

Abstract
Bile acids are synthesized from cholesterol in the liver and subjected to multiple metabolic biotransformations in hepatocytes, including oxidation by cytochromes P450 (CYPs) and conjugation with taurine, glycine, glucuronic acid, and sulfate. Mice and rats can hydroxylate chenodeoxycholic acid (CDCA) at the 6β-position to form α-muricholic acid (MCA) and ursodeoxycholic acid (UDCA) to form β-MCA. However, MCA is not formed in humans to any appreciable degree and the mechanism for this species difference is not known. Comparison of several Cyp-null mouse lines revealed that α-MCA and β-MCA were not detected in the liver samples from Cyp2c-cluster null (Cyp2c-null) mice. Global bile acid analysis further revealed the absence of MCAs and their conjugated derivatives, and high concentrations of CDCA and UDCA in Cyp2c-null mouse cecum and feces. Analysis of recombinant CYPs revealed that α-MCA and β-MCA were produced by oxidation of CDCA and UDCA by Cyp2c70, respectively. CYP2C9-humanized mice have similar bile acid metabolites as the Cyp2c-null mice, indicating that human CYP2C9 does not oxidize CDCA and UDCA, thus explaining the species differences in production of MCA. Because humans do not produce MCA, they lack tauro-β-MCA, a farnesoid X receptor antagonist in mouse that modulates obesity, insulin resistance, and hepatosteatosis.
Keywords: chenodeoxycholic acid, cytochrome P450, Cyp2c70, enzyme kinetics, liver, muricholic acid, ursodeoxycholic acid
Bile acids are synthesized from cholesterol in the liver and secreted through the biliary tract into the small intestine, where they aid in the absorption of lipids and fat-soluble vitamins (1, 2). Greater than 90% of bile acids produced in the liver are reabsorbed in the small intestine in the process of enterohepatic circulation. Bile acid synthesis and transport in the liver and intestine is regulated by the farnesoid X receptor (FXR, NR1H4), a member of the nuclear receptor superfamily (3, 4). Bile acids are subject to multiple metabolic biotransformations in hepatocytes, including conjugation with taurine, glycine, glucuronic acid, and sulfate (5). Mice and rats can hydroxylate chenodeoxycholic acid (CDCA) at the 6β-position to form α-muricholic acid (MCA) and ursodeoxycholic acid (UDCA) to form β-MCA. MCA is produced in both mouse and rat liver, but is not formed at significant levels in human liver, thus indicating a species difference in MCA synthesis. In addition, mice mainly produce taurine conjugates of bile acids, while humans produce mostly glycine conjugates and some taurine conjugates (6); rats also carry out both taurine and glycine conjugation. This is of particular interest because the taurine conjugate of β-MCA, tauro- (T-)β-MCA, is an antagonist of FXR in the ileum that controls FXR signaling and metabolic disease in mouse models of obesity (7–9).
While the hepatic cytochromes P450 (CYPs) play a central role in the metabolism of drugs, toxins, and carcinogens, they also carry out key metabolic reactions in steroid hormone and bile acid synthesis. A number of CYPs participate in the synthesis of bile acid metabolites, with cholesterol 7α-hydroxylase (CYP7A1) generally considered to be the rate limiting enzyme in bile acid synthesis (10). Cyp7a1 is under control of FXR in the liver, and by FXR in the intestine through modulation of fibroblast growth factor 15 produced in the intestine that suppresses Cyp7a1 expression in the liver (11). The mammalian CYPs have remarkable diversity between species, with 57 CYP genes identified in humans, and more than 100 putatively functional Cyp genes described in the mouse (12). For example, while the CYP1A/Cyp1a genes are conserved between mice and humans, the CYP2C, CYP2D, and CYP3A gene clusters have markedly diverged between the two species (13). To overcome the differences that exist in the substrate specificity and multiplicity of CYPs between species, significant efforts have been made to develop and characterize Cyp-null and CYP-humanized mice, with the aim to determine the metabolic functions of CYPs and to provide mouse models that better predict human pathways of metabolism (14, 15). Viable knockout models of the mouse Cyp3a (16, 17), Cyp2d (18), Cyp1a (19), and Cyp2c (20) gene clusters were described, which are associated with metabolic differences in the metabolism of xenobiotics as compared with wild-type controls.
In the present study, Cyp-null mice were used to determine which CYPs are responsible for the differences in production of bile acid metabolites between humans and mice, notably the hepatic synthesis of MCA. Individual bile acid concentrations were determined in wild-type and Cyp-null mice leading to the identification of Cyp2c70 as the CYP responsible for MCA synthesis in mice. This was confirmed by recombinant Cyp2c70 expression and Cyp2c70 siRNA inhibition in primary mouse hepatocytes.
MATERIALS AND METHODS
Animal maintenance and treatments
All animal studies and procedures were carried out in accordance with Institute of Laboratory Animal Resources guidelines and approved by the National Cancer Institute Animal Care and Use Committee. Mice were housed in a pathogen-free animal facility under a standard 12 h light/dark cycle and given pelleted NIH-31 chow diet and water ad libitum. Male mice between 8 and 12 weeks of age were used for isolation of primary hepatocytes and preparation of liver microsomes. Liver tissue, fecal samples, and ileum/cecum contents for metabolomic analysis were collected in Dundee and shipped to the National Cancer Institute for analysis. Adult Cyp1a-cluster null (Cyp1a-null) (19), Cyp2c-cluster null (Cyp2c-null) (20), Cyp2d-cluster null (Cyp2d-null) (18), Cyp3a-cluster null (Cyp3a-null) (16), and CYP2C9-humanized (hCYP2C9) (20) mice were housed singly in open-top cages with ad libitum 24 h access to food and water. RM1A chow diet (Special Diet Services, Witham, UK) was removed for the final 4 h before euthanization. After 24 h, fecal pellets were collected, mixed, and 500 mg snap-frozen and stored at −80°C. Mice were euthanized by CO2 asphyxiation and blood collected by cardiac puncture. Tissues were excised and immediately frozen in liquid nitrogen and serum and tissues were stored at −80°C until use.
