

Abstract
Aromatase (CYP19A1) the key enzyme of estrogen biosynthesis, is often deregulated in breast cancer patients. It catalyzes the conversion of androgen to estrogen, thus responsible for production of estrogen in human body. However, it causes over-production of estrogen which eventually leads to proliferation of breast cancer cells. Identification of new small molecule inhibitors targeted against CYP19A1 therefore, facilitates to increase drug sensitivity of cancer cells. In this scenario, the present study aims to identify new molecules which could block or suppress the activity of aromatase enzyme by molecular docking studies using Schrödinger-Maestro v9.3. In this study we used in silico approach by modeling CYP19A1 protein the strcture was subjected to protein preparation wizard; to add hydrogen and optimize the protonation states of Thr310 and Ser478 and Asp309 residues. Active site of the CYP19A1 protein was identified using SiteMap tool of Scchrodinger package. We further carried out docking studies by means of Glid, with various ligands. Based on glid score, potential ligands were screeened and their interaction with CYP19A1 was identified. The best hits were further screened for Lipinski’s rule for drug-likeliness and bioactivity scoring properties. Thus, we report two rubivivaxin and rhodethrin compounds that have successfully satisfied all in silico parameters, necessitating further in vitro and in vivo studies.
Keywords: aromatase inhibitors, anticancer drug, breast cancer, molecular docking
Background
Breast cancer is the most prevailing malignancy among females, and the second most common cause of cancer-related deaths in women in the United States [1]. An estimated 235,030 new cases of invasive breast cancer are diagnosed among both males and females in the US during 2014 withan estimated death of 40,430 people [2]. Breast cancer is a complex and heterogeneous diseasewith varying clinical outcomes, disease progression, and responses to specific treatmentsattributed by a wide array of elements ranging from tumorintrinsic genetic factorstoextrinsic tumor micro-environmental factors [3]. Gene expression studies using DNA microarrays have identified several distinct breast cancer subtypes based on an intrinsic gene list that includes 496 genes that differentiate breast cancers into separate groups based only on gene expression patterns [4]. Estrogens are important players in breast cancer tumorio-genesis. The estrogen-bound Estrogen Receptor (ER) complex regulates the transcriptome of breast cancer cells by interaction with different transcription factors.Despite the plethora of physiological and pathophysiological functions of estrogen, a large number of studies support the role ofestrogens in growth and development of ER positive breast cancer [5]. The production of estrogens in human body is meditated by the enzyme Cytochrome P450 aromatase, commonly known as estrogen synthase [6].
Cytochrome P450 aromatase (CYP19A1; EC 1.14.14.1) catalyzes the rate limiting step of estrogen biosynthesis, by the aromatization of C-19 aliphatic androgens to C-18 aromatic estrogens [7,8]. It is encoded by CYP19A1gene [9] located on chromosome 15q21.1.The 57.9 kDA molecular weight protein,is madeof a single polypeptide chain of 503 amino-acid residues and a prosthetic heme group at its active site [10]. It is widely expressed in many tissues including the gonad of both sexes [11]. In the male gonads, it is generally expressed in the testis and accessory glands wherein it maintains high levels of estradiol needed for normal spermiogenesis, maturation, and motility of sperm [12]. In females, it is mostly expressed in the ovaries of premenopausal women, and in the placenta of pregnant women. Furthermore, it is also expressed in the peripheral adipose tissues of men and postmenopausal women.
Since ER plays a critical role in breast cancer [13,14], several therapies have been developed over the past few decades among which endocrine therapy is the foremost treatment for ERPositive breast cancer patients [15]. The therapy includes (AIs) aromatase inhibitors, (SERMs) selective estrogen receptor modulators and (SERDs) selective estrogen receptor down regulators. These agents function by either reducing circulating estrogen levels or competing with estrogen for binding to its receptor [16]. AIs bind and block the active site of aromatase thus preventing the conversion of androgens to estrogens. Hence they are widely utilized for inhibition of growth and proliferation of breast cancer cells [17] and serve as first-line therapy for metastatic breast cancer.
