

Significance
Some lipids such as sphingosine and diacylglycerol are potent signaling effectors. However, comprehensive investigations of their bioactive actions are often hampered by a lack of tools that can be used in living cells. Here, we present chemically modified lipids that allow investigation of acute lipid signaling, lipid metabolism, lipid−protein interactions, and lipid localization by using a single probe for each target lipid. Equipped with a caging group, the lipid probe is biologically inactive, until activated by a flash of light. A second photoreaction cross-links the probe to protein interactors that may subsequently be analyzed by mass spectrometry or fluorescence/electron microscopy. We envision that this versatile design will be central to unraveling complex lipid signaling networks.
Keywords: lipid−protein interaction, sphingosine, diacylglycerol, caged lipids, Niemann−Pick disease type C
Abstract
Lipid-mediated signaling events regulate many cellular processes. Investigations of the complex underlying mechanisms are difficult because several different methods need to be used under varying conditions. Here we introduce multifunctional lipid derivatives to study lipid metabolism, lipid−protein interactions, and intracellular lipid localization with a single tool per target lipid. The probes are equipped with two photoreactive groups to allow photoliberation (uncaging) and photo–cross-linking in a sequential manner, as well as a click-handle for subsequent functionalization. We demonstrate the versatility of the design for the signaling lipids sphingosine and diacylglycerol; uncaging of the probe for these two species triggered calcium signaling and intracellular protein translocation events, respectively. We performed proteomic screens to map the lipid-interacting proteome for both lipids. Finally, we visualized a sphingosine transport deficiency in patient-derived Niemann−Pick disease type C fibroblasts by fluorescence as well as correlative light and electron microscopy, pointing toward the diagnostic potential of such tools. We envision that this type of probe will become important for analyzing and ultimately understanding lipid signaling events in a comprehensive manner.
The roles of lipids in cells go far beyond providing the structural backbone of cellular membranes. Certain lipid species are powerful signaling molecules. Examples include the roles of sphingosine (Sph) and the diacylglycerol (DAG) variant, stearoyl-arachidonylglycerol (SAG) in intracellular calcium signaling (1, 2). The study of such signaling lipids is often complicated by the fact that they are under tight metabolic control and that they occur only in very low concentrations. Overexpression of metabolic enzymes for manipulation of signaling lipid levels is a slow process compared with the rapid turnover of those lipids and may therefore produce not only the target lipid but also multiple downstream metabolites. Chemical dimerizer and optogenetic approaches are options to manipulate lipid contents more rapidly, but they depend on cytosolic lipid-metabolizing enzymes. In the past, many applications therefore focused on phosphoinositides (3, 4). A more general way to rapidly increase lipid concentration is the use of caged lipids. These are equipped with a photocleavable protecting group (caging group), which blocks biological activity and renders them resistant to metabolic turnover before the active lipid is released using a flash of light (2, 5–7). The sudden increase in target lipid concentration facilitates analysis of downstream lipid signaling events as well as lipid metabolism within living cells in pulse−chase experiments. To correctly interpret such signaling events, underlying processes such as lipid−protein interactions, intracellular lipid localization, and kinetics of lipid metabolism need to be considered. To date, lipid metabolism is typically monitored using isotope-labeled or alkyne-modified lipids (8–10). Fluorescent lipids, lipid-binding antibodies, or lipid biosensors are mainly used to study lipid localization (11, 12). Most assays for studying lipid−protein interactions rely on reconstituted membranes/liposomes and are therefore largely restricted to soluble proteins (13–16). The plethora of methods used to investigate these different processes makes it difficult to compare or validate their respective results. A promising approach to integrate the study of lipid metabolism, lipid localization, and lipid−protein interactions has emerged in recent years; bifunctional lipids feature a small diazirine group to allow photo–cross-linking with interacting proteins in the intact cellular environment and a terminal alkyne for subsequent functionalization (17). Biotinylation of cross-linked lipid−protein conjugates enables their enrichment and identification of lipid-interacting proteins. To date, bifunctional lipids are one of the few methods to screen for lipid−protein interactions in living cells (18–21). Alternatively, bifunctional lipids can be used to visualize lipid localization by click reaction with a fluorophore (1, 18, 20). The application of the bifunctional lipid principle to signaling lipids, however, is handicapped by their tight metabolic control. Any precursor is rapidly incorporated into downstream lipids, complicating the interpretation of resulting data. The ability to liberate a single, well-defined signaling lipid species within cells and to immediately capture its interacting partners, investigate downstream signaling, and study its subcellular localization would enable much-needed insight into the regulation of lipid-dependent signaling. Here, we present “trifunctional” lipids as tools, combining the advantages of caged and bifunctional lipids in a single molecule to allow for a wide range of studies in living cells with tight temporal control. Applied to Sph and DAG, we show that trifunctional lipids enable (i) acute alteration of signaling lipid concentration, (ii) measurement of lipid metabolism on a population-wide as well as on a single-cell level, (iii) screening for lipid−protein interactions, and (iv) direct visualization of lipid localization by light and correlative light and electron microscopy (CLEM) in comparable experimental settings.
Results
Synthesis of Trifunctional Sphingosine, Trifunctional Diacylglycerol, and a Trifunctional Fatty Acid.
The previously reported (20) bifunctional sphingosine 4 was equipped with a coumarin caging group via carbamate linkage at the amino group giving the trifunction sphingosine (TFS 1) in 96% yield (Fig. 1A). Synthesis of trifunctional diacylglycerol (TFDAG 2) started by para-methoxybenzyl (PMB) protection of the free hydroxyl group of S-isopropylidene glycerol followed by removal of the isopropylidene group to give diol 6. Dimethoxytrityl (DMT) protection of the primary hydroxyl group and attachment of arachidonic acid to the secondary hydroxyl group gave intermediate 7. The PMB-protected diacylglycerol 8 was obtained by mild DMT deprotection using FeCl3 in MeOH/dichloromethane and carbodiimide-mediated coupling of the bifunctional fatty acid 9. The PMB group was removed by brief exposure to acidic conditions at 0 °C, effectively preventing acyl migration as the only possible alternative regioisomer was not detected by NMR. Lastly, N,N-diethylaminocoumarin was linked to the primary hydroxyl of 10, giving the TFDAG 2 (Fig. 1B). The trifunctional fatty acid (TFFA 3) was obtained by standard carbodiimide-mediated attachment of the coumarin group (Fig. 1C).
Fig. 1.

Synthesis of trifunctional lipids 1 to 3 featuring a photocage, a cross-linkable diaziridine group, and an alkyne. (A) Synthesis of trifunctional sphingosine (TFS, 1). (B) Synthesis of trifunctional diacylglycerol (TFDAG, 2). (C) Synthesis trifunctional fatty acid (TFFA, 3). (D) Aliphatic section of the 1H NMR spectrum of the TFDAG. Samples were irradiated for 2 min with >400-nm light and for 2 min with >345-nm light. The latter reaction produces a ketone. A spectrum of bifunctional DAG (BFDAG, 10, bottom) is shown for comparison.
In Vitro Validation of Sequential Photoreactions.
We established that the photoreactions used for uncaging and cross-linking are indeed orthogonal and may be carried out in a sequential manner. To this end, a 1 mM solution of TFDAG 2 in MeOH-d4 was irradiated using a UV mercury arc source equipped with 400-nm and 355-nm high-pass filters. The uncaging reaction was performed at >400 nm, whereas photo–cross-linking the diazirine group required 355-nm light. 1H NMR spectra were acquired after each irradiation step and compared with the spectrum of the corresponding pure compounds 2 and 10 (Fig. 1D). We found that uncaging at >400 nm quantitatively removed the coumarin group, as signals stemming from the protons in the glycerol backbone (highlighted in blue and red) reverted to the pattern exhibited by bifunctional DAG (BFDAG) upon illumination. The diazirine moiety (green signals) is unaffected by uncaging and is only activated by UV light of >355 nm. Crucially, the alkyne group remained intact through all illumination steps (cyan signals in Fig. 1D), enabling subsequent functionalization.
Uncaging of Trifunctional Lipids in Living Cells.
After establishing photochemical suitability, we applied trifunctional lipids to living cells to investigate their biocompatibility and signaling functionality. Intracellular Sph was reported to induce cytosolic calcium increase by release from acidic stores (1), whereas elevated DAG levels trigger rapid translocation of C1-domain-containing proteins to the plasma membrane (2). As expected, local uncaging of TFS led to Ca2+ transients for 40 to 80 s as measured by the cytosolic calcium dye Fluo-4. The kinetics of calcium release were comparable to previously demonstrated uncaging of native Sph (1). Importantly, the negative control compound dihydrosphingosine (dhSph) was unable to elicit calcium release under these conditions (Fig. 2 A and B). The effects of DAG elevation were monitored by the fluorescent biosensor C1-GFP. Adding the bifunctional DAG probe 10 to the medium of HeLa cells expressing C1-GFP led to translocation of the protein to the plasma membrane over the course of 2 min (Fig. 2C, Upper). Simple addition of TFDAG, on the other hand, failed to induce C1-GFP translocation, demonstrating its biological inertness in the caged form. Only 405-nm uncaging of TFDAG through the microscope objective induced translocation (Fig. 2C, Lower). It is important to note that illumination through the microscope objective only uncages a small fraction of the probe compared with biochemical bulk experiments that make use of strong UV lamps. The simultaneous uncaging of all probe molecules in such experiments can be used to investigate probe metabolism by TLC (Fig. S1). Here, cells labeled with 3 µM TFS or 50 µM TFDAG were subjected to pulse experiments; the resulting metabolites were visualized by click reaction (8) and separated on TLC. Unfortunately, the TFDAG probe partially fragmented during lipid extraction and click reaction thus preventing analysis by TLC (Fig. S1 C and D). TFS, on the other hand, was stable in cells (Fig. S1A) and was readily incorporated into sphingolipids as well as phospholipids upon uncaging (Fig. S1B), in accordance with previous studies using bifunctional sphingosine (20). Together with the results obtained by live-cell microscopy, this confirms that the diazirine and alkyne moieties do not impact on the biological activity of Sph and DAG, respectively, and that acute signaling events can be triggered by uncaging of trifunctional lipids.
Fig. 2.

Trifunctional lipids for the study of cellular signaling. (A) Time-lapse confocal microscopy images of HeLa cells labeled with Fluo-4 (a cytosolic calcium indicator) and 2 µM TFS 1. Uncaging was performed by irradiation of a circular area within the cells (indicated by the white circle) for 3 s at t = 10 s. (B) Quantification of mean Fluo-4 fluorescence of cells loaded with 2 µM caged Sph, 2 µM caged dihydro-Sph, or 2 µM TFS. Traces represent mean values, with the SEM plotted as error bars. (C) Time-lapse montage of HeLa cells expressing C1-GFP and treated with 100 µM bifunctional SAG (BFDAG) 10 at t = 0:30 min (Upper) or treated with 100 µM TFDAG 2 at t = 0:30 min and uncaged by scanning the entire field of view with a 405-nm laser once at t = 4:00 min (Lower). (D) Response rates for C1-GFP translocation upon SAG and TFDAG uncaging. (E) Mean translocation traces for SAG and TFDAG uncaging with SEM plotted as error bars. (F) Individual traces after a quality control step with a representative trace and the corresponding biexponential fit highlighted. (G) Single-cell half-life times for DAG turnover for SAG and TFDAG.
