

Abstract
Non-small cell lung cancer (NSCLC) is the most dominating and lethal type of lung cancer triggering more than 1.3 million deaths per year. The most effective line of treatment against NSCLC is to target epidermal growth factor receptor (EGFR) activating mutation. The present study aims to identify the novel anti-lung cancer compounds form nature against EGFR 696-1022 T790M by using in silico approaches. A library of 419 compounds from several natural resources was subjected to pre-screen through machine learning model using Random Forest classifier resulting 63 screened molecules with active potential. These molecules were further screened by molecular docking against the active site of EGFR 696-1022 T790M protein using AutoDock Vina followed by rescoring using X-Score. As a result 4 compounds were finally screened namely Granulatimide, Danorubicin, Penicinoline and Austocystin D with lowest binding energy which were -6.5 kcal/mol, -6.1 kcal/mol, -6.3 kcal/mol and -7.1 kcal/mol respectively. The drug likeness of the screened compounds was evaluated using FaF-Drug3 server. Finally toxicity of the hit compounds was predicted in cell line using the CLC-Pred server where their cytotoxic ability against various lung cancer cell lines was confirmed. We have shown 4 potential compounds, which could be further exploited as efficient drug candidates against lung cancer.
Keywords: lung cancer, natural compounds, EGFR, machine learning, molecular docking, prediction
Background
The lung cancer accounts for approximately 1.59 million deaths per year worldwide and imposes major threat to human health. There are two subtypes of lung cancer, small cell lung cancer (SCLC) (occurrence-10-15%) and non-small cell lung cancer (NSCLC) (occurrence-85-90%) [1]. NSCLC is mainly caused by mutations like in-frame deletions or amino acid substitutions, clustered in the vicinity with ATP-binding pocket of the tyrosine kinase domain present in the epidermal growth factor receptor (EGFR) [2]. The epidermal growth factor receptor family of tyrosine kinases has been defined in four types: EGFR (HER1), ErbB2 (HER2), ErbB3 (HER3) and ErbB4 (HER4) [3]. Studies show that mutations in the EGFR gene occur more often in women (37.5%) than in men (13.0%), in non-smokers (50.8%) than in smokers (9.0%), and in patients of East Asian area (29.1%) than in patients from the United States (9.5%) [4]. These statistics prove that targeting EGFR is a promising way of attacking against NSCLC to cure a patient and some Tyrosine kinase inhibitors (TKIs) for instance Gefitinib and Erlotinib are extensively being used in clinical treatment of NSCLC [5]. HKI-272 (irreversible inhibitor of the EGFR and HER-2 receptors) is a second-generation TKI that exploits both the strategies of covalent binding and multi-targeting [6]. Similar other inhibitors in this context are EKB-569 (irreversible inhibitor of EGFR TK), CI-1033 (irreversible inhibitor of EGFR, HER-2 and ErbB-4), ZD6474 (dual-kinase inhibitor of VEGFR-2 and EGFR) etc. Nevertheless, the clinical effectiveness of these inhibitors is restricted due to emergence of mutations causing drug resistance, including the gatekeeper T790M mutation. There has been a limited success in strategies targeting EGFR T790M with irreversible inhibitors and associated with toxicity because of concurrent inhibition of wild-type EGFR. To extend the finding of better TKI inhibitors, natural compounds are better due to various diverse classes of features. The numerous natural compounds have been reported to have many biological activities as anti-viral, antibacterial, and anti-cancer [7]. The natural products and their derivatives mimic over 50% of all drugs that are being used clinically. The present work aimed in silico investigation of natural inhibitors against EGFR696-1022 T790M from diverse natural sources namely plants, fungi, lichen, bacteria, cyanobacteria, vegetables and nutritional food. The hit compounds obtained from this study could play an important role in designing personalized therapy against lung cancer patients and innovative drug discovery against NSCLC.
