

Abstract
Background
Major depressive disorder (MDD) is highly debilitating, difficult to treat, has a high rate of recurrence, and negatively impacts the individual and society as a whole. One emerging potential treatment for MDD is n‐3 polyunsaturated fatty acids (n‐3PUFAs), also known as omega‐3 oils, naturally found in fatty fish, some other seafood, and some nuts and seeds. Various lines of evidence suggest a role for n‐3PUFAs in MDD, but the evidence is far from conclusive. Reviews and meta‐analyses clearly demonstrate heterogeneity between studies. Investigations of heterogeneity suggest differential effects of n‐3PUFAs, depending on severity of depressive symptoms, where no effects of n‐3PUFAs are found in studies of individuals with mild depressive symptomology, but possible benefit may be suggested in studies of individuals with more severe depressive symptomology.
Objectives
To assess the effects of n‐3 polyunsaturated fatty acids (also known as omega‐3 fatty acids) versus a comparator (e.g. placebo, anti‐depressant treatment, standard care, no treatment, wait‐list control) for major depressive disorder (MDD) in adults.
Search methods
We searched the Cochrane Depression, Anxiety and Neurosis Review Group’s Specialised Registers (CCDANCTR) and International Trial Registries over all years to May 2015. We searched the database CINAHL over all years of records to September 2013.
Selection criteria
We included studies in the review if they: were a randomised controlled trial; provided n‐3PUFAs as an intervention; used a comparator; measured depressive symptomology as an outcome; and were conducted in adults with MDD. Primary outcomes were depressive symptomology (continuous data collected using a validated rating scale) and adverse events. Secondary outcomes were depressive symptomology (dichotomous data on remission and response), quality of life, and failure to complete studies.
Data collection and analysis
We used standard methodological procedures as expected by Cochrane.
Main results
We found 26 relevant studies: 25 studies involving a total of 1438 participants investigated the impact of n‐3PUFA supplementation compared to placebo, and one study involving 40 participants investigated the impact of n‐3PUFA supplementation compared to antidepressant treatment.
For the placebo comparison, n‐3PUFA supplementation results in a small to modest benefit for depressive symptomology, compared to placebo: standardised mean difference (SMD) ‐0.30 (95% confidence interval (CI) ‐0.10 to ‐0.50; 25 studies, 1373 participants, very low quality evidence), but this effect is unlikely to be clinically meaningful (an SMD of 0.30 represents a difference between groups in scores on the HDRS (17‐item) of approximately 2.1 points (95% CI 0.7 to 3.5)). The confidence intervals include both a possible clinically important effect and a possible negligible effect, and there is considerable heterogeneity between the studies. Although the numbers of individuals experiencing adverse events were similar in intervention and placebo groups (odds ratio (OR) 1.24, 95% CI 0.95 to 1.62; 19 studies, 1207 participants; very low‐quality evidence), the confidence intervals include a significant increase in adverse events with n‐3PUFAs as well as a small possible decrease. Rates of remission and response, quality of life, and rates of failure to complete studies were also similar between groups, but confidence intervals are again wide.
The evidence on which these results are based is very limited. All studies contributing to our analyses were of direct relevance to our research question, but we rated the quality of the evidence for all outcomes as low to very low. The number of studies and number of participants contributing to all analyses were low, and the majority of studies were small and judged to be at high risk of bias on several measures. Our analyses were also likely to be highly influenced by three large trials. Although we judge these trials to be at low risk of bias, they contribute 26.9% to 82% of data. Our effect size estimates are also imprecise. Funnel plot asymmetry and sensitivity analyses (using fixed‐effect models, and only studies judged to be at low risk of selection bias, performance bias or attrition bias) also suggest a likely bias towards a positive finding for n‐3PUFAs. There was substantial heterogeneity in analyses of our primary outcome of depressive symptomology. This heterogeneity was not explained by the presence or absence of comorbidities or by the presence or absence of adjunctive therapy.