Methodology
Despite remarkable success of endocrine therapy, resistance toward these therapies has been detected [18,19]. In particular, long-time exposure of targeted agents leads to development of resistant malignant cells [20]. Moreover aromatase inhibitors increase the risk of cardiovascular diseases and musculoskeletal symptoms like arthralgia, osteoporosis and decreased bone mineral density (BMD) in post-menopausal women [[21, 22] & [23]]. As a result, there is a need for improved and effective therapies which could increase therate of patient survival and in turn improve the quality of life. An important solution towards solving this puzzle would be discoveryof new inhibitors and assessing the potential of currently utilized drugs for treating diversekinds of cancer. Consequently, the present study was conducted to search for novel biomoleculesin-silico which could suppress or inhibit the activity of aromatase enzyme. In this study, we have evaluated various biomolecules by performing molecular docking using Maestro (Schrödinger) [24] focused on novel therapeutic targets may throw light into more moleculear targeted therapies that could specifically and potentially bring down for aromatase enzyme inhibitors.
A comparative molecular docking approach was followed to trace out the most potent inhibitors of aromatase enzyme. We have performedthe molecular screening to filter out and identify most relevant aromatase inhibitors. The identified and screened inhibitor molecules were then further processed for docking studies. Rhodethrin [32] and rubrivivaxin [33] are produced by purple non sulfar photosynthetic bacterium Rubrivivax benzoatilyticus JA2has biological significance in cancer cell lines. Hence, we chose rhodethrin and rubrivivaxin for the molecular docking studies and comparatively analysed the docking results with those of the most prevalent aromatase inhibitors such as troglitazone, toremifene, testolactoneand danazol. Most of the molecular docking analysis was performed using Maestro (Schrödinger).
Selection of docking molecules
A total set of 45 drug molecules were identified by Transfacand KEGG databases which target and metabolize aromataseenzyme. Out of 45 identified drugs, 42 were downloaded from drug bank [25] and KEGG Drug [26] databases for carrying out docking studies. The remaining 3 drugs (p-Bromophenol, rhodethrin and rubrivivaxin) were sketched in ChemDrawUltra 13.0 (http://www.cambridgesoft.com/EnsembleforChemistry/ ChemBio3D/ChemBio3DUltra13.0Suite/Default.aspx) and saved in MDL molfiles. Subsequently, all the ligands were prepared using LigPrep (Schrödinger) by modifying the torsions of the ligands and assigning them appropriate protonation states (Glide-Schrödinger), a single sterochemical structure was generated per ligand with possible states at target pH 7.0 ± 2.0 using Ionizer and Epik by adding metal binding states, tautomerized, desalted and optimized by producing lowenergy 3D conformation for the ligand under the Optimized Potentials for Liquid Simulations (OPLS-2005) force field while retaining the specified chiralities of the input ligand.
Preparation of target protein
The molecular structure of human aromatase enzyme (PDB ID: 3S79) was retrieved from Protein Data Bank (PDB) with a resolution of 2.75 Å. Before docking the ligands into the protein's active site, the protein was prepared using Schrödinger’s molecular docking software, Maestro9.3. The protein was imported to Maestro, in order to investigate the heme-prosthetic group as binding pocket of the enzyme adopted by a ligand in the binding cleft for depiction of both ligand and recepetor was a key importance of all hetero groups using protein preparation wizard. Hydrogen atoms were added to the protein to define correct ionization and tautomeric states of amino acid residues. Finally energy minimization was performed using default constraint of 0.3 Å of Root Mean Square Deviation (RMSD) and OLPS-2005 force field.
Induced Fit Docking (IFD) and Extra Precision (XP) Protocol
IFD and XP were performed using the module IFDof Schrödinger-Maestro v9.3 [19]. Initially Glide docking was carried out for each ligand. The sample ring conformations of ligands were selected and the side chains were trimmed. The prime side chain prediction and minimization was carried out in which residues were refined within 6.0 Å of ligand poses and side chains were optimized. This led tocreation of a ligand structure and conformation that is IF to each pose of the receptor structure. Finally, Glide XP redocking was carried out as per default conditions. The ligand was rigorously docked into the induced-fit receptor structure (IFRS) and the results yielded by IFD score for each output pose.
