

Abstract
A new and versatile method is described to engineer precisely defined protein/peptide–polymer therapeutics by a modular approach that consists of three steps: (1) fuse a protein/peptide of interest with an elastin–like–polypeptide that enables facile purification and high yield; followed by (2) installation of a clickable group at the C–terminus of the recombinant protein/peptide with close to complete conversion by enzyme–mediated ligation; and subsequently (3) attachment of a polymer by a click reaction with near quantitative conversion. We demonstrate that this modular approach is applicable to various protein/peptide drugs and used it to conjugate structurally diverse water-soluble polymers that prolong the plasma circulation duration of proteins. These protein/peptide–polymer conjugates exhibit significantly improved pharmacokinetics and improved therapeutic effects over the native protein/peptide after administration to mice. The studies reported here provide a facile methodology for synthesis of protein/peptide-polymer conjugates for therapeutic use and other applications.
Keywords: Protein modification, Prodrugs, Click chemistry, Pharmacokinetics, Diabetes
Graphical Abstract
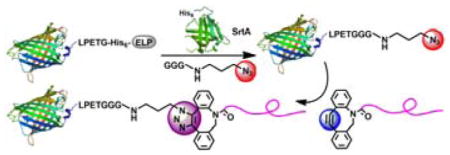
A modular method is reported to engineer precisely defined protein/peptide–polymer therapeutics with high yield that are site–specific and stoichiometric by a combination of recombinant expression, enzyme–mediated ligation and click chemistry; this method is suitable for conjugation of structurally diverse polymers to various protein and peptide drugs.
With more than 200 approved biopharmaceuticals marketed in the United States and/or EU and many more in preclinical pipeline since the early 1980s,[1] the field of protein/peptide therapeutics is booming,[2] because of their high biological activity and specificity.[3] Unfortunately, a major barrier to their clinical adoption stems from the fact that parenteral injection remains the mainstay of protein and peptide drugs, but it leads to rapid clearance due to their short in vivo half–life and poor stability,[4] which in turn requires frequent injections, leading to reduced patient compliance and undesirable side effects.[5] Covalently conjugating protein and peptide drugs with “stealth” polymers that prevent opsonization of the protein and provide extended blood circulation is an effective strategy to overcome some of these limitations.[6]
The most common approach for the synthesis of protein-polymer conjugates involves reaction of semitelechelic polymers with the reactive side chains of protein/peptide residues, which is typically carried out by the separate synthesis of protein, polymer, and linker, and sequential conjugation of the three entities to create the protein/peptide–polymer conjugate.[7] Unfortunately, most such methods provide limited control of the site and grafting ratio because most proteins/peptides contain numerous chemically reactive residues, which results in heterogeneous protein/peptide–polymer conjugates with poorly controlled stoichiometry and unacceptably low biological activity.[8] The limitations of these methods is further exacerbated when the polymer to be conjugated is a “stealth” polymer because these polymers, by definition, are designed to evade interactions with proteins, so that the yield of conjugation is typically low.[9] The residue-specific incorporation of unnatural amino acids (UAAs)–especially biorthogonal “clickable” residues–into recombinant proteins/peptides provides an elegant alternative to site-specifically attach polymers with high yield. However, UAAs are expensive, especially at the concentration required for genetically encoded incorporation, and the expression yield of recombinant UAA-containing proteins/peptides is typically much lower than the native protein, and the site of UAA incorporation affects the yield in unexpected ways that are impossible to predict, so that this methodology, while conceptually attractive, requires significant amount of up-front optimization for each protein/peptide of interest.[10] Hence, methods to synthesize stealth protein/peptide–polymer conjugates are still needed that are: (i) useful for protein/peptide drugs without case-by-case optimization, (ii) with structurally diverse polymers, and that allow (iii) site–specific and (iv) stoichiometric conjugation with (v) high yield.