Measurement of mRNAs
Total RNA of liver was extracted using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA). Quantitative (q)PCR was performed using cDNA generated from 1 μg total RNA with qScript™ cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD). qPCR reactions were carried out using SYBR green qPCR master mix (Biotools, Houston, TX) in a QuanStudioTM 7 Flex system. The primer pairs were designed using Primer-BLAST (National Center for Biotechnology Information) and are shown in supplemental Table S1. Values were quantified with the comparative CT method and normalized to peptidylprolyl isomerase A (Ppia).
Quantification of bile acid metabolites
Twenty milligrams of liver, cecum, and feces were homogenized with 200 μl of 100% acetonitrile containing 1 μM d5-taurocholate (Sigma-Aldrich) as an internal standard and centrifuged twice at 15,000 g for 25 min at 4°C for removal of precipitated proteins and other particulates. The supernatant was diluted by an equal volume of HPLC grade water (Thermo Fisher Scientific, Waltham, MA) containing 0.1% formic acid. Quantification of bile acid metabolites was measured as described previously (7, 9). LC-MS was performed on a Waters Acquity H-Class ultra-performance LC (UPLC) system using a Waters Acquity BEH C18 column (2.1 × 100 mm) coupled to a Waters Xevo G2 quadrupole (Q)TOF-MS. UPLC was performed by the following protocol: A, 0.1% formic acid in water; B, 0.1% formic acid in acetonitrile. An initial gradient of 80% A for 4 min, to 60% A at 15 min, to 40% A at 20 min, to 10% A at 21 min, followed by flushing for 1 min, then equilibration under the initial conditions for 4 min. The flow rate was 0.4 ml/min and the column temperature was maintained at 45°C. A Waters Xevo G2 QTOF was operated in negative mode, scanning m/z 50–1,200 at a rate of 0.3 s/scan. The following instrument conditions were used: 1.5 kV capillary voltage, 150°C source temperature, 30 V sampling cove, and a desolvation gas flow rate of 850 l/h at 500°C. The chromatograms showing the separation of the various bile acid isomers are included in supplemental Figs. S1, S2.
CDCA and UDCA oxidation activity
The CDCA and UDCA oxidation was determined as follows. A typical incubation mixture (final volume of 0.1 ml) contained 100 mM potassium phosphate buffer, pH 7.4, and various enzyme sources (0.4 mg/ml mouse microsomal protein, 1.0 mg/ml S9 from liver tissues of Cyp2c-null, Cyp3a-null, and wild-type mice). In a preliminary study, the rate of formation of α-MCA and β-MCA was found to be linear with respect to the protein concentrations (up to 0.5 mg/ml mouse microsomal protein and incubation time for 30 min). Chenodeoxycholic acid-2,2,4,4-d4 (CDCA-d4; Toronto Research Chemicals, Inc., Toronto, Canada), ursodeoxycholic acid-2,2,4,4-d4, (UDCA-d4; Cambridge Isotope Laboratories, Inc., Andover, MA), and 7-ethoxyresorufin (7-ER; Sigma-Aldrich) were dissolved in DMSO, and the final concentration of DMSO in the incubation mixture was 0.1%. The reaction was initiated by the addition of 4–100 μM CDCA, 40–1,000 μM UDCA, and 1 μM 7- ER, after 7 min preincubation at 37°C. Following a 20 min incubation at 37°C, the reaction was terminated by the addition of 0.1 ml of ice-cold acetonitrile. After removal of the protein by centrifugation at 15,000 g, 4°C, 15 min, an equal volume of HPLC grade water containing 0.1% formic acid was added followed by centrifugation at 15,000 g, 4°C, 15 min. Ten microliters of the supernatant was subjected to UPLC-QTOF-MS. The ions for α-MCA-d4 and β-MCA-d4 were m/z 411.3069 in the negative ion mode.
Transfection of plasmids expressing mouse Cyp2c29 and Cyp2c70
HepG2 cells were maintained in RPMI 1640 medium with L-glutamine containing 10% fetal bovine serum with 5% CO2 at 37°C. The cells were transfected in a 12-well plate with 1 μg of Cyp2c29 expression vector (MGC Premier Expression-Ready cDNA clone for Cyp2c29, pCS6, BC019908; transOMIC Technologies, Huntsville, AL), Cyp2c70 expression vector (MGC Premier Expression-Ready cDNA clone for Cyp2c70, pCS6, BC016494; transOMIC Technologies), and mock vector using Lipofectamine 3000 (Thermo Fisher Scientific). After incubation for 48 h, the cells were treated with 25 μM CDCA-d4 or 400 μM UDCA-d4.
