

Abstract
The design of drug delivery systems capable of efficiently delivering poorly soluble drugs to target sites still remains a major challenge. Such materials require several different functionalities; typically, these materials should be biodegradable and nontoxic, non-immunogenic, responsive to their environment, and soluble in aqueous solution while retaining the ability to solubilize hydrophobic drugs. Here, we report on a polypeptide-polymer hybrid of elastin-like polypeptides (ELPs) and poly(2-oxazoline)s (POx). This paper describes the chemical synthesis, physical characteristics, and drug loading potential of these novel hybrid macromolecules. To this end, we introduce a novel method for terminal functionalization of POx with protected maleimide moieties. Following recovery of the maleimide group via a retro Diels-Alder reaction, the consecutive Michael addition of thiol-functionalized ELPs yielded the desired protein-polymer conjugate. These conjugates formed nanoparticles in aqueous solution capable of solubilizing the anti-cancer drug paclitaxel with up to 8 wt% loading.
Keywords: drug delivery systems, bioconjugation, elastin-like polypeptides, ring-opening polymerization, paclitaxel
Graphical Abstract
We describe a novel method to incorporate maleimide functionality into poly(2-oxazoline)s via nucleophilic termination with the Diels-Alder adduct of furan and maleimide. With this functionality we create a new peptide-polymer hybrid of hydrophilic cysteine-bearing Elastin-like Polypeptides and hydrophobic poly(2-oxazoline)s. These conjugates spontaneously form aggregates in water and are used in first experiments for loading of anti-cancer drug paclitaxel.

1. Introduction
Many active pharmaceutical ingredients (API) are hydrophobic molecules that exhibit poor bioavailability and a short circulatory half-life. In the field of nanomedicine, drug delivery systems (DDS) are designed to increase the biological effectiveness of the API. This is typically accomplished by increasing the solubility and bioavailability of the API, increasing its concentration in diseased tissue while decreasing toxicity in healthy tissue, and lengthening its circulation half-life.[1] Ideally, the carrier should also be biocompatible, biodegradable, easily excreted, and non-immunogenic.
Paclitaxel (PTX) is a highly potent cancer therapeutic that is notoriously difficult to incorporate into stable formulations at high drug loading.[2–4] Recent efforts to efficiently formulate PTX include utilization of a poly(2-oxazoline) (POx) triblock copolymer, which demonstrated high drug loading and solubility, excellent shelf-stability, as well as low toxicity and complement activation.[5–9] POx exhibits several characteristics desirable in DDS. It can be synthesized with a high degree of control due to the living nature of the cationic ring opening polymerization (LCROP).[10] The hydrophilic-hydrophobic balance can be precisely tuned through the choice of the POx monomer side-chain and the composition of the block-copolymer,[11] which has recently been shown to be a key component in formulating PTX.[9] Poly(2-methyloxazoline) (PMeOx) has been shown to display an immunological stealth effect,[12–15] and in contrast to poly(ethylene glycol) functionalized systems, POx shows the same pharmacokinetic behavior over multiple injections,[16] though a contradictory report has been published recently.[17] A particularly effective POx for formulating PTX consists of two hydrophilic PMeOx blocks flanking one central hydrophobic poly(2-butyloxazoline) (PBuOx) block. In an aqueous environment, this tri-block copolymer forms stable micelles with a hydrophobic, yet polar inner compartment capable of solubilizing a variety of hydrophobic drugs.[5–7] This system exhibits exceptionally high loading capacity for PTX, indicating that the polymer architecture provides an ideal balance of polarity and hydrophobicity.[7,9]
A second approach to solubilize hydrophobic compounds is to chemically link the drug to the DDS. This has been used extensively with both hydrophilic polymers such as PEG, and biopolymers such as elastin-like polypeptides (ELPs).[18,19] ELPs are bioinspired, artificial polypeptides derived from the hydrophobic domain of tropoelastin.[20] ELPs are biocompatible and biodegradable, and the polypeptide as well as the degradation products show very low toxicity.[21,22] The most commonly used motif in ELPs is comprised of a pentapeptide repeat unit (Val-Pro-Gly-Xaa-Gly), where Xaa is a guest residue that can be any amino acid except proline.[23] As the biopolymer consists exclusively of amino acids, the repeat sequence can be genetically encoded and expressed in Escherichia coli.[24] The genetically encoded synthesis of ELPs yields uniform products that can be designed — at their gene level — to incorporate specific guest residue in the repeating pentamer or at the amino or carboxy terminus of the ELP. ELPs also display a characteristic lower critical solution temperature phase transition behavior; below a transition temperature (Tt) the polymer is water soluble, and above this temperature, the polymer phase separates into an insoluble ELP-rich phase. The Tt can be tuned by altering the composition of the amino acids in the guest residue position, or by changing the salt concentration, pH, or redox potential of the solvent.[25,26] These properties make ELPs an enticing candidate for delivering therapeutics. For instance, recent reports show that doxorubicin (Dox) and paclitaxel derivatives linked to the inner core of ELP micelles are able to efficiently abolish tumors in mice with a single injection.[27–29] In this system, Dox and PTX derivatives with an acid sensitive linker and a terminal maleimide moiety were coupled to cysteine residues on one end of an ELP chain, which drove the formation of soluble micelles with a hydrophilic ELP shell and a hydrophobic, drug-rich core. The release of the drug was then triggered by the acidic cleavage of the linker between the drug and the ELP.
We hypothesized that a POx-ELP hybrid could combine the excellent solubilization capacity of amphiphilic POx block copolymers for PTX, with the defined nature and biodegradability of the ELP. In this paper, we describe the synthesis of a new maleimide-functionalized POx, the subsequent thiol-end coupling to cysteine residues on an ELP chain, and the size distribution and drug loading potential of the hybrid structure.
2. Experimental
2.1. Materials and general methods
Furan, maleimide, glutathione, dioxane, methanol (MeOH), diethyl ether, dichloromethane (DCM), chloroform, 2-amino ethanol, valeronitrile, trifluoroacetic acid (TFA) and cadmium acetate dihydrate were of analytical grade and used without further purification. Acetonitrile (ACN) and toluene were of anhydrous grade and sealed with a septum. The monomers 2-phenyl, 2-methyl oxazoline and the initiator methyl triflate were distilled under inert gas atmosphere and over CaH2, prior to use. All reagents were purchased from Sigma-Aldrich (Weinheim, Germany).