Fig. S1.

TFS and TFDAG metabolism in cells by TLC. (A and C) HeLa cells were labeled with (A) 3 µM TFS or (C) 50 μM TFDAG for the indicated times without UV irradiation. Cellular lipids were extracted, labeled with 3-azido-7-hydroxycoumarin by click reaction, and separated on a TLC plate. (B and D) HeLa cells were labeled with (B) 3 μM TFS or (D) 50 μM TFDAG for 15 min, washed, and irradiated with >400-nm UV light for 2.5 min, and incubated for the indicated times. Cellular lipids were extracted, labeled with 3-azido-7-hydroxycoumarin by click reaction, and separated on a TLC plate.
Quantification of DAG Turnover at the Plasma Membrane.
Being able to trigger C1-GFP translocation to the plasma membrane in a controlled fashion allowed us quantify lipid turnover on a population-wide as well as on a single-cell level, thereby accounting for the inherent heterogeneity of cell populations. Compared with standard biochemical experiments, such live-cell uncaging experiments also offer superior temporal resolution; however, they do not measure pure metabolism, as the signal acquired through biosensors accounts for both lipid transport and metabolic transformations. Here, we performed a series of uncaging experiments using TFDAG and compared it to caged SAG (2) (Movies S1 and S2). Changes in plasma membrane DAG levels were quantified using the ratio between plasma membrane-bound and cytosolic fractions of C1-GFP as calculated using a recently developed algorithm (22) (SI Materials and Methods for details). Most cells exhibited significant C1-GFP translocation to the plasma membrane, with similar response rates for TFDAG and SAG, respectively (TFDAG 78%, SAG 71%; Fig. 2D). Kinetic traces were measured for all cells (Fig. S2), and responding cells were included in the kinetic analyses. Mean translocation traces (Fig. 2E) showed an initial rapid increase of the C1-GFP plasma membrane/cytosol ratio, reflective of the rapid increase of DAG concentration caused by uncaging. After reaching a maximum ∼100 s after illumination, the observed signal declined with comparable slopes for both lipids, indicating ongoing DAG turnover. To characterize their kinetics more quantitatively, we analyzed and fitted the responses on a single-cell level. A simple biexponential model featuring terms for C1-GFP recruitment to the plasma membrane and DAG turnover was fitted to the individual traces (Fig. S3 and SI Materials and Methods for details). After quality control (SE estimate below 0.06 and a positive turnover rate; see Supporting Information), a set of 31 and 43 traces were obtained for SAG and TFDAG, respectively (Fig. 2F). The determined half-life times for DAG turnover (Fig. 2G) revealed striking differences on a cell-to-cell level, as individual cells exhibited DAG turnover rates varying over an order of magnitude. These data suggest that heterogeneity of signaling lipid turnover might be an underrated aspect in lipid signaling events.
Fig. S2.

Individual traces of C1-GFP translocation. The fluorescence ratio of C1-GFP at the plasma membrane and the cytosol was recorded for HeLa cells treated with 100 μM caged SAG (Left) and 100 μM TFDAG (Right). Uncaging was carried out by scanning the entire field of view as indicated by the arrow. Cells were then classified into responder and nonresponder (gray traces).
Fig. S3.

Individual, fitted C1-GFP translocation traces for cells that matched the quality control test. Each cell was fitted with a biexponential model (see SI Materials and Methods). The resulting half-times of DAG metabolism are displayed in seconds next to the traces.
Chemoproteomic Profiling of Lipid−Protein Complexes.
A further application of trifunctional lipids is the systematic mapping of lipid−protein interactions by mass spectrometry. Owing to the two orthogonal photoreactions, which enable a “release and catch” approach, trifunctional lipids allow profiling the protein interactome of single lipid species. Due to the reduced complexity of the bait compared with bifunctional lipids, this screening technology is more sensitive to low-abundance lipids and should pick up scarce lipid−protein interactions. HeLa cells were labeled with 6 µM TFS and 100 µM TFDAG or TFFA, which resulted in comparable loading of the cells, owing to their different uptake kinetics (Fig. S4). After uncaging and photoaffinity labeling, the protein−lipid conjugates were reacted with biotin azide using copper-catalyzed click chemistry and subsequently enriched via neutravidin beads. The preparation of peptides for proteomic analysis was performed according to a recent protocol optimized for ultrasensitive analysis of complex biological samples (23). Using this method, a total of 3,263 proteins were identified. For further analysis, only proteins that were identified in both screens using either TFS or TFDAG were considered. The peptide spectral matches of these high-confidence proteins are displayed as a heat map in Fig. 3A. As noted in previous studies using bifunctional lipids (20, 21), a high overlap in the interactome of different lipids was observed; this may be attributed to unspecific cross-linking of highly abundant proteins with either probe or background binding to the beads during the pull-down procedure, highlighting the need for control lipids. By grouping the resulting proteins according to the ratio of their peptide spectral matches over control lipids, we were able to identify two subsets of proteins, which interacted preferentially with either TFS (Fig. 3A, top left group) or TFDAG (Fig. 3A, bottom right group); a sample 55 hits are displayed for TFS and TFDAG, respectively. The full list of proteins arranged as in Fig. 3A as well as lists for Sph- and DAG-interacting proteins can be found in Dataset S1. Reassuringly, we found proteins previously reported to interact with Sph [ceramide synthase 2, which uses Sph as a substrate (24), and cathepsin B, which is a mediator of Sph-induced apoptosis (25)] or with DAG [phosphatidylinositol 4,5-bisphosphate phosphodiesterase delta 3, a DAG producing enzyme (26)] among our top hits. We then compared the list of putative Sph-interacting proteins from this screen with results from a previous screen performed with bifunctional Sph (20) (Fig. S5A). Fourteen proteins were already detected using bifunctional Sph, further strengthening them as bona fide Sph binders. However, the majority of these proteins were not previously identified. Here, we benefited from the major advantage of trifunctional lipids to circumvent lipid metabolism and identify scarcer or very short-lived lipid−protein interactions. In the absence of previous proteomic screens with DAG as bait, we compared proteins identified using TFDAG to hits from previous screens performed with bifunctional endocannabinoid probes (21). These probes contain arachidonic acid just as TFDAG does. Reassuringly, only 17 of 130 putative DAG-interacting proteins were previously identified using the endocannabinoid probes (Fig. S5B), which further confirms that proteins identified with TFDAG are specifically interacting with DAG.
Fig. S4.

Subcellular distribution of trifunctional lipids. Shown are confocal images of HeLa cells incubated with the indicated concentrations of trifunctional lipids.
Fig. 3.

Mass spectrometric identification of Sph- and SAG-binding proteins. (A) Heat map of high-confidence proteins identified in both screens. Peptide spectral matches are color-coded according to the legend on the top. Proteins are arranged such that preferential TFS interactors are displayed on the top (the gene symbols for the first 55 proteins are displayed on the left) and TFDAG interactor are grouped near the bottom (55 proteins are displayed on the right). (B) Putative Sph- and DAG-binding were analyzed according to their cellular compartment (CC) GO terms. (C) Confocal microscopy images of HeLa cells labeled with TFS (Left) and TFDAG (Right) under the conditions used in the proteomic experiments. Cells were fixed with methanol, non–cross-linked lipids were washed away, and the remaining cross-linked lipids were clicked to Alexa488-azide.
Fig. S5.

(A) Overlap of TFS interactors with previous screens. Proteins identified as unique interactors using TFS were compared with proteins identified in a previous screen using pacSph as bait (20).The spectral counts of proteins that were either not present in the previous screen (not previously identified, left column), identified with pacSph (pacSph, top right column), pacFA only (pacFA, middle right column), or identified with both pacFA and pacSph (pacFA/pacSph, bottom right column) are displayed using the same color code as in Fig. 3. (B) Overlap of TFDAG interactors with previous screens. Proteins identified as unique interactors using TFDAG were compared with proteins identified in a previous screen using arachidonic acid-containing lipids in HEK293-T cells (21). The spectral counts of proteins that were either not present in the previous screen (not previously identified, left columns), identified with AEA-DA (AEA-DA, center column), identified with A-DA (A-DA, middle right column), or identified with both AEA-DA and A-DA (AEA.DA/A-DA, bottom left column) are displayed using the same color code as in Fig. 3.
Subcellular Localization of Lipids and Lipid-Interacting Proteins.
To further analyze these new, putative Sph and DAG-interacting proteins, we accessed their gene ontology (GO) terms and compared the annotated subcellular localizations of TFS and TFDAG hits (Fig. 3B). A larger percentage of TFS interactors were annotated to the secretory pathway [endoplasmic reticulum (ER), Golgi, vesicles, endosomes, lysosomes, and exosomes] compared with TFDAG hits, which are mainly annotated as cytosolic proteins. This result hints toward a different subcellular localization of the trifunctional lipids at the time of cross-linking. Before uncaging, trifunctional lipids are indiscriminately localized to internal membranes and the cytoplasm as visualized by fluorescence of the coumarin caging group (Fig. S6A, Upper). To investigate their localization at the time of cross-linking, HeLa cells were treated in the same way as for the proteomic screens, but followed by fixing and staining of the lipid−protein complexes with Alexa488-azide (Fig. 3C and Fig. S6A, Lower). We observed that each lipid now localized to distinct cellular compartments. Strikingly, the staining of TFDAG resulted in 52 to 59% weaker fluorescence compared with TFS and TFFA, respectively (Fig. S6B). Cytosolic and nuclear proteins are known to be extracted by methanol fixation (27), which is necessary for this protocol, as it removes all non–cross-linked lipids. This may explain the reduced fluorescence intensity in these regions and confirms the GO-term analysis of DAG-interacting proteins, which listed surprisingly many cytosolic proteins. We speculate that the high proportion of cytosolic proteins likely reflects a highly efficient DAG transporting machinery, which requires extraction of DAG from membranes, thereby potentially exposing the cross-linkable group. For example, extended synaptotagmins (E-Syts) were recently shown to extract DAG from the plasma membrane (28). Accordingly, we identified both E-Syt1 and E-Syt2 in screens performed with TFDAG. However, E-Syt1 was also found using TFS and TFFA, and E-Syt2 was identified with TFS but not with TFFA, hinting at a broader lipid specificity of these proteins. TFS, on the other hand, was found to be predominantly localized to the perinuclear ER, Golgi, and, mainly, endosomes/lysosomes (Fig. S6 C, E, and F for colocalization). We quantified the amount of Sph labeling in LAMP1-stained compartments to 40% of total fluorescence, whereas the more uniform labeling of TFDAG only localized 22% of total fluorescence to areas also marked by LAMP1 (Fig. S6D).