Only one study was available for the antidepressant comparison, involving 40 participants. This study found no differences between treatment with n‐3PUFAs and treatment with antidepressants in depressive symptomology (mean difference (MD) ‐0.70 (95% CI ‐5.88 to 4.48)), rates of response to treatment or failure to complete. Adverse events were not reported in a manner suitable for analysis, and rates of depression remission and quality of life were not reported.
Authors' conclusions
At present, we do not have sufficient high quality evidence to determine the effects of n‐3PUFAs as a treatment for MDD. Our primary analyses suggest a small‐to‐modest, non‐clinically beneficial effect of n‐3PUFAs on depressive symptomology compared to placebo; however the estimate is imprecise, and we judged the quality of the evidence on which this result is based to be low/very low. Sensitivity analyses, funnel plot inspection and comparison of our results with those of large well‐conducted trials also suggest that this effect estimate is likely to be biased towards a positive finding for n‐3PUFAs, and that the true effect is likely to be smaller. Our data, however, also suggest similar rates of adverse events and numbers failing to complete trials in n‐3PUFA and placebo groups, but again our estimates are very imprecise. The one study that directly compares n‐3PUFAs and antidepressants in our review finds comparable benefit. More evidence, and more complete evidence, are required, particularly regarding both the potential positive and negative effects of n‐3PUFAs for MDD.
Keywords: Adult; Humans; Antidepressive Agents; Antidepressive Agents/adverse effects; Antidepressive Agents/therapeutic use; Depressive Disorder, Major; Depressive Disorder, Major/drug therapy; Fatty Acids, Omega‐3; Fatty Acids, Omega‐3/adverse effects; Fatty Acids, Omega‐3/therapeutic use; Randomized Controlled Trials as Topic
Omega‐3 fatty acids for depression in adults
Why is this review important?
Major depressive disorder (MDD) is characterised by depressed mood and/or a markedly decreased pleasure or interest in all activities. It has negative impacts on the individual and on society, often over the long term. One possible treatment for MDD is n‐3 polyunsaturated fatty acids (n‐3PUFAs), also known as omega‐3 oils, naturally found in fatty fish, some other seafood and some nuts and seeds. Various lines of evidence suggests that n‐3PUFAs may impact on depressive symptoms, but a lot of studies have different findings, making it difficult to draw conclusions.
Who will be interested in this review?
Health professionals, including general practitioners, mental health and psychiatric specialists; individuals with MDD, more mild or additional depressive disorders; and the people around them.
What questions does this review aim to answer?
Do n‐3PUFAs, compared to an alternative, have an effect on depressive symptoms, negative side effects, rates of recovery, quality of life, and rates of dropout from studies, in individuals with a diagnosis of MDD?
Which studies were included in the review?
We searched scientific databases for all randomised controlled trials in adults with a diagnosis of MDD, where individuals received either n‐3PUFAs or an alternative, that were carried out up to May 2015.
We found 26 relevant studies: 25 studies involving 1438 people compared the impact of n‐3PUFAs with that of placebo, and one study involving 40 people compared the impact of n‐3PUFAs with that of antidepressants. All studies were of direct relevance to our review, but we considered the quality of the evidence to be low to very low.
What does the evidence from the review tell us?
At present, we do not have enough high quality evidence to determine the effects of n‐3PUFAs as a treatment for MDD. We found a small‐to‐modest positive effect of n‐3PUFAs compared to placebo, but the size of this effect is unlikely to be meaningful to people with depression, and we considered the evidence to be of low or very low quality, with many differences between studies. There was also insufficient high quality evidence to determine the effects of n‐3PUFAs on negative side effects or numbers failing to complete trials.
What should happen next?
We need more evidence, particularly to explain the differences between study findings, e.g. by looking at individuals who may and may not benefit from n‐3PUFAs. Future studies should also compare n‐3PUFAs with usual antidepressant treatment, and investigate the way these treatments may work.