Drug-likeness, total drug-score and toxicity risk of compounds
Calculation for various drugs properties, such as mutagenic, tumorigenic, irritant nature and an adverse effect of the compounds on the reproductive system was performed using Molinspiration (http://www.molinspiration.com/cgibin/ properties) and Osiris Property Explorer (http://www.organic-chemistry.org/prog/peo). The tools also give the drug-likeness and total drug-score of the compounds based on the ‘‘Lipinski rule of five’’. The overall drug score of a compound is the drug-likeness, i.e. logarithm of the partition coefficient between 1-octanol and water, for the hydrophilicity of the compound, where low log P refers to high absorption or permeation, value less than 5 (c log P) and a unit stripped logarithm (log S) for the aqueous solubility of a compound in mol l_1. The molecular weight and toxicity risk were calculated by Osiris Property Explorer (OPE) for each compound, thus having an overall potential to qualify for a drug.
Results
The purification and crystallization of human CYP19A1 protein has greatly facilitated the identification of compounds which could inhibit its expression, thus providing therapeutic treatments for ERpositive breast cancer patients. Discovery of novel inhibitors is primarily based on computational techniques, among which IFD plays a vital role in understanding the molecular interaction between ligand and the active site of protein. In view of this, all the ligands were docked into the active site of aromatase enzyme (Table 1). The active site of protein is occupied by its natural substrate androstenedione (ASD). It forms H-bonds with backbone residues metal Asp309 (1.689 Å) and Met374 (1.888 Å) (Figure 1).
Table 1. Induced Fit docking results of drug molecules and reference ligand ASD showing Glide gScore, Glide energy and IFD score with CYP19A1.
| Drug Bank ID | Ligand | Glide gScore (Kcal/mol) | Glide Energy (Kcal/mol) | IFD Score (Kcal/mol) |
| DB00197 | Troglitazone | -13.24 | -63.51 | -981.61 |
| DB00539 | Toremifene (SERM) | -10.97 | -52.09 | -976.98 |
| DB000894 | Testolactone | -10.85 | -50.76 | -976.54 |
| DB01406 | Danazol | -10.88 | -60.43 | -975.58 |
| Nil | Rubivivaxin | -11.04 | -49.96 | -975.58 |
| DB01536 | Androstenedione (ASD) | -11.06 | -50.31 | -974.5 |
| DB04839 | Cyproterone | -10.32 | -47.21 | -974.36 |
| DB00882 | Clomifene | -10.79 | -52.1 | -974.21 |
| DB00367 | Levonorgestrel | -11.21 | -43.14 | -974.09 |
| DB00675 | Tamoxifen (SERM) | -8.95 | -59.11 | -974.08 |
| DB03784 | Liarozole hydrochloride | -10.54 | -50.28 | -973.99 |
| DB06710 | Methyltestosterone | -11.07 | -57.21 | -973.85 |
| DB03749 | Plome stane | -9.78 | -43.75 | -973.17 |
| DB00269 | Chlorotrianisene | -10.44 | -49.88 | -972.83 |
| DB03781 | Liarozolefumarate | -9.32 | -50.27 | -972.83 |
| Nil | Rhodethrin | -9.53 | -42.34 | -972.74 |
| DB00351 | Megestrol | -10.75 | -52.38 | -972.68 |
| DB00255 | Diethylstilbestrol | -9.21 | -44.85 | -972.03 |
| DB01234 | Dexamethasone | -11.83 | -51.78 | -971.93 |
| DB00531 | Cyclophosphamide | -6.38 | -33.83 | -971.68 |
| DB00987 | Cytarabine | -8.87 | -47.05 | -969.89 |
| DB01168 | Procarbazine | -7.01 | -35.71 | -969.9 |
| DB02451 | Fadrozole hydrochloride | -7.43 | -40.94 | -969.4 |
| DB03778 | Fadrozole hydrochloride hydrate | -7.46 | -41.7 | -969.37 |
| DB01101 | Capecitabne | -10.84 | -59.63 | -968.72 |
| DB00201 | Caffeine | -6.2 | -33.91 | -966.93 |
| Nil | p-Bromophenol | -6.46 | -26.26 | -966.91 |
| DB00441 | Gemcitabine | -6.95 | -36.03 | -966.63 |
| DB01169 | Arsenic trioxide | -3.82 | -25.69 | -964.89 |
| DB00958 | Carboplatin | -5.7 | -32.73 | -964.89 |
| DB00544 | Fluorouracil | -4.36 | -14.75 | -961.49 |
Figure 1.