Fig. S6.

Subcellular localization of trifunctional lipids. (A) Confocal images of HeLa cells treated with (Left) 6 μM TFS for 5 min or (Center) 100 μM TFDAG or (Right) 100 μM TFFA for 15 min. Lipid localization before uncaging as visualized by coumarin fluorescence (Upper). Uncaged, cross-linked, and fixed lipids are visualized using Alexa488-azide (Lower). (B) Quantification of total fluorescence intensity of Alexa488-labeled TFS, TFDAG, and TFFA. (C) Confocal images of HeLa cells treated with 6 μM TFS (Upper) and 100 µM TFDAG (Lower). Alexa488-azide-conjugated lipids are shown in gray (left-hand image) and green (merged image). Late endosomes/lysosomes were stained using LAMP1 antibody and are displayed in gray (middle image) and red (merged image). (Scale bars, 20 µm.) (D) Quantification of fluorescence intensity in lysosomes. LAMP1 immunofluorescence was used to identify regions that mark lysosomes. The combined integrated density of these regions in the lipid channel was divided by the integrated density of the whole cell to obtain the ratio displayed on the y axis. (E and F) Confocal images of HeLa cells treated with 6 μM TFS (Upper) and 100 μM TFDAG (Lower). Alexa488-azide-conjugated lipids are shown in gray (left-hand image) and green (merged image). Golgi apparatus (E) and ER (F) were stained using GM130 and p72 antibody, respectively, and are displayed in gray (middle image) and red (merged image). (Scale bars, 20 μm.)
Sphingosine Localization in Niemann−Pick Disease Type C.
To highlight the potential of trifunctional lipids for the visualization of subcellular lipid localization, we chose as an example the study of Niemann−Pick disease type C (NPC) cells. NPC is a rare lysosomal storage disease caused mainly by mutation of the gene encoding for the NPC1 protein (29). In diseased cells, Sph is known to accumulate alongside other lipids such as sphingomyelin, cholesterol, and higher glycosphingolipids (30, 31). The function of NPC1 is not fully understood, but NPC cells exhibit a trafficking defect at the late endosomal/lysosomal stage, as observed for cholesterol by using Filipin staining (32). Accumulation of Sph and lactosylceramide was previously visualized using fluorescent lipid analogs (33, 34). Here, we used TFS to visualize Sph localization and trafficking. To create a cellular model of NPC, HeLa cells were either treated with the cationic amphiphilic drug U18666A, which acts as an NPC1 inhibitor (35), or with siRNA targeted to NPC1. Both treatments produced an NPC phenotype, as confirmed by Filipin staining (Fig. S7A). Using TFS on these cell models, a noticeable increase of fluorescence was observed in late endosomal/lysosomal vesicles (Fig. 4A; for colocalization, Fig. S7B), indicative of Sph storage.
Fig. S7.

(A) Filipin staining of HeLa cell models of NPC. Confocal images of HeLa cells treated with U18666A (2 μg/mL for 24 h) or siRNA against NPC1. Cells were fixed with PFA and stained using Filipin complex (from Staphylococcus aureus, 50 μg/mL). (B) TFS colocalization with endosomes/lysosomes in NPC cell models. Confocal images of HeLa cells treated with U18666A (2 μg/mL for 24 h) or siRNA against NPC1; 6 μM TFS was added for 5 min, and the cells were washed and subsequently uncaged and cross-linked. The time between uncaging and cross-linking was varied from 0 min to 30 min. Cells were fixed and washed, and the cross-linked lipids were functionalized with Alexa488. Late endosomes/lysosomes are visualized via LAMP1 antibody in gray (middle image) and red (merged image), and Pearson’s correlation coefficient is shown in the top left corner of the merged image. (Scale bars, 20 μm.)
Fig. 4.

Sph localization and transport in NPC models. (A) Confocal images of Sph stained with Alexa488-azide in HeLa cells in control conditions or upon induction of the NPC cellular phenotype by U18666A (2 µg/mL for 24 h) or by siRNA-mediated knockdown of NPC1. Pulse−chase experiments were performed by varying the time between uncaging and cross-linking from 0 min to 30 min. (B) Confocal images of Sph stained with Alexa488-azide in human fibroblasts derived from a healthy donor (control) or from NPC patients with varying genotypes and severity scores (NPC22, NPC17, NPC25). Pulse−chase experiments were performed as in A. (C) Quantification of human fibroblasts by automated image analysis. Skewness values for each cell were extracted and plotted according to cell line and time after uncaging. (D) Workflow for CLEM of NPC fibroblasts. EM, electron microscopy; FM, fluorescence microscopy. (E) CLEM images of NPC fibroblast labeled with TFS according to the procedure in D.
Sphingosine Transport in NPC.
We then investigated whether the transport of Sph out of the acidic compartment was affected as well. Here, the main advantages of trifunctional lipids became obvious: By using two photoreactions (uncaging/cross-linking), it was possible to bypass the uptake and possible retention of Sph in the endocytic pathway and to set up pulse−chase experiments. The accumulation of bifunctional Sph 4 in acidic compartments after uncaging set a precise starting point for quantifying transport of Sph and its metabolites out of the acidic compartment. In control cells, Sph was rapidly (<10 min) cleared from the vesicles, as exemplified by a drop in Pearson’s correlation coefficient with the late endosomal/lysosomal marker LAMP1 (Fig. S7B). Both NPC models (U18666A and siRNA), on the other hand, retained most Sph in the late endosomal/lysosomal vesicles even after 30-min postuncaging (Fig. 4A), as expected for cells with lipid transport defect. Next, we investigated Sph transport in skin fibroblasts derived from three NPC patients with varying disease severity. Cells derived from the patient with the mildest phenotype were able to export most of the lysosomal Sph within 10 min, whereas the more severe patients still showed marked lysosomal Sph accumulation after 30 min (Fig. 4B). We quantified this retention by investigating the skewness of the pixel distribution in each cell through automated image processing (Fig. 4C). High skewness values represent vesicular staining, whereas lower values are indicative of an even distribution throughout the cells as achieved by ER or internal membrane staining.
Ultrastructural Localization of Sphingosine.
To further examine this accumulation on an ultrastructural level, we subjected NPC patient fibroblasts to high-precision CLEM. Briefly, cross-linked lipids were functionalized with Alexa594-azide followed by high-pressure freezing (HPF) and thin sectioning. Fluorescent and electron dense fiducial marker beads (36) were used to correlate fluorescence images and electron tomograms (Fig. 4D). We found that Sph localized to intraluminal vesicles of late endosomes/multivesicular bodies, which strengthens the hypothesis of a trafficking block at the late endosomal stage.
Discussion
We have developed a photochemical probe type featuring two sequential photoreactions, which allows different aspects of lipid biology to be studied while using the same molecule. Importantly, trifunctional lipids constitute a unique way to investigate single lipid species in a live-cell setting, which is especially important when looking at active signaling lipid species. We successfully used these probes to quantify cellular signaling after uncaging by live-cell imaging of downstream effects including C1-domain translocation and changes of calcium levels. Trifunctional lipids have furthermore proven useful in the unbiased identification of novel, putative Sph and DAG-interacting proteins by proteomic analysis. Some of the identified, high-confidence interactors open up exciting avenues for further study: For example, we found beta-hexosaminidase A and B (HexA and HexB) to interact with Sph. It is interesting to speculate that Sph might act as regulator of lipid-catabolizing enzymes such as HexA, the activity of which is known to depend on the lipid composition of the substrate membranes (37). Such a potential regulation of HexA by Sph might thereby further contribute to lipid storage diseases such as NPC, which accumulate glycosphingolipids. A further application of trifunctional lipids lies in the visualization of their subcellular localization by fluorescence microscopy and CLEM. Reassuringly, the annotated cellular compartments of the identified proteins corresponded well with their observed localization. Taking this further, we set up an assay to investigate the transport of Sph in NPC disease and observed a trafficking block at the late endosomal stage. For the future, we imagine that this trafficking assay could serve as a valuable diagnostic tool complementing the established Filipin staining analysis, as it is independent of cholesterol accumulation but assays the capability of the cells to move Sph out of late endosomes/lysosomes. In the three NPC cell lines investigated, the observed Sph transport correlated well with patients’ severity scores. However, further tests on a larger number of patient samples will be necessary to confirm that Sph retention in the late endosomes/lysosomes is a marker for disease severity.
In conclusion, we demonstrated the practicality of trifunctional lipids for studying different aspects of lipid biology in the context of living cells, and we envision that this design will lead to the generation of more trifunctional lipids and studies of the relevant signaling networks.
Materials and Methods
Chemical Synthesis and Cell Culture.
Detailed protocols for the synthesis of trifunctional lipids and their characterization data (Dataset S2) as well as information about cell lines and their culture conditions can be found in SI Materials and Methods.
Confocal Uncaging Experiments.
HeLa cells were transfected with C1-GFP 24 h before experiments. For calcium imaging, cells were labeled with 100 μL of 5 μM Fluo-4 AM for 30 min before the experiment. Cells were treated with a 100 μM solution of TFDAG/caged SAG or a 2 μM solution of TFS in imaging buffer. Confocal time lapses were acquired at 37 °C, and uncaging was performed by either scanning the entire field of view with a 405-nm laser or by spot-uncaging for 3 s at 2 μs per pixel. For more detailed protocols, please refer to SI Materials and Methods.
Lipid Analysis by TLC.
Briefly, cells were labeled with 3 µM TFS or 50 µM TFDAG for the indicated times and subjected to UV-induced uncaging, as indicated. Cellular lipids were extracted, labeled with 3-azido-7-hydroxycoumarin via click reaction, separated on a TLC plate, and visualized via the coumarin fluorescence.
Proteomic Screens.
A detailed procedure can be found in SI Materials and Methods. Briefly, HeLa cells were labeled with trifunctional lipids and UV-irradiated for 2.5 min at >400 nm and for another 2.5 min >345 nm. The lysate was subjected to click reaction with biotin azide, and protein−lipid complexes were enriched using NeutrAvidin. Proteins were digested according to a recently developed protocol (23) and subjected to high-pH fractionation (38). Peptides were separated using the nanoAcquity ultra performance liquid chromatography (UPLC) system coupled directly to a linear trap quadrupole (LTQ) OrbitrapVelos Pro using the Proxeon nanospray source.
Visualization of Lipid−Protein Complexes.
A detailed protocol for visualizing lipid localization in cells by fluorescent microscopy as well as by correlated light and electron microscopy can be found in SI Materials and Methods.