Summary of findings
Summary of findings for the main comparison.
n‐3PUFAs compared to placebo for depression in adults
| n‐3PUFAs compared to placebo for depression in adults | |||||
| Patient or population: adult patients with depression Settings: Clinical and community settings Intervention: n‐3PUFAs Comparison: Placebo | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Placebo | N‐3PUFAs | ||||
| Depressive symptomology (continuous) HDRS where possible; higher scores indicate greater symptomology Follow‐up: 4 ‐ 16 weeks | The mean depressive symptomology (continuous) in the intervention groups was 0.30 standard deviations lower (0.50 to 0.10 lower). This represents a difference between groups in scores on the HDRS (17‐item) of approximately 2.1 points (95% CI 0.7 to 3.5). | 1373 (25 studies) | ⊕⊝⊝⊝ very low1,2,3,4,5 | ||
| Adverse events Study reports Follow‐up: 0 ‐ 16 weeks | Study population | OR 1.24 (0.95 to 1.62) | 1207 (19 studies) | ⊕⊝⊝⊝ very low3,4,5,6,7 | |
| 482 per 1000 | 536 per 1000 (469 to 601) | ||||
| Moderate | |||||
| 208 per 1000 | 246 per 1000 (200 to 298) | ||||
| Depressive symptomology (dichotomous ‐ remission) Depressive symptomology rating scale as used by authors Follow‐up: 4 ‐ 16 weeks | Study population | OR 1.38 (0.87 to 2.2) | 426 (6 studies) | ⊕⊕⊝⊝ low3,4,6,7,8,9 | |
| 238 per 1000 | 301 per 1000 (214 to 407) | ||||
| Moderate | |||||
| 216 per 1000 | 275 per 1000 (193 to 377) | ||||
| Depressive symptomology (dichotomous ‐ response) Depressive symptomology rating scale as used by authors Follow‐up: 4 ‐ 16 weeks | Study population | OR 1.39 (0.95 to 2.04) | 611 (15 studies) | ⊕⊕⊝⊝ low3,4,6,7,8,9 | |
| 328 per 1000 | 404 per 1000 (317 to 499) | ||||
| Moderate | |||||
| 235 per 1000 | 299 per 1000 (226 to 385) | ||||
| Quality of life Validated scales as used by authors, CGI where possible, higher scores indicate poorer quality of life Follow‐up: 4‐16 weeks | The mean quality of life in the intervention groups was 0.47 standard deviations lower (0.99 lower to 0.06 higher) | 383 (9 studies) | ⊕⊝⊝⊝ very low3,4,6,8,9,10 | ||
| Failure to complete Study reports Follow‐up: 0‐16 weeks | Study population | OR 0.84 (0.62 to 1.14) | 1344 (21 studies) | ⊕⊝⊝⊝ very low3,4,5,6,7 | |
| 192 per 1000 | 166 per 1000 (128 to 213) | ||||
| Moderate | |||||
| 200 per 1000 | 174 per 1000 (134 to 222) | ||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1Quality of the evidence downgraded by one level for study limitations. Judgements of high risk of bias in all studies, and different effects when comparing analyses including only those studies with judgements of low risk of selection bias (allocation concealment), performance bias (blinding of participants and personnel), or attrition bias (incomplete outcome data), and analyses including all studies 2Quality of the evidence downgraded by one level for inconsistency. Evidence of high heterogeneity between studies. Heterogeneity not well explained by the subgroup analyses 3No serious concerns regarding indirectness. All evidence is directly related to the research question 4Quality of the evidence downgraded by one level for imprecision. Moderate to wide confidence intervals 5Quality of the evidence downgraded by one level for publication bias. Strong suspicion of publication bias based on visual inspection of the funnel plot 6Quality of the evidence downgraded by one level for study limitations. Judgements of high risk of bias in all studies included in this analysis 7No serious concerns regarding inconsistency. Limited evidence of heterogeneity between studies 8Selected studies only were available to be included in this analysis 9Funnel plots were not created for this analysis, due to the low numbers of studies involved 10Quality of the evidence downgraded by one level for inconsistency. High heterogeneity between studies.