Motivated by this rationale, we report a new and versatile method to synthesize precisely defined protein/peptide–polymer conjugates that are site–specific, stoichiometric, and proceed with high yield and should be generally applicable to diverse protein and peptide drugs (Figure 1). This modular approach has three steps: (1) recombinantly express a protein/peptide of interest that is fused to an elastin–like–polypeptide (ELP), followed by a LPETG peptide that is a substrate for the transpeptidase–sortase A[11] at the C–terminus of the protein or peptide; (2) quantitatively install a clickable group at the protein/peptide by the canonical sortase A–mediated native peptide ligation at the C-terminus of the protein/peptide; and subsequently (3) attach a polymer by strain–promoted azide–alkyne click reaction. We show that all three steps involved in this method can be carried out with near quantitative conversion to enable conjugation of structurally diverse “stealth” polymers to green fluorescent protein (GFP). We further show the therapeutic utility of a zwitterionic polymer conjugate of a therapeutic exendin–4, a peptide clinically used to treat type–2 diabetes.[12] The exendin–zwitterionic polymer conjugate reduced blood glucose levels for up to 3 days in fed mice after a single subcutaneous (s.c.) injection, 18 times longer than an injection of the unmodified peptide drug. We believe that this highly efficient and modular approach to synthesize precisely defined protein/peptide–polymer therapeutics may be applicable to a large subset of protein/peptide drugs and to diverse water soluble polymers, and provides a general methodology for clinical development of polymer conjugates as biologic drugs.
Figure 1.

(a) GFP–polymer conjugates were synthesized by recombinant expression of a quaternary fusion protein, GFP–srt–His6–ELP, followed by site–specific installation of an azido group at the C–terminus of GFP by sortase A–catalyzed native peptide ligation and subsequent copper–free click reaction with DBCO–terminated polymers. (b) Synthetic route of DBCO–terminated PEG–like biodegradable polycarbonate, nucleic acid–like biodegradable polyphosphoesters, phospholipid–like zwitterionic polymethacrylates, and PEG.
A quaternary fusion protein, abbreviated as “GFP–srt–His6–ELP”, was recombinantly expressed to serve as the sortase A substrate (Figure 1a), where the “srt” stands for the LPETG pentapeptide that is the recognition sequence for sortase A,[13] and ELP refers to a thermally responsive elastin–like polypeptide that is introduced to enable facile purification of the quaternary fusion protein by inverse transition cycling (ITC).[14] The “LPETG” peptide sequence was deliberately located between GFP and ELP, so that transpeptidation by sortase A allows attachment of a clickable group to the C-terminus of GFP and subsequent liberation of the purification tag. The GFP–srt–His6–ELP was obtained at high purity with a yield of ~300 mg L−1 in shaker flask culture of E. coli transformed with an expression plasmid that encodes the fusion protein. A short linker (GGGN3), consisting of an N–terminal triglycine (Gly3) motif linked with a clickable azido group (N3), was synthesized to serve as the nucleophile for the native peptide ligation catalyzed by sortase A[15] (Supporting Information, Figure S1). Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) analysis of the sortase A–catalyzed installation of azido group showed almost quantitative conversion after 5 h incubation at 37 °C as seen by the near complete disappearance of the band corresponding to GFP–srt–His6–ELP at ~67 kDa, and the appearance of two bands at ~28 and ~39 kDa, corresponding to the azido–attached GFP (abbreviated as GFP–C–N3) and the cleaved ELP (Figure 2a). A control reaction was also performed using Gly3 as the nucleophile, to yield GFP–C–Gly3 as a negative control for the subsequent click reaction. Pure GFP–C–N3 was easily obtained with a yield of ~85% by immobilized metal affinity chromatography (IMAC). Matrix assisted laser desorption ionization–mass (MALDI–MS) spectrometry analysis of the purified GFP–C–N3 showed a single peak at 28,021.6 Da, which closely agrees with the theoretical mass of 28,034.1 Da (Error: 0.04%) for GFP–C–N3 (Figure 2b). Fourier transform infrared (FT-IR) spectrum of GFP–C–N3 displayed a peak at 2100 cm−1, corresponding to the stretching band of the azido group (Figure S2a), further indicating the successful attachment of the azido group to GFP. To prove the site–specificity of azido installation, GFP–C–N3 was subjected to trypsin digestion and the peptide fragments were analyzed by MALDI–MS. The C–terminal peptide fragment was detected at 3,796.7 Da that agrees well with the theoretical mass of 3,799.1 Da (Error: 0.06%), providing strong evidence that the azido group was solely attached at the C–terminus of GFP (Figure S2b).
Figure 2.