Methodology
Library of compounds
An in house library of 419 compounds from various natural resources (Plants-105, fungi-51, lichen- 50, bacteria- 111, cyanobacteria- 52, vegetables- 50 and nutritional food-51) [See Supplementary data] was constructed. The 3D structures of these compounds were retrieved from PubChem, ZINC and Chemspider database. The structures, which were not available in databases, were drawn by using ChemSketch [8] software in mol2 format.
Molecular descriptors and data editing
The molecular descriptors are properties that describe a molecule on the basis of either some physico-chemical property like twodimensional (2D) fingerprint. The geometrical or 3D descriptors are resulted based on x, y and z coordinates, which provide rich information regarding a molecule’s orientation in space. All these calculated descriptors were used as features to build machinelearning model. Total 3500 1d, 2d, 3d and fingerprint descriptors were calculated for each molecule to prepare the data set using PaDEL-Descriptor v2.20 [9], popular software used for calculation of descriptors in chemo informatics. After calculating the descriptors, the WEKA software was used to edit resultant data in .csv format. CfsSubsetEval evaluator was used to remove nonsignificant features from data [10], which ensures accuracy, completeness, consistency, timeliness, believability and interpretability of the data.
Machine learning models
A data set comprising of 400 compounds were collected from PubChem bioassay database [AID: 977608] with known IC50 values. The IC50 values (the concentration of an inhibitor required for 50% inhibition of its target) of the highest 100 compounds were used as active to make a training set for current study. Descriptors for training set were calculated by above mention method. Using this data set and WEKA tool kit three machine learning models based on Random Forest, J48, and Bayes Net were built and compared statistically to select the best predictive model.
Molecular docking
Receptor preparation
Co-crystal structure of EGFR 696-1022 T790M mutant covalently attached to WZ4002 (PDB NO: 3IKA) was retrieved from the Protein Data Bank [11]. The structure was imported into Chimera software for protein preparation. The water molecules and heat atoms (OUN i.e. WZ4002) were deleted from the protein. Dock Prep function was then used to fix the protein.
Docking
The computational docking of all molecules and reference ligand were performed by AutoDock Vina program using PyRx graphic user interface (GUI). The binding coordinates were automatically decided by PyRx, which correlated with the co-ordinate of experimentally bound ligand OUN. They are center_x = -10.7299, center_y = 27.9715, center_z = 36.4637, size_x = 25, size_y = 25 and size_z = 25.
Lamarckian Genetic Algorithm (LGA) was used during docking with standard docking protocols. In LGA the orientation/conformation of the ligand with respect to the receptor is represented through a chromosome. The constructed chromosome consists of a number of variables for ligand translation/rotation that was the same for all ligands and a number of variables for ligand flexibility that is specific to each ligand. The sum of the chromosome signifies the genotype of a ligand. Its 3D coordinates represent the phenotype of a ligand after applying all the transformations in the genotype. Fitness of the ligands is calculated from its phenotype using any of the standard docking scoring functions. AutoDock uses a modification on the AMBER'95 force field [12] with terms empirically determined by linear regression analysis from a set of protein-ligand complexes with already known binding constants [13]. Free energy (Gibbs, ΔG) is denoted by a master equation that contains six terms to model dispersion/repulsion, internal ligand torsional constraints, electrostatic interactions, deviation from the covalent geometry, hydrogen bonding and desolvation effects: ΔG= ΔGvdw + Ghbond+ Gelec+ Gconform+ Gtor+ Gsol
Revalidation of docking results
The docking results of screened ligands were revalidated using XScore a “scoring function” [14] to re-score the selected ligands with EGFR 696-1022 T790M protein. FAF-Drug3 [15] server was used to check their drug likeness, which is originally, based on the free chemo-informatics toolkit Frowns [16].
Pharmacophore elucidation and Cytotoxicity prediction on cell lines
The common pharmacophores for the hit molecules and nine active molecules (used as reference) obtained from bioassay (AID: 256664) were predicted using PharmaGist server [17-18]. Finally the hit molecules were annotated to check their cytotoxic effect on tumor and normal cell-lines by using CLC-Pred [19] web services, which predicts cytotoxicity of molecules for tumor and normal cell-lines based on the structural formula.
