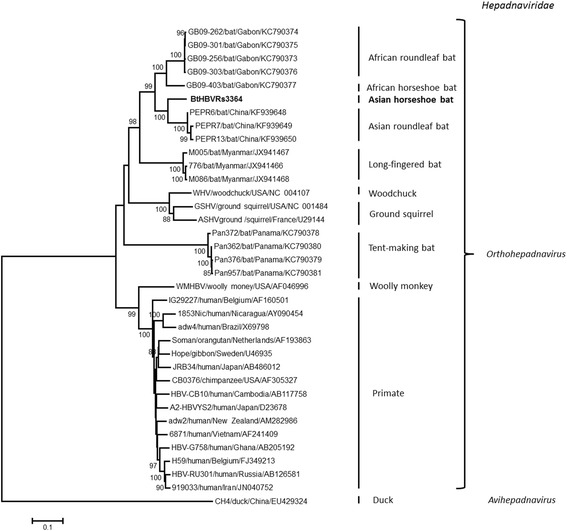
Phylogenetic analysis of bat hepadnavirus based on the full-length genomic sequences. Maximum likelihood phylogenetic tree was constructed based on the complete genomes of BtHBVRs3364 (in bold) and representative members of the family Hepadnaviridae using the Hasegawa-Kishino-Yano substitution model and complete deletion option in MEGA version 7. The values at the nodes indicate the bootstrap values (using 1,000 replications). The branches are labeled with the strain designation, the host species, and the GenBank accession number. The classification of the family Hepadnaviridae is indicated on the right
Fig. 2.
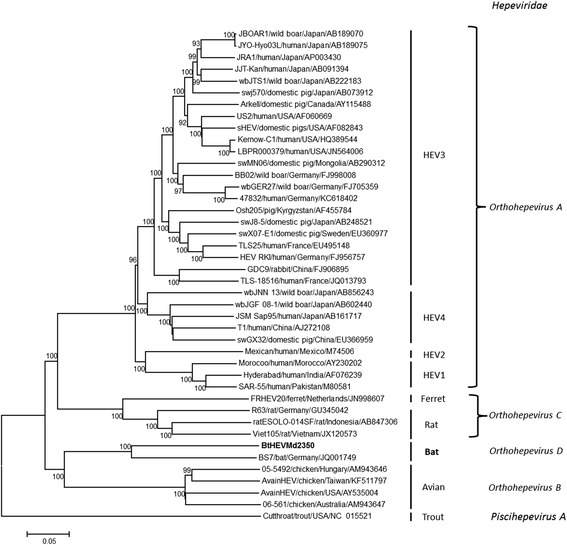
Phylogenetic analysis of bat hepevirus based on the full-length genomic sequences. Neighbor joining phylogenetic tree was constructed based on the alignment of the complete genomes of BtHEVMd2350 (in bold) and representative members of the family Hepeviridae using nucleotide percentage distance substitution matrix and complete deletion option implemented in MEGA version 7. The values at nodes indicate the bootstrap values (using 1,000 replications). The branches are labeled with the strain designation, the host species, and the GenBank accession number. The classification of the family Hepeviridae is indicated on the right
Quantification real-time PCR
Virus load of bat hepevirus and hepadnaviruese of different tissues was measured by using photometrically quantified in vitro RNA transcripts and specific real-time RT-PCR primers (Table 3). Quantification was done by using 5 μL of RNA extract, 300 nM each primer, using the One Step SYBR PrimeScript™ PLUS RT-PCR Kit (TaKaRa, Japan). Cycling in a Biorad CFX Connect instrument involved the following steps: 42 °C for 5 min, 95 °C for 10 s, and 40 cycles of 95 °C 5 s and 60 °C 20 s with measurement of fluorescence.