NMR spectra were recorded on a Varian Unity - 500 MHz multinuclear NMR (1H-NMR: 500 MHz and 13C-NMR: 125 MHz). Gel permeation chromatography (GPC) was performed on a PL-GPC-120 (Polymer Laboratories) with dimethyl acetamide (DMAc) with 5 g/L LiBr and 1 vol% H2O as eluent. The sample was filtered through 0.2 µm PTFE syringe filter prior to injection and a calibration based on poly(methyl methacrylate) (PMMA) calibration standards. Matrix assisted laser desorption ionization time of flight mass spectroscopy (MALDI-ToF-MS) data were obtained on a Bruker biflex IV. A 20 g/L solution of dithranol in chloroform (1 % TFA) was used as matrix and the ratio of analyte (1 g/L) to matrix was 1:5 (v/v), respectively. Melting points were determined with a Büchi Melting Point B-545. Dialysis tubes were purchased from Roth with a molecular weight cut off from 12–14 kDa.
Dynamic light scattering was carried out on DynaPro Protein Solutions (Wyatt Technology Corp) and the data were analyzed with Dynamics V6 (Version 6.10.1.2) from the same manufacturer. Typically, a 25 µM solution of freeze-dried product in PBS was prepared and filtered through one of several syringe filters: 0.22 µm (PVDF), 0.45 µm (PVDF), or 1 µm pore size (PTFE). The samples were measured in a 16 µL cuvette at 25 °C with a radius cut off from 2–1000 µm.
HPLC was carried out on a Shimadzu system with an OHpak KB-803 gel permeation column from Shodex. The HPLC mobile phase was a mixture of 70:30 MilliQ/ACN with 0.05 % formic acid. The mixture was filtered before use. The HPLC was run in isocratic mode with a flow rate of 1 mL/min. The peaks were detected with an UV/Vis detector at 228 nm.
2.2. Synthesis
2.2.1. 10-Oxa-4-aza-tricyclo[5.2.1.02.6]dec-8-ene-3,5-dione (furan-maleimide adduct)
Maleimide (1 g, 10.3 mmol) was added to a saturated furan-water solution, which contained 1.052 g (15.45 mmol) furan. The solution was heated in a 250 mL round bottom flask to 90 °C overnight. The solution was allowed to cool to room temperature and was then placed in a 4 °C fridge overnight. The precipitated white solid was collected, washed three times with cold diethyl ether, and dried in vacuo. The solid was recrystallized from hot water, pestled and triturated in dry toluene three times. The product was then dried in vacuo and stored under inert gas to yield 767 mg of a colorless solid (4.70 mmol, 45.1 %).
Tmp: 160 °C (literature: 162 °C[30])
1H-NMR (DMSO-d6, 500 MHz):
δ (ppm): 3.23 (s, 2 H, HC), 5.94 (s, 2 H, HC-O), 6.91 (s, 2 H, HC=C), 11.54 (s, 1 H, HNH)
13C-NMR (MeOD, 125 MHz):
δ (ppm): 50.09 (CC), 82.2 (CC-O), 137.63 (CC=C), 179.82 (CC=O)
2.2.2. 2-Butyl-2-oxazoline (BuOx)
The synthesis of the monomer 2-butyl-2-oxazoline was performed as described previously.[31]
2.2.3. Poly(2-phenyl-2-oxazoline)s
The polymerization procedure for poly(2-oxazoline) using a microwave has been extensively described.[32,33] Briefly, in a typical experiment, 5 mL ACN, 50 mg (0.30 mmol) methyl triflate and 886 mg (6.02 mmol) 2-phenyl oxazoline were added to a 10 mL microwave vessel under dry nitrogen atmosphere. The vessel was sealed with a crimp cap and placed into a microwave. The polymerization proceeded at 150 W and 140 °C for 33 min. After the reaction was completed, 152 mg (0.920 mmol) of the Diels-Alder protected maleimide and 133 mg (0.962 mmol) K2CO3 were added to the reaction solution under inert atmosphere. The vessel was sealed and the polymerization was terminated at 50 W and 40 °C for 90 min in the microwave. The reaction mixture was filtered, the residue washed with MeOH. The resulting solution was precipitated through a 0.2 µm PTFE syringe filter into a tenfold excess of deionized water and centrifuged (10,000 RPM, 22 °C, 12 min). The supernatant was discarded and the pellet was dried in vacuo. The obtained product was freeze-dried from dioxane.
1H-NMR (CDCl3, 500 MHz): δ (ppm): 2.50–3.90 (br, 80 H, -N-CH2-CH2-N-), 5.20 (d, 2 H, -C-CH-O-CH-C), 6.45 (s, 2 H, -C-CH=CH-C-), 6.90–7.50 (br, 95 H, phenyl)
2.2.4. Retro Diels-Alder reaction
In a 10 mL microwave vessel, 262 mg (0.0838 mmol) of the protected polymer were dissolved in 3 mL of a 2:1 (v/v) MeOH/MilliQ. Deprotection was performed under microwave irradiation at 150 W and 120 °C for 25 min. The polymer was then precipitated in 40 mL deionized water and the suspension was centrifuged (10,000 RPM, 22 °C, 12 min). The supernatant was discarded, the pellet was dried in vacuo, and the product was freeze-dried from dioxane. The product was obtained as a white solid in a quantitative yield. The efficiency of the rDA reaction was 87 % as determined by H-NMR.
1H-NMR (CDCl3, 500 MHz): δ (ppm): 2.50–3.90 (br, 80 H, -N-CH2-CH2-N-), 6.63 (d, 2 H, -CO-CH=CH-CO-), 6.90–7.50 (br, 95 H, phenyl)
2.2.5. Synthesis and purification of elastin-like polypeptides (ELPs)
The gene encoding the ELP was designed from custom ordered oligomers (Integrated DNA Technologies, Inc.) using recursive directional ligation as described previously,[24] cloned into a pET-25b(+) expression vector (Novagen, Madison, WI), and transformed into BLR (DE3) cells (EdgeBio, Gaithersburg, MD). These cells were used to inoculate a 50 mL starter culture containing TBDry media (MOBIO, Carlsbad, CA) supplemented with 100 µg/mL ampicillin, which was then incubated overnight at 37 °C and 200 RPM. The following day, the cultures were pelleted (3,000 g, 4 °C, 10 min), resuspended in 12 mL PBS, and used to inoculate 12 4-L flasks containing 1 L of TBDry media supplemented with 100 µg/mL ampicillin. The cultures were incubated in a shaker at 37 °C and 200 RPM for 6–10 h, induced with 0.2 mM IPTG (Gold Biotechnology, Inc, St. Louis, MO), and grown overnight. The cells were concentrated by centrifugation in 1 L bottles (3,000 g, 4 °C, 10 min), resuspended in 10 mL cold PBS, and lysed by 3 min of sonication (10 s on, 40 s off; S-4000 Misonix Sonicator; Farmingdale, NY). Polyethylenimine (PEI) was added to the lysate to a final concentration of 1 % w/v, and the solution was centrifuged (14,000 g, 4 °C, 10 min) to remove cell debris and nucleic acid contaminants. The ELP was then purified by inverse transition cycling as described previously.[34] Briefly, the supernatant was heated to 37 °C, and NaCl was added to the solution to a final concentration of 1–3 M to trigger the ELP phase transition. The ELP was precipitated by centrifugation (14,000 g, 25 °C, 10 min), resuspended in cold PBS containing 20 mM TCEP (pH 7.4), and centrifuged again (14,000 g, 4 °C, 10 min) to remove remaining insoluble contaminants. This cycle was repeated 2–4 times to yield the final product. The ELP was then dialyzed with a molecular weight cut off (MWCO) of 12–14,000 Da overnight against deionized water and lyophilized.