Preparation and treatment of mouse primary hepatocytes
Primary hepatocytes were isolated from C57BL/6J mice as described previously (21). Briefly, after euthanization of mice by CO2 asphyxiation, the abdomen was incised and mesentery and intestine moved to expose the portal vein. A cannula was inserted into the portal vein and the liver was perfused with 40 ml of HBSS without magnesium or calcium (Thermo Fisher Scientific) containing 1 mM EDTA at 4 ml/min. Blood was extravasated by cutting the inferior vena cava. After perfusion of the entire liver using 50 ml of HBSS containing collagenase I and II (0.6 mg/ml each; Thermo Fisher Scientific) and calcium chloride dehydrate (5 mM) at the speed of 4 ml/min, the digested liver was removed and placed in a sterile 10 cm Petri dish with 10 mM PBS. The hepatic capsule was torn by fine-tip forceps and dispersed cells were filtered through a 70 μm cell strainer (Becton, Dickinson, and Company) into a 50 ml tube and centrifuged at 200 g at 4°C for 2 min. Hepatocytes were further washed and purified by gradient centrifugation using Percoll Plus (GE Healthcare, Buckinghamshire, UK). After washing with HBSS and trypan blue staining, the number of hepatocytes were counted and then seeded in collagen-coated 12-well plates (Becton, Dickinson, and Company) at a density of 4 × 105 cells/well. Primary hepatocytes were cultured in William’s E medium (Thermo Fisher Scientific) with 10% fetal bovine serum and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin). Four to six hours after seeding, the cells were treated with 10 nM Cyp2c29 Silencer® Select Predesigned siRNA (Thermo Fisher Scientific), Cyp2c70 Silencer® Select Predesigned siRNA (Thermo Fisher Scientific), and Silencer® Select Negative Control #1 (Thermo Fisher Scientific) using Lipofectamine® RNAiMAX (Thermo Fisher Scientific). Twenty-four hours after siRNA treatment, the cells were treated with medium containing CDCA-d4 or UDCA-d4 for 4 h. At the prescribed time points, medium and cells were harvested and subjected to measurement of bile acids using QTOF-MS and qPCR, respectively.
Statistical analysis
Statistical analysis was performed with Prism version 6 (GraphPad Software). Appropriate statistical analysis was applied, assuming a normal sample distribution. When comparing two groups, statistical significance was determined using two-tailed Student’s t-test. When more than two groups were investigated, one-way ANOVA followed by Tukey’s post hoc correction was applied for comparisons. P < 0.05 was considered as significant difference. Results are expressed as the mean and SD values.
RESULTS
MCA and conjugated-MCA in the liver of Cyp-null mice
To determine the influence of CYPs on the formation of α-MCA and β-MCA, bile acids were measured in the livers of Cyp1a-null, Cyp2c-null, Cyp2d-null, and Cyp3a-null mice. α-MCA and β-MCA were not detected in the livers from Cyp2c-null mice, although they were present in other Cyp-null mouse lines (Fig. 1A). Moreover, T-α-MCA and T-β-MCA were also not detected in Cyp2c-null mouse livers (Fig. 1B). These results suggest that a mouse Cyp2c enzyme might produce α-MCA and β-MCA.
Fig. 1.
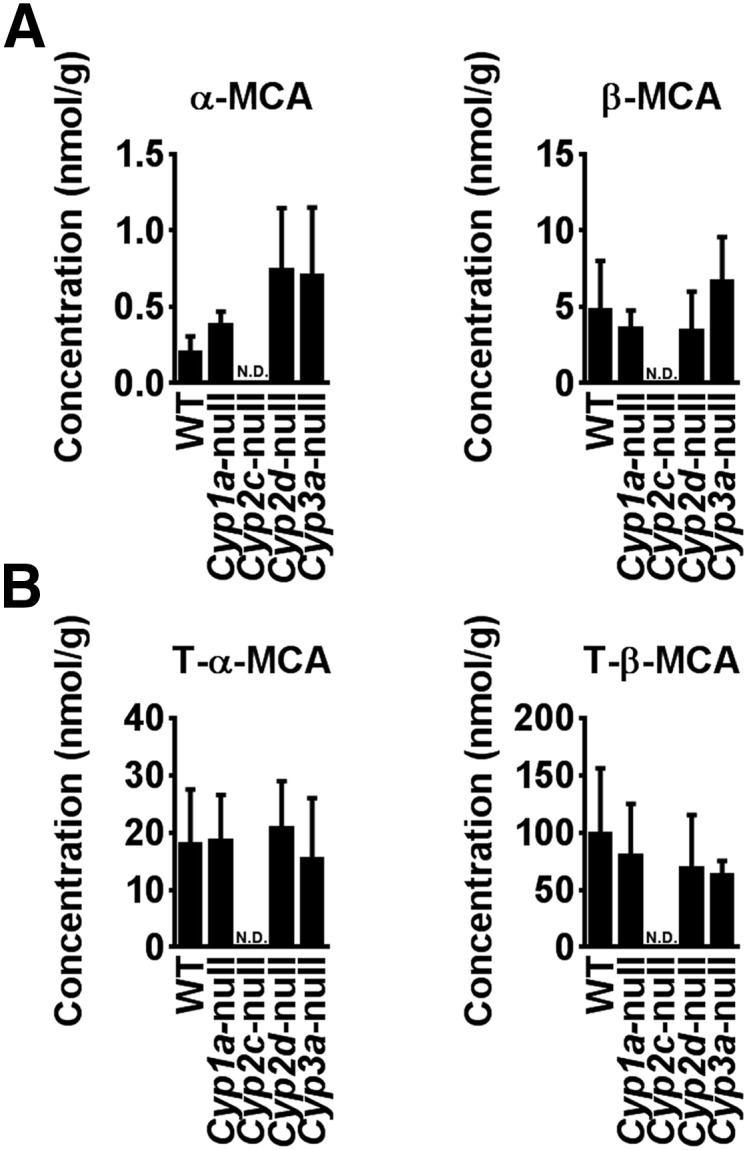
Determination of MCA in various Cyp-null mice. Concentrations of MCA and conjugated-MCA in the liver extracts from Cyp1a-, Cyp2c-, Cyp2d-, Cyp3a-, and wild-type mice. The mice, on a RM1A chow diet, were euthanized in the late morning after a 4 h fast. A: α-MCA and β-MCA in liver samples. B: T-α-MCA and T-β-MCA in liver samples. Data are presented as the mean ± SD (n = 4–5). N.D., not detected.