SI Materials and Methods
The chemicals used were purchased from commercial sources (Acros, Sigma-Aldrich, Enzo, Lancaster, or Merck) at the highest available grade and were used without further purification. Solvents for chromatography (HPLC grade) were obtained from VWR, and dry solvents were obtained from Sigma-Aldrich. Deuterated solvents were purchased from Deutero. Alexa488-azide and Alexa694-azide were obtained from Life Technologies (Thermo Fisher Scientific), and 3-azido-7-hydroxycoumarin was obtained from baseclick (#BCFA-047-1). Biotin-azide was purchased from Sigma-Aldrich (#762024), and Neutravidin-agarose was purchased from Life Technologies (#29204). The protease inhibitor mixture tablets were obtained from Roche (#11873580001).
Chemical Synthesis.
Reaction monitoring was performed via thin layer chromatorgraphy (TLC) on plates of silica gel (60 F254; Merck) and visualized under UV light (254 or 366 nm) or using a solution of phosphomolybdic acid in EtOH (10% wt/vol). Preparative column chromatography was carried out using Merck silica gel 60 (grain size 0.063 nm to 0.200 mm) under pressure (<1.5 bar). The 1H NMR and 13C NMR spectroscopy was performed on a 400-MHz Bruker UltraShield spectrometer at 25 °C. Chemical shifts are given in parts per million, referenced to the residual solvent peak. J values are given in Hertz, and splitting patterns are designated using s (single), d (doublet), t (triplet), q (quartet), m (multiplet), and b (broad signal). High-resolution mass spectra were recorded on a Finnigan LCQ quadrupole ion trap at the Organic Chemistry Institute and the Institute of Pharmacy and Molecular Biotechnology of the University of Heidelberg.
Compounds 4, 6, 9, S1, and S3 as well as caged SAG were synthesized according to literature (2, 18, 20, 39). Compound 6 was equipped with a DMT protecting group using a procedure described by Sato et al. (40). Detailed procedures for the synthesis of all other new compounds are given below.
(2S,3R,E)-2-amino-(7′-(diethylamino)-coumarin-4′-yl)-methoxycarbonyl)-13-(3-(pent-4′-yn-1′-yl)-3H-diazirin-3-yl)tridec-4-ene-1,3-diol (1, TFS)
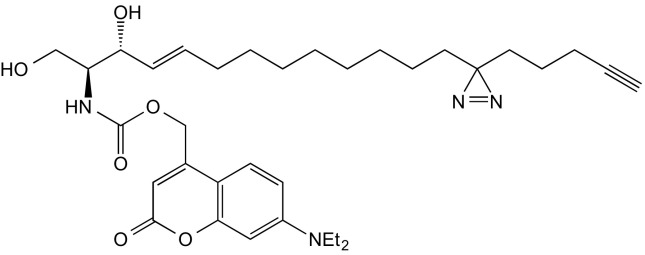
A solution of 7-diethylamino-4-hydroxymethylene-coumarin (48 mg, 194 μmol) in 2 mL of dry THF was cooled to 0 °C. Diisopropylethylamine (DIEA) (0.1 μL, 575 μmol) and phosgene (300 μL, 610 μmol) were added dropwise and stirred in the dark for 2 h at 0 °C. The reaction mixture was extracted with EtOAc/H2O (1:1, 75 mL), the layers were separated, and the organic layer was washed with brine and dried using Na2SO4. The solvent was removed under reduced pressure, and the resulting 7-(diethylamino)-coumarin-4-yl)-methyl chloroformate was immediately used without further purification.
Fifty-two microliters of DIEA (0.3 mmol) was added to a solution of 20 mg (59.7 μmol) photoactivatable and clickable sphingosine (20) in 1.5 mL THF, cooled to 0 °C; [7-(diethylamino)-coumarin-4-yl)-methyl chloroformate (28 mg, 0.09 mmol) in 1 mL of dry THF was added and stirred at room temperature for 1 h. The product was extracted with 30 mL of EtOAc and 30 mL of citric acid (5% wt/vol) and washed twice with 30 mL of citric acid, once with NaHCO3, and once with brine. The organic phase was dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (first column: eluent: cyclohexane/EtOAc 1:1; second column: eluent: DCM/MeOH 14:1), which gave the title compound as a yellow oil (35 mg, 57.5 mmol, 96% yield).
1H NMR (500 MHz, CDCl3) δ = 7.29 (d, J = 9.0, 1H), 6.58 (dd, J = 8.9, 2.2, 1H), 6.50 (d, J = 2.2, 1H), 6.14 (s, 1H), 5.89 (d, J = 6.8, 1H), 5.84 to 5.75 (m, 1H), 5.55 (dd, J = 15.2, 6.1, 1H), 5.23 (s, 2H), 4.39 (s, 1H), 4.02 (dd, J = 11.2, 2.8, 1H), 3.77 (dd, J = 11.5, 3.0, 1H), 3.69 (dd, J = 7.8, 3.3, 1H), 3.66 to 3.59 (m, 1H), 3.41 (q, J = 7.0, 4H), 2.56 (s, 2H), 2.15 (td, J = 6.9, 2.6, 2H), 2.05 (dd, J = 13.9, 7.0, 3H), 1.94 (t, J = 2.6, 1H), 1.48 (dd, J = 9.1, 6.6, 3H), 1.39 to 1.29 (m, 9H), 1.28 to 1.15 (m, 28H), 1.10 to 1.02 (m, 5H), 0.91 to 0.77 (m, 4H).
13C NMR (126 MHz, CDCl3) δ = 162.34, 156.17, 155.63, 150.63, 134.39, 128.68, 124.40, 108.87, 105.92, 97.91, 83.49, 77.28, 77.03, 76.77, 74.72, 68.88, 62.12, 61.88, 55.67, 44.83, 32.85, 32.24, 31.85, 29.71, 29.31, 29.28, 29.17, 29.10, 29.02, 28.46, 23.81, 22.77, 17.97, 14.12, 12.43.
HRMS (m/z) calculated for C34H49N4O6+: 609.36521, found: 609.36508
2-O-Arachidonyl-3-O-(7-(diethylamino)-coumarin-4-yl)-methoxycarbonyl-1-O-(3′(4′′-pentyn-1′′-yl)-H-diazirine-3′-octanoyl)-sn-glycerol (2, TFDAG)
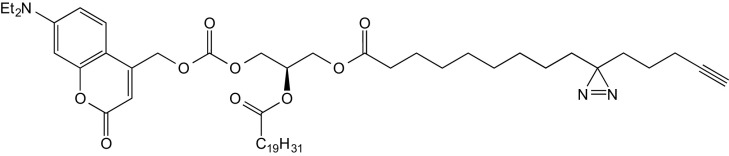
A solution of 10 (55 mg, 90 μmol, 1.0 eq) in a mixture of dry DCM and dry pyridine (4:1, 2 mL) was subsequently treated with DIEA (0.08 mL, 816 μmol, 9.0 eq) and a solution of freshly prepared coumarin chloroformate (2) S1 in DCM (2 mL) at 0 °C under inert conditions. Stirring was continued, and the reaction mixture was allowed to reach room temperature overnight. The reaction mixture was then poured into a mixture of EtOAc (100 mL) and H2O, and the layers were separated. The organic layer was washed with brine (100 mL) and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by repeated flash chromatography (FC) (1. eluent: CyHex/EtOAc 4:1; 2. eluent DCM/acetone 97:3) and the title compound obtained as yellow oil (28 mg, 32 μmol, 36% yield).
1H NMR (400 MHz, CDCl3) δ = 7.28 (d, J = 8.8, 1H), 6.58 (dd, J = 9.0, 2.0, 1H), 6.51 (d, J = 2.0, 1H), 6.15 (s, 1H), 5.45 to 5.24 (m, 11H), 4.42 (dd, J = 11.7, 3.8, 1H), 4.37 to 4.26 (m, 2H), 4.18 (dd, J = 12.0, 5.6, 1H), 3.41 (q, J = 7.1, 4H), 2.86 to 2.76 (m, J = 11.3, 5.5, 6H), 2.39 to 2.28 (m, 4H), 2.19 to 2.00 (m, 6H), 1.95 (t, J = 2.3, 1H), 1.76 to 1.65 (m, 2H), 1.64 to 1.55 (m, 4H), 1.53 to 1.45 (m, 2H), 1.40 to 1.16 (m, 22H), 1.12 to 1.02 (m, J = 7.3, 2H), 0.88 (t, J = 6.7, 3H) ppm.
13C NMR (101 MHz, CDCl3) δ = 173.18, 172.66, 161.64, 156.32, 154.42, 150.72, 148.36, 130.50, 128.99, 128.79, 128.60, 128.28, 128.12, 127.86, 127.54, 124.36, 108.72, 106.77, 105.76, 97.90, 68.89, 68.63, 66.34, 65.00, 61.62, 44.79, 33.96, 33.52, 32.83, 31.82, 31.52, 29.33, 29.19, 29.12, 29.08, 29.02, 27.22, 26.46, 25.62, 24.77, 24.68, 23.80, 22.75, 22.58, 17.96, 14.09, 12.43 ppm. Signals at 29.4 to 29.0 ppm are partially not resolved due to very similar 13C chemical shifts.
HRMS (m/z): [M+H]+ calc. for: C53H76N3O9, 898.55761, found: 898.55777
7-(diethylamino)-coumarin-4-yl)-3-(4′-pentyn-1′-yl)-H-diazirine-3-octanoate (3, TFFA)
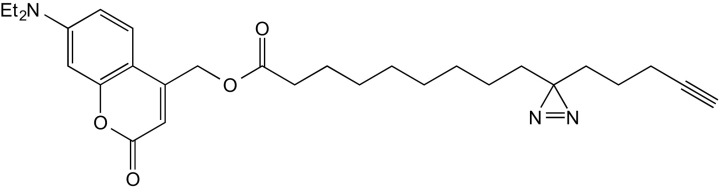
A solution of 9 (100 mg, 348 µmol) in a mixture of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (0.14 g, 731 µmol) and 4-(dimethylamino)pyridine (DMAP) (6.38 mg, 62.2 µmol) in dry DCM was treated with a solution of coumarin alcohol (89.31 mg, 382.8 µmol) in DCM under inert conditions. The reaction mixture was stirred at 0 °C for 20 min and allowed to reach room temperature overnight. The reaction mixture was then poured into a mixture of EtOAc (150 mL) and H2O, and the layers were separated. The organic layer was washed with brine (150 mL) and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by repeated FC (1. eluent: CyHex/EtOAc 9:1; 2. eluent CyHex/EtOAc 3:1). The title compound was obtained as yellow oil (0.17 g, 344.6 μmol, 97% yield).
1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 9.0 Hz, 1H), 6.58 (dd, J = 9.0, 2.6 Hz, 1H), 6.52 (d, J = 2.6 Hz, 1H), 6.13 (s, 1H), 5.22 (s, 2H), 3.41 (q, J = 7.1 Hz, 4H), 2.43 (t, J = 7.5 Hz, 2H), 2.16 (td, J = 6.9, 2.6 Hz, 2H), 1.94 (t, J = 2.6 Hz, 1H), 1.73 to 1.60 (m, J = 7.3 Hz, 2H), 1.52 to 1.45 (m, 2H), 1.42 (s, 4H), 1.39 to 1.16 (m, 1H), 1.12 to 1.02 (m, J = 7.3 Hz, 2H).