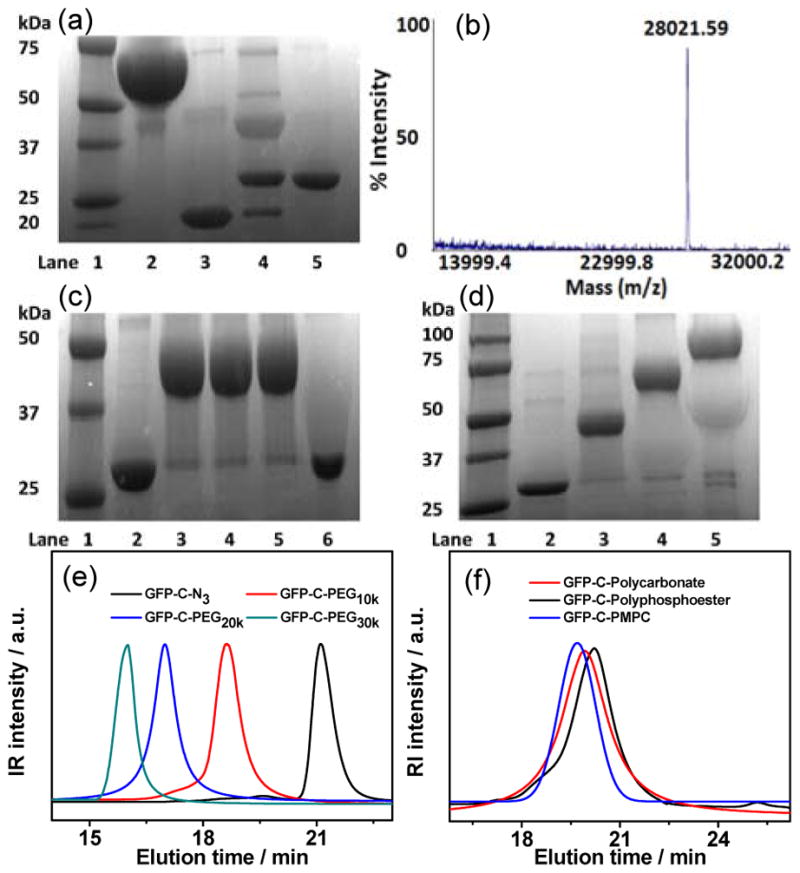
(a) SDS–PAGE analysis of azido installation by sortase A. Lane 1: MW marker, lane 2: GFP–srt–His6–ELP, lane 3: sortase A, lane 4: reaction mixture after 5 h of incubation, lane 5: purified GFP–C–N3. (b) MALDI–TOF of GFP–C–N3. (c) SDS-PAGE of click reaction between GFP–C–N3 and DBCO–PEG10k. Lane 1: MW marker, lane 2: GFP–C–N3, lane 3 to 5: reaction mixture of GFP–C–N3 and DBCO–PEG10k after 10, 30 and 180 min of reaction, lane 6: mixture of GFP–C–Gly3 and DBCO–PEG10k after 180 min of reaction. (d) SDS-PAGE of click reaction between GFP–C–N3 and DBCO–PEG with different MW. Lane 1: MW marker, lane 2: GFP–C–N3, lane 3 to 5: reaction mixture of GFP–C–N3 and DBCO–PEG with MW of 10, 20 and 30 kDa after reaction for 60 min. GPC curves of (e) GFP–C–PEG and (f) GFP–C–Polycarbonate, GFP–C–Polyphosphoester and GFP–C–PMPC, respectively.
The installation of the azido group at the C–terminus of a protein is a generalizable and convenient method for site-specific attachment of structurally diverse polymers with an alkyne end-group via a click reaction.[16] A set of four novel cyclooctyne–terminated water–soluble polymers, including a biodegradable polycarbonate and a polyphosphoester as well as a nondegradable polymethacrylate and polyether were synthesized to test the generality of this approach. We chose these polymers because they represent are commonly used for protein conjugation. As shown in Figure 1b, the poly(ethylene glycol)-like (PEG–like) polycarbonate (DBCO–polycarbonate) and nucleic acid–like polyphosphoester (DBCO–polyphosphoester) were synthesized by in–situ organocatalyzed ring–opening polymerization (ROP) using DBCO–PEG4–OH as the initiator. A phospholipid–like zwitterionic poly[2-methacryloyloxyethyl phosphorylcholine] (DBCO–PMPC) was synthesized by atom transfer radical polymerization (ATRP) using an N–hydroxysuccinimide (NHS) ATRP initiator and subsequent reaction with DBCO–PEG4–amine. PEGs (DBCO–PEG) with different molecular weights were obtained by reaction of DBCO–PEG4–amine with NHS ester terminated PEGs of different MWs. All these clickable polymers were obtained with narrow polydispersity and adjustable molecular weight (MW). Details of the synthesis and characterization of these polymers are described in the Supporting Information.