Kinetic analysis of MCA production in vitro
To investigate the formation of MCA from CDCA and UDCA, the activities of α-MCA and β-MCA production were measured using liver S9 fractions of wild-type, Cyp2c-null, and Cyp3a-null mice at 50 μM CDCA-d4 or 500 μM UDCA-d4 (Fig. 2A, B). α-MCA-d4 was detected in CDCA-d4-treated S9 from wild-type and Cyp3a-null mice, but was not detected in S9 from Cyp2c-null mice (Fig. 2A). β-MCA-d4 was not detected in any of the CDCA-d4-treated groups. α-MCA-d4 was not detected in the UDCA-d4 treated-group, while β-MCA was detected in S9 from wild-type and Cyp3a-null mice, but was undetected in Cyp2c-null mice (Fig. 2B). As a control, it was confirmed that liver S9 fractions from all of mouse lines showed 7-ER O-deethylase activity, a marker activity for mouse Cyp1a2 (Fig. 2C). Kinetic analyses using different concentrations of CDCA and UDCA were performed using liver microsomes to determine the Km and Vmax values for α-MCA and β-MCA productions from CDCA and UDCA, respectively. The Km and Vmax values for α-MCA production were 8.19 ± 0.24 μM and 0.58 ± 0.01 nmol/min/mg, and those for β-MCA production were 321 ± 46 μM and 0.418 ± 0.03 nmol/min/mg, respectively (Fig. 2D). These data indicate that the catalytic efficiency for α-MCA production is higher than that for β-MCA production.
Fig. 2.

Kinetic analyses of MCA oxidation activities in mouse liver S9 and microsomes. A: α-MCA-d4 and β-MCA-d4 production from CDCA-d4 by liver S9 from Cyp2c- and Cyp3a-null mice and WT mice. B: α-MCA-d4 and β-MCA-d4 production from UDCA-d4 by liver S9 from Cyp2c- and Cyp3a-null mice, and WT mice. C: Resorufin production from 7-ER by liver S9 from Cyp2c- and Cyp3a-null mice, and WT mice. D: Kinetics of α-MCA-d4 and β-MCA-d4 production from CDCA-d4 and UDCA-d4 by mouse liver microsomes, respectively. The kinetic parameters were estimated from the fitted curve using the computer program GraphPad Prism designed for nonlinear regression analysis. Each data point represents the mean ± SD of triplicate determinations. N.D., not detected; conc., concentration.
Levels of individual Cyp2c mRNAs in wild-type mice
There are 16 Cyp2c genes in mice, including Cyp2c53, a pseudogene, and their proteins are mainly expressed in the liver. The expression of each Cyp2c mRNA in liver was determined by RT-qPCR. The levels of Cyp2c29, Cyp2c50, Cyp2c67, Cyp2c69, and Cyp2c70 mRNAs were 5.3-, 1.8-, 3.0-, 1.5-, and 1.9-fold that of Cyp2c44 mRNA, which is the only Cyp2c gene not deleted in the Cyp2c-null mice (Fig. 3). The mouse Cyp2c cluster, with the exception of Cyp2c44, which is located 4 Mb from the main Cyp2c gene cluster, was flanked with Cre recombinase recognition (loxP) sites using two consecutive rounds of targeting in mouse ES cells (20).
Fig. 3.

Expression levels of mouse Cyp2c mRNA. Relative mRNA expression levels of mouse Cyp2c mRNAs in liver were determined by RT-qPCR analysis. Levels are relative to Cyp2c44 mRNA. Each column represents the mean ± SD (n = 5).
CDCA and UDCA oxidation activity by recombinant mouse Cyp2c70
To investigate which mouse Cyp2c isoform produces α-MCA from CDCA, Cyp2c29 and Cyp2c70 were transiently expressed in HepG2 cells, as revealed by mRNA expression (Fig. 4A). Recombinant Cyp2c70 generated α-MCA-d4 from CDCA, while Cyp2c29 had no activity. β-MCA was not detected in these groups (Fig. 4B). In contrast, α-MCA-d4 was not detected when UDCA-d4 was used as a substrate. Cyp2c70-expressing cells also produced β-MCA from UDCA (Fig. 4C). These results suggest that Cyp2c70 catalyzed the oxidation of CDCA and UDCA to α-MCA and β-MCA, respectively.
Fig. 4.

MCA production in HepG2 cells expressing recombinant Cyp2c70. A: Cyp2c29 and Cyp2c70 mRNA levels in HepG2 cells transfected with Cyp expression vectors. B: α-MCA-d4 and β-MCA-d4 production in HepG2 cells transfected with Cyp2c29 and Cyp2c70 expression vectors and the cells were incubated with 50 μM CDCA-d4. C: α-MCA-d4 and β-MCA-d4 production in HepG2 cells transfected with Cyp2c29 and Cyp2c70 expression vectors. Cells were incubated with 500 μM UDCA-d4. Data are presented as the mean ± SD (n = 3).
Knockdown of Cyp2c70 reduces MCA production in mouse primary hepatocytes
To further confirm whether Cyp2c70 produces MCA from CDCA or UDCA, siRNA against Cyp2c29 (Si-Cyp2c29) and Cyp2c70 (Si-Cyp2c70) were transfected to mouse primary hepatocytes. Si-Cyp2c29 and si-Cyp2c70 significantly decreased Cyp2c29 and Cyp2c70 mRNA levels, respectively, and did not affect Cyp2c44 mRNA levels (Fig. 5A). In the presence of CDCA, α-MCA levels were specifically decreased in the si-Cyp2c70-treated cells, while β-MCA production was lowered in mouse primary hepatocytes in the presence of UDCA. This result revealed that Cyp2c70 is responsible for MCA production from CDCA and UDCA in mouse liver.