13C NMR (100 MHz, CDCl3) δ = 206.87, 172.99, 161.83, 156.24, 150.62, 149.57, 129.00, 128.20, 125.27, 124.39, 108.67, 106.43, 106.07, 97.86, 83.45, 68.88, 61.14, 44.77, 34.08, 32.82, 31.81, 30.90, 29.15, 24.81, 23.77, 22.75, 17.94, 12.42.
HRMS (m/z): [M+H]+ calc. for: C29H39N3NaO4, 516.28328; found: 516.28345.
2-O-Arachidonyl-1-O-dimethoxytrityl-3-O-(4′-methoxybenzyl)-sn-glycerol (7)
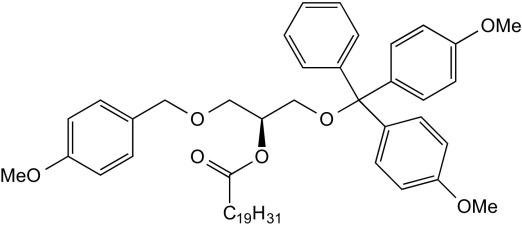
A solution of EDC (2.00 g, 10.4 mmol, 1.78 eq) and DMAP (200 mg, 1.64 mmol, 0.3 eq) in dry DCM (15 mL) was treated with arachidonic acid (2.00 g) and stirred for 10 min under inert conditions. A solution of 1-O-dimethoxytrityl-3-O-(4′-methoxybenzyl)-sn-glycerol (3.00 g, 5.84 mmol, 1.0 eq) in dry DCM (8 mL) was added slowly, and the reaction mixture was stirred at room temperature for an additional 3.5 h. The reaction mixture was transferred onto a mixture of EtOAc (200 mL), water (100 mL), and brine (100 mL). The layers were separated, and the organic layer was washed with brine (100 mL) and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by FC (eluent: CyHex/EtOAc 4:1). The title compound was obtained as colorless oil (3.30 g, 4.12 mmol, 71% yield).
1H NMR (400 MHz, CDCl3) δ = 7.44 to 7.39 (m, 1H), 7.34 to 7.14 (m, 10H), 6.92 to 6.78 (m, 6H), 5.46 to 5.30 (m, 8H), 5.25 (m, 1H), 5.09 to 4.99 (m, 1H), 4.54 to 4.38 (m, 2H), 3.86 to 3.76 (m, 11H), 3.68 to 3.58 (m, 2H), 2.88 to 2.77 (m, 6H), 2.37 (t, J = 6.8, 2H), 2.17 to 2.02 (m, 4H), 1.78 to 1.67 (m, 2H), 1.40 to 1.24 (m, 6H), 0.95 to 0.85 (m, J = 1.7, 3H) ppm. Broadened signals were observed, potentially two conformers in equilibrium. Cleavage of the DMT group in the next step removed this issue.
13C NMR (100 MHz, CDCl3) δ = 173.00, 158.46, 147.34, 144.83, 139.48, 135.98, 130.53, 130.50, 130.05, 129.37, 129.22, 129.15, 129.02, 128.95, 128.89, 128.84, 128.62, 128.59, 128.28, 128.24, 128.20, 128.14, 127.90, 127.87, 127.78, 127.57, 127.09, 126.74, 113.89, 113.74, 113.18, 113.07, 85.91, 73.13, 73.03, 72.75, 71.74, 68.73, 68.52, 62.81, 62.31, 55.19, 33.94, 33.75, 31.53, 29.34, 27.23, 26.93, 26.62, 26.53, 25.63, 24.90, 24.80, 22.59, 14.09 ppm. Signals at 130.5 to 127.5 ppm and 27 to 25 ppm are partially not resolved due to very similar 13C chemical shifts. Signal duplications in the glycerol and aromatic region were observed. Cleavage of the DMT group in the next step removed this issue.
HRMS (m/z): [M+Na]+ calc. for: C52H64NaO7, 823.45443, found: 823.45465.
2-O-Arachidonyl-3-O-(4′-methoxybenzyl)-sn-glycerol (S2)

A freshly prepared solution of FeCl3 (5mM) in MeOH/DCM (3:1, 20 mL) was used to dissolve 7 (950 mg, 1.19 mmol). The reaction mixture was stirred at room temperature for 2 h and subsequently transferred onto a mixture of H2O and EtOAc (1:1, 200 mL). The organic layer was washed with brine and dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by FC (eluent: CyHex/EtOAc 4:1) and the title compound obtained as colorless oil (383 mg, 769 μmol, 66%, repeated twice, average yield given).
1H NMR (400 MHz, CDCl3) δ = 7.24 (d, J = 8.6, 2H), 6.88 (d, J = 8.6, 2H), 5.44 to 5.30 (m, 8H), 5.03 (p, J = 4.8, 1H), 4.48 (q, J = 11.7, 1H), 3.83 to 3.78 (m, 5H), 3.68 to 3.58 (m, 2H), 2.88 to 2.76 (m, 6H), 2.37 (t, J = 7.6, 2H), 2.15 to 2.00 (m, 4H), 1.71 (p, J = 7.5, 2H), 1.40 to 1.23 (m, 6H), 0.89 (t, J = 6.8, 3H) ppm.
13C NMR (101 MHz, CDCl3) δ = 173.44, 159.38, 130.52, 129.68, 129.37, 128.95, 128.88, 128.61, 128.28, 128.15, 127.87, 127.54, 113.88, 73.12, 73.03, 68.71, 62.79, 55.28, 33.74, 31.53, 29.33, 27.23, 26.52, 25.62, 24.79, 22.59, 14.09 ppm.
HRMS [M+Na]+calc. for: C31H46NaO5, 521.3243, found: 521.3237.
2-O-Arachidonyl-1-O-(3′(4′′-pentyn-1′′-yl)-H-diazirine-3′-octanoyl)-3-O-(4′-methoxybenzyl)-sn-glycerol (8)
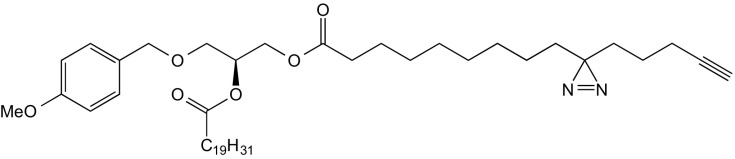
A solution of 9 (250 mg, 1.00 mmol, 1.0 eq), EDC (300 mg, 1.56 mmol, 1.6 eq) and DMAP (50 mg, 409 μmol, 0.4 eq) in dry DCM was stirred for 10 min under inert conditions. A solution of compound 8 (500 mg, 1.00 mmol, 1.0 eq) was added subsequently, and stirring was continued for an additional 4 h. The reaction mixture was transferred onto a mixture of water (50 mL), brine (50 mL), and EtOAc (100 mL), the layers were separated, and the organic layer was washed with brine (100 mL) and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by FC (eluent: CyHex/EtOAc 9:1) and the title compound obtained as colorless oil (230 mg, 314 μmol, 31% yield).
1H NMR (400 MHz, CDCl3) δ = 7.23 (d, J = 8.5, 2H), 6.87 (d, J = 8.5, 2H), 5.44 to 5.29 (m, 8H), 5.26 to 5.19 (m, 1H), 4.53 to 4.41 (m, J = 11.7, 2H), 4.33 (dd, J = 11.9, 3.8, 1H), 4.17 (dd, J = 11.9, 6.4, 1H), 3.81 (s, 3H), 3.58 to 3.52 (m, 2H), 2.82 (dt, J = 9.1, 5.2, 6H), 2.34 (t, J = 7.6, 2H), 2.26 (t, J = 7.5, 2H), 2.20 to 2.01 (m, J = 30.5, 14.0, 4.8, 6H), 1.95 (t, J = 2.6, 1H), 1.70 (p, J = 7.5, 2H), 1.63 to 1.53 (m, 2H), 1.49 (dd, J = 9.3, 6.5, 2H), 1.40 to 1.17 (m, 16H), 1.13 to 1.02 (m, J = 7.8, 2H), 0.89 (t, J = 6.8, 3H).
13C NMR (101 MHz, CDCl3) δ = 173.38, 172.88, 159.27, 130.52, 129.73, 129.33, 128.91, 128.89, 128.61, 128.27, 128.14, 127.86, 127.54, 113.82, 72.97, 70.12, 68.90, 67.86, 62.71, 55.28, 34.05, 33.71, 32.83, 31.82, 31.53, 29.34, 29.21, 29.14, 29.11, 29.04, 27.23, 26.51, 25.61, 24.81, 23.82, 22.75, 22.59, 17.96, 14.10 ppm. Signals at 29.4 to 29.0 ppm are partially not resolved due to very similar 13C chemical shifts.
HRMS (m/z): [M+Na]+ calc. for: C46H68N2NaO6, 767.49696, found: 767.49715.
2-O-Arachidonyl-1-O-(3′(4′′-pentyn-1′′-yl)-H-diazirine-3′-octanoyl)-sn-glycerol (10)
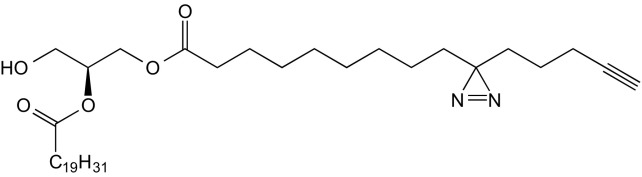
A solution of 9 (96 mg, 131 μmol, 1.0 eq) in DCM (3 mL) was treated with 20% (vol/vol) TFA in DCM (3 mL) at 0 °C. The reaction mixture was stirred for 3 min and then poured into saturated Na2CO3 solution (100 mL). The mixture was diluted with EtOAc (100 mL), and the layers were separated. The organic layer was washed with brine (100 mL) and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by repeated FC (1. eluent: CyHex/EtOAc 4:1; 2. eluent DCM/acetone 97:3) and the title compound obtained as colorless oil (56 mg, 92 μmol, 70% yield).
1H NMR (400 MHz, CDCl3) δ = 5.46 to 5.30 (m, 8H), 5.09 (p, J = 4.9, 1H), 4.32 (dd, J = 11.9, 4.6, 1H), 4.28 to 4.19 (m, 2H), 3.77 to 3.67 (m, 2H), 2.88 to 2.76 (m, 6H), 2.41 to 2.28 (m, 4H), 2.21 to 2.01 (m, 6H), 1.95 (t, J = 2.6, 1H), 1.77 to 1.67 (m, 2H), 1.65 to 1.54 (m, 4H), 1.52 to 1.45 (m, 2H), 1.41 to 1.18 (m, 16H), 1.13 to 1.02 (m, 2H), 0.89 (t, J = 6.7, 3H).