Results
Detection of four hepadnaviruses and a hepevirus in bat liver samples
Among the 78 bat liver samples, four were positive for hepadnavirus from Jinning city, Yunnan province and only one was positive for hepevirus from Xianning city, Hubei province (Fig. 3). However, none were positive for hepatovirus or hepacivirus. The nucleotide sequences of the four novel hepadnaviruses and the hepevirus described in this study are available from GenBank under the accession numbers KX513949–KX513953.
Fig. 3.
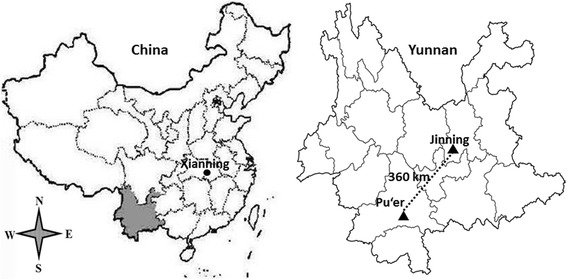
Representation map of China and Yunnan province. Circle indicates the sampling site where the hepevirus (BtHEVMd2350) was detected. Triangles indicates the sampling sites where the hepadnaviruses were detected
Sequence analysis of the bat hepadnavirus
All four of the hepadnavirus-positive samples were from horseshoe bats, two each from R. sinicus (designated BtHBVRs3364 and BtHBVRs3366) and R. affinis (designated BtHBVRa4325 and BtHBVRa4328) (Table 2). The four partial polymerase gene sequences had 92.1–97.5% nucleotide sequence identity and they were found to be closely related to the roundleaf bat hepadnavirus from Yunnan province, China, with nucleotide identities of 88.8–95.5% [9].
The full-length genomic sequence of a sample from R. sinicus (designated bat HBV Rs3364, or BtHBVRs3364) was determined and it was found to have a length of 3,272 nucleotides. The virus has an identical genomic organization to other hepadnaviruses, with four open reading frames (ORFs) encoding the surface (S), polymerase (P), core (C), and X proteins. In addition, the typical direct repeat (DR) sequences for viral genome replication and the secondary structure Ɛ–loops for viral reverse transcription were present in the BtHBVRs3364 genome. A detailed comparison of the full-length genomic sequence and the ORFs of the virus with other known hepadnaviruses is shown in Table 4. The results showed that the four genes of BtHBVRs3364 have the highest degree of identity with the roundleaf bat hepadnavirus from Yunnan province, at both the nucleotide and the amino acid levels [9]. Notably, we found large differences between BtHBVRs3364 and other hepadnaviruses from the African horseshoe and roundleaf bats, the long-fingered bat from Myanmar, and the tent-making bat from Panama.
Table 4.
Nucleotide and amino acid sequence identity between BtHBVRs3364 and representative orthohepadnavirus strainsa
| Hepadnavirus (no. of strains compared) | Degree of identity (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Full genome | S gene (1–672) | X gene (1217–1645) | C gene (1657–2301) | P gene (2147–1475) | |||||
| Nucleotides | Nucleotides | Amino acids | Nucleotides | Amino acids | Nucleotides | Amino acids | Nucleotides | Amino acids | |
| Asian roundleaf bat hepadnavirus (3) | 81.2–81.9 | 90.5–93.9 | 83.5–90.2 | 86.7 | 75.4 | 85.2–85.3 | 91.2–91.7 | 80.1–80.9 | 77.4–77.9 |
| African horseshoe bat hepadnavirus (1) | 72.2 | 89.0 | 84.8 | 78.8 | 65.5 | 75.2 | 77.9 | 71.8 | 65.9 |
| African roundleaf bat hepadnavirus (4) | 72.0–72.1 | 89.0–89.2 | 86.2 | 81.8–82.1 | 70.4–71.1 | 76.9–77.1 | 77.9 | 72.3–72.4 | 65.6–66.0 |
| Long-fingered bat hepadnavirus (3) | 69.0–69.2 | 79.7–80.1 | 67.4 | 69.2–70.2 | 49.3–51.4 | 74.2–74.5 | 75.6–76.0 | 68.1–68.3 | 61.5–61.8 |