Fig. 5.

MCA production in siRNA for Cyp2c70-transfected mouse primary hepatocytes. A: Cyp2c29 and Cyp2c70 mRNA levels in mouse primary hepatocytes transfected with siRNAs against Cyp2c29 (Si-Cyp2c29) and Cyp2c70 (Si-Cyp2c70). B: α-MCA-d4 and β-MCA-d4 production in mouse primary hepatocytes transfected with Si-Cyp2c29 and Si-Cyp2c70. Hepatocytes were incubated with 50 μM CDCA-d4. C: α-MCA-d4 and β-MCA-d4 production in mouse primary hepatocytes transfected with Si-Cyp2c29 and Si-Cyp2c70. Hepatocytes were incubated with 500 μM UDCA-d4. Data are presented as the mean ± S. D. (n = 3).
Bile acids in the liver, cecum, and feces from Cyp2c-null mice and hCYP2C9 mice
To determine the consequence of loss of Cyp2c70 in the Cyp2c-null and hCYP2C9 mice, bile acids were measured in liver, cecum, and feces of wild-type, Cyp2c-null, and hCYP2C9 mice. hCYP2C9 mice were generated from the Cyp2c-deleted ES cells described above by further Cre-mediated insertion of an expression cassette in which the human CYP2C9 gene is under control of the liver-specific mouse albumin promoter (20). In this study, the hCYP2C9 mice were used for the human model because among human CYP2C enzymes, CYP2C9 is most abundant in the liver and is involved in the metabolism of various endogenous and exogenous compounds. α-MCA and β-MCA and their taurine conjugates were not detected in the liver, cecum, and feces of Cyp2c-null and hCYP2C9 mice (Fig. 6). Moreover, the contents of CDCA and UDCA, which are precursors of α-MCA and β-MCA, in Cyp2c-null and hCYP2C9 mice, respectively, were significantly higher than those in wild-type mice. The hepatic, cecum, and feces contents of T-CDCA and T-UDCA were also higher in Cyp2c-null and hCYP2C9 mice. In cecum and feces, lithocholic acid (LCA) was also significantly highly detected in Cyp2c-null and hCYP2C9 mice as compared with wild-type mice. In addition, T-LCA was highly detected in these mice.
Fig. 6.

Bile acid profile in the liver, cecum, and feces from WT, Cyp2c-null mice, and hCYP2C9 mice. A: Unconjugated-bile acids and taurine-conjugated-bile acids in liver samples of Cyp2c-null mice, hCYP2C9 mice, and WT mice. B: Unconjugated-bile acids and taurine-conjugated-bile acids in cecum samples of Cyp2c-null mice, hCYP2C9 mice, and WT mice. C: Unconjugated-bile acids and taurine-conjugated-bile acids in feces samples of Cyp2c-null mice, hCYP2C9 mice, and WT mice. Data are presented as the mean ± SD (n = 3). N, not detected.
DISCUSSION
An earlier study revealed that mice with hepatocyte-specific deletion of NADPH-CYP reductase (22), which decreased all CYP activities, were found to have marked differences in bile acid compositions, including lower MCA and taurine-conjugated MCA as compared with their wild-type counterparts (23). However, this study could not distinguish which CYP isoform produced MCA. The current work demonstrates that mouse Cyp2c70 is responsible for production of MCA from CDCA or UDCA. Cyp2c-null mice and hCYP2C9 mice did not produce any MCA, in contrast to wild-type mice and mouse lines lacking expression of Cyp1a, Cyp2d, and Cyp3a isoforms. Cyp2c-null mice also have a high concentration of CDCA and UDCA (and their taurine conjugates), the substrates for α-MCA and β-MCA, respectively.
LCA is produced from CDCA in the human and mouse large intestine. LCA is present in only trace levels in mouse liver, likely due to the low concentrations of CDCA. LCA, and T-LCA, which are present at very low levels in wild-type mice, were found at significant concentrations in Cyp2c-null and hCYP2C9 mouse livers. The hepatic Cyp7a1 and Cyp8b1 mRNA levels were not significantly different between wild-type, Cyp2c-null, and hCYP2C9 mice (supplemental Fig. S3). The increased concentrations of CDCA in livers of Cyp2c-null mice led to the production of LCA through a 7-dehydroxylation reaction by gut bacteria (24). LCA is then reabsorbed to the liver where it is conjugated with taurine. This is of particular interest because LCA is considered to be a toxic bile acid (25). Thus, the lack of MCA production indirectly causes an increase in LCA. However, LCA is efficiently conjugated in mice, leading to decreased potential for hepatotoxicity. Indeed, the homozygous hCYP2C9 and Cyp2c-null mice appeared normal and could not be distinguished from wild-type mice; they had normal body weights, liver weights, and fertility (supplemental Fig. S4A). The Cyp2c-null mice showed no evidence of liver damage as compared with wild-type mice, although an increase in aspartate transaminase and alanine transaminase was observed in some mice due to inter-animal variability, this change was not significant. The only significant phenotypic change in the hCYP2C9 mice was a decrease in alkaline phosphatase activity, while the Cyp2c-null mice exhibited a similar change and a small but significant decrease in plasma high-density lipoprotein and cholesterol (20).