13C NMR (101 MHz, CDCl3) δ = 173.74, 173.16, 130.53, 129.03, 128.78, 128.63, 128.32, 128.10, 127.84, 127.53, 72.19, 68.89, 61.98, 61.53, 34.04, 33.64, 32.83, 31.83, 31.53, 29.33, 29.18, 29.11, 29.07, 29.02, 27.23, 26.49, 25.65, 24.81, 24.75, 23.79, 22.75, 22.58, 17.96, 14.08 ppm. Signals at 29.4 to 29.0 ppm are partially not resolved due to very similar 13C chemical shifts.
HRMS (m/z): [M+H]+ calc. for: C38H60N2O5, 625.4575; found: 625.4575.
Cell Culture.
HeLa cells (human cervical adenocarcinoma cells, No. CCL-2; ATCC/LGC Standards GmbH) were cultured in DMEM (1 g/L glucose; Gibco/Life Technologies) supplemented with 10% FCS (Lot 032M3395; Sigma-Aldrich) and 1% primocin (InvivoGen). Control and NPC human fibroblasts (for genotypes, see table below) were obtained from F.D.P. and were cultured in DMEM (4.5 g/L glucose; Gibco/Life Technologies) supplemented with 10% FCS (Lot 032M3395; Sigma-Aldrich), 2 mM l-glutamine (Gibco/Life Technologies), and 1% penicillin/streptomycin (Gibco/Life Technologies). All cells were kept in a humidified incubator at 37 °C with 5% CO2 and were passaged two to three times per week.
| Cell line | Protein change | cDNA change |
| NPC17 | I1061T, 10bp deletion in exon 19 at codon 962 = fs(exon19) | c.3182T > C, c.2884–93delATCACTGACC |
| NPC22 | R978C, IVS21-2 A > G | c.2932C > T, c.3246–2A > G |
| NPC25 | fs(exon20), N701K | c.2979dupA | C2103C > G |
Sph Uncaging.
HeLa cells in eight-well Labteks at 70 to 80% confluence were labeled with 100 μL of 5 μM Fluo-4 AM (Molecular Probes) solution in imaging buffer [20 mM Hepes, 115 mM NaCl, 1.8 mM CaCl2, 1.2 mM MgCl2, 1.2 mM K2HPO4, and 0.2% (wt/vol) glucose] at 37 °C for 30 min. Fifteen minutes before the start of the experiment, caged Sph, caged dhSph, or trifunctional Sph 1 were added to a final concentration of 2 μM. The cells were then washed and kept in imaging buffer at 37 °C for the duration of the experiment.
The fluorescence of the calcium indicator Fluo-4 was monitored on a dual scanner confocal laser scanning microscope (Olympus FluoView 1200) using a 63× oil objective at 488-nm excitation and emission settings between 500 and 550 nm at an interval of 1 s per frame. A baseline of 10 frames (=10 s) was captured before photoactivation (uncaging) in a circular region (10 pixel units diameter, 8.9 μm2) inside the cells using the tornado function of the Olympus software. Coumarin uncaging was carried out using the 405-nm laser line set to 50% intensity for 3 s at 2 μs per pixel. The time-lapse images were analyzed using Fiji software (W. Rasband, National Institutes of Health, Bethesda) with the FluoQ macro (41) set to the following parameters: background subtraction method: mean of an interactively selected ROI; noise reduction/smoothing method: none; threshold method: interactively with ImageJ’s built-in threshold window; ROI segmentation: semiautomatically with binary mask modification; calculate amplitude changes: using maximum observed amplitude change.
The resulting intensity series/amplitude values represent mean values of whole cells and were loaded in R and grouped according to lipids (caged Sph vs. TFS). Single-cell traces belonging to the same groups were summarized using the R function called summarySE, which calculated the mean as well as the SEM of all traces for every time point. Line and bar graphs were generated using the ggplot2 package (42) in R.
DAG Uncaging.
DAG uncaging experiments were carried out using HeLa cells transiently transfected with a C1-GFP fusion protein as DAG biosensor. Cell culture conditions and transfection protocols were identical to those reported earlier (2). Cells were seeded in eight-well Lab-TekTM dishes and transfected 24 h before uncaging experiments. Confocal time lapses were acquired using an Olympus FV1200 confocal microscope with a 63× oil objective. Cells were treated with a 100-μM solution of the respective caged DAG (TFDAG or SAG) in glucose (20 mM) containing imaging buffer and allowed to equilibrate at 37 °C for 5 to 10 min. Suitable clusters of C1-GFP-expressing cells were chosen for DAG uncaging to ensure high cell numbers. Images were acquired at a frame rate of ∼7 s per frame using 488-nm excitation light, and fluorescence was detected between 500 and 580 nm. Uncaging was performed by scanning the entire field of view with a 405-nm laser set to 40% laser intensity after acquiring a baseline of 10 frames. The cellular responses were monitored for an additional 50 frames. A maximum of two movies was acquired per single well to minimize batch effects.
Time lapses were analyzed using the ImageJ macro PM/background ratio-calculator Macro reported earlier (22), with plasma membrane width defined at five pixels. Single pm/cytosol-ratio traces were copied into an Excel file and loaded into R. During this process, traces were manually classified into responding and nonresponding cells. Furthermore, single-cell traces were normalized by dividing each time point by the average of the first 10 time points (time points before uncaging). Averaged traces of SAG and TFDAG were plotted using the ggplot2 packages (42). All responding cells were fitted using the nlsLM function of the minpack.lm package in R. The following formula was used to fit each normalized single-cell trace:
where kacc is the accumulation rate constant, kmet is the apparent metabolism rate constant, and DAGex is the height of the trace.
As a quality criterion, fits were only used for the statistical analysis if the metabolism constant was above zero and the average difference between the fit and the original values was found to be below 0.06.
Thin-Layer Chromatographic Analysis of Trifunctional Lipids.
HeLa cells were grown in 60-cm dishes to 85 to 95% confluence and labeled with 3 µM TFS or 50 µM TFDAG for the indicated times. After washing with PBS, the dishes were transferred onto an ice block and UV-irradiated for 2.5 min using a 450- to 1,000-W high-pressure mercury lamp (Newport) equipped with a 400-nm high-pass filter. The cells were washed again and scraped in 300 µL of PBS and mixed with 600 µL of MeOH and 150 µL of CHCl3. The mixture was vortexed and centrifuged at 14,000 rpm for 3 min, and the supernatant was transferred into a new vial. Then, 300 µL of CHCl3 and 600 µL of acetic acid [0.1% (vol/vol) in water] were added, the mixture was again vortexed and centrifuged (14,000 rpm, 4 min), and the aqueous phase was discarded. The organic phase was dried in a speed-vac at 30 °C for 15 min. The lipids were dissolved in 7 µL of CHCl3, and 30 µL of click mixture (5 µL of 44.5 mM 3-azido-7-hydroxycoumarin, 500 µL of 10 mM [acetonitrile]4CuBF4, and 2 mL of EtOH) was added. The reaction vial was vortexed and incubated in a heating block at 42 °C with no shaking for 3 h to 4 h until all solvent was condensed under the lid. The tube was then vortexed again, and the mixture was applied onto a 10 × 10 cm high-performance thin-layer chomotography (HPTLC) Silica 60 glass plate (VWR) using the automatic Camaq system. TLC plates were developed using CHCl3/MeOH/H2O/AcOH 65:25:4:1 for 6 cm and then cyclohexane/ethylacetate 1:1 for 9 cm. Lipids containing the fluorescent coumarin group were visualized using a geldoc system.
Proteomic Screens.
Cell labeling and cross-linking.
HeLa cells were grown in three 10-cm dishes for each condition and labeled with 4 mL of 100 μM TFFA for 15 min, 100 μM TFDAG for 15 min, and 6 μM TFS probe for 5 min. The cells were then UV-irradiated for 2.5 min at wavelengths above 400 nm followed by a second irradiation at wavelengths above 345 nm and washed with ice-cold PBS. The cells were scraped off in 2 mL of PBS and centrifuged at 3,000 rpm for 10 min at 4 °C. The cell pellet was resuspended in 300 μL of PBS, 600 μL of MeOH and 150 μL of CHCl3 were added, and the solution was vortexed and centrifuged at 14,000,rpm for 3 min. The protein pellet was resuspended in 100 μL of PBS, and 100 μL of lysis buffer were added, and the solution was shaken at 95 °C for 5 min. After cooling down, 2 μL of benzonase was added, and the solution was incubated for 30 min at 37 °C. Next, 100 μL of 200 mM DTT (in 200 mM Hepes) was added, and the solution was incubated at 45 °C for 30 min, and then 20 μL of 400 mM iodoacetamide (in 200 mM Hepes) was added and shaken at 24 °C for 30 min. Finally, 20 μL of 200 mM DTT were added to quench the reaction. The proteins were precipitated by addition of 600 μL of MeOH and 150 μL of CHCl3 and resuspended in 152.5 μL of PBS/1% (wt/vol) SDS/2× protein inhibitor cocktail (PIC) by shaking at 37 °C for >1 h. Then 5 μL of the solution was removed for protein determination; the three lysates belonging to the same condition were pooled and stored at −20 °C.
Protein determination using Amido Black.
A serial dilution of BSA was prepared with 0, 2.5, 5, 7.5, 10, 25, and 50 μg BSA in 100 μL of water. The samples were diluted to 100 μL. To all samples, 400 μL of amido black solution was added, vortexed, incubated at room temperature for 5 min, and centrifuged at 14,000 rpm for 5 min. The supernatant was aspirated, and the pellets were washed twice with 500 μL of MeOH/AcOH 10/1. The pellets were dissolved in 300 μL of 0.1 N NaOH, and 150 μL were spotted onto a 96-well plate. The absorbance at 550 nm was recorded on a Synergy 4 microplate reader (BioTek).
Biotinylation and pull-down assay.
The volume of the lysate was adjusted to 1 mg of protein and diluted to 630 μL. Then 30 μL of each reagent for click reaction (2.5 mM TBTA in DMSO, 25 mM CuSO4 in water, 25 mM biotin azide in water, and 25 mM ascorbic acid in water) was added, and the mixture was shaken at 37 °C for >3 h. The proteins were precipitated twice using CHCl3/MeOH (1:4), redissolved in 160 μL of PBS/1% SDS and diluted with 240 μL of PBS to reach a final SDS concentration of 0.4% (wt/vol). After centrifugation at 1,000 rpm for 3 min, the supernatant was transferred into a new vial and mixed with 10 μL of Neutravidin-agarose resin (Thermo Fisher Scientific), which were previously washed three times with 180 μL of PBS/0.2% SDS. The lysate was incubated with the resin for 1 h at room temperature, and the supernatant was removed. The resin was washed 15 times with PBS/1% SDS, and the proteins were eluted with 50 μL of elution buffer [100 mM Tris, pH 6.8, 4% (wt/vol) SDS, 4% (vol/vol) β-mercaptoethanol] at room temperature for 1 h. The supernatant was taken after heating to 95 °C for 30 min.