Results and Discussion
Initially an in house library of 419 natural compounds from several diverse sources was constructed [See Supplementary data]. The entire library was screened to find compounds against EGFR 696-1022 T790M receptor. Initially machine learning models were built using three classifiers; Random Forest, J48 and Bayes Net and analyzed for their performance by comparing True Positive Rate (TPR) which is ratio of predicted true actives to real number of actives and False Positive Rate (FPR) that is ratio of predicted false actives to actual number of inactive. The accuracy of these models was measured in terms of sensitivity, which predicts the test’s ability to identify positive results, and specificity that evaluates the test’s ability to identify negative results. Finally Random Forest classifier model (with highest sensitivity and specificity) was applied on the data set of test and library compounds, which resulted 63 compounds with active potential. Subsequent to machine learning approach molecular docking was used for screening using AutoDockVina. Ligand receptor interactions of the docked molecules were visualized by using LigPlot+ software (Figure 1) [20]. Ligplot analysis was done to investigate in-depth interaction pattern (hydrophobic interactions, hydrogen bonding pattern) between docked ligand and active site residues of the protein. Finally four molecules were screened by docking process namely Granulatimide, Daunomycin, Penicinoline, Austocystin D with binding energy -8.8 kcal/mol, -9.4 kcal/mol, -7.7 kcal/mol, - 8.6 kcal/mol which showed comparable binding energy with reference ligand i.e. -8.4 kcal/mol. Rescoring of docked molecules was carried out by estimation of binding free energies of screened molecules with EGFR 696-1022 T790M using X–Score program. All compounds showed acceptable range of negative binding energies with comparable results. After that Faf Drug3 server was used to check their druglikeness. In Faf Drug 3 server more than twentythree molecular properties, which are important for evaluating drug likeness of a molecule like number of violations of Lipinski’s RO5, Veber Rule, Egan Rule, and Topological Polar Surface Area etc., were calculated. All of molecules showed good binding pattern in Ligplot analysis and accepted in terms of Drug likeness by FaF-Drug3 (Table 1). PharmaGist server was then used to elucidate pharmacophores for the 4 hit compounds where their common pharmacophore properties were compared with the common pharmacophore properties of 8 potential anti EGFR molecules obtained from PubChem bioassay (AID: 256664). Pharmacophores were detected by extracting common chemical features from 3D structures of the active ligand set that are representative of essential interactions between the ligands and EGRF. The results of pharmacophores analysis predict physicochemical properties namely, hydrogen-bond acceptor/donor atom; a set of atoms of an aromatic ring and adjacent hydrophobic atoms which are important for specific biological activity. The active compounds showed 3 hydrogen bond acceptor, 2 hydrogen bond donor and 1 hydrophobic group whereas the common pharmacophore from 4 hit molecules showed 3 hydrogen bond acceptor, 1 hydrogen bond donor and 1 hydrophobic group. The final annotation was done using CLC-Pred server where all of the hits were evaluated for their cytotoxicity on cell lines (Table 1). The results showed these 4 hit molecules have high potential to inhibit lung cancer by targeting EGFR. All of these molecules where already reported for their anti-cancer properties. Granulatimide is an aromatic alkaloid with G2 checkpoint inhibitory activity [21]. Daunomycin (daunorubicin) was the primary anthracycline compound to be demonstrated structurally and stereo chemically. Danorubicin is used against acute lymphoblastic and myeloblastic leukaemias [22]. Penicinoline is a pyrrolyl 4-quinolinone alkaloid with an extraordinary ring system, isolated from a mangrove endophytic fungus. Penicinoline has strong in vitro cytotoxicity toward 95-D and HepG2 cell lines [23]. Austocystin D is found in A. pseudoustus has been reported for inhibiting growth of human colon carcinoma LS174T cells in mice and tumor cell lines that overexpress the multidrug resistance protein. Additionally the compound can inhibit a number of cancer cell lines: SR, U-87, MCF- 7, MDA-MB-231, PC-3, SW-620, HCT-15, and MX-2 [24,25].