Background
Description of the condition
Major depressive disorder (MDD) is characterised by: depressed mood; markedly diminished pleasure or interest in all activities; significant weight loss or weight gain, or decrease or increase in appetite; insomnia or hypersomnia; psychomotor agitation or retardation; fatigue or lethargy; feelings of worthlessness or inappropriate guilt; disruptions to concentration and decision making; and recurrent thoughts of death (APA 2013). Diagnosis is achieved by: the presence of four or more symptoms (as above) plus depressed mood or markedly diminished pleasure or interest in all activities, for a consecutive period of two weeks; significant distress or impairment in functioning as a result of symptoms; and an inability to attribute symptoms to the physiological effects of a substance or another medical condition (APA 2013). MDD is currently estimated to affect approximately 7% of western populations, with resulting impact both at an individual and a societal level (APA 2013). MDD can be highly debilitating; can affect all areas of an individual's life; can be difficult to treat, with a high rate of recurrence; and often exists in combination with other conditions and disorders, such as cardiovascular disease and anxiety disorders (APA 2013). Recent figures (2011) published by the World Health Organization estimate major depressive disorders to account for 3% of global ill health in terms of disability‐adjusted life years (WHO 2014), and projections for 2030 suggest an increase to 6% or 7% (WHO 2014). Given this increasing trend, there is an urgent need for effective treatments and strategies for prevention.
Description of the intervention
One emerging potential treatment for MDD is n‐3 polyunsaturated fatty acids (n‐3PUFAs), also known as omega‐3 fatty acids.
n‐3PUFAs are a family of polyunsaturated fatty acids, named as such because of the positioning of the first double carbon bond on the third atom from the methyl end of the acyl chain. All members of the family are derived from parent fatty acid 18:3n‐3 (Alpha‐linolenic acid (ALA)), via desaturation and elongation. ALA, however, can not be synthesised by humans, and thus must be obtained from the diet (Haag 2003; Ruxton 2005). Longer‐chain n‐3PUFAs can be formed in humans, but biological conversion is slow and inefficient, making diet an important source for these fatty acids as well (Ma 1995). Dietary sources of ALA include certain nuts and seeds, such as walnuts, flaxseed and rapeseed (canola) oil. Dietary sources of the longer n‐3PUFAs eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) include fatty fish, some white fish, shellfish and other seafood such as seaweed, and certain eggs and animal products, depending on the animal's diet (BNF 1999; James 2000; Ruxton 2005; Simopolous 1999).
Links between n‐3PUFAs and MDD were suggested following recognition of a reduction in the dietary consumption of n‐3PUFAs in recent decades and an increase in depressive conditions (Simopolous 1999). Coupled with the reduction in n‐3PUFA intakes, intakes of n‐6 long chain polyunsaturated fatty acids (n‐6PUFAs) have also increased. Closely related to n‐3PUFAs, n‐6PUFAs (named from the positioning of the first double bond on the sixth carbon atom from the methyl end of the acyl chain) are derived from parent essential fatty acid 18:2n‐6 (linoleic acid (LA)), and for synthesis, share the same desaturases and elongases as n‐3PUFAs. n‐3PUFAs and n‐6PUFAs thus compete for synthesis from their parent fatty acids. Dietary sources of LA and n‐6PUFAs include plant and vegetable seeds and oils, as found in margarines and many processed foods (James 2000; Simopolous 1999). Our traditional diet is thought to have contained approximately equal amounts of energy from n‐3PUFAs and n‐6PUFAs (Simopolous 1999). By comparison, a current western diet is estimated to contain approximately five to 20 times more energy from n‐6PUFAs than from n‐3PUFAs (Gregory 2000; Simopolous 1999).
Early work investigating population consumption levels of n‐3PUFAs and n‐3PUFA‐rich foods, such as fish, suggested links with population levels of MDD and various psychiatric conditions (Hibbeln 1998; Noaghiul 2003; Peet 2004), and studies since have found similar associations. Within countries, n‐3PUFA intakes have been negatively associated with depressive illness (e.g. Silvers 2002; Tanskanen 2001). In clinical studies, low levels of n‐3PUFAs have been found in individuals diagnosed with MDD (e.g. Edwards 1998; Peet 1998) and depressive disorders (e.g. Garland 2007), and reporting high levels of depressed mood (e.g. Mamalakis 2002; Mamalakis 2006), compared to controls. Continuous relationships between n‐3PUFA status and depressive symptoms have also been found (e.g. Edwards 1998). In randomised controlled trials (RCTs), beneficial effects of supplementation with n‐3PUFAs compared to placebo have been reported for MDD (e.g. Nemets 2002; Su 2003) and depressive disorders (e.g. Frangou 2006; Stoll 1999).