| Tent-making bat hepadnavirus (4) | 56.7–56.8 | 73.5–74.2 | 61.0–61.9 | 58.9–59.3 | 43.4–44.1 | 51.6 | 49.5 | 56.8–57.1 | 49.1–49.2 |
| Primate HBV (15) | 59.5–61.1 | 76.5–77.8 | 65.0–66.8 | 62.1–64.9 | 46.5–52.1 | 61.0–67.9 | 63.0–66.5 | 57.0–59.7 | 50.2–52.0 |
| Woolly monkey hepadnavirus (1) | 61.5 | 75.3 | 67.1 | 63.2 | 50.0 | 64.2 | 61.4 | 60.3 | 51.9 |
| Ground squirrel hepadnavirus (2) | 64.0–65.1 | 78.7–78.8 | 67.9–68.3 | 60.6–63.9 | 45.1–46.5 | 71.1–71.4 | 70.9–74.0 | 58.9–59.1 | 49.3–50.2 |
| Woodchuck hepadnavirus (1) | 59.8 | 77.9 | 67.4 | 63.9 | 47.2 | 69.8 | 72.5 | 58.8 | 50.9 |
| Duck hepadnavirus (1) | 31.1 | 44.1 | 28.7 | NA | NA | 31.3 | 20.9 | 35.1 | 23.2 |
aThe sequences were aligned using MAFFT. The evolutionary analyses were conducted using MEGA version 7. The GenBank accession numbers are as follows: KF939648, KF939649, and KF939650 for the Asian roundleaf bat hepadnaviruses; KC790377 for the African horseshoe bat hepadnavirus; KC790374, KC790375, KC790373, and KC790376 for the African roundleaf bat hepadnaviruses; KC790378, KC790379, KC790380, and KC790381 for the Tent-making bat hepadnaviruses; AF160501, AY090454, X69798, AF193863, U46935, AB486012, AF305327, AB117758, D23678, AM282986, AF241409, AB205192, FJ349213, AB126581, and JN040752 for the Primate HBVs; AF046996 for the woolly monkey hepadnavirus; NC 001484 and U29144 for the ground squirrel hepadnaviruses; NC_004107 for the woodchuck hepadnavirus; and EU429324 for the duck hepadnaviruses. NA not available
Phylogenetic analysis of the bat hepadnavirus
A phylogenetic tree was constructed based on the alignment of the full-length genomic sequence of BtHBVRs3364 with those of representative hepadnavirus strains available in GenBank. As shown in Fig. 1, the previously reported bat hepadnaviruses formed three clusters, with clear specificities for particular hosts. Although BtHBVRs3364 clustered with the bat hepadnaviruses, it formed an independent branch. Interestingly, the BtHBVRs3364 detected in the horseshoe bat is phylogenetically closer to viruses from the Asian roundleaf bat compared to viruses from the African horseshoe bat, despite the fact that it was found in an Asian horseshoe bat.
Sequence analysis of the bat hepevirus
One sample found in the whiskered bat, M. davidii, from Hubei province was positive for hepevirus (designated bat HEV Md2350, or BtHEVMd2350). The genomic sequence of BtHEVMd2350 was found to have a length of 6,607 nucleotides (excluding the poly(A) tail at the 3′ end). This is slightly shorter than BS7 (which has a genomic sequence length of 6,671 nucleotides), the only reported bat hepevirus with a fully sequenced genome, which was identified from the serotine bat, Eptesicus serotinus, in Germany [7]. BtHEVMd2350 was found to have a 5′ untranslated region (UTR) of 33 nucleotides and a 3′ UTR of 76 nucleotides. The three unique ORFs that are found in other members of the family Hepeviridae were also found in BtHEVMd2350: ORF1 encodes a nonstructural polyprotein that includes the RdRp, ORF2 encodes the capsid protein, and ORF3 encodes a multifunctional protein. Notably, most of the elements and domains characterized in BS7 could be found in BtHEVMd2350, but with a high level of divergence (Table 5).