Molecular interaction of natural substrate Androstenedione (ASD) with CYP19A1 protein.
The side chains of residues Thr310 and Ser478 make polar interactions with the bound substrate while positive charge interaction was found with Arg115. The residues Ile133, Phe134, Phe221, Trp224, Ile305, Ala306, Val370, Leu372, Val373 and Leu 477 make hydrophobic interactions with the substrate.The ranking of ligands was based on the IFD score. More negative IFD score indicates better interaction of inhibitor with the target protein (Table 1). A Comparison of IFD score of drug molecules with AIs suggested that the drug molecules namely troglitazone, toremifene, testolactone, danazol, rubivivaxin and rhodethrin showed fairly potent activity against CYP19A1 with more negative IFD Score value. Thus, these drug molecules could prove to be more potent than the available third-generation aromatase inhibitors (Table 2). To understand the in-depth interaction pattern between the ligands and CYP19A1 protein, Maestro Ligand interaction 2-D diagram was used. The moleculesrubivivaxinand rhodethrin shown the drug-likeness and toxicity risk values are less than with troglitazone, toremifene, testolactone, danazol, it indicates that, the molecules has an overall potential to qualify as lead molecules (Table 3).
Table 2. Molecular Docking Interaction of drug molecules with CYP19A1.aNumber of hydrogen bonds formed between the CYP19A1 binding domain and the drug compounds. bThe interacting active site residues of CYP19A1 protein with the drug molecules in the ligand-receptor complex.
| Ligand-enzyme complex | No.of H-bonds | Interacting residuesb | Atom of the ligand | Bond length A0 |
| Troglitazone-CYP19A1 | 2 | Leu 372, Met 374 | H of NH, O of C=O | 1.932, 1.654 |
| Toremifene-CYP19A1 | Nil | Nil | Nil | Nil |
| Testolactone--CYP19A1 | 2 | Ash 309, Met 374 | O of C=O, O of C=O | 1.689, 1.845 |
| Danazol-CYP19A1 | 2 | Ash 309, Met 375 | N of , O of -OH | 1.990, 2.071 |
| Rubivivaxin-CYP19A1 | 6 | Arg 115, Ala 306, Ala 306, Thr 310, Leu 372, Met 374 | O of -COOH, H of -OH, H of -OH, O of -OH, H of -OH, O of -COOH | 1.834, 1.906, 1.575, 2.136, 1.773, 1.705 |
| Androstenedione-CYP19A1 | 2 | Ash 309, Met 374 | O of C=O, O of C=O | 1.689, 1.888 |
| Cyproterone-CYP19A1 | 2 | Met 374, Hem 600 | O of C=O, H of -OH | 1.71 |
| Clomifene-CYP19A1 | Nil | Nil | Nil | Nil |
| Levonorgestrel-CYP19A1 | 2 | Thr 310, Met 374 | H of -OH, O of C=O | 1.895, 1.635 |
| Tamoxifen-CYP19A1 | 1 | Ash 309 | H of N-H | 1.687 |
| Liarozole hydrochloride-CYP19A1 | 2 | Leu 477, Hem 600 | H of NH, H of N-H | 1.771 |
| Methyltestosterone-CYP19A1 | 3 | Ala 306, Thr 310, Met 374 | H of -OH, O of -OH, O of C=O | 1.938, 1.787, 1.759 |
| Plome stane-CYP19A1 | 1 | Met 374 | O of C=O | 1.976 |