How the intervention might work
The positive effects of n‐3PUFAs on depressive illness are thought to occur as a result of changes to cell membrane structure and function, impacting particularly on cell communication, inflammatory processes and neurotransmitter activities (Haag 2003; James 2000; Ruxton 2005). Further details are available in Appendix 1. Disrupted and abnormal cell signalling, inflammatory processes and neurotransmitter system activities have all been implicated in MDD (Parker 2006b; Stahl 2008).
Why it is important to do this review
n‐3PUFAs are known to be important in brain development and function, and have been linked to depression in a variety of studies, see Appendix 2. Not all studies, however, report beneficial effects (see Appendix 2), and reviews and meta‐analyses clearly demonstrate variability between studies (e.g. Appleton 2006; Appleton 2008b; Appleton 2010; Lin 2007; Parker 2006b; Smith 2011; Stahl 2008). Meta‐analyses reveal some small benefit of n‐3PUFAs for depressive disorders (Appleton 2006; Lin 2007), but investigations of the heterogeneity also suggest differential effects of n‐3PUFAs, depending primarily on severity of depressive symptoms at baseline (Appleton 2010). Sensitivity analyses based on severity of depressive symptoms at baseline suggest no benefits of n‐3PUFAs for individuals with mild depressive symptoms or without a diagnosis of depression, but provide some evidence of benefits in individuals with severe depressive symptoms or with depressive diagnoses (Appleton 2010). These findings suggest a possible benefit of n‐3PUFAs for MDD. This review investigates a role for n‐3PUFAs as a treatment for MDD.
Other reviews investigating a role for n‐3PUFAs in depressive disorders have recently been conducted (e.g. Bloch 2012; Grosso 2014; Martins 2011; Sublette 2011). These reviews typically use a very broad definition of depression to include a variety of depressive disorders and conditions, in a number of populations, including children. This review considers solely major or unipolar depressive disorder, and focuses on adults.
Various reviews of other treatments for MDD and other depressive disorders are also available. A recent search of the Cochrane Library revealed 407 completed reviews or reviews in progress focusing on treating or preventing depression. The majority of these reviews investigate pharmacological (e.g. antidepressant) or psychological (e.g. cognitive behavioural therapy) treatments for depressive conditions, or focus on specific clinical populations, e.g. people with stroke or people with diabetes mellitus. Only two of these reviews include n‐3PUFAs, both focusing on antenatal and postnatal depression. One review investigates 'dietary supplements for preventing postnatal depression' (Miller 2013), and includes one study of n‐3PUFAs. This study found no preventive impact of n‐3PUFAs on the presence of postnatal depression. The other review (Dennis 2013) includes two trials investigating the use of n‐3PUFAs for antenatal depression, and reports a beneficial effect on depression in one trial and no benefit in the other. One further review also focuses on a herbal treatment (St John's Wort) for depression (Linde 2008), but the active component of this plant‐based treatment is unrelated to n‐3PUFAs or other fatty acids.
Objectives
To assess the effects of n‐3 polyunsaturated fatty acids (n‐3PUFAs) (also known as omega‐3 fatty acids) versus a comparator (e.g. placebo, antidepressant treatment, standard care, no treatment, wait‐list control) for major depressive disorder in adults.
Methods
Criteria for considering studies for this review
Types of studies
Only randomised controlled trials (RCTs) were eligible, as the best study design for assessing an intervention. We included all suitable RCTs, regardless of quality, but we also recorded measures of risk of bias. We also included cross‐over and cluster‐RCTs where suitable. We excluded observational and case‐control studies. Our aim was to include as many relevant studies as possible to avoid limitations and bias.