2.3. Coupling reactions
2.3.1. Coupling reaction to glutathione (GSH)
In a typical experiment, 50.4 mg (0.0165 mmol) of maleimide-PPhOx and 6 mg (0.02 mmol) GSH were dissolved in 2 mL DMSO. The solution was stirred and heated to 60 °C for 1 min, followed by stirring at room temperature for 1.5 h. The solution was precipitated in a tenfold excess of deionized water and centrifuged (10,000 RPM, 22 °C, 12 min). The supernatant was discarded and the pellet was dried in vacuo. The product was obtained as a white solid (38 mg, 69 %).
1H-NMR (CDCl3, 500 MHz): δ (ppm): 2.10 (s, 2 H, -C-CH2-C-, glutamic acid), 2.29 (s, 2 H, -N-CO-CH2-C-, glutamic acid), 2.50–3.90 (br, 80 H, -N-CH2-CH2-N-), 4.69 (s, 1 H, -CO-CH-N-, cysteine), 6.90–7.50 (br, 95 H, phenyl)
2.3.2. Coupling reaction of POx to ELP
In a typical experiment, 10 mg (0.1636 µmol) of freeze-dried ELP were dissolved in 4.5 mL of a 2:1 (v/v) MeOH/MilliQ. The POx solution (500 µL of 1 g/L in 2:1 (v/v) MeOH/MilliQ, 0.1884 µmol) was added, and the mixture was stirred for 2 h at room temperature.
The reaction solution was transferred to a dialysis tube (MWCO 12–14,000 Da). The solution was dialyzed overnight against 2 L acetonitrile/deionized water 30:70 (v/v). The dialysis medium was changed to pure deionized water and the dialysis proceeded overnight. After freeze-drying, the product was obtained as a white powder (8.1 mg, 81 %).
1H-NMR (DMSO-d6, 500 MHz): δ (ppm): 0.90 (m, 1020 H, CH3-C-, valine), 1.20 (m, 240 H, -N-CH2-CO-, alanine), 2.50–3.90 (br, 80 H, -N-CH2-CH2-N-), 6.63 (d, 2 H, -CO-CH=CH-CO-), 6.90–7.50 (br, 95 H, phenyl)
2.4. Solubilization of paclitaxel (PTX)
Solubilization of paclitaxel (PTX) by the polymer-peptide conjugates was achieved via the thin-film method. In a typical experiment, 3 mg of freeze-dried conjugate and 3 mg of PTX were codissolved in 6 mL MeOH in a 50 mL round bottom flask. PTX was dissolved completely, whereas the conjugate formed a film. The solvent was removed under reduced pressure on a rotary evaporator within 30 min. To the dry film on the flask wall, 1 mL MilliQ water was added slowly and the solution was mixed vigorously with a pipette. Subsequently, the solution was filtered through a syringe filter (0.45 µm PVDF or 1 µm PTFE) prior to further analysis.
3. Results and Discussion
3.1. Maleimide functionalization
POx-peptide hybrids have already shown promising results regarding unaffected antibody activity[35] or extended solubility of the peptide in organic media.[36] There are multiple approaches to covalently link both moieties. Velander et al. directly terminated living POx chains by protein amine moieties by addition of proteins to the reaction mixture. We have introduced RGD motifs into alkyne-bearing poly(2-oxazoline)s using click chemistry.[37,38] Also aldehyde-amine/hydroxylamine coupling has been reported to form POx based peptide conjugates and hydrogels.[38–40] One of the most commonly employed reactions in bioconjugation is the Michael-addition of thiols and double bonds.[41] Hoogenboom and Dargaville recently introduced RGD motifs into POx hydrogels using thiol-ene coupling.[42] Thiol groups, in the form of cysteine side chains, can be found in numerous peptides and the double bond moiety can be introduced to POx in various forms. Schmitz et al. used 2-butenyl- or 2-decenyl oxazoline units to click a thiol functionalized thiazolidine to the POx side chains.[43] The resulting cysteine containing POx was then coupled to a peptide with native chemical ligation. To circumvent the UV-mediated thiol-ene reaction from Schmitz et al. one can also use activated double bonds. Polymers, including POx, that utilize maleimide-thiol click reactions have been previously described in detail.[44] However, the maleimide functionality was incorporated after side-chain hydrolysis and polymer analogue modification of the resulting secondary amines using 3-maleimido propionic acid.[45]
To the best of our knowledge, the combination of a living cationic ring opening polymerization (LCROP) of POx with a maleimide functionality has not been described until now. Protected maleimide functionalities may be considered via the initiator, a functional monomer or the termination reaction. The introduction of the maleimide functionality at the living terminus of the growing POx chain has some distinct advantages over previous approaches. The termination reaction can be carried out at ambient or slightly elevated temperatures, at which the protected maleimide is stable. In contrast to this, the polymerization of 2-oxazolines is carried out under elevated temperatures, where the protection group is likely to be cleaved and side reactions could occur.
To make the maleimide more nucleophilic, the imide proton can be readily removed by sufficiently strong bases, such as K2CO3. However, maleimide cannot be used as a termination reagent as it readily reacts with the 2-oxazoline monomer and the propagating species.[46] To protect the functionality, the [4+2] Diels-Alder (DA) reaction between furan and maleimide was chosen (Scheme 1a). After washing, recrystallization, and drying of the product, the reaction yielded white, crystalline 3,6-Endo/oxo-tetrahydrophthalhide.