On-bead tryptic digest [adapted from Hughes et al. (23)].
Twenty microliters of hydrophilic and hydrophobic Sera-Mag Speed beads (Thermo Fermo Scientific) were mixed and kept in 100 μL of water. The protein lysate was mixed with 5 μL of fomic acid [5% (vol/vol) in water] and 5 μL of beads. Acetonitrile was added immediately to a final concentration of greater than 50%. The mixture was incubated for 8 min at room temperature and was then placed on a magnetic rack for a further 2 min. The supernatant was removed and discarded. The beads were washed twice with 200 μL of EtOH [70% (vol/vol) in water] and once with 180 μL of acetonitrile. The dry beads were then reconstituted in 10 μL of digestion solution [50 mM Hepes pH 8.0 + 1 μg trypsin/LysC (mass spectrometry grade; Promega)] and incubated for 14 h at 37 °C. The peptides were recovered by adding 190 μL of acetonitrile to the beads, incubating at room temperature for 8 min and for a further 2 min on the magnetic rack. The supernatant was removed and discarded. The beads were washed with 180 μL of acetonitrile and 200 μL of EtOH [90% (vol/vol) in water]. Beads were reconstituted in 9 μL of DMSO [4% (vol/vol) in water] and sonicated for 5 min. The supernatant was removed and transferred to a deactivated glass vial containing 1 μL of formic acid [10% (vol/vol) in water].
High-pH reverse-phase offline fractionation [adapted from Bock et al. (38)].
The peptide mixture was basified by addition of 10 μL of 200 mM NH4OH and 2 μL of ammonium formate and injected into an Agilent 1200 Infinity HPLC system equipped with a Peltier-cooled autosampler and fraction collector (both set at 10 °C for all samples). The column was a Gemini C18 column (3 μm, 110 Å, 100 × 1.0 mm; Phenomenex) with a Gemini C18, 4 × 2.0 mm SecurityGuardcartrigdge (Phenomenex) as a guard column. The solvent system consisted of two mobile phases: Phase A was 20 mM ammonium formate (pH 10.0), and phase B was acetonitrile (100%). Separation was achieved at a flow rate of 0.1 mL/min using the following linear gradient: 100% A for 2 min, from 100% A to 35% B in 59 min, and 100% A and reequilibration for 13 min. Thirty-two fractions were collected, which were then nonsequentially pooled into 10 fractions and dried under vacuum centrifugation and reconstituted in 10 μL of 0.1% formic acid.
Peptide cleanup and enrichment.
An Oasis HLB microelution plate (Waters) was washed twice with 200 μL of 80% acetonitrile/0.05% formic acid and then equilibrated twice with 200 μL of formic acid [0.05% (vol/vol) in water] per well. The samples were loaded, and the flow-through analysis was discarded. Samples were then washed twice with 200 μL of formic acid [0.05% (vol/vol) in water]. Elution was achieved with 50 μL of 80% acetonitrile/0.05% formic acid. The eluate was dried under vacuum centrifugation and reconstituted in DMSO [4% (vol/vol) in water] + 1% formic acid.
Liquid Chromatography-MS/MS.
Peptides were separated using the nanoAcquity UPLC system (Waters) fitted with a trapping (nanoAcquity Symmetry C18, 5 μm, 180 μm × 20 mm) and an analytical column (nanoAcquity BEH C18, 1.7 μm, 75 μm × 200 mm). The outlet of the analytical column was coupled directly to an LTQ OrbitrapVelos Pro (Thermo Fisher Scientific) using the Proxeon nanospray source. Solvent A was water, 0.1% formic acid, and solvent B was acetonitrile, 0.1% formic acid. The samples (7 μL) were loaded with a constant flow of solvent A at 5 μL/min onto the trapping column. Trapping time was 6 min. Peptides were eluted via the analytical column at a constant flow of 0.3 μL/min. During the elution step, the percentage of solvent B increased in a linear fashion from 3 to 7% in 10 min, then increased to 25% in 20 min and finally to 40% in a further 10 min. The peptides were introduced into the mass spectrometer via a Pico-Tip Emitter 360 μm OD × 20 μm ID; a 10-μm tip (New Objective) and a spray voltage of 2.2 kV were applied. The capillary temperature was set at 300 °C. Full-scan MS spectra with mass range 300 to 1700 m/z were acquired in profile mode in the Fourier transform with resolution of 30,000. The filling time was set at a maximum of 500 ms with limitation of 106 ions. The most intense ions (up to 15) from the full-scan MS were selected for fragmentation in the LTQ. Normalized collision energy of 40% was used, and the fragmentation was performed after accumulation of 3 × 104 ions or after filling time of 100 ms for each precursor ion (whichever occurred first). MS/MS data were acquired in centroid mode. Only multiply charged (2+, 3+, 4+) precursor ions were selected for MS/MS. The dynamic exclusion list was restricted to 500 entries with a maximum retention period of 30 s and relative mass window of 10 ppm. To improve the mass accuracy, a lock mass correction using a background ion (445.12003 m/z) was applied.
Data analysis.
Proteome Discoverer 1.4 (version 1.4.1.14; Thermo Fisher Scientific) was used as raw data postprocessing interface with the possibility to select scan events for peptide/protein identification. Identification was performed using a species-specific Uniprot database (Homo sapiens taxonomy, 2012, 86,945 entries). Mascot (version 2.2.07; Matrix Sciences) was used as a search engine. Variable amino acid modification was oxidized methionine. Carbamidomethylation of cysteines was set as fixed modification. Trypsin was selected as the enzyme, with one potential missed cleavage. Peptide and fragment ion tolerance was, respectively, 10 ppm and 0.5 Da. False discovery rates of 5% (relaxed) and 1% (strict), validated based on q value, were calculated by Proteome Discoverer based on the search against the corresponding randomized database. Results were filtered on upload for only high peptide confidence, peptide length greater than 6 amino acids, and a minimum mascot peptide ion score of 20. The resulting table was read by R, sorted according to peptide spectral match ratios of TFS and TFDAG over control lipids, and visualized as a heat map by using the ggplot2 package (42). To generate Fig. 3B, the GO-term annotations of putative TFS-binding proteins (top 60 hits) and TFDAG-binding proteins (bottom 129 hits) were retrieved from the ensemble database using the biomaRt package (43) for R.
Visualization of Protein−Lipid Complexes in Cells.
Cells were seeded onto 11-mm coverslips placed in wells of a 24-well plate and labeled with 3 μM TFS or 50 μM TFDAG or TFFA in imaging buffer for 10 min. Cells were washed, overlaid with 1 mL of imaging buffer, and UV-irradiated on ice for 2.5 min at wavelengths of >400 nm and for a further 2.5 min at wavelengths of >355 nm. Cells were immediately fixed with MeOH at −20 °C for 20 min. Non–cross-linked lipids were extracted by washing three times with 1 mL of CHCl3/MeOH/AcOH 10:55:0.75 (vol/vol) at room temperature. Cells were then incubated with 50 μL of click mixture (1 mM ascorbic acid, 100 μM TBTA, 1 mM CuSO4, and 2 μM Alexa 488 azide in PBS) for 1 h at room temperature in the dark. Cells were then washed with PBS and incubated with 50 μL of primary antibody (rabbit α-LAMP1, Cell Signaling, 1:100, mouse α-GM130, Abcam, 1:200, and rabbit α-p72, Thermo Fisher Scientific, 1:100 in PBS supplemented with 4% BSA and 0.02% Triton X-100) overnight at 4 °C. Coverslips were again washed in PBS and incubated with secondary antibody (α-rabbit conjugated to AlexaFluor555, α-mouse conjugated to AlexaFluor555, Cell Signaling, 1:800) for 1 h, washed, and mounted in DAPI-containing mounting medium (Vectashield; Vector Laboratories). Microscopy images were captured at room temperature using a confocal laser scanning microscope (Zeiss LSM780) with a 63× oil objective. Settings were as follows: DAPI-channel: 405-nm excitation (ex), 409- to 475-nm emission (em); green channel: 488-nm ex, 489- to 550-nm em; red channel: 561-nm ex, 569- to 655-nm em. Images were further processed using Fiji software (fiji.sc/). The Skewness value for analysis of pixel distribution within cells was directly extracted from the raw images by ticking the Skewness check box in Analyze > Set Measurements.
Visualization of Sph via Correlated Light and Electron Microscopy.
NPC patient cells were seeded on carbon-coated Sapphire discs (3 mm diameter, thickness 0.05 mm, Engineering Office of M. Wohlwend at GmbH, Sennwald, Switzerland) for 24 h. Uncaging and cross-linking was performed as described. Cells were then fixed in 4% (vol/vol) paraformaldehyde (PFA) in PHEM buffer for 10 min and permeabilized with 0.001% saponin in PHEM buffer for 5 min before click reaction for 1 h in the dark. The samples were processed within 1 h for HPF using the HPM 010 (Bal-Tec), placed between two aluminum carriers and a gold slot grid spacer (Plano GmbH).
The freeze substitution was performed in a temperature-controlling device (AFS2; Leica) in acetone supplemented with 0.1% (wt/vol) uranyl acetate for 24 h at −90 °C (44, 45). The temperature was raised to −45 °C within 10 h, held at that temperature, washed three times with acetone, and infiltrated by Lowicryl HM20 (Polysciences Europe GmbH) for 4-h periods at increasing concentrations (10, 25, 50, and 75%). The temperature was subsequently raised to −25 °C (5 °C/h), and the final Lowicryl concentration of 100% was held and washed every 10 h three times. The blocks were finally polymerized by UV light incubation for 48 h at 20 °C.
Thick sections (300 nm) were cut using a Leica Utracut UCT microtome mounted with a diamond knife (Diatome). Each section was placed on a carbon-coated copper grid (Plano GmbH) and incubated for 8 min in a drop of 1:200 with 50-nm fluorescent microspheres in PBS (TetraSpecks; Life Technologies) followed by washing three times in water. The grids were sandwiched between two coverslips (No. 1; Menzel-Gläser) filled with 30 μL of water and sealed by vacuum grease. Samples were imaged using a Metamorph (Molecular Devices) operated wide-field fluorescence microscope (Olympus IX81) equipped with an X-Cite 120PC light source (EXFO Life 2 Sciences), an Olympus PlanApo 100× 1.45 NA oil immersion objective, an Orca-ER camera (Hamamatsu Photonics), and electronic shutters and filter wheels (Sutter Instruments Co.); 470/22- and 556/20-nm excitation filters were used in combination with emission filters 520/35 nm for Tetraspecks and 624/40 nm for Alexa594.