| Chlorotrianisene-CYP19A1 | 1 | Met 374 | O of | 2.058 |
| Liarozolefumarate-CYP19A1 | 2 | Leu 372, Ser 473 | H of NH, H of N-H | 1.94 |
| Rhodethrin-CYP19A1 | 3 | Thr 310, Leu 477, Hem 600 | O of -COOH, H of NH, H of -OH | 1.940, 2.011, 2.458 |
| Megestrol-CYP19A1 | 2 | Leu 372, Met 374 | H of -OH, O of C=O | 1.938, 1.804 |
| Diethylstilbestrol-CYP19A1 | 2 | Glu 302, Leu 477 | H of -OH, H of -OH | 1.787, 1.548 |
| Dexamethasone-CYP19A1 | 3 | Ash 309, Met 374, Ser 478 | O of -OH, O of C=O, H of -OH | 2.193, 1.712, 1.805 |
| Cyclophosphamide-CYP19A1 | 1 | Met 374 | O of O=P | 2.132 |
| Cytarabine-CYP19A1 | 4 | Ala 306, Thr 310, Leu 372, Leu 477 | H of -OH, O of -OH, H of NH2, H of NH2 | 1.870, 1.861, 1.960, 1.907 |
| Procarbazine-CYP19A1 | 2 | Hem 600, Hem 600 | N of N-NH2, N of NH | 2.003 |
| Fadrozole hydrochloride-CYP19A1 | 1 | Met 374 | N of C=N | 1.876 |
| Fadrozole hydrochloride hydrate-CYP19A1 | 2 | Met 374, Hem 600 | N of C=N, N of N-H | 1.961 |
| Capecitabne-CYP19A1 | 2 | Leu 372, Met 374 | H of -OH, O of | 2.100, 1.859 |
| Caffeine-CYP19A1 | 1 | Met 374 | O of C=O | 2.011 |
| p-Bromophenol-CYP19A1 | 2 | Arg 115, Met 374 | H of -OH, O of -OH | 2.045 |
| Gemcitabine-CYP19A1 | 3 | Thr 310, Met 374, Leu 477 | H of NH2, O of -OH, H of -OH | 2.307, 2.081, 1.722 |
| Arsenic trioxide-CYP19A1 | 2 | Arg 115, Met 374 | O of O=As, O of O=As | 1.988, 1.654 |
| Carboplatin-CYP19A1 | 1 | Met 374 | O of C=O | 1.911 |
| Fluorouracil-CYP19A1 | 1 | Met 374 | N of | 2.017 |
Table 3. Molinspiration and Osiris results of compounds.
| Compounds | cLogP | Solubility | Mol.Weight | TPSA | Drug-Likeness | Drug-Score | Toxicity Risk |
| Troglitazone | 4.39 | -5.46 | 441 | 110.1 | 3.55 | 0.47 | No Risk |
| Toremifene | 4.95 | -4.8 | 405 | 12.47 | 4.33 | 0.15 | High Risk |
| Testolactone | 2.71 | -3.74 | 300 | 43.37 | -2.73 | 0.43 | No Risk |
| Danazol | 3.46 | -4.88 | 337 | 46.26 | 2.07 | 0.62 | No Risk |
| Rubivivaxin | 1.03 | -1.73 | 298 | 124.2 | 3.46 | 0.55 | No Risk |
| Rhodethrin | 1.55 | -2.62 | 263 | 82.55 | -1.21 | 0.56 | No Risk |
Binding mode of Troglitazone with the ligand binding region of Aromatase
Docking results showed that the ligand troglitazone occupied the ligand binding region of CYP19A1 with an IFD Score of -981.60, Glide gScore of -13.23 and the Glide energy is -63.51 Kcal/mol. Hydrogen bond interactions were identified with the backbone amino acid residuesLeu372 and Met374.Twelve hydrophobic interactions with the amino acid residues Ile133, Phe134, Phe221, Trp224, Ile305, Ala306, Ala307, Val369, Val370, Val373, Cys437 andLeu477; one positive charge interaction with Arg115 and two polar interaction with the amino acid residuesThr310 and Ser478 in the ligand binding region of CYP19A1 were observed (Figure 4).