Scheme 1.

a) Protection of maleimide via [4+2] Diels-Alder reaction with furan. b) General polymerization procedure of poly(2-oxazoline)s and termination with the Diels-Alder adduct of maleimide and furan. c) Cleavage of the maleimide protection group. d) Michael-addition reaction to couple the terminal group of poly(2-oxazoline)s to thiol bearing residues.
The formation of the desired product was confirmed by 1H-NMR spectroscopy (Figure S1), which displays signals that are consistent with formation of the desired educt, 3,6-endo/oxo-tetrahydrophthalide. The integral intensities of a – d correspond to the expected 1:2:2:2 ratios. According to the reaction mechanism, the applied conditions[30] favor the exo-isomer and the 1H-NMR as well as the melting point of 160 °C (Lit. 162 °C) indicate successful synthesis of the product.
In the following reaction, the DA product is used as a termination agent in LCROP of various 2-oxazoline monomers. We selected a range of monomers (2-methyl-, 2-butyl- and 2-phenyl-2-oxazoline; MeOx, BuOx and PhOx, respectively) to generate polymers of hydrophilic and hydrophobic character.
A general reaction route is shown in Scheme 1b. In order to increase the nucleophilicity of the maleimide, a base was added to the termination step to deprotonate the imide. K2CO3 was sufficient for this purpose. It should be noted that the base must be non-nucleophilic in order to avoid interference in the termination reaction. The efficiency of the termination was determined by correlating the integrated signals from the initiator to the signals from the coupled DA product. This was performed for MeOx because the signals for the initiator overlay the signals from the backbone in the case of PhOx. The efficiency for the K2CO3 assisted termination was found to be ~ 56 %. Although it would be desirable to increase it, the high selectivity of the subsequent Michael-addition obviated the need for further efforts to improve reaction efficiency at this stage. Optimization should be possible by variation of the base used in the reaction and other termination conditions. After termination, the maleimide group was recovered by the retro-Diels-Alder reaction (rDA) (Scheme 1c) as previously reported.[47] The rDA was confirmed by 1H-NMR (Figure S2). Signals that can be assigned to the protection group are observed at 5.2 ppm and 6.45 ppm. The third proton pair at the connection of the two rings is obscured by the broad polymer backbone peak. Following cleavage of the maleimide protection group, signals of the protection group were greatly diminished, whereas the signal assignable to the recovered double bond arises as a duplet at 6.6 ppm. From these three signals, the rDA efficiency was calculated to be ~ 85 %.
Further confirmation of successful termination and deprotection reactions was obtained by MALDI-ToF-MS measurements. In Figure 1a, two main distributions with a Δ m/z = 147 can be observed, which corresponds to the mass of one PhOx monomer unit. The peak with the highest intensity can correspond to two different Na+ doped species. One is the DA-terminated PPhOx chain with 20 repeat units (Figure 1b, distribution β), which is in excellent agreement with the [M0]/[I0] ratio. The other is the OH-terminated PPhOx with a block length of 21 repeating units. A further differentiation is difficult, since both species are theoretically only separated by Δ m/z = 0.2. The signals of the second distribution are spaced by Δ m/z = 69 to the most intense one, this corresponds to the molar mass of furan. Thus, the second main distribution represents the Na+ doped form of the PPhOx chain with a cleaved protection group (δ). In this distribution, the peak with the highest intensity also corresponds to a polymer chain with 20 repeat units. Furthermore, a distribution with Δ m/z = 16 with respect to the Na+ doped signals is also observed, which can be assigned to the polymer chains with K+ as the counter ion (γ, ɛ) (Figure 1b). Since no evidence of the rDA product was found in 1H-NMR spectra, it is reasonable to assume that the rDA is partially triggered under the laser irradiation during spectra acquisition.
Figure 1.
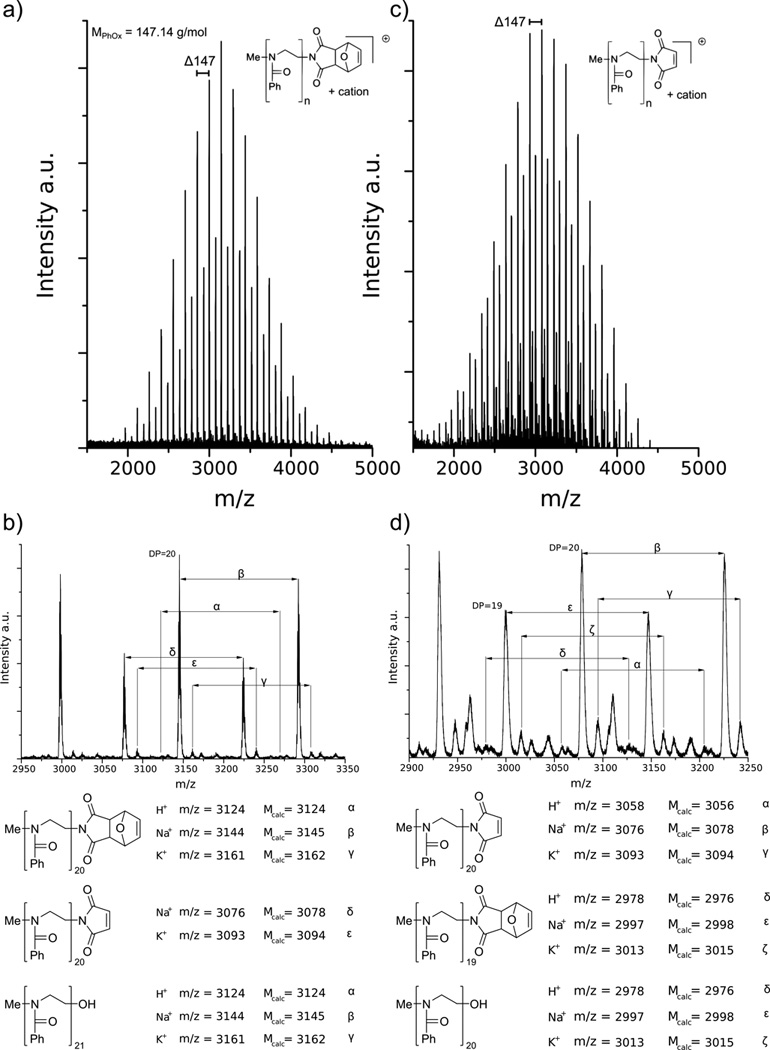
MALDI-ToF spectrum of PPhOx before (a) and after (c) cleavage of the maleimide protection group. The displayed masses equal the Na+ doped molecule peak of the desired polymer with 20 repeat units. Detailed MALDI-ToF spectra of PPhOx b) before and d) after cleavage of the maleimide protection group. The assigned distributions are represented with polymer structures below the spectra.