T-β-MCA was identified as a potent FXR antagonist in mice (9, 26). Inhibition of intestinal FXR resulted in the alleviation of metabolic disease, including obesity, insulin resistance, and hepatic steatosis (7–9). However, the question arises whether human bile acids would have similar effects on FXR because humans do not produce MCA and, thus, T-β-MCA, due to a lack of a CYP2C enzyme activity similar to mouse Cyp2c70. It is also noteworthy that UDCA is elevated in the Cyp2c-null mice and shows potential FXR antagonist activity in humans (27). FXR signaling in mice is dependent on the relative local concentration of endogenous agonist and antagonist that result from bile acid metabolism in the liver and intestine. Body weights and hepatic lipid concentrations (triglyceride, total cholesterol, phospholipid, and nonesterified fatty acid) were not significantly changed in wild-type, Cyp2c-null, and hCYP2C9 mice (supplemental Fig. S4B). The impact of these changes on the susceptibility to metabolic disease in the Cyp2c-null mice is an area of great interest and will require analysis of high-fat diet-induced obesity, insulin resistance, and fatty livers (7, 9).
The mouse and the human CYP2C cluster differ significantly in their genomic organization, with 15 functional genes described in mice compared with only four genes in humans. Analysis of hepatic mRNAs revealed that Cyp2c29 is the most abundant while Cyp2c50, Cyp2c67, Cyp2c69, and Cyp2c70 mRNAs are expressed at similar levels (12). Humans do not have an obvious homolog of Cyp2c70, at least at the level of primary amino acid sequence comparison (CYP2C8, 76%; CYP2C9, 79%; CYP2C18, 79%; CYP2C19, 79%) (Table 1). However, there is significant protein homology between Cyp2c70 and CYP2C22 which is the rat homolog of Cyp2c70 (Table 2). This is noteworthy because rats also have MCA similar to mice (28). Murine Cyp2c37/38/39 and Cyp2c40 genes are endogenous female-specific Cyp2c genes. However, a much smaller gender difference in expression is noted with Cyp2c29 and Cyp2c70 when comparing wild-type males and wild-type females (29). These findings support the view that Cyp2c70 is a primary enzyme responsible for MCA production. No other specific substrates for Cyp2c70 have been reported.
TABLE 1.
Primary amino acid sequence comparison between mouse Cyp2c and human CYP2C
| Mouse | Protein Homology (%) | Human |
| Cyp2c29 | 83 | CYP2C8 |
| Cyp2c37 | — | — |
| Cyp2c38 | 84 | CYP2C8 |
| Cyp2c39 | 73 | CYP2C8 |
| Cyp2c40 | — | — |
| Cyp2c44 | — | — |
| Cyp2c50 | — | — |
| Cyp2c54 | — | — |
| Cyp2c55 | 88 | CYP2C18 |
| Cyp2c65 | 86 | CYP2C9 |
| Cyp2c66 | 85 | CYP2C9 |
| Cyp2c67 | — | — |
| Cyp2c68 | — | — |
| Cyp2c69 | — | — |
| Cyp2c70 | — | — |
TABLE 2.
Primary amino acid sequence comparison between mouse Cyp2c and rat CYP2C
| Mouse | Protein Homology (%) | Rat |
| Cyp2c29 | 82 | CYP2C7 |
| Cyp2c37 | — | — |
| Cyp2c38 | 82 | CYP2C7 |
| Cyp2c39 | 82 | CYP2C7 |
| Cyp2c40 | — | — |
| Cyp2c44 | 95 | CYP2C23 |
| Cyp2c50 | — | — |
| Cyp2c54 | — | — |
| Cyp2c55 | 95 | CYP2C24 |
| Cyp2c65 | 84 | CYP2C11 |
| Cyp2c66 | 84 | CYP2C11 |
| Cyp2c67 | — | — |
| Cyp2c68 | — | — |
| Cyp2c69 | — | — |
| Cyp2c70 | 94 | CYP2C22 |
In mice, β-MCA and T-β-MCA appear to be more abundant than α-MCA and T-α-MCA. However, kinetic studies revealed a higher affinity of Cyp2c70 for CDCA than UDCA. These results suggest that α-MCA production is higher than that of β-MCA due to increased catalytic efficiency toward CDCA in mouse. The microbiota is involved in the metabolism of bile acids, particularly dehydroxylation and deconjugation reactions (9, 30, 31). Epimerization from α-MCA to β-MCA might occur in metabolism by mouse intestine microbiota through enzymes other than CYPs. Oxidation and epimerization of the 7-hydroxy groups of bile acids in the intestine are carried out by hydroxysteroid dehydrogenase expressed by intestinal bacteria (32).
Some studies have investigated whether bile acid concentrations might be useful to differentiate among various liver diseases (33–35). Bile acids also act as agonists or antagonists for nuclear receptors such as FXR, pregnane X receptor (NR1I2), vitamin D receptor (NR1I1), and the G protein-coupled bile acid receptor, TGR5 (36). However, it is known that bile salt composition markedly differs between various species (37). As noted above, T-β-MCA is an FXR antagonist in mouse intestine, but humans do not produce MCA (26).
In addition, humans make glycine conjugates of bile acids while mice only make taurine conjugates. Thus, mice are a poor model to investigate and predict the influence of bile acid metabolites in human disease. Mice lacking expression of the Cyp2c cluster showed similar bile acid profiling to humans, but still made taurine conjugates. Perhaps humanizing the bile acid conjugating enzymes, bile acid-CoA ligase and bile acid-CoA:amino acid N-acyltransferase, would make a more complete bile acid-humanized mouse line. Further, the bile acid profiles indicate that the gut microbiota of the Cyp2c-null mice may not optimally hydrolyze the taurine conjugates of CDCA and LCA because T-CDCA and T-LCA accumulate in the cecum and feces. Because mouse gut microbiota seldom encounter these conjugates, the Cyp2c-null mice may not be an accurate model for human bile acid metabolism. Therefore, a mouse model that is optimal to study human diseases related to bile acids may have to be colonized with human gut microbiota. Taken together, the present study revealed that Cyp2c70 is the principal enzyme involved in MCA production and is responsible for the differences in bile acid metabolite profile between humans and mice.