Figure 4.

Close view, Binding mode and interacted ligands of Troglitazone with CYP19A1. A. Close view of Troglitazone with CYP19A1. B. Binding mode of Troglitazone with CYP19A1. C. Interaction with ligands.
Binding mode of Toremifene with the ligand binding region of Aromatase
Docking results showed that the ligand toremifene occupied the ligand binding region of CYP19A1 with an IFD Score of -976.97, Glide gScore of -10.97 and the Glide energy is -52.09 Kcal/mol. Trp224 was involved in the π-π stacking interaction with ligand.Sixteen hydrophobic interactions with the amino acid residues Met127, Ile133, Phe134, Phe148, Ile217, Tyr220, Phe221, Met303, Ile305, Ala306, Val369, Val370, Leu372, Val373, Met374 andLeu477;one positive charge interaction with Arg115; one negative charge interaction with Glu302 and two polarinteraction with the amino acid residues Thr310 and Ser478 in the ligand binding region of CYP19A1 were observed (Figure 5).
Figure 5.

Close view, Binding mode and interacted ligands of Toremifene with CYP19A1. A. Close view of Toremifene with CYP19A1. B. Binding mode of Toremifene with CYP19A1929. C. Interaction with ligands 5929.
Binding mode of Testolactone with the ligand binding region of Aromatase
The ligand testolactone occupied the ligand binding region of CYP19A1 with an IFD Score of -976.54, Glide gScore of -10.85 and the Glide energy is -50.76Kcal/mol. Hydrogen bond interactions were identified with the backbone amino acid residue Met374 and metal Ash309.Ten hydrophobic interactions with the amino acid residues Ile133, Phe134, Phe221, Trp224, Ile305, Ala306, Val370, Leu372, Val373 and Leu477; onepositive charge interaction with Arg115; one negative charge interaction with Glu302 and two polar interaction with the amino acid residues Thr310 and Ser478 in the ligand binding region of CYP19A1 were observed (Figure 6).
Figure 6.

Close view, Binding mode and interacted ligands of Testolactone with CYP19A1. A. Close view of Testolactone with CYP19A1. B. Binding mode of Testolactone with CYP19A1. C. Interaction with ligands.
Binding mode of danazol with the ligand binding region of Aromatase
Docking results showed that the ligand danazol occupied the ligand binding region of CYP19A1 with an IFD Score of -975.58, Glide gScore of -10.88 and the Glide energy is -60.43 Kcal/mol. Hydrogen bond interactions were identified with the backbone amino-acid residue Met374 and metal Ash309. Phe221 was involved in the π-π stacking interaction with ligand. Ten hydrophobic interactions with the amino acid residues Ile133, Phe134, Trp224, Ile305, Ala306,Val370, Leu372, Val 373, Ile398 and Leu477; one positive charge interaction with Arg115 and two polar interactions with the amino acid residues Thr310 and Ser478 in the ligand binding region of CYP19A1 were observed (Figure 7).
Figure 7.
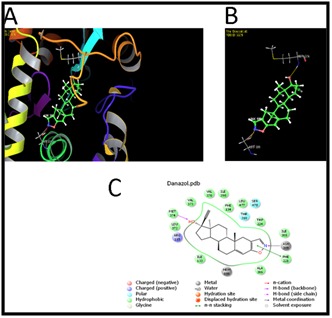
Close view, Binding mode and interacted ligands of Danazol with CYP19A1.A. Close view of Danazol with CYP19A1 B. Binding mode of Danazol with CYP19A1. C. Interaction with ligands.
Binding mode of rubrivivaxin with the ligand binding region of Aromatase
The ligand rubrivivaxinoccupied the ligand binding region of CYP19A1 with an IFD Score of -975.58, Glide gScore of -11.04 and the Glide energy is -49.96Kcal/mol. Hydrogen bond interactions were identified with the backbone amino acid residues Ala306, Leu372, Met374 and side chain amino acid residues Arg115 and Thr310. Ten hydrophobic interactions with the amino acid residues Ile133, Phe134, Phe221, Trp224, Ile305, Ala307, Val370, Val373, Cys437and Leu477, and one polar interaction with the amino acid residue Ser478 in the ligand binding region of CYP19A1 were observed (Figure 3).