Types of participants
Participant characteristics
We included studies regardless of participant demographics (e.g. gender, country of residence), although we considered only studies involving adults (18 years and over).
Diagnosis
We only included studies that enrolled participants with a primary diagnosis of major or unipolar depressive disorder, from a trained professional or using a validated rating scale, or studies that included a subgroup of these individuals. If a subgroup was used, we included only the data from the subgroup in the review, and only if the subgroup was defined and distinguished prior to randomisation. If data from diagnosed and non‐diagnosed individuals were mixed, we did not include these studies and data. We excluded studies that enrolled participants without MDD, but with a primary diagnosis of an alternative depressive disorder, e.g. bipolar disorder, postpartum depression (APA 2013), or any other psychiatric condition. We also excluded studies that describe a diagnosis of MDD that was given only during or in relation to pregnancy. If diagnoses were unclear, we did not include these studies or these data. We included studies in the review only if we were certain that all data relevant to our review were gained from participants with MDD.
Comorbidities
We included studies regardless of the inclusion of participants with other comorbid conditions (physical conditions, e.g. congestive heart disease, or psychiatric conditions, e.g. anxiety). The inclusion of studies involving participants with comorbid conditions was due to the high likelihood of existing comorbidities in the MDD population (APA 2013), and a desire to make the review as generalisable as possible. We investigated any effects due to existing comorbidities in subgroup analyses.
Adjunctive Therapy
We also included studies regardless of participant use of adjunctive therapy. We included studies that recruited participants with concomitant adjunctive therapy due to the high likelihood of adjunctive therapy use in the MDD population (APA 2013), and a desire to make the review as generalisable as possible. We recorded adjunctive therapies as part of the review, and also investigated these in subgroup analyses.
Setting
We included studies regardless of setting, provided they used a clinical diagnosis or equivalent depressive rating score.
Types of interventions
Experimental intervention
We included studies if they used an exposure of n‐3PUFAs as the sole or as an adjunctive therapy. We included studies regardless of: the type and source of n‐3PUFA provided (pure ALA, EPA, DHA or any combination of these, fish, flaxseed, rapeseed, etc); the dose of n‐3PUFA or duration of supplementation; and the mode of provision (i.e. supplement capsules, supplemented foods). We kept records of these differences, and used sensitivity analyses to investigate effects based on n‐3PUFA type. We included studies if details of the type of n‐3PUFA, dose, and ratio were not available, as mechanisms for action remain unknown. We accepted studies with a 'lead‐in' phase to allow for spontaneous remission or placebo responding in participants, and recorded use of the 'lead‐in' phase.
Comparator intervention
We included studies regardless of the comparator used, but there had to be a comparator. We counted waiting‐list controls, no treatment or standard care as possible comparators. We recorded all comparators. We conducted separate analyses, depending on the comparator used, to allow clear combination of like with like.
Types of outcome measures
We included studies that met the above criteria, regardless of whether they reported on all of the following outcomes.
Primary outcomes
1. Depressive symptomology (continuous data): We assessed depressive symptomology using any continuous validated measure. The most commonly used validated rating scales are the Beck Depression Inventory (BDI) (Beck 1987), the Montgomery‐Asberg Depression Rating Scale (MADRS) (Montgomery 1979), and the Hamilton Depression Rating Scale (HDRS) (Hamilton 1960), but we also included studies using other scales.
2. Adverse events: We recorded measures of adverse events where possible. We recorded the number and type (e.g. gastrointestinal, psychiatric) of adverse events experienced, as reported in studies. We used the number of individuals suffering, rather than the number of events, in analyses. Where adverse events were not reported, we recorded this.
Secondary outcomes
3. Depressive symptomology (dichotomous data): We also assessed depressive symptomology using remission or improvement as assessed using clinical diagnoses by a trained professional or a validated rating scale, where provided.
4. Quality of life (continuous data): We assessed quality of life using any continuous validated measure.
5. Failure to complete: We recorded the number of individuals leaving each study early, and the reasons for early dropout.
Timing of outcome assessment
Where studies used multiple time points, we used only data from the longest follow‐up period for analyses. Previous work suggests that effects are likely to increase over time (Calder 2003; Ruxton 2005).