The MALDI-ToF-MS spectrum of the deprotected polymer also shows both main distributions (Figure 1c, d). In this case, the most intense distribution corresponds to the cleaved DA adduct (β), as expected. However, the intensity of the distribution with the intact DA end group (ɛ) is higher than one would expect from the 85 % cleavage efficiency, as determined by 1H-NMR. Therefore, it is most likely that in this case this species is the OH-terminated PPhOx, although it is possible that DA-adduct terminated PPhOx contributes to the signal intensity of the distribution.
3.2. Coupling reaction to glutathione
Glutathione (GSH), a tripeptide consisting of glutamic acid, cysteine and glycine, was selected as a model compound to evaluate the thiol-ene coupling reaction. Solubility tests indicated that GSH and POx were sufficiently soluble in DMSO, DMF, and a 2:1 (v/v) mixture of MeOH and deionized water. The successful conjugation of GSH to PPhOx was confirmed by 1H-NMR (Figure 2).
Figure 2.

1H-NMR spectrum (in CDCl3) of PPhOx with malemide end function a) and PPhOx coupled to glutathione (GSH) b). The inset in shows the signals of the PPhOx end functionality from c) protected maleimide, to d) recovered maleimide and e) GSH coupled PPhOx.
The area between 2 and 5 ppm is shown as well as the area between 5 and 6.8 ppm. After coupling in DMSO, three signals attributed to GSH appear in between 2 and 5 ppm, along with residual DMSO peaks (observed between 2.5 and 2.8 ppm). Further, the inset shows the development of the PPhOx end group from a) the DA functionality to b) the cleaved maleimide and c) the Michael-addition of GSH. One can clearly see that the rDA was not quantitative but after the coupling the signals from the maleimide double bond disappear. GPC reveals a small but noticeable shift of the elution peak towards a larger molar mass; this shift is expectedly small as the small GSH should not influence the hydrodynamic volume of the polymer significantly (Figure S3). The MALDI-ToF-MS spectrum of the conjugate shows multiple distributions (Figure 3a, b). The most intense signal distribution can represent again either the Na+ doped PPhOx terminated with the intact DA adduct or the OH-terminated PPhOx with one additional repeating unit (α). The distribution with the next highest intensity can be attributed to the desired glutathione conjugate, in which the glutamic acid residue was cleaved from the peptide chain (γ). This distribution has a Δ m/z = 179 with respect to deprotected PPhOx, which is the third most intense species (ɛ) (Figure 3b). Within all three distributions, the signals are spaced by Δ m/z = 147, which equals one PhOx monomer unit. To all of the three mentioned species distributions shifted by Δ m/z = 16, corresponding to a K+ counter ion, are observed (β, δ, ζ).
Figure 3.

MALDI-ToF-MS spectrum of PPhOx-GSH conjugate. a) Overview of the polymer species distributions with the most intense corresponding to PPhOx with DA termination and the conjugated PPhOx marked with *. b) Detailed view of the most intense species, along with polymer structures representing the different assigned distributions.
The expected signal distribution of the maleimide terminated PPhOx coupled to GSH is observed as a distribution of low intensity with a shift of Δ m/z = 307 (η) with respect to the ɛ distribution. The low intensity of η can be attributed to fragmentation of the peptide component of the GSH-POx conjugate, resulting in the adduct of PPhOx and the dipeptide of cysteine and glycine. Such a fragmentation was reported by Murphy et al.[48] for GSH conjugates, although they were referring primarily to high energy collisions. Other than the cleavage of the glutamic acid, no other fragments of the desired product were observed.
The differences in signal intensity between the conjugated and unconjugated polymers could be a result of different ionization energies (especially regarding the furan protected maleimide). We assume this is the case because the PPhOx sample used in this coupling reaction displays the same ratio of protected to deprotected chain ends as in Figure 1. However, successful coupling of GSH to maleimide functionalized PPhOx was evidenced by NMR and MALDI-ToF-MS.
Further coupling reactions to other poly(2-oxazoline)s show that the accessibility of the maleimide end group is possible independent of the nature of the monomer side chain (Table 1).
Table 1.
Analysis data from POx homopolymers with maleimide end functionality
| Polymer | [M]0/[I]0a | DP | Mn,theor. [g/mol] |
Mnc [g/mol] |
Đc,e | coupling reaction to maleimide |
|---|---|---|---|---|---|---|
| PMeOx-MI | 25 | 20b | 2239 | 2350 | 1.20 | ✓ |
| PBuOx-MI | 20 | 29b | 2654 | 2379 | 1.12 | ✓ |
| PPhOx-MI | 20 | 20d | 3055 | 2752 | 1.14 | ✓ |
Initial monomer/initiator ratio.
Determined by end group analysis from 1H NMR spectra.
Determined by gel permeation chromatography (GPC).
Determined by MALDI-TOF-MS
Đ=Mw/Mn
3.3. Coupling reaction to elastin-like polypeptides
After the successful coupling of POx to GSH, maleimide functionalized PPhOx was reacted with two different ELPs. Both ELPs are comprised of the Val-Pro-Gly-X-Gly)160 sequence, with the guest residues X=Val, Gly and Ala at a ratio of 1:7:8, respectively.[24] The ELPs only differ in their terminal sequence; ELP-WPC contains a single cysteine at the terminus, while ELP-WPC-(GGC)7 contains eight cysteine residues for multiple thiol-ene coupling. These polypeptides have similar molecular weights of 61.1 kg/mol and 62.6 kg/mol, respectively. Similar to the POx-GSH model reaction, ELP and POx were mixed in either DMSO or a 2:1 (v/v) mixture of MeOH and water, and were reacted at room temperature. POx was added in a 1.15 times molar excess over ELP in the case of ELP-WPC and 8.64 times molar excess for ELP-WPC-(GGC)7. After dialysis to remove the solvent and excess of POx, the products were lyophilized and characterized by 1H-NMR (Figure 4) and GPC.
Figure 4.

1H-NMR (in DMSO-d6) of spectra of ELP-WPC before (a) and after (b) coupling to maleimide functionalized PPhOx20. Spectra of ELP-WPC-(GGC)7 before (c) and after (d) coupling to maleimide functionalized PPhOx20.