Figure 1.
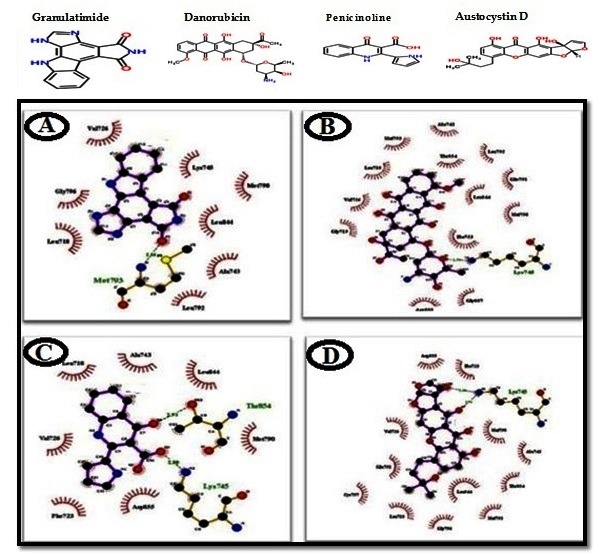
Binding conformation of four hit molecules (A= Granulatimide, B= Daunomycin, C= Penicinoline, D= Austocystin D) with target protein
Table 1. CLC- Pred (Cell-Line Cytotoxicity Predictor) and binding energy of four hit molecules.
| Compounds Name | Pa | Pi | Cell line | Cell line full Name | Tissue | Tumor type | FAF-Drug3 |
| Granulatimide | 0.553 | 0.103 | NCI-H226 | Non-small cell lung carcinoma cells | Lung | Carcinoma | Accepted |
| 0.586 | 0.071 | L2987 | Lung adenocarcinoma cells | Lung | Adenocarcinoma | ||
| 0.61 | 0.042 | Lu1 | Lung carcinoma cells | Lung | Carcinoma | ||
| 0.643 | 0.05 | HOP-62 | Non-small cell lung carcinoma cells | Lung | Carcinoma | ||
| 0.847 | 0.03 | NCI-H522 | Non-small cell lung carcinoma cells | Lung | Carcinoma | ||
| Danorubicin | 0.583 | 0.073 | NCI-H128 | Small cell lung cancer | Lung | Carcinoma | Accepted |
| 0.651 | 0.01 | SPC-A4 | Lung Adenocarcinoma | Lung | Adenocarcinoma | ||
| 0.895 | 0.002 | NCI-H157 | Non-small cell lung carcinoma cells | Lung | Carcinoma | ||
| 0.926 | 0.013 | NCI-H1975 | Bronchoalveolar carcinoma cells | Lung | Carcinoma | ||
| Penicinoline | 0.509 | 0.076 | HOP-62 | Non-small cell lung carcinoma cells | Lung | Carcinoma | Accepted |
| 0.568 | 0.05 | NCI-H1975 | Bronchoalveolar carcinoma cells | Lung | Carcinoma | ||
| Austocystin D | 0.662 | 0.005 | NCI-H157 | Non-small cell lung carcinoma cells | Lung | Carcinoma | Accepted |
| 0.688 | 0.028 | NCI-H460 | Non-small cell lung carcinoma | Lung | Carcinoma | ||
| 0.798 | 0.042 | NCI-H226 | Non-small cell lung carcinoma cells | Lung | Carcinoma | ||
| 0.811 | 0.033 | NCI-H1299 | Non-small cell lung carcinoma | Lung | Carcinoma | ||
| 0.843 | 0.017 | NSCLC | Non-small cell lung carcinoma cells | Lung | Carcinoma | ||
| 0.993 | 0.007 | NCI-H1975 | Bronchoalveolar carcinoma cells | Lung | Carcinoma |
Conclusion:
Currently the clinical efficacy of TKIs (gefitinib and erlotinib) to treat NSCLC is becoming limited because of the emergence of drug resistance conferred by a mutation: substitution of threonine 790 with methionine (T790M). The amino acid residue of EGFR Threonine 790 holds a key location at the entrance of a hydrophobic pocket in the back of the ATP binding cleft of EGFR protein. When this residue is substituted with a bulky methionine, the protein becomes resistant to TKIs, including gefitinib and erlotinib by steric interference. So targeting this mutant EGFR is a promising approach for designing drugs against lung cancer. In this context the findings of present study represent four hit molecules namely Granulatimide, Daunomycin, Penicinoline, Austocystin D, that may have potential as novel EGFR 696-1022 T790M inhibitors. Granulatimide and Daunomycin bind with EGFR by one hydrogen bond where as Penicinoline and Austocystin D by two hydrogen bonds respectively as compare with the reference ligand that makes two hydrogen bonds. The results also showed that these molecules have efficient drug like properties and have also the cytotoxic capacity against lung cancer cell lines. Therefore these molecules have future prospect in drug development with low cost and less side effects (as obtain from nature). Natural compounds constitute a higher percentage of marketed drugs, which indicates a significantly higher rate of return per molecule against NSCLC.