Search methods for identification of studies
We identified suitable studies for inclusion by searching databases, international trials registers and published review articles, and by contacting authors of published trials.
Electronic searches
The Cochrane Depression, Anxiety and Neurosis Review Group's Specialised Register (CCDANCTR)
The Cochrane Depression, Anxiety and Neurosis Group (CCDAN) maintain two clinical trials registers at their editorial base in Bristol, UK: a references register and a studies‐based register. The CCDANCTR‐References Register contains over 39,000 reports of RCTs in depression, anxiety and neurosis. Approximately 60% of these references have been tagged to individual, coded trials. The coded trials are held in the CCDANCTR‐Studies Register and records are linked between the two registers through the use of unique Study ID tags. Coding of trials is based on the EU‐Psi coding manual, using a controlled vocabulary (please contact the CCDAN Trials Search Co‐ordinator for further details). Reports of trials for inclusion in the Group's registers are collated from routine (weekly), generic searches of MEDLINE (1950‐), EMBASE (1974‐) and PsycINFO (1967‐); quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review‐specific searches of additional databases. Reports of trials are also sourced from international trials registers through the World Health Organization's trials portal (the International Clinical Trials Registry Platform (ICTRP)), pharmaceutical companies, the handsearching of key journals, conference proceedings and other (non‐Cochrane) systematic reviews and meta‐analyses. Details of CCDAN's generic search strategies (used to identify RCTs) can be found on the Group's website.
1. We searched the CCDANCTR (Studies and References Registers) using the following terms: (depress* or dysthymi* or “affective disorder*” or “affective symptom*” or “mood disorder*” or "mental health") AND (dha or docosahex* or eicosapent* or epa or “fatty acid*” or *fish* or *linolenic* or *omega* or n‐3 or w‐3 or *PUFA* or “cod liver oil”)
2. We also conducted complementary searches of the bibliographic database Cumulative Index to Nursing & Allied Health (CINAHL) (1982 to 19th Sept. 2013), using relevant subject headings (controlled vocabularies) and search syntax; the search strategy listed in Appendix 3. This database yielded no unique studies to September 2013 (only secondary references were identified by CINAHL), and we therefore excluded it from subsequent searches to May 2015.
3. We searched international trial registries via the World Health Organization’s trials portal (ICTRP) and ClinicalTrials.gov to identify unpublished or ongoing studies.
There were no restrictions on date, language or publication status applied to the searches. We ran our most recent database searches on 4th May 2015.
Searching other resources
We checked the reference lists of all included studies and relevant reviews to identify additional studies missed from the original electronic searches. We also contacted authors of included studies for information on unpublished or ongoing studies or to request additional trial data.
Data collection and analysis
We downloaded search results into Endnote. We downloaded selected studies into Review Manager 5 (RevMan 2014). We detail the number of search results at each stage of the search and selection process in the Results section.
Selection of studies
Two review authors (RP, HS) independently screened the titles and abstracts of all studies identified by the search, and coded them as 'retrieve' (eligible or potentially eligible/unclear) or 'do not retrieve'. We retrieved the potentially‐relevant full‐text study reports/publications and two review authors (RP, HS) independently screened the full text, identified studies for inclusion, and recorded reasons for exclusion of the ineligible studies. We resolved disagreements through discussion or consultation with a third author (KA). We identified and excluded duplicate records, and we collated multiple reports that related to the same study, so that each study rather than each report was the unit of interest in the review. We included in the list and obtained titles or abstracts which were potentially relevant, but where relevance was not clear. We obtained and translated articles in foreign languages. We recorded the selection process in sufficient detail to complete a PRISMA flow diagram and Characteristics of excluded studies tables.
Data extraction and management
We used a data collection form to extract study characteristics and outcome data. We developed the form specifically for this work, and piloted it on two studies in the review, prior to use for all studies. Two review authors (HS and KA or RP) extracted the following study characteristics and outcome data from included studies:
Methods: study design, total duration of study, details of any 'lead‐in' period, use of several study centres, study location, study setting, and date of study.