PPhOx was selected for the reaction because, unlike for PBuOx, the 1H-NMR signal from the repeating amino acid units did not interfere with the signal from the polymer side chains. The aromatic signals from PPhOx are observed between 6.80 and 7.60 ppm for each of the ELP-POx hybrids. Additionally, the signals from the POx backbone appear at roughly 2.70 ppm, but are partially overlaid by the signals from DMSO and water at 2.50 and 3.33 ppm, respectively. The broad nature of the PPhOx signals complicates a straightforward integration of this section of the NMR spectrum. However, a rough estimation of the coupling efficiency was performed by using the methyl proton signals of valine and alanine of ELP. They are present at 0.90 ppm and 1.20 ppm, respectively, and are used as a reference. In total, 170 valines and 80 alanines contribute to both NMR spectra and are equal to the intensities of the 1020 and 240 proton signals. Taking this into account, the broad aromatic signals of PPhOx yield an integral of 123 protons for ELP-WPC and 805 protons for ELP-WPC-(GGC)7 (data not shown). As the theoretical values are 100 and 800 protons, respectively, this suggests that a residual amount of unbound PPhOx remained following the work up.
To confirm that PPhOx is covalently bound to the ELP, GPC was performed on the compounds before and after coupling. The elution profiles of the products show a shift towards higher molar mass, which is more pronounced with higher cysteine content, as expected (Figure 5). The GPC traces also show a peak at 18.2 min that corresponds to unconjugated PPhOx, showing that removal of the residual PPhOx by dialysis was not complete. Nevertheless, the analytical results show that reaction between the maleimide terminus of PPhOx and the cysteine(s) in the ELPs was successful.
Figure 5.

Gel permeation chromatography elution profiles before and after coupling reactions. a) ELP-WPC (dashed line) and ELP-WPC-PPhOx20 (solid line). b) ELP-WPC-(GGC)7 (dashed line) and ELP-WPC-(GGC)7-PPhOx20 (solid line). The insets are the region of 12–15 min retention time in detail.
3.4. Drug Loading of POx-ELP conjugates
After the successful conjugation of PPhOx to ELPs by Michael-addition, we investigated the potential for single and multigrafted PBuOx-ELP hybrids to solubilize the hydrophobic PTX in drug loading experiments. Dynamic light scattering (DLS) measurements indicated the formation of aggregates with sizes of 151.2±3.9 nm up to 196.2±3.8 nm for single grafted hybrids in aqueous solution, before incorporation of the drug. For ELP-WPC-(GGC)7, the aggregate sizes changes with the molar excess of PBuOx that is used for the coupling. If 2 eq of PBuOx are used the aggregates are 87.3±9.4 nm, for 4 eq they are 96.3±3.0 nm and for 8 eq the aggregates are on average 80.9±4.5 nm in diameter. One has to be careful with the absolute values because we also observe a wide spread of particle sizes for the single grafted hybrids, but hybrids with more than one PBuOx chain seem to form smaller aggregates. However, a clear trend within the multi grafted hybrids cannot be seen.
These results suggest that non-covalent drug solubilization into the hydrophobic core should be possible.
PTX loading was performed by the thin-film method.[5] In this procedure, the DDS and the drug are dissolved in a common volatile solvent. As the solvent evaporates, it leaves behind a thin film on the flask wall containing both components. Aggregates containing the loaded drug spontaneously form upon addition of aqueous solvent. This procedure was performed with MeOH, as it was able to dissolve both the ELP-POx fusion and the PTX. Millipore water was then added to the dried film, and the solution was filtered prior to injection into the HPLC. HPLC measurements were carried out in a 70:30 (v/v) mixture of water/ACN to dissociate the micelles and release any encapsulated drug. The ability of the method to differentiate between the single compounds was tested with pure compounds. Uncoupled ELP in aqueous solution as well as POx and PTX in methanol were sufficiently separated under the given conditions in the HPLC, as shown in Figure 6a.
Figure 6.

a) HPLC elution profiles for PTX (blue), PBuOx (red) and the uncoupled ELP with one cysteine residue (black). b) HPLC elugram for a PTX loaded ELP-POx conjugate, with the ELP bearing one cysteine residue. Displayed are three different samples with 3.3 wt% (black), 4.1 wt% (red) and 5.0 wt% (black) loading capacity. The first peak at 7.5 min can be attributed to the ELP-POx conjugate, while the second signal at 11 min represents the PTX.
The loading capacity (LC) was calculated as the ratio of the PTX concentration to the sum of the conjugate and PTX concentrations (Equation 1).
| (1) |
With this method, we were able to load PTX into ELPs with a single cysteine residue conjugated to PBuOx (Figure 6b). As the maximum solubility of free PTX in aqueous solution is only 1 µg/mL,[5] the determined amount of in average 65 µg/mL PTX implies that PTX must have been solubilized by the presence of the ELP-POx conjugate. However, reproducibility seems to be a major issue at this point. In consecutive experiments the values for LC averaged at 3 % but ranged from 1 to 5 % (Table 2). Therefore, this issue should be assessed with particular scrutiny in future work.[49]
Table 2.