Table 5.
Nucleotide and amino acid sequence identity between BtHEVMd2350 and representative hepevirus strainsa
| Hepevirus (no. of strains compared) | Degree of identity (%) | ||||||
|---|---|---|---|---|---|---|---|
| Full genome | ORF1 (genome positions 56–4699) | ORF2 (genome positions 4700–6607) | ORF3 (genome positions 4779–5192) | ||||
| Nucleotides | Nucleotides | Amino acids | Nucleotides | Amino acids | Nucleotides | Amino acids | |
| Bat hepevirus (1) | 67.8 | 65.5 | 72.0 | 70.9 | 79.2 | 73.2 | 44.3 |
| Avian hepevirus (4) | 50.9–51.7 | 48.2–49.0 | 44.3–44.5 | 46.0–47.3 | 44.4–44.9 | 19.9–21.1 | 5.3–6.1 |
| HEV genotype 3 (22) | 44.6–46.0 | 43.4–45.5 | 39.1–40.9 | 47.4–49.9 | 44.6–47.4 | 30.7–31.8 | 14.8–18.9 |
| HEV genotype 4 (5) | 45.2–46.0 | 44.9–45.9 | 40.4–41.0 | 47.5–48.2 | 45.8–47.1 | 30.0–31.6 | 14.6–16.3 |
| HEV genotype 1 (3) | 45.6–45.9 | 45.0–45.5 | 40.5–41.1 | 48.7–49.3 | 46.6–46.9 | 31.9–32.6 | 17.1 |
| HEV genotype 2 (1) | 45.6 | 45.3 | 40.8 | 49.7 | 47.1 | 31.6 | 17.1 |
| Rodent hepevirus (3) | 44.0–44.4 | 45.9–46.1 | 41.6–42.0 | 47.9–48.4 | 45.4–46.7 | 25.2–27.4 | 9.0 |
| Ferret hepevirus (1) | 44.0 | 46.4 | 42.6 | 48.3 | 46.0 | 25.5 | 8.0–12.8 |
| Trout hepevirus (1) | 33.7 | 34.9 | 24.5 | 30.2 | 15.5 | 21.1 | 9.8 |
aThe sequences were aligned using MAFFT. The evolutionary analyses were conducted using MEGA version 7. The GenBank accession numbers are as follows: JQ001749 for the bat hepevirus; AM943646, AM943647, KF511797, and AY535004 for the avian hepeviruses; AB189070, AB189075, AP003430, AB091394, AB222183, AB073912, AY115488, AF060669, AF082843, HQ389544, JN564006, AB290312, FJ998008, FJ705359, KC618402, AF455784, AB248521, EU360977, EU495148, FJ956757, FJ906895, and JQ013793 for the HEVs genotype 3; AB856243, AB602440, AB161717, AJ272108, and EU366959 for the HEVs genotype 4; AY230202, AF076239, and M80581 for the HEVs genotype 1; M74506 for HEV genotype 2; GU345042, AB847306, and JX120573 for the rodent hepeviruses; JN998607 for the ferret hepevirus; and NC_015521 for the trout hepevirus
Phylogenetic analysis of the bat hepevirus
A phylogenetic tree was constructed based on the alignment of the full-length genomic sequence of BtHEVMd2350 with those of representative full-length hepevirus genomic sequences (Fig. 2). The results showed that bat hepeviruses (BtHEVMd2350 and BS7) cluster into a separate monophyletic clade within the family Hepeviridae.
Quantification of novel viruses
Viral load detected by qPCR in different tissues were presented in the Fig. 4. The highest viral load of the BtHEVMd2350 was found in the liver (1.9 × 1010 RNA copies per gram of tissue) and followed by spleen (7.3 × 108 RNA copies per gram of tissue), intestine and kidney, but not detectable in the brain. For bat hepadnavirus, the highest viral load was found in the liver of BtHBVRs3364 (2.0 × 1010 RNA copies per gram of tissue), the virus load of tissues of BtHBVRs3366, BtHBVRa4325, and BtHBVRa4328 were relatively similar (medien, 6.2 × 106 RNA copies per gram of tissue; range, 4.9 × 105 to 2.7 × 1010 RNA copies per gram of tissue).