We next studied the click reaction by simply mixing GFP–C–N3 with the cyclooctyne–terminated polymers at a GFP–C–N3/polymer ratio of 1:5 in phosphate-buffered saline (PBS) without adding any catalysts and organic solvents. SDS–PAGE analysis showed that near quantitative conversion of the reaction between GFP–C–N3 and DBCO–PEG10k was achieved only after 10 min incubation at room temperature (Figure 2c), indicating the extremely high efficiency of this catalyst–free click reaction. As expected, no reaction occurred when GFP–C–Gly3 was added to the reaction mixture as a negative control, which suggests the reaction occurs selectively between the azido group at the C–terminus of GFP-C-N3 and the cyclooctyne group at the chain-end of the PEG, forming a site–specific (C–terminus) and stoichiometric (1:1) conjugate (GFP-C-PEG). We next investigated the reaction between GFP–C–N3 and DBCO–PEG with a range of MW to understand the impact of polymer size, and potentially the steric hindrance imposed by it, on the reaction yield. As shown in Figure 2d, almost complete conversion was observed for both DBCO–PEG20k and DBCO–PEG30k, which suggests that at least in the MW range of 10–30 kDa, there is no dependent on reaction yield on MW, and that different MW conjugates can be easily synthesized without the need to optimize the conjugation conditions. The quantitative conversion of GFP–C–N3 also facilitated the purification of GFP–C–PEG with a high yield of >80%, which compares favorably with the 10–20% overall yield often seen in conventional PEGylation processes.[17] Gel permeation chromatography (GPC) curves showed monomodal and symmetric elution peaks for GFP–C–PEG that exhibited a clear shift to a larger size with increasing MW of DBCO–PEG (Figure 2e). A cyclooctyne terminated polycarbonate, polyphosphoester, and PMPC were separately conjugated to the C–terminus of GFP by using identical reaction procedures (Figure 2f and Table S1).
We further investigated the blood circulation of these polymer conjugates (Figure 3a). The GFP–C–PEG and GFP–C–PMPC conjugates were chosen for pharmacokinetic studies because of they are “stealth” polymers whose protein conjugates are known to provide long plasma circulation.[9] As expected, native GFP was rapidly cleared from blood with a high total body clearance (CL) rate of 7 mL/h and a rapid terminal elimination phase (T1/2β=4 h) (Table S3). In contrast, the CL rate of GFP–C–PEG decreased to 0.72 mL/h, and its T1/2β was prolonged to 12 h. These pharmacokinetic differences resulted in a 17–fold increase in the area under the curve (AUC) of GFP–C–PEG (240%h/mL) compared to native GFP (14%h/mL). Comparable blood exposure of GFP was observed for GFP–C–PMPC with a similar hydrodynamic radius (Rh). These results reveal that C–terminal site–specific conjugation of a “stealth” polymer significantly improved the cumulative blood exposure of the protein.
Figure 3.