Analysis data from PTX loaded ELP-PBuOx conjugates
| Polymer | ELP trailer |
[conjugate] [g/l] |
[PTX] [mg/l] |
LC [%] |
N(PTX) per PBuOx block |
|---|---|---|---|---|---|
| PBuOx single graft (average) | 1 Cys | 2.0±1.0 | 65.4±37.5 | 3.2±1.2 | 2.5±1.0 |
| PBuOx multi graft (1:2) | 8 Cys | 1.1 | 44.2 | 3.9 | 3.1a |
| PBuOx multi graft (1:4) | 8 Cys | 0.7 | 44.7 | 5.9 | 4.7a |
| PBuOx multi graft (1:8) | 8 Cys | 0.3 | 23.5 | 7.9 | 6.4a |
| PMeOx37-PBuOx23-PMeOx37 (P2)b | - | 10.0 | 8200 | 45 | 9.6 |
PTX per conjugate due to unkown grafting efficiency
Data from Ref. 5
We then investigated the LC of ELP-WPC-(GGC)7 by modulating the molar ratio of ELP to PBuOx. As the molar ratio of POx to ELP increased (1:2, 1:4 and 1:8), we observed a clear trend of increasing PTX solubilization (from 3.9 wt%, to 5.9 wt% to 7.9 wt%, respectively) (Table 2, Figure 7). The PTX drug loading was clearly much lower compared to the 45 wt% loading achieved with PMeOx-PBuOx-PMeOx triblock copolymer. Also, the overall solubility appears to negatively correlate with the LC, which we did not observe in the case of PMeOx-PBuOx-PMeOx triblock copolymer PTX formulation.[7] The relatively low LC may be attributed to the relatively low content of hydrophobic polymer with respect to the total mass of the POx-ELP conjugate. Even in the conjugate bearing 8 PBuOx chains, the overall hydrophobic content is only 25 wt.%, which is lower than the previously studied PMeOx-PBuOx-PMeOx triblock copolymer (approx. 32 wt.%). With respect to aqueous solubility, it is clear that the ELP is less hydrophilic than the PMeOx, which appears to negatively influence the overall solubility of the drug loaded ELP-POx hybrids. For a better comparison between PBuOx-ELP conjugates and PMeOx-PBuOx-PMeOx, the relative amount of PTX molecules to PBuOx is calculated for both. On average, aggregates of ELPs grafted with a single PBuOx were able to incorporate 2.5 PTX per hydrophobic side chain. For ELP-WPC-(GGC)7 the calculation is less straightforward, since the average grafting efficiency of PBuOx chains is unknown. When the amount of PTX is calculated per ELP chain (Table 2) a trend towards higher solubilization rates for increasing amount of PBuOx can be observed. However, when the values are divided by the intended amount of PBuOx grafts (2, 4 and 8) this trend is reversed. The ELP to PBuOx ratios of 1:2, 1:4 and 1:8 would than result in 1.5, 1.2 and 0.8 PTX per PBuOx chain, respectively. This could possibly be explained by the space within the hydrophobic core. Since the particle sizes showed to be similar for the different grafting ratios, the ability for one PBuOx to solubilize PTX molecules might be sterically limited by neighboring PBuOx chains. But more data needs to be evaluated to test this hypothesis.
Figure 7.

HPLC elution profile for PTX loaded ELP-WPC-(GGC)7-PPhOx20. Displayed are different ELP-POx coupling ratios with normalized intensities. The molar ratios of ELP and POx are 1:2 (black), 1:4 (blue) and 1:8 (red). The first peak at around 7.5 min can be attributed to the ELP-POx conjugate, while the second signal at 11 min retention time represents PTX. With increasing amount of POx, PTX solubilization increases from 3.9 wt% to 5.9 wt% and 7.9 wt%. All samples were filtered with a 1 µm filters prior to analysis.
The POx tri-block P2 shows a tremendously higher solubilization potential for PTX (a 126-fold increase) and is itself more soluble by a factor of 5 when compared to the average singly grafted ELP.
4. Conclusion
We report a facile and general procedure to successfully incorporate a maleimide end group into different poly(2-oxazoline) homopolymers via the Diels-Alder product of maleimide and furan. The termination of the POx with this Diels-Alder adduct, as well as the recovery of the maleimide groups were tracked by 1H-NMR spectroscopy and MALDI-ToF-MS. However, this procedure is not restricted to POx homopolymers; we anticipate it could be applied to any polymer chain that can be functionalized by a nucleophile. Furthermore, we demonstrated that this functionalized polymer is accessible for the Michael addition of small molecules like glutathione, as well as macromolecules such as ELPs with thiol-bearing end groups. To the best of our knowledge, this represents the first reported formation of such polymer-peptide hybrids. These conjugates have successfully demonstrated the ability to solubilize the hydrophobic anti-cancer drug PTX with a loading capacity that positively correlates with the amount of the hydrophobic POx chains.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This research was supported by grants from the NIH to A.C. (R01 EB000188, R01 EB007205, and R01 GM61232).
J.F.N. was partially funded by the PROMOS scholarship from DAAD.
Contributor Information
Jonas F. Nawroth, Professur für Makromolekulare Chemie, Department Chemie, Technische Universität Dresden, Mommsenstr. 4, 01069 Dresden, Germany
Jonathan R. McDaniel, Department of Biomedical Engineering, Duke University, Durham, North Carolina, 27708-0281, USA.
Ashutosh Chilkoti, Department of Biomedical Engineering, Duke University, Durham, North Carolina, 27708-0281, USA.
Rainer Jordan, Professur für Makromolekulare Chemie, Department Chemie, Technische Universität Dresden, Mommsenstr. 4, 01069 Dresden, Germany.
Robert Luxenhofer, Professur für Makromolekulare Chemie, Department Chemie, Technische Universität Dresden, Mommsenstr. 4, 01069 Dresden, Germany.
References
- 1.Langer R. Science. 1990;249:1527. doi: 10.1126/science.2218494. [DOI] [PubMed] [Google Scholar]
- 2.Hennenfent KL, Govindan R. Ann. Oncol. 2006;17:735. doi: 10.1093/annonc/mdj100. [DOI] [PubMed] [Google Scholar]