Fig. 4.
Tissue distribution of bat hepevirus and hepadnaviruses. Virus concentrations assessed by virus-specific one-step real-time RT-PCR using quantified in vitro-transcribed RNA controls
Conclusions and discussion
Since the discovery of genetically diverse hepatitis virus-related sequences in bats, bats have been considered to be important natural reservoirs for hepatitis viruses, and potential sources of human diseases [10]. However, these hypotheses need to be proved by screening more bat samples from across the globe for hepatitis viruses. In this study, we screened for hepatitis viruses in bats from China and discovered four novel hepadnaviruses circulating in two species of horseshoe bat in Jinning city, Yunnan province and one hepevirus in the whiskered bat M. davidii in Xianning city, Hubei province. The full-length genomic sequences of one of the two hepadnaviruses from R. sinicus and the hepevirus from M. davidii were determined.
The phylogenetic analysis indicates that the bat hepadnavirus found in this study is closely related to roundleaf bat hepadnaviruses, which were discovered in Pu’er city, Yunnan province in 2011 [9], but shows remarkable divergence when compared to the African horseshoe bat, despite the fact that it was found in an Asian horseshoe bat. A similar phylogenetic relationship was found between hepadnaviruses from the African roundleaf bat and the African horseshoe bat [6], indicating the separate evolution of these viruses and their hosts.
Regarding the bat hepevirus, the phylogenetic analysis indicates that the known bat hepeviruses are highly divergent from other mammalian hepeviruses and that they form an independent branch in the family Hepeviridae. According to the latest proposal of the ICTV in 2016, amino acid distances of concatenated ORF1 and ORF2 (lacking hypervariable regions) greater than 0.088 could then act as threshold to demarcate intra- and inter- genotype distances [15]. The hepevirus detected in the whiskered bat, M. davidii, and that found in the German serotine bat, E. serotinus (the only reported bat hepevirus with a full-length genome) shared significant diversity from both nucleotide and amino acid levels, we propose that they can be grouped into the species Orthohepevirus D which is divided into two genotypes: D1 and D2.
Our results provide further evidence to support the theory regarding the long-term co-evolution of hepadnaviruses and hepeviruses with their hosts, and the theory that bats act as major natural reservoirs for these hepatitis viruses. Our results have limitations due to the small sample size used, which was a result of the protection of bat populations in China, as bats play important roles in the pollination of plants and in pest control, as they feed on insects. However, based on our discovery of hepatitis viruses in bats, it is expected that there are many more hepatitis viruses circulating in numerous bat species and in various geographic regions. In order to obtain larger sample sizes, non-invasive methods of virus detection should be considered for future studies.
Acknowledgements
We appreciate Wei Zhang, Bei Li and Yu-Tao Gao (all Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan, China) for the excellent technical assistance.
Funding
This work was jointly funded by the National Natural Science Foundation of China (81290341), China Mega-Project for Infectious Disease (2014ZX10004001), the Scientific and Technological Basis Special Project (2013FY113500) and Funds for Environment Construction & Capacity Building of GDAS’ Research Platform (2016GDASPT-0215). BW was supported by the China Scholarship Council (CSC), Beijing, China.
Availability of data and materials
Not applicable.