Figure 3.

Close view, Binding mode and interacted ligands of Rubrivivaxin with CYP19A1. A. Close view of Rubrivivaxin with CYP19A1. B. Binding mode of Rubrivivaxin with CYP19A1. C. Interaction with ligands
Binding mode of rhodethrin with the ligand binding region of Aromatase
The ligand rhodethrin occupied the ligand binding region of CYP19A1 with an IFD Score of -972.74, Glide gScore of -9.53 and the Glide energy is -42.34 Kcal/mol.Hydrogen bond interactions were identified with the backbone amino acid residues Leu477 and Hem600 and side chain amino acid residue Thr310. Eleven hydrophobic interactions with the amino acid residues Ile133, Phe134, Phe221, Trp224, Ile305, Ala306, Val370, Leu372, Val373, Met374, Leu477; one positive charge interaction with Arg115; one negative charge interaction with Glu302 and two polar interaction with the amino acid residue Thr310 and Ser478 in the ligand binding region of CYP19A1 were observed (Figure 2).
Figure 2.

Close view, Binding mode and interacted ligands of Rhodethrin with CYP19A1. A. Close view of Rhodethrin with CYP19A1. B. Binding mode of Rhodethrin with CYP19A1. C. Interaction with ligands.
Discussion
Molecular docking is considered as one of the best methods to identify the protein-ligandinteractions during in silico phases of drug discovery [27]. Previous studies suggest that aromatase activity can be suppressed in the presence of troglitazone [28], toremifene [29], testolactone [30] and danazol [31]. Hence, we have selected these four different aromatase inhibitors and novel drug targets rubrivivaxinand rhodethrinfor our docking studies to comparatively investigate binding efficiency withCYP19A1. The results of docking were verified by considering some top clusters of conformations in to the best scored one. The Novel rubivivaxin and rhodethrin shown thedrug-likeness and toxicity risk values are less than with troglitazone, toremifene, testolactone, danazol, it indicates that, the novel drugs has an overall potential to qualify for a drug. The docking results have shown that troglitazone has relatively lesser binding energy followed by rubivivaxin and toremifene.Thus, our results suggest that the novel compoundrubivivaxin and rhodethrin which are shown a lesser binding energy in comparison with the other molecules may prove potential in structure based drug design to make a major impact on anticancer chemotherapy.
Conclusion:
Aromatase (CYP19A1) is associated with and rosteroidine human breast cancer. Hence, it is important to develop improved inhibitors for Aromatase enzyme. In this study, we described in silico docking studies to identify inhibitors and performed to analyze the inhibition of aromatase activity.Two biomolecues namely rubivivaxin and rhodethrin showed good binding affinity with Aromatase enzyme inhibition. Consequently, the results obtained from this study may be worthwhile, to carry out further in vitro and in vivo studies to design novel and potential inhibitors against Aromatase (CYP19A1).
Conflict of interests
The authors confirmed that this research article content has no conflict of interest.
Acknowledgments
We are highly thankful to Schrodinger for providing the free version of software to carry out the entire project. Also, we are grateful to Dr. Ravi Kumar Muttineni for his technical assistance at various stages of the project. This work has been supported by DST-SERB, Govt. of India and also APK was supported by grants from National Medical Research Council of Singapore [R-713-000- 177-511], the NCIS Yong Siew Yoon Research Grant through donations from the Yong Loo Lin Trust and the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centers of Excellence initiative to Cancer Science Institute of Singapore, National University of Singapore.
Edited by P Kangueane
Citation: Kumavath et al. Bioinformation 12(6): 324-331 (2016)
References
- 1.Jemal, et al. CA Cancer J Clin. 2010;60:277. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2. http://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2014/cancer-facts-and-figures-2014.pdf.