- 3.Matsumura Y, Kataoka K. Cancer Sci. 2009;100:572. doi: 10.1111/j.1349-7006.2009.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang T, Cui FD, Choi MK, Cho JW, Chung SJ, Shim CK, Kim DD. Int. J. Pharm. 2007;338:317. doi: 10.1016/j.ijpharm.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Luxenhofer R, Schulz A, Roques C, Li S, Bronich TK, Batrakova EV, Jordan R, Kabanov AV. Biomaterials. 2010;31:4972. doi: 10.1016/j.biomaterials.2010.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han Y, He Z, Schulz A, Bronich TK, Jordan R, Luxenhofer R, Kabanov AV. Mol. Pharm. 2012;9:2302. doi: 10.1021/mp300159u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schulz A, Jaksch S, Schubel R, Wegener E, Di Z, Han Y, Meister A, Kressler J, Kabanov AV, Luxenhofer R, Papadakis CM, Jordan R. ACS Nano. 2014;8:2686. doi: 10.1021/nn406388t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He Z, Schulz A, Wan X, Seitz J, Bludau H, Alakhova DY, Darr DB, Perou CM, Jordan R, Ojima I, Kabanov AV, Luxenhofer R. J. Control. Release. 2015;208:67. doi: 10.1016/j.jconrel.2015.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seo Y, Schulz A, Han Y, He Z, Bludau H, Wan X, Tong J, Bronich TK, Sokolsky M, Luxenhofer R. Polym. Advan. Technol. 2015;26:837. [Google Scholar]
- 10.Saegusa T, Ikeda H, Fujii H. Macromolecules. 1972;5:359. [Google Scholar]
- 11.Saegusa T, Ikeda H. Macromolecules. 1973;6:805. [Google Scholar]
- 12.He Z, Miao L, Jordan R, S-Manickam D, Luxenhofer R, Kabanov AV. Macromol. Biosci. 2015;15:1004. doi: 10.1002/mabi.201500021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang N, Pompe T, Amin I, Luxenhofer R, Werner C, Jordan R. Macromol. Biosci. 2012;12:926. doi: 10.1002/mabi.201200026. [DOI] [PubMed] [Google Scholar]
- 14.Bauer M, Lautenschlaeger C, Kempe K, Tauhardt L, Schubert US, Fischer D. Macromol. Biosci. 2012;12:986. doi: 10.1002/mabi.201200017. [DOI] [PubMed] [Google Scholar]
- 15.Gaertner FC, Luxenhofer R, Blechert B, Jordan R, Essler M. J. Control. Release. 2007;119:291. doi: 10.1016/j.jconrel.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Ishida T, Kiwada H. J. Control. Release. 2007;119:236. doi: 10.1016/j.jconrel.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 17.Kierstead PH, Okochi H, Venditto VJ, Chuong TC, Kivimae S, Fréchet JM, Szoka FC. J. Control. Release. 2015;213:1. doi: 10.1016/j.jconrel.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Etrych T, Sírová M, Starovoytova L, Ríhová B, Ulbrich K. Mol. Pharm. 2010;7:1015. doi: 10.1021/mp100119f. [DOI] [PubMed] [Google Scholar]
- 19.Pang X, Du HL, Zhang HQ, Zhai YJ, Zhai GX. Drug. Discov. Today. 2013;18:1316. doi: 10.1016/j.drudis.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Vrhovski B, Weiss AS. Eur. J. Biochem. 1998;258:1. doi: 10.1046/j.1432-1327.1998.2580001.x. [DOI] [PubMed] [Google Scholar]
- 21.Urry DW. Trends. Biotechnol. 1999;17:249. doi: 10.1016/s0167-7799(99)01306-2. [DOI] [PubMed] [Google Scholar]
- 22.Bidwell GL, Fokt I, Priebe W, Raucher D. Biochem. Pharmacol. 2007;73:620. doi: 10.1016/j.bcp.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 23.Urry DW. Prog. Biophys. Mol. Biol. 1992;57:23. doi: 10.1016/0079-6107(92)90003-o. [DOI] [PubMed] [Google Scholar]
- 24.Meyer DE, Chilkoti A. Biomacromolecules. 2002;3:357. doi: 10.1021/bm015630n. [DOI] [PubMed] [Google Scholar]
- 25.Urry DW. J. Phys. Chem. B. 1997;101:11007. [Google Scholar]
- 26.Urry DW. Angew. Chem. Int. Edit. 1993;32:819. [Google Scholar]
- 27.MacKay JA, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A. Nat. Mater. 2009;8:993. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mastria EM, Chen M, McDaniel JR, Li X, Hyun J, Dewhirst MW, Chilkoti A. J. Control. Release. 2015;208:52. doi: 10.1016/j.jconrel.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhattacharyya J, Bellucci JJ, Weitzhandler I, McDaniel JR, Spasojevic I, Li X, Lin CC, Chi JT, Chilkoti A. Nat. Commun. 2015;6:7939. doi: 10.1038/ncomms8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwart H, Burchuk I. J. Am. Chem. Soc. 1952;74:3094. [Google Scholar]
- 31.Huber S, Jordan R. Colloid. Polym. Sci. 2008;286:395. [Google Scholar]
- 32.Luxenhofer R, Jordan R. Macromolecules. 2006;39:3509. [Google Scholar]
- 33.Hoogenboom R, Wiesbrock F, Huang H, Leenen MA, Thijs HM, van Nispen SF, van der Loop M, Fustin C-A, Jonas AM, Gohy J-F. Macromolecules. 2006;39:4719. [Google Scholar]
- 34.Meyer DE, Chilkoti A. Nat. Biotechnol. 1999;17:1112. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 35.Velander WH, Madurawe RD, Subramanian A, Kumar G, Sinai-Zingde G, Riffle JS. Biotechnol. Bioeng. 1992;39:1024. doi: 10.1002/bit.260391006. [DOI] [PubMed] [Google Scholar]
- 36.Konieczny S, Krumm C, Doert D, Neufeld K, Tiller JC. J. Biotechnol. 2014;181:55. doi: 10.1016/j.jbiotec.2014.03.035. [DOI] [PubMed] [Google Scholar]
- 37.Luxenhofer R, López-García M, Frank A, Kessler H, Jordan R. PMSE Prepr. 2006;95:283. [Google Scholar]
- 38.Luxenhofer R. PhD thesis. TU München; 2007. Garching. [Google Scholar]
- 39.Taubmann C, Luxenhofer R, Cesana S, Jordan R. Macromol. Biosci. 2005;5:603. doi: 10.1002/mabi.200500059. [DOI] [PubMed] [Google Scholar]
- 40.Legros C, De Pauw-Gillet M-C, Tam KC, Lecommandoux S, Taton D. Eur. Polym. J. 2015;62:322. doi: 10.1039/c5sm00313j. [DOI] [PubMed] [Google Scholar]
- 41.Mather BD, Viswanathan K, Miller KM, Long TE. Prog. Polym. Sci. 2006;31:487. [Google Scholar]
- 42.Farrugia BL, Kempe K, Schubert US, Hoogenboom R, Dargaville TR. Biomacromolecules. 2013;14:2724. doi: 10.1021/bm400518h. [DOI] [PubMed] [Google Scholar]
- 43.Schmitz M, Kuhlmann M, Reimann O, Hackenberger CPR, Groll J. Biomacromolecules. 2015;16:1088. doi: 10.1021/bm501697t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tolstyka ZP, Kopping JT, Maynard HD. Macromolecules. 2008;41:599. [Google Scholar]
- 45.Chujo Y, Sada K, Saegusa T. Macromolecules. 1990;23:2636. [Google Scholar]
- 46.Simionescu CI, Grigoras M, Bîcu E, Onofrei G. Polym. Bull. 1985;14:79. [Google Scholar]
- 47.Sánchez A, Pedroso E, Grandas A. Org. Lett. 2011;13:4364. doi: 10.1021/ol201690b. [DOI] [PubMed] [Google Scholar]
- 48.Murphy CM, Fenselau C, Gutierrez PL. J. Am. Soc. Mass. Spectr. 1992;3:815. doi: 10.1016/1044-0305(92)80004-5. [DOI] [PubMed] [Google Scholar]
- 49.Luxenhofer R. Nanomedicine. 2015;10:3109. doi: 10.2217/nnm.15.139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.