Supplementary Material
Acknowledgments
The authors wish to acknowledge the assistance of Julia Carr, Aileen McLaren, and Tania Frangova for performing the animal studies in University of Dundee.
Footnotes
Abbreviations:
- CDCA
- chenodeoxycholic acid
- CDCA-d4
- chenodeoxycholic acid-2,2,4,4-d4
- CYP
- cytochrome P450
- Cyp1a-null
- Cyp1a-cluster null
- Cyp2c-null
- Cyp2c-cluster null
- Cyp2d-null
- Cyp2d-cluster null
- Cyp3a-null
- Cyp3a-cluster null
- Cyp7a1
- cholesterol 7α-hydroxylase
- 7-ER
- 7-ethoxyresorufin
- FXR
- farnesoid X receptor
- hCYP2C9
- CYP2C9-humanized
- LCA
- lithocholic acid
- MCA
- muricholic acid
- QTOF-MS
- quadrupole TOF-MS
- .T-
- tauro-
- UDCA
- ursodeoxycholic acid
- UDCA-d4
- ursodeoxycholic acid-2,2,4,4-d4
- UPLC
- ultra-performance LC
This work was supported by the National Cancer Institute Intramural Research Program, Center for Cancer Research, and National Institutes of Health Grant U54 ES16015 (F.J.G), and a Programme Grant support from Cancer Research UK, C4639/A10822 to C.R.W. S.T. was supported by a Japanese Society for the Promotion of Science Research Fellowship for Japanese Biomedical and Behavioral Researchers at the National Institutes of Health (KAITOKU-NIH). T.F. and Y.M. were supported by a program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers (S2601) from the Japanese Society for the Promotion of Sciences. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The online version of this article (available at http://www.jlr.org) contains a supplement.
REFERENCES
- 1.Russell D. W. 2003. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 72: 137–174. [DOI] [PubMed] [Google Scholar]
- 2.Hofmann A. F. 2009. The enterohepatic circulation of bile acids in mammals: form and functions. Front. Biosci. (Landmark Ed.). 14: 2584–2598. [DOI] [PubMed] [Google Scholar]
- 3.Matsubara T., Li F., and Gonzalez F. J.. 2013. FXR signaling in the enterohepatic system. Mol. Cell. Endocrinol. 368: 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzalez F. J. 2012. Nuclear receptor control of enterohepatic circulation. Compr. Physiol. 2: 2811–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deo A. K., and Bandiera S. M.. 2008. Identification of human hepatic cytochrome p450 enzymes involved in the biotransformation of cholic and chenodeoxycholic acid. Drug Metab. Dispos. 36: 1983–1991. [DOI] [PubMed] [Google Scholar]
- 6.Hardison W. G. 1978. Hepatic taurine concentration and dietary taurine as regulators of bile acid conjugation with taurine. Gastroenterology. 75: 71–75. [PubMed] [Google Scholar]
- 7.Jiang C., Xie C., Li F., Zhang L., Nichols R. G., Krausz K. W., Cai J., Qi Y., Fang Z. Z., Takahashi S., et al. . 2015. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Invest. 125: 386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang C., Xie C., Lv Y., Li J., Krausz K. W., Shi J., Brocker C. N., Desai D., Amin S. G., Bisson W. H., et al. . 2015. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat. Commun. 6: 10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li F., Jiang C., Krausz K. W., Li Y., Albert I., Hao H., Fabre K. M., Mitchell J. B., Patterson A. D., and Gonzalez F. J.. 2013. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 4: 2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russell D. W., and Setchell K. D.. 1992. Bile acid biosynthesis. Biochemistry. 31: 4737–4749. [DOI] [PubMed] [Google Scholar]
- 11.Inagaki T., Choi M., Moschetta A., Peng L., Cummins C. L., McDonald J. G., Luo G., Jones S. A., Goodwin B., Richardson J. A., et al. . 2005. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2: 217–225. [DOI] [PubMed] [Google Scholar]
- 12.Nelson D. R., Zeldin D. C., Hoffman S. M., Maltais L. J., Wain H. M., and Nebert D. W.. 2004. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics. 14: 1–18. [DOI] [PubMed] [Google Scholar]
- 13.Scheer N., McLaughlin L. A., Rode A., Macleod A. K., Henderson C. J., and Wolf C. R.. 2014. Deletion of 30 murine cytochrome p450 genes results in viable mice with compromised drug metabolism. Drug Metab. Dispos. 42: 1022–1030. [DOI] [PubMed] [Google Scholar]
- 14.Cheung C., and Gonzalez F. J.. 2008. Humanized mouse lines and their application for prediction of human drug metabolism and toxicological risk assessment. J. Pharmacol. Exp. Ther. 327: 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheer N., and Wolf C. R.. 2014. Genetically humanized mouse models of drug metabolizing enzymes and transporters and their applications. Xenobiotica. 44: 96–108. [DOI] [PubMed] [Google Scholar]
- 16.van Herwaarden A. E., Wagenaar E., van der Kruijssen C. M., van Waterschoot R. A., Smit J. W., Song J. Y., van der Valk M. A., van Tellingen O., van der Hoorn J. W., Rosing H., et al. . 2007. Knockout of cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism. J. Clin. Invest. 117: 3583–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasegawa M., Kapelyukh Y., Tahara H., Seibler J., Rode A., Krueger S., Lee D. N., Wolf C. R., and Scheer N.. 2011. Quantitative prediction of human pregnane X receptor and cytochrome P450 3A4 mediated drug-drug interaction in a novel multiple humanized mouse line. Mol. Pharmacol. 80: 518–528. [DOI] [PubMed] [Google Scholar]