- 3.Bertos NR, Park M, J Clin Invest. 2011;121(10):3789. doi: 10.1172/JCI57100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perou CM, et al. Nature. 2000;406(6797):747. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 5.Marta K, Halina M, Adv Clin Exp Med. 2012;21(4):535. [Google Scholar]
- 6.Ackerman GE, Carr BR, Endocr Metab Disord. 2002;3:225. doi: 10.1023/a:1020024409888. [DOI] [PubMed] [Google Scholar]
- 7.Simpson, et al. Endocr Rev. 1994;15:342. doi: 10.1210/edrv-15-3-342. [DOI] [PubMed] [Google Scholar]
- 8.Brueggemeier, et al. Endocrine Reviews. 2005;26(3):331. doi: 10.1210/er.2004-0015. [DOI] [PubMed] [Google Scholar]
- 9.Sebastian S, Bulun SE, J.Clin Endocrinol Metab. 2001;86:4600. doi: 10.1210/jcem.86.10.7947. [DOI] [PubMed] [Google Scholar]
- 10.Suvannang, et al. Molecules. 2014;16:3597. [Google Scholar]
- 11.Bulun, et al. Pharmacol Rev. 2005;57:359. doi: 10.1124/pr.57.3.6. [DOI] [PubMed] [Google Scholar]
- 12.Carreau, et al. Adv Med Sci. 2008;53:139. doi: 10.2478/v10039-008-0022-z. [DOI] [PubMed] [Google Scholar]
- 13.Russo, et al. J. Steroid. Biochem. Mol. Biol. 2003;87:1. doi: 10.1016/s0960-0760(03)00390-x. [DOI] [PubMed] [Google Scholar]
- 14.Cui, et al. J. Clin Oncol. 2005;23:7721. doi: 10.1200/JCO.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Rowan TCCG, arnet LA, J Natl Cancer Inst. 2012;104:517. [Google Scholar]
- 16.Adam Brufsky M. Community Oncology. 2011;8(8):343. [Google Scholar]
- 17.Altundag K, Ibrahimb NK, The Oncologist. 2006;11:553. doi: 10.1634/theoncologist.11-6-553. [DOI] [PubMed] [Google Scholar]
- 18.Selma, et al. J Steroid Biochem. Mol. Bio. 2010;18(4-5):277. [Google Scholar]
- 19.Cynthie W, Shiuan C, Journal of Steroid Biochemistry & Molecular Biology. 2012;131:83. doi: 10.1016/j.jsbmb.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DowsettM, et al. J Steroid Biochem Mol Bio. 2005;l95:167. [Google Scholar]
- 21.Henry, et al. Breast Cancer Res Treat. 2008;111(2):365. doi: 10.1007/s10549-007-9774-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kittaneh, Glück, Breast Cancer Basic and Clinical Research. 2011;5:209. doi: 10.4137/BCBCR.S6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomao, et al. Expert Rev Anticancer Ther. 2011;11(8):1253. doi: 10.1586/era.11.96. [DOI] [PubMed] [Google Scholar]
- 24. http://www.schrodinger.com/citations#Maestro.
- 25.Wishart DS, et al. Nucleic Acids Res. 2008;36:D901.. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa M, Goto S, Nucleic Acids Res. 2000;28:27. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meng XY, et al. Current computer-aided drug design, 2011;7(2):146. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown KA, Simpson ER, In Obesity and Breast Cancer Springer, New York, USA. 2014:37. [Google Scholar]
- 29.Ogata H, et al. Cancer & chemotherapy. 2013;40(6):749. [PubMed] [Google Scholar]
- 30.Schweikert HU, TunnUW, Steroids. 1987;50(1):191. doi: 10.1016/0039-128x(83)90071-5. [DOI] [PubMed] [Google Scholar]
- 31.Murakami K, et al. Fertility and sterility. 2006;86(2):291. doi: 10.1016/j.fertnstert.2005.12.074. [DOI] [PubMed] [Google Scholar]
- 32.Ranjith NK, et al. Biotechnol. Lett. 2007;29(9):1399. doi: 10.1007/s10529-007-9413-7. [DOI] [PubMed] [Google Scholar]
- 33.Kumavath RK, et al. World Journal of Microbiology and Biotechnology. 2011;27:11. [Google Scholar]