Supplementary material
Acknowledgments
The authors are thankful to the Head, Botany Department, SSJ Campus, Kumaun University, Almora, for providing necessary facilities to carry out the present study.
Edited by P Kangueane
Citation: Nand et al. Bioinformation 12(6): 311-317 (2016)
References
- 1.Petersen I. Dtsch Arztebl Int. 2011;108:531. doi: 10.3238/arztebl.2011.0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang ZF. Nat Genet. 2012;44:852. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Azab AS, et al. Eur. J. MedChem. 2010;45:4198. [Google Scholar]
- 4.Franklin WA, et al. J. Semin Oncol. 2002;29:14. [Google Scholar]
- 5.Kris MG, et al. JAMA. 2003;290:2158. [Google Scholar]
- 6.Fukuoka M, et al. J Clinical Oncology. 2003;21:2246. [Google Scholar]
- 7.Harvey AL. Discovery Today. 2008;894 doi: 10.1016/j.drudis.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 8. http://www.acdlabs.com.
- 9.Liew HY, et al. Journal of Computer Chem. 2012;4:15. [Google Scholar]
- 10.Hall M, et al. ACM SIGKDD Explore News. 2009;11:11. http://dl.acm.org. [Google Scholar]
- 11. http://www.pdb.org.
- 12.Morris GM, et al. Journal of Computer-Aided Mol. Design. 1996;304 doi: 10.1007/BF00124499. [DOI] [PubMed] [Google Scholar]
- 13.Kuntz D, et al. Journal of Molecular Biology. 1982;161:288. [Google Scholar]
- 14.Wang RJ, et al. Computer Aided Mol. Des. 2002;16:26. doi: 10.1023/a:1016357811882. [DOI] [PubMed] [Google Scholar]
- 15.Miteva MA, et al. J Nucleic Acids Res. 2006;22:744. [Google Scholar]
- 16. http://frowns.sourceforge.net.
- 17. http://bioinfo3d.cs.tau.ac.il/PharmaGist.
- 18.Schneidman-Duhovny Nucleic Acids Research. 2008;36:W223. doi: 10.1093/nar/gkn187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. http://www.pharmaexpert.ru/PASS.
- 20.Wallace C, et al. Protein Engineering. 1995;8:134. [Google Scholar]
- 21.Roberto GS, et al. J. Org. Chem. 1998;63:9856. http://pubs.acs.org. [Google Scholar]
- 22.Shao B, et al. Med Chem Lett. 2010;3284:3286. doi: 10.1016/j.bmcl.2010.04.043. [DOI] [PubMed] [Google Scholar]
- 23.Tanja T, et al. Molecule. 2013;18:11376. [Google Scholar]
- 24.Yadav, PK, et al. Bioinformation. 2013;9(3):158. doi: 10.6026/97320630009158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farmer R, et al. Bioinformation. 2010;4(7):290. doi: 10.6026/97320630004290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.