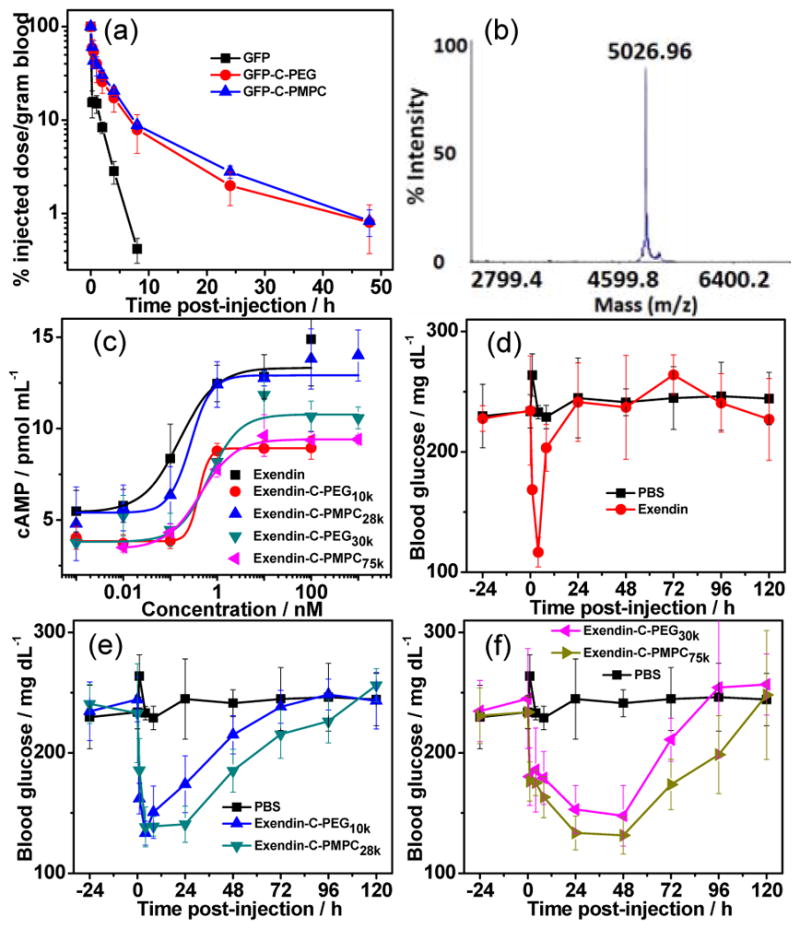
(a) Blood concentration as a function of time post–injection. (b) MALDI–TOF of exendin–C–N3. (c) Cyclic adenosine monophosphate (cAMP) response of exendin–4 conjugates in baby hamster kidney (BHK) cells expressing the GLP–1R. Fed blood glucose levels in 6–week–old male C57Bl/6J mice (n=5) maintained on a 60 kCal% diet before and after a single s.c. injection of (d) unmodified exendin–4, (e) exendin–C–PEG10k and exendin–C–PMPC28k, (f) exendin–C–PEG30k and exendin–C–PMPC75k. Exendin–4 and conjugates were administered at a dose of 25 nmol/kg and PBS was injected as control at equivalent volume at t=0 h. Glucose levels of blood sampled from the tail of mouse were measured at given time points.
Having established with GFP that these stealth polymer conjugates show significantly improved pharmacokinetics compared to the protein, we next investigated the utility of this methodology to improve the in vivo efficacy of a therapeutic peptide, exendin–4, that is a highly potent drug for treatment of type-2 diabetes but has a notoriously short half-life of ~2 h in blood circulation.[18] Exendin–C–N3 (Figure 3b) and two sets of exendin–C–PEG and exendin–C–PMPC conjuates with different polymer MWs (Table S1 and Figure S10d) but almost the same Rh were similarly synthesized and tested in vitro and in vivo.
The in vitro potency of native exendin–4 and its conjugates were assessed by quantifying intracellular cyclic adenosine monophosphate (cAMP) release as a result of GLP–1R activation in baby hamster kidney (BHK) cells that were stably transfected with rat GLP–1R.[19] As shown in Figure 3c, unmodified exendin–4 activates GLP–1R with a half–maximal effective concentration (EC50) of 0.16 ± 0.07 nM. Grafting PEG to the C–terminus of exendin–4 increased the EC50 to 0.40–0.66 nM while the PMPC conjugates had an EC50 of 0.27–0.44 nM (Table S2). The slightly lower potency of the conjugates, seen by their ~2-fold greater EC50, is consistent with results observed for other polymer conjugates because of the steric hindrance imposed by the polymer.[20]
Next, the in vivo efficacy of these conjugates was assessed in 6–week–old male C57BL/6J mice that were maintained on a 60 kCal% fat diet, so as to develop a diabetic phenotype.[21] The PEG and PMPC conjugates were administered into mice (n=5) by a single s.c. injection at a dose of 25 nmol/kg mouse body weight, and fed glucose levels in blood were then measured at various time points post–injection. Unmodified exendin–4 and an equivalent volume of PBS were separately injected as a positive and negative control. We found that all conjugates significantly extended the glucose–lowering effect of exendin–4. The exendin–C–PEG10k conjugate provided extended glucose reduction for up to 24 h (P<0.05, Figure 3e), while the larger exendin–C–PEG30k conjugate reduced blood glucose level for up to 48 h (P<0.001, Figure 3f). These results are in stark contrast with the very temporally limited activity of native exendin–4 that only lowers glucose levels for 4 h following s.c. injection (Figure 3d). Remarkably, a single s.c. injection of exendin–C–PMPC28k resulted in blood glucose reduction for up to 48 h (P<0.05, Figure 3e), and the larger exendin–C–PMPC75k lowered glucose for up to 72 h (P<0.05, Figure 3f). Clearly both PEG and a phosphorylated “stealth” polymer markedly extended the duration of efficacy of exendin-4. In addition, a similar trend was observed for the mouse body weights that decreased with glucose levels (Figure S12), which is consistent with the weight-lowering benefit of exendin–4.[22] It is worth pointing out that these data demonstrate, for the first time, that attaching a zwitterionic polymer to the C–terminus significantly improves the therapeutic efficacy of a peptide drug and is at least as good as the “gold standard” PEG.
