

Abstract
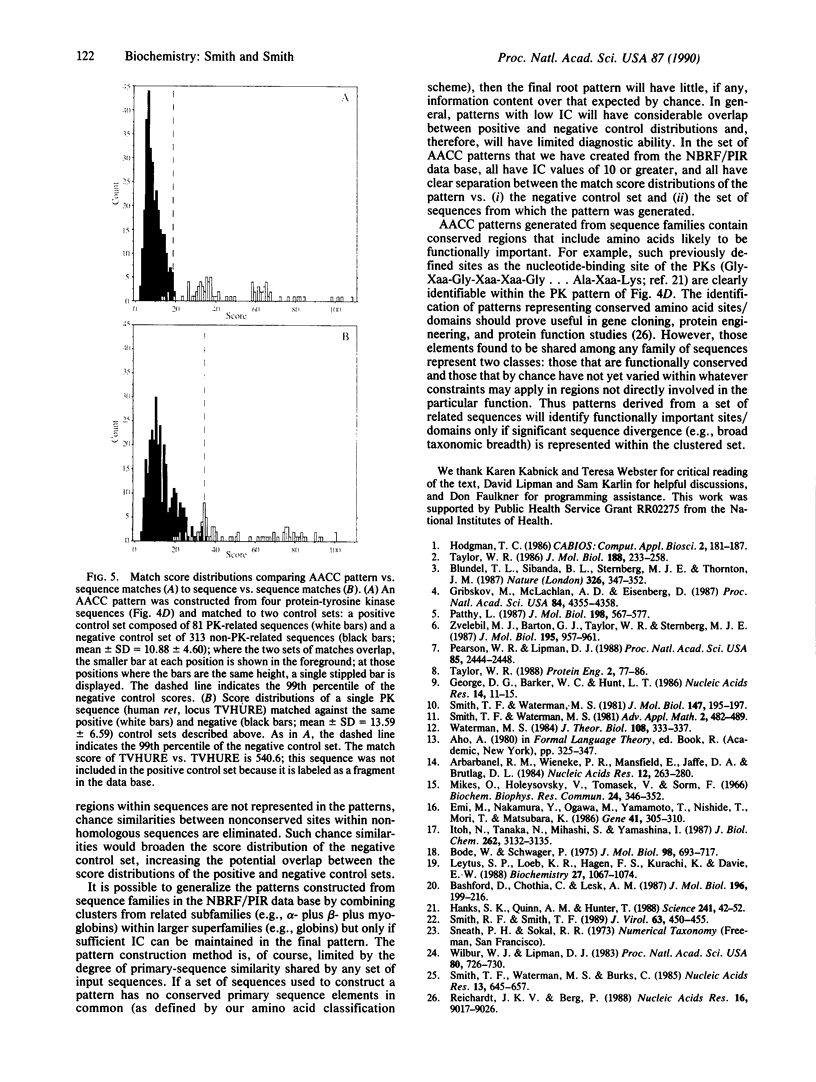
We have developed a computer algorithm that can extract the pattern of conserved primary sequence elements common to all members of a homologous protein family. The method involves clustering the pairwise similarity scores among a set of related sequences to generate a binary dendrogram (tree). The tree is then reduced in a stepwise manner by progressively replacing the node connecting the two most similar termini by one common pattern until only a single common "root" pattern remains. A pattern is generated at a node by (i) performing a local optimal alignment on the sequence/pattern pair connected by the node with the use of an extended dynamic programming algorithm and then (ii) constructing a single common pattern from this alignment with a nested hierarchy of amino acid classes to identify the minimal inclusive amino acid class covering each paired set of elements in the alignment. Gaps within an alignment are created and/or extended using a "pay once" gap penalty rule, and gapped positions are converted into gap characters that function as 0 or 1 amino acid of any type during subsequent alignment. This method has been used to generate a library of covering patterns for homologous families in the National Biomedical Research Foundation/Protein Identification Resource protein sequence data base. We show that a covering pattern can be more diagnostic for sequence family membership than any of the individual sequences used to construct the pattern.