- 18.Scheer N., Kapelyukh Y., McEwan J., Beuger V., Stanley L. A., Rode A., and Wolf C. R.. 2012. Modeling human cytochrome P450 2D6 metabolism and drug-drug interaction by a novel panel of knockout and humanized mouse lines. Mol. Pharmacol. 81: 63–72. [DOI] [PubMed] [Google Scholar]
- 19.Dragin N., Uno S., Wang B., Dalton T. P., and Nebert D. W.. 2007. Generation of ‘humanized’ hCYP1A1_1A2_Cyp1a1/1a2(-/-) mouse line. Biochem. Biophys. Res. Commun. 359: 635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scheer N., Kapelyukh Y., Chatham L., Rode A., Buechel S., and Wolf C. R.. 2012. Generation and characterization of novel cytochrome P450 Cyp2c gene cluster knockout and CYP2C9 humanized mouse lines. Mol. Pharmacol. 82: 1022–1029. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka N., Takahashi S., Zhang Y., Krausz K. W., Smith P. B., Patterson A. D., and Gonzalez F. J.. 2015. Role of fibroblast growth factor 21 in the early stage of NASH induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta. 1852: 1242–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu J., Weng Y., Zhang Q. Y., Cui H., Behr M., Wu L., Yang W., Zhang L., and Ding X.. 2003. Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J. Biol. Chem. 278: 25895–25901. [DOI] [PubMed] [Google Scholar]
- 23.Cheng X., Zhang Y., and Klaassen C. D.. 2014. Decreased bile-acid synthesis in livers of hepatocyte-conditional NADPH-cytochrome P450 reductase-null mice results in increased bile acids in serum. J. Pharmacol. Exp. Ther. 351: 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fedorowski T., Salen G., Tint G. S., and Mosbach E.. 1979. Transformation of chenodeoxycholic acid and ursodeoxycholic acid by human intestinal bacteria. Gastroenterology. 77: 1068–1073. [PubMed] [Google Scholar]
- 25.Hofmann A. F. 2004. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab. Rev. 36: 703–722. [DOI] [PubMed] [Google Scholar]
- 26.Sayin S. I., Wahlstrom A., Felin J., Jantti S., Marschall H. U., Bamberg K., Angelin B., Hyotylainen T., Oresic M., and Backhed F.. 2013. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 17: 225–235. [DOI] [PubMed] [Google Scholar]
- 27.Mueller M., Thorell A., Claudel T., Jha P., Koefeler H., Lackner C., Hoesel B., Fauler G., Stojakovic T., Einarsson C., et al. . 2015. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J. Hepatol. 62: 1398–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katagiri K., Nakai T., Hoshino M., Hayakawa T., Ohnishi H., Okayama Y., Yamada T., Ohiwa T., Miyaji M., and Takeuchi T.. 1992. Tauro-beta-muricholate preserves choleresis and prevents taurocholate-induced cholestasis in colchicine-treated rat liver. Gastroenterology. 102: 1660–1667. [DOI] [PubMed] [Google Scholar]
- 29.Löfgren S., Baldwin R. M., Carlerös M., Terelius Y., Fransson-Steen R., Mwinyi J., Waxman D. J., and Ingelman-Sundberg M.. 2009. Regulation of human CYP2C18 and CYP2C19 in transgenic mice: influence of castration, testosterone, and growth hormone. Drug Metab. Dispos. 37: 1505–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greathouse K. L., Harris C. C., and Bultman S. J.. 2015. Dysfunctional families: Clostridium scindens and secondary bile acids inhibit the growth of Clostridium difficile. Cell Metab. 21: 9–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buffie C. G., Bucci V., Stein R. R., McKenney P. T., Ling L., Gobourne A., No D., Liu H., Kinnebrew M., Viale A., et al. . 2015. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 517: 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirano S., and Masuda N.. 1981. Epimerization of the 7-hydroxy group of bile acids by the combination of two kinds of microorganisms with 7 alpha- and 7 beta-hydroxysteroid dehydrogenase activity, respectively. J. Lipid Res. 22: 1060–1068. [PubMed] [Google Scholar]
- 33.de Aguiar Vallim T. Q., Tarling E. J., Ahn H., Hagey L. R., Romanoski C. E., Lee R. G., Graham M. J., Motohashi H., Yamamoto M., and Edwards P. A.. 2015. MAFG is a transcriptional repressor of bile acid synthesis and metabolism. Cell Metab. 21: 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seeley R. J., Chambers A. P., and Sandoval D. A.. 2015. The role of gut adaptation in the potent effects of multiple bariatric surgeries on obesity and diabetes. Cell Metab. 21: 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones R. D., Lopez A. M., Tong E. Y., Posey K. S., Chuang J. C., Repa J. J., and Turley S. D.. 2015. Impact of physiological levels of chenodeoxycholic acid supplementation on intestinal and hepatic bile acid and cholesterol metabolism in Cyp7a1-deficient mice. Steroids. 93: 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas C., Gioiello A., Noriega L., Strehle A., Oury J., Rizzo G., Macchiarulo A., Yamamoto H., Mataki C., Pruzanski M., et al. . 2009. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 10: 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Botham K. M., and Boyd G. S.. 1983. The metabolism of chenodeoxycholic acid to beta-muricholic acid in rat liver. Eur. J. Biochem. 134: 191–196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.