In summary, we report a new and general method to engineer precisely defined protein/peptide–polymer therapeutics that are site–specific, stoichiometric, and with high yield by a combination of recombinant expression, enzyme–mediated ligation and click chemistry. This modular approach is significant because: (1) it can be used with diverse various protein/peptide drugs; (2) it is suitable for conjugation of structurally diverse polymers; (3) all steps involved in this method exhibit almost quantitative conversion and avoid use of any organic solvent and catalyst; (4) the synthesized protein/peptide–polymer conjugates significantly improved pharmacokinetics and therapeutic effects. This conjugation method is likely to be useful in a manufacturing setting, as it provides a high yield with low reaction times, and is experimentally robust in its tolerance to structurally diverse proteins and polymers. We believe that this methodology represents an important expansion of the methods available for polymer conjugation to proteins, with attributes that make it suitable for clinical and commercial development of polymer conjugates that improve the delivery of protein/peptide therapeutics.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R01-DK092665) to A.C.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Walsh G. Nat Biotech. 2014;32:992–1000. doi: 10.1038/nbt.3040. [DOI] [PubMed] [Google Scholar]
- 2.Leader B, Baca QJ, Golan DE. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 3.Lee VHL. Peptide and protein drug delivery. Mercel Dekker; New York: 1991. [Google Scholar]
- 4.Werle M, Bernkop-Schnurch A. Amino Acids. 2006;30:351–367. doi: 10.1007/s00726-005-0289-3. [DOI] [PubMed] [Google Scholar]
- 5.Banga AK. Therapeutic Peptides and Proteins. CRC; Boca Raton, FL: 2005. Parenteral controlled delivery and pharmacokinetics of therapeutic peptides and proteins; pp. 177–227. [Google Scholar]
- 6.a) Hamidi M, Azadi A, Rafiei P. Drug Deliv. 2006;13:399–409. doi: 10.1080/10717540600814402. [DOI] [PubMed] [Google Scholar]; b) Bansal R, Post E, Proost JH, de Jager-Krikken A, Poelstra K, Prakash J. J Controlled Release. 2011;154:233–240. doi: 10.1016/j.jconrel.2011.05.027. [DOI] [PubMed] [Google Scholar]; c) Duncan R. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]; d) Kim TH, Jiang HH, Lim SM, Youn YS, Choi KY, Lee S, Chen X, Byun Y, Lee KC. Bioconjugate Chem. 2012;23:2214–2220. doi: 10.1021/bc300265n. [DOI] [PubMed] [Google Scholar]; e) Liu M, Johansen P, Zabel F, Leroux JC, Gauthier MA. Nat Commun. 2014;5:5526–5533. doi: 10.1038/ncomms6526. [DOI] [PubMed] [Google Scholar]; f) Keefe AJ, Jiang SY. Nat Chem. 2012;4:59–63. doi: 10.1038/nchem.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Broyer RM, Grover GN, Maynard HD. Chem Commun. 2011;47:2212–2226. doi: 10.1039/c0cc04062b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gaberc-Porekar V, Zore I, Podobnik B, Menart V. Curr Opin Drug Discov Devel. 2008;11:242–250. [PubMed] [Google Scholar]; c) Le Droumaguet B, Nicolas J. Polym Chem. 2010;1:563–598. [Google Scholar]; d) Sumerlin BS. ACS Macro Lett. 2012;1:141–145. doi: 10.1021/mz200176g. [DOI] [PubMed] [Google Scholar]; e) Gauthir MA, Klok H. Chem Commun. 2008:2591–2611. doi: 10.1039/b719689j. [DOI] [PubMed] [Google Scholar]; f) Schulz JD, Patt M, Basler S, Kries H, Hilvert D, Gauthier MA, Leroux Jean-Christophe. Adv Mater. 2016;28:1455–1460. doi: 10.1002/adma.201504797. [DOI] [PubMed] [Google Scholar]