Full text
PDF




Images in this article
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Abarbanel R. M., Wieneke P. R., Mansfield E., Jaffe D. A., Brutlag D. L. Rapid searches for complex patterns in biological molecules. Nucleic Acids Res. 1984 Jan 11;12(1 Pt 1):263–280. doi: 10.1093/nar/12.1part1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashford D., Chothia C., Lesk A. M. Determinants of a protein fold. Unique features of the globin amino acid sequences. J Mol Biol. 1987 Jul 5;196(1):199–216. doi: 10.1016/0022-2836(87)90521-3. [DOI] [PubMed] [Google Scholar]
- Blundell T. L., Sibanda B. L., Sternberg M. J., Thornton J. M. Knowledge-based prediction of protein structures and the design of novel molecules. 1987 Mar 26-Apr 1Nature. 326(6111):347–352. doi: 10.1038/326347a0. [DOI] [PubMed] [Google Scholar]
- Bode W., Schwager P. The refined crystal structure of bovine beta-trypsin at 1.8 A resolution. II. Crystallographic refinement, calcium binding site, benzamidine binding site and active site at pH 7.0. J Mol Biol. 1975 Nov 15;98(4):693–717. doi: 10.1016/s0022-2836(75)80005-2. [DOI] [PubMed] [Google Scholar]
- Emi M., Nakamura Y., Ogawa M., Yamamoto T., Nishide T., Mori T., Matsubara K. Cloning, characterization and nucleotide sequences of two cDNAs encoding human pancreatic trypsinogens. Gene. 1986;41(2-3):305–310. doi: 10.1016/0378-1119(86)90111-3. [DOI] [PubMed] [Google Scholar]
- George D. G., Barker W. C., Hunt L. T. The protein identification resource (PIR). Nucleic Acids Res. 1986 Jan 10;14(1):11–15. doi: 10.1093/nar/14.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribskov M., McLachlan A. D., Eisenberg D. Profile analysis: detection of distantly related proteins. Proc Natl Acad Sci U S A. 1987 Jul;84(13):4355–4358. doi: 10.1073/pnas.84.13.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks S. K., Quinn A. M., Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988 Jul 1;241(4861):42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Hodgman T. C. The elucidation of protein function from its amino acid sequence. Comput Appl Biosci. 1986 Sep;2(3):181–187. doi: 10.1093/bioinformatics/2.3.181. [DOI] [PubMed] [Google Scholar]
- Itoh N., Tanaka N., Mihashi S., Yamashina I. Molecular cloning and sequence analysis of cDNA for batroxobin, a thrombin-like snake venom enzyme. J Biol Chem. 1987 Mar 5;262(7):3132–3135. [PubMed] [Google Scholar]
- Leytus S. P., Loeb K. R., Hagen F. S., Kurachi K., Davie E. W. A novel trypsin-like serine protease (hepsin) with a putative transmembrane domain expressed by human liver and hepatoma cells. Biochemistry. 1988 Feb 9;27(3):1067–1074. doi: 10.1021/bi00403a032. [DOI] [PubMed] [Google Scholar]
- Mikes O., Holeysovský V., Tomásek V., Sorm F. Covalent structure of bovine trypsinogen. The position of the remaining amides. Biochem Biophys Res Commun. 1966 Aug 12;24(3):346–352. doi: 10.1016/0006-291x(66)90162-8. [DOI] [PubMed] [Google Scholar]
- Patthy L. Detecting homology of distantly related proteins with consensus sequences. J Mol Biol. 1987 Dec 20;198(4):567–577. doi: 10.1016/0022-2836(87)90200-2. [DOI] [PubMed] [Google Scholar]
- Pearson W. R., Lipman D. J. Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A. 1988 Apr;85(8):2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt J. K., Berg P. Conservation of short patches of amino acid sequence amongst proteins with a common function but evolutionarily distinct origins: implications for cloning genes and for structure-function analysis. Nucleic Acids Res. 1988 Sep 26;16(18):9017–9026. doi: 10.1093/nar/16.18.9017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. F., Smith T. F. Identification of new protein kinase-related genes in three herpesviruses, herpes simplex virus, varicella-zoster virus, and Epstein-Barr virus. J Virol. 1989 Jan;63(1):450–455. doi: 10.1128/jvi.63.1.450-455.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T. F., Waterman M. S., Burks C. The statistical distribution of nucleic acid similarities. Nucleic Acids Res. 1985 Jan 25;13(2):645–656. doi: 10.1093/nar/13.2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T. F., Waterman M. S. Identification of common molecular subsequences. J Mol Biol. 1981 Mar 25;147(1):195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Taylor W. R. Identification of protein sequence homology by consensus template alignment. J Mol Biol. 1986 Mar 20;188(2):233–258. doi: 10.1016/0022-2836(86)90308-6. [DOI] [PubMed] [Google Scholar]
- Taylor W. R. Pattern matching methods in protein sequence comparison and structure prediction. Protein Eng. 1988 Jul;2(2):77–86. doi: 10.1093/protein/2.2.77. [DOI] [PubMed] [Google Scholar]
- Waterman M. S. Efficient sequence alignment algorithms. J Theor Biol. 1984 Jun 7;108(3):333–337. doi: 10.1016/s0022-5193(84)80037-5. [DOI] [PubMed] [Google Scholar]
- Wilbur W. J., Lipman D. J. Rapid similarity searches of nucleic acid and protein data banks. Proc Natl Acad Sci U S A. 1983 Feb;80(3):726–730. doi: 10.1073/pnas.80.3.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvelebil M. J., Barton G. J., Taylor W. R., Sternberg M. J. Prediction of protein secondary structure and active sites using the alignment of homologous sequences. J Mol Biol. 1987 Jun 20;195(4):957–961. doi: 10.1016/0022-2836(87)90501-8. [DOI] [PubMed] [Google Scholar]