Authors’ contributions
BW conducted the experiments and drafted the manuscript. X-LY, WL and YZ conducted molecular studies. BW, X-LY, X-YG, L-BZ, and Y-ZZ performed the sampling. L-ZS devised the study design and provided scientific oversight. The manuscript was revised by X-LY, C-TB, and L-ZS with input from all the contributing authors. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The study protocol was reviewed and approved by the Ethics Committee of the Yunnan Institute of Endemic Disease Control and Prevention. All the animals were treated in strict accordance with the Guidelines for the Use and Care of Laboratory Animals from the Chinese CDC and the Rules for the Implementation of Laboratory Animal Medicine (1998) from the Ministry of Health, China. The protocols followed for the use of the animals were approved by the National Institute for Communicable Disease Control and Prevention, China. All surgery was performed under ether anesthesia, and all efforts were made to minimize suffering.
Abbreviations
- C
Core
- HAV
Hepatitis A virus
- HBV
Hepatitis B virus
- HCV
Hepatitis C virus
- HDV
Hepatitis D virus
- HEV
Hepatitis E virus
- ORFs
Open reading frames
- P
Polymerase
- RT-PCR
Reverse transcription polymerase chain reaction
- S
Surface
References
- 1.Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Daszak P. Global trends in emerging infectious diseases. Nature. 2008;451:990–993. doi: 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi ZL. Emerging infectious diseases associated with bat viruses. Sci China-Life Sci. 2013;56:678–682. doi: 10.1007/s11427-013-4517-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wibbelt G, Moore MS, Schountz T, Voigt CC. Emerging diseases in Chiroptera: why bats? Biol Lett. 2010;6:438–440. doi: 10.1098/rsbl.2010.0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drexler JF, Corman VM, Lukashev AN, van den Brand JM, Gmyl AP, Brunink S, Rasche A, Seggewibeta N, Feng H, Leijten LM, et al. Evolutionary origins of hepatitis A virus in small mammals. Proc Natl Acad Sci U S A. 2015;112:15190–15195. doi: 10.1073/pnas.1516992112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drexler JF, Geipel A, Konig A, Corman VM, van Riel D, Leijten LM, Bremer CM, Rasche A, Cottontail VM, Maganga GD, et al. Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes. Proc Natl Acad Sci U S A. 2013;110:16151–16156. doi: 10.1073/pnas.1308049110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drexler JF, Seelen A, Corman VM, Tateno AF, Cottontail V, Zerbinati RM, Gloza-Rausch F, Klose SM, Adu-Sarkodie Y, Oppong SK, et al. Bats worldwide carry hepatitis E virus-related viruses that form a putative novel genus within the family Hepeviridae. J Virol. 2012;86:9134–9147. doi: 10.1128/JVI.00800-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He B, Fan Q, Yang F, Hu T, Qiu W, Feng Y, Li Z, Li Y, Zhang F, Guo H, et al. Hepatitis virus in long-fingered bats, Myanmar. Emerg Infect Dis. 2013;19:638–640. doi: 10.3201/eid1904.121655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He B, Zhang F, Xia L, Hu T, Chen G, Qiu W, Fan Q, Feng Y, Guo H, Tu C. Identification of a novel Orthohepadnavirus in pomona roundleaf bats in China. Arch Virol. 2015;160:335–337. doi: 10.1007/s00705-014-2222-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rasche A, Souza BF, Drexler JF. Bat hepadnaviruses and the origins of primate hepatitis B viruses. Curr Opin Virol. 2016;16:86–94. doi: 10.1016/j.coviro.2016.01.015. [DOI] [PubMed] [Google Scholar]
- 11.Quan PL, Firth C, Conte JM, Williams SH, Zambrana-Torrelio CM, Anthony SJ, Ellison JA, Gilbert AT, Kuzmin IV, Niezgoda M, et al. Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc Natl Acad Sci U S A. 2013;110:8194–8199. doi: 10.1073/pnas.1303037110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C, et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503:535–538. doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith DB, Simmonds P, members of the International Committee on the Taxonomy of Viruses Hepeviridae Study G. Jameel S, Emerson SU, Harrison TJ, Meng XJ, Okamoto H, Van der Poel WH, Purdy MA. Consensus proposals for classification of the family Hepeviridae. J Gen Virol. 2015;96:1191–1192. doi: 10.1099/vir.0.000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.