- 8.a) Roberts MJ, Bentley MD, Harris JM. Adv Drug Deliv Rev. 2002;54:459–476. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]; b) Qi Y, Chilkoti A. Polym Chem. 2014;5:266–276. [Google Scholar]
- 9.Lewis A, Tang Y, Brocchini S, Choi J-W, Godwin A. Bioconjugate Chem. 2008;19:2144–2155. doi: 10.1021/bc800242t. [DOI] [PubMed] [Google Scholar]
- 10.Johnson JA, Lu YY, Van Deventer JA, Tirrell DA. Curr Opin Chem Biol. 2010;14:774–780. doi: 10.1016/j.cbpa.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Popp MW, Antos JM, Ploegh HL. Curr Protoc Protein Sci. 2009;56:15.13.11–15.13.19. doi: 10.1002/0471140864.ps1503s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Hepatology. 2006;43:173–181. doi: 10.1002/hep.21006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boekhorst J, de Been MW, Kleerebezem M, Siezen RJ. J Bacteriol. 2005;187:4928–4934. doi: 10.1128/JB.187.14.4928-4934.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer DE, Chilkoti A. Nat Biotechnol. 1999;14:1112–1115. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 15.Mao H, Hart SA, Schink A, Pollok BA. J Am Chem Soc. 2004;126:2670–2671. doi: 10.1021/ja039915e. [DOI] [PubMed] [Google Scholar]
- 16.a) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem, Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2002;114:2708–2711. [Google Scholar]; b) Debets MF, van Berkel SS, Schoffelen S, Rutjes FPJT, van Hest JCM, van Delft FL. Chem Commun. 2010;46:97–99. doi: 10.1039/b917797c. [DOI] [PubMed] [Google Scholar]; c) Gaetke LM, Chow CK. Toxicology. 2003;189:147–163. doi: 10.1016/s0300-483x(03)00159-8. [DOI] [PubMed] [Google Scholar]
- 17.Youn YS, Na DH, Lee KC. J Control Release. 2007;117:371–379. doi: 10.1016/j.jconrel.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 18.a) Bond A. Proc (Bayl Univ Med Cent) 2006;19:281–284. doi: 10.1080/08998280.2006.11928181. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Drucker DJ. Diabetes. 1998;47:159–169. doi: 10.2337/diab.47.2.159. [DOI] [PubMed] [Google Scholar]; c) Parkes DG, Mace KF, Trautmann ME. Expert Opinion Drug Deliv. 2012;8:219–244. doi: 10.1517/17460441.2013.741580. [DOI] [PubMed] [Google Scholar]; d) Kothare PA, Linnebjerg H, Isaka Y, Uenaka K, Yamamura A, Yeo KP, Pena A, Teng CH, Mace K, Fineman M, Shigeta H, Sakata Y, Irie S. J Clin Pharmacol. 2008;48:1389–1399. doi: 10.1177/0091270008323750. [DOI] [PubMed] [Google Scholar]
- 19.Goke R, Fehmann H-C, Linn T, Schmidt H, Krause M, Eng J, Goke B. J Biol Chem. 1993;268:19650–19655. [PubMed] [Google Scholar]
- 20.Amiram M, Luginbuhl KM, Li XH, Feinglos MN, Chilkoti A. Proc Natl Acad Sci U S A. 2013;110:2792–2797. doi: 10.1073/pnas.1214518110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winzell MS, Ahren B. Diabetes. 2004;53:S215–S219. doi: 10.2337/diabetes.53.suppl_3.s215. [DOI] [PubMed] [Google Scholar]
- 22.Mack CM, Moore CX, Jodka CM, Bhavsar S, Wilson JK, Hoyt JA, Roan JL, Vu JL, Laugero KD, Parkes DG, Young AA. Int J Obes. 2006;30:1332–1340. doi: 10.1038/sj.ijo.0803284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.