The grids were incubated with gold fiducial markers (15 nm) and placed into a single-tilt holder on a Tecnai TF30 microscope (FEI) at 300 kV using Serial EM for electron tomography (36). Images were acquired at −60° to +60° (1° increments) recorded by a 4K Eagle camera (FEI) at a pixel size of 5.068 nm and reconstructed using IMOD (versions 4.1.4) (46). Fluorescent and electron tomography images were correlated based on the fluorescent and gold fiducials using a MATLAB script.
Supplementary Material
Acknowledgments
We thank the Advanced Light Microscopy Facility and the Proteomic Core Facility of the European Molecular Biology Laboratory (EMBL). This research has received funding from the European Union Seventh Framework Programme under Grant Agreement 289278—“Sphingonet”—as well as Transregio 83 funded by the Deutsche Forschungsgemeinschaft. This work was partially supported by the intramural research program of the National Institute of Child Health and Human Development. M.S. is a fellow of the EMBL Interdisciplinary Postdoc Program, cofunded by the European Union.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1611096114/-/DCSupplemental.
References
- 1.Höglinger D, et al. Intracellular sphingosine releases calcium from lysosomes. eLife. 2015;4:e10616. doi: 10.7554/eLife.10616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nadler A, et al. The fatty acid composition of diacylglycerols determines local signaling patterns. Angew Chem Int Ed Engl. 2013;52(24):6330–6334. doi: 10.1002/anie.201301716. [DOI] [PubMed] [Google Scholar]
- 3.Feng S, et al. A rapidly reversible chemical dimerizer system to study lipid signaling in living cells. Angew Chem Int Ed Engl. 2014;53(26):6720–6723. doi: 10.1002/anie.201402294. [DOI] [PubMed] [Google Scholar]
- 4.Idevall-Hagren O, Dickson EJ, Hille B, Toomre DK, De Camilli P. Optogenetic control of phosphoinositide metabolism. Proc Natl Acad Sci USA. 2012;109(35):E2316–E2323. doi: 10.1073/pnas.1211305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Höglinger D, Nadler A, Schultz C. Caged lipids as tools for investigating cellular signaling. Biochim Biophys Acta. 2014;1841(8):1085–96. doi: 10.1016/j.bbalip.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 6.Mentel M, Laketa V, Subramanian D, Gillandt H, Schultz C. Photoactivatable and cell-membrane-permeable phosphatidylinositol 3,4,5-trisphosphate. Angew Chem Int Ed Engl. 2011;50(16):3811–3814. doi: 10.1002/anie.201007796. [DOI] [PubMed] [Google Scholar]
- 7.Hövelmann F, et al. Optotaxis: Caged lysophosphatidic acid enables optical control of a chemotactic gradient. Cell Chem Biol. 2016;23(5):629–634. doi: 10.1016/j.chembiol.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 8.Thiele C, et al. Tracing fatty acid metabolism by click chemistry. ACS Chem Biol. 2012;7(12):2004–2011. doi: 10.1021/cb300414v. [DOI] [PubMed] [Google Scholar]
- 9.Matyas GR, Beck Z, Karasavvas N, Alving CR. Lipid binding properties of 4E10, 2F5, and WR304 monoclonal antibodies that neutralize HIV-1. Biochim Biophys Acta. 2009;1788(3):660–65. doi: 10.1016/j.bbamem.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 10.Alvarez-Vasquez F, et al. Simulation and validation of modelled sphingolipid metabolism in Saccharomyces cerevisiae. Nature. 2005;433(7024):425–430. doi: 10.1038/nature03232. [DOI] [PubMed] [Google Scholar]
- 11.Kuerschner L, et al. Polyene-lipids: A new tool to image lipids. Nat Methods. 2005;2(1):39–45. doi: 10.1038/nmeth728. [DOI] [PubMed] [Google Scholar]
- 12.Nishioka T, et al. Rapid turnover rate of phosphoinositides at the front of migrating MDCK cells. Mol Biol Cell. 2008;19(10):4213–4223. doi: 10.1091/mbc.E08-03-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manifava M, et al. Differential binding of traffic-related proteins to phosphatidic acid- or phosphatidylinositol (4,5)- bisphosphate-coupled affinity reagents. J Biol Chem. 2001;276(12):8987–8994. doi: 10.1074/jbc.M010308200. [DOI] [PubMed] [Google Scholar]
- 14.Gallego O, et al. A systematic screen for protein−lipid interactions in Saccharomyces cerevisiae. Mol Syst Biol. 2010;6(1):430. doi: 10.1038/msb.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saliba A-E, et al. A quantitative liposome microarray to systematically characterize protein-lipid interactions. Nat Methods. 2014;11(1):47–50. doi: 10.1038/nmeth.2734. [DOI] [PubMed] [Google Scholar]
- 16.Ceccato L, et al. PLIF: A rapid, accurate method to detect and quantitatively assess protein-lipid interactions. Sci Signal. 2016;9(421):rs2. doi: 10.1126/scisignal.aad4337. [DOI] [PubMed] [Google Scholar]
- 17.Haberkant P, Holthuis JCM. Fat & fabulous: Bifunctional lipids in the spotlight. Biochim Biophys Acta. 2014;1841(8):1022–1030. doi: 10.1016/j.bbalip.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Haberkant P, et al. In vivo profiling and visualization of cellular protein-lipid interactions using bifunctional fatty acids. Angew Chem Int Ed Engl. 2013;52(14):4033–4038. doi: 10.1002/anie.201210178. [DOI] [PubMed] [Google Scholar]
- 19.Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, Cravatt BF. Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat Methods. 2013;10(3):259–264. doi: 10.1038/nmeth.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haberkant P, et al. Bifunctional sphingosine for cell-based analysis of protein-sphingolipid interactions. ACS Chem Biol. 2016;11(1):222–230. doi: 10.1021/acschembio.5b00810. [DOI] [PubMed] [Google Scholar]
- 21.Niphakis MJ, et al. A global map of lipid-binding proteins and their ligandability in cells. Cell. 2015;161(7):1668–1680. doi: 10.1016/j.cell.2015.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nadler A, et al. Exclusive photorelease of signalling lipids at the plasma membrane. Nat Commun. 2015;6:10056. doi: 10.1038/ncomms10056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes CS, et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol. 2014;10(757):757. doi: 10.15252/msb.20145625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy M, Futerman AH. Mammalian ceramide synthases. IUBMB Life. 2010;62(5):347–356. doi: 10.1002/iub.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kågedal K, Zhao M, Svensson I, Brunk UT. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem J. 2001;359(Pt 2):335–343. doi: 10.1042/0264-6021:3590335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pawelczyk T, Matecki A. Phospholipase C-delta3 binds with high specificity to phosphatidylinositol 4,5-bisphosphate and phosphatidic acid in bilayer membranes. Eur J Biochem. 1999;262(2):291–298. doi: 10.1046/j.1432-1327.1999.00388.x. [DOI] [PubMed] [Google Scholar]
- 27.Hoetelmans RW, et al. Effects of acetone, methanol, or paraformaldehyde on cellular structure, visualized by reflection contrast microscopy and transmission and scanning electron microscopy. Appl Immunohistochem Mol Morphol. 2001;9(4):346–351. doi: 10.1097/00129039-200112000-00010. [DOI] [PubMed] [Google Scholar]
- 28.Saheki Y, et al. Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat Cell Biol. 2016;18(5):504–515. doi: 10.1038/ncb3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanier MT. Niemann−Pick disease type C. Orphanet J Rare Dis. 2010;5:16. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.te Vruchte D, et al. Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport. J Biol Chem. 2004;279(25):26167–26175. doi: 10.1074/jbc.M311591200. [DOI] [PubMed] [Google Scholar]
- 31.Lloyd-Evans E, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14(11):1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 32.Sokol J, et al. Type C Niemann-Pick disease. Lysosomal accumulation and defective intracellular mobilization of low density lipoprotein cholesterol. J Biol Chem. 1988;263(7):3411–3417. [PubMed] [Google Scholar]
- 33.Sun X, et al. Niemann-Pick C variant detection by altered sphingolipid trafficking and correlation with mutations within a specific domain of NPC1. Am J Hum Genet. 2001;68(6):1361–72. doi: 10.1086/320599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blom T, Li Z, Bittman R, Somerharju P, Ikonen E. Tracking sphingosine metabolism and transport in sphingolipidoses: NPC1 deficiency as a test case. Traffic. 2012;13(9):1234–1243. doi: 10.1111/j.1600-0854.2012.01379.x. [DOI] [PubMed] [Google Scholar]
- 35.Lu F, et al. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. eLife. 2015;4:1–16. doi: 10.7554/eLife.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kukulski W, et al. Correlated fluorescence and 3D electron microscopy with high sensitivity and spatial precision. J Cell Biol. 2011;192(1):111–119. doi: 10.1083/jcb.201009037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sandhoff K. Neuronal sphingolipidoses: Membrane lipids and sphingolipid activator proteins regulate lysosomal sphingolipid catabolism. Biochimie. 2016;130:146–151. doi: 10.1016/j.biochi.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Bock T, et al. An integrated approach for genome annotation of the eukaryotic thermophile Chaetomium thermophilum. Nucleic Acids Res. 2014;42(22):13525–13533. doi: 10.1093/nar/gku1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimada Y, Usuda K, Okabe H, Suzuki T, Matsumoto K. Deracemization of 1,2-diol monotosylate derivatives by a combination of enzymatic hydrolysis with the Mitsunobu inversion using polymer-bound triphenylphosphine. Tetrahedron Asymmetry. 2009;20(24):2802–2808. [Google Scholar]
- 40.Sato S, et al. Occurrence of a bacterial membrane microdomain at the cell division site enriched in phospholipids with polyunsaturated hydrocarbon chains. J Biol Chem. 2012;287(29):24113–24121. doi: 10.1074/jbc.M111.318311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stein F, Kress M, Reither S, Piljić A, Schultz C. FluoQ: A tool for rapid analysis of multiparameter fluorescence imaging data applied to oscillatory events. ACS Chem Biol. 2013;8(9):1862–1868. doi: 10.1021/cb4003442. [DOI] [PubMed] [Google Scholar]
- 42.Wickham H. 2009. ggplot2: Elegant Graphics for Data Analysis (Springer, New York)
- 43.Smedley D, et al. The BioMart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015;43(W1):W589–W598. doi: 10.1093/nar/gkv350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Avinoam O, Schorb M, Beese CJ, Briggs JAG, Kaksonen M. Endocytic sites mature by continuous bending and remodeling of the clathrin coat. Science. 2015;348(6241):1369–1372. doi: 10.1126/science.aaa9555. [DOI] [PubMed] [Google Scholar]
- 45.Kukulski W, et al. Precise, correlated fluorescence microscopy and electron tomography of lowicryl sections using fluorescent fiducial markers. Methods Cell Biol. 2012;111:235–257. doi: 10.1016/B978-0-12-416026-2.00013-3. [DOI] [PubMed] [Google Scholar]
- 46.Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116(1):71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.