Participants: N, mean age, age range, gender, severity of condition, diagnostic criteria, inclusion criteria, and exclusion criteria, withdrawals.
Interventions: intervention, comparator, concomitant therapies, and comorbidities.
Outcomes: primary and secondary outcomes, and time points reported.
Notes: funding for trial, and notable conflicts of interest of trial authors.
Where multiple reports of the same study were available, we abstracted data from all reports on separate data extraction forms and subsequently combined them. We resolved discordances by independent abstraction and then by discussion with a third author (RP or KA, respectively). We also contacted corresponding authors directly for relevant information.
We have noted data that were not usable for analyses in the Characteristics of included studies tables (Notes section). Two review authors (HS, RP) transferred all data into the Review Manager 5 (RevMan 2014) file, and double‐checked that we had entered data correctly by comparing the data presented in the review with the study reports. A third review author (KA) also checked study characteristics for accuracy against the trial reports.
Main comparisons
n‐3PUFAs versus comparator. Analyses are conducted by comparator type.
Assessment of risk of bias in included studies
Three review authors (KA, HS, RP) independently assessed the risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved disagreements by discussion. We assessed the risk of bias according to the following domains.
Random sequence generation.
Allocation concealment.
Blinding of participants and personnel.
Blinding of outcome assessment.
Incomplete outcome data.
Selective outcome reporting.
Other bias.
We judged each potential source of bias as high, low or unclear risk, using the criteria provided in Appendix 4, and have provided a supporting quotation from the study report together with a justification for our judgement in each 'Risk of bias' table. The review authors (KA, HS, RP) agreed the criteria for judging risk of bias following some experience of the literature, but prior to formal data abstraction. We have summarised the risk of bias judgements across different studies for each of the domains listed. Where information on risk of bias relates to unpublished data or correspondence with a trialist, we have noted this in the 'Risk of bias' table. We have taken account when considering treatment effects of the risk of bias for the studies that contribute to each outcome.
Measures of treatment effect
Continuous data
We recorded depressive symptomology and quality of life using all scales as used in each study, after ensuring comparable direction. We conducted analyses on data from only one scale per study. For depressive symptomology, we used the scale most commonly used in all studies (the HDRS: Hamilton 1960), where possible. For quality of life, we used the scale most commonly used in all studies reporting quality of life (the CGI: Guy 1976), where possible.
We collected continuous data in the form of N, mean, and standard deviation per intervention group at baseline and at the end of each intervention, as required for meta‐analysis. If data were only provided in other forms, e.g. as medians, change from baseline, we contacted study authors and requested appropriate data.
We analysed continuous data as a standardised mean difference (SMD) with a 95% confidence interval (CI). We undertook meta‐analyses only where this was meaningful, i.e. where treatments, participants and the underlying clinical question were similar enough for pooling to make sense. Where multiple trial arms were reported in a single trial, we included only the relevant arms in each analysis.
Dichotomous data
Data on adverse events were reported by the number of individuals suffering, as opposed to the number of events. We collected dichotomous data in the form of N per intervention group. We analysed dichotomous data as Mantel‐Haenszel odds ratios (ORs) with 95% CIs. We also recorded reasons where possible.
We recorded depressive remission and response as provided.
Data on failure to complete were reported as the number of individuals failing to complete each trial, and reasons given for non‐completion.
Unit of analysis issues
Cross‐over RCTs
No cross‐over RCTs were included.
Cluster RCTs
No cluster RCTs were included.
Studies with multiple treatment groups
Where studies used multiple treatment groups, we treated each group independently and included them in all appropriate analyses. In these cases, we used the same comparator for all treatment groups, and split the data from comparison groups across treatment groups, as equally as possible for analysis. Where insufficient numbers required numbers of individuals with events either to be rounded up or rounded down, the number of individuals was rounded to err on the side of no effect as opposed to an effect. Assuming individuals took part in only one treatment/comparator group, groups are independent. No studies involved individuals in more than one treatment or comparison group.
Dealing with missing data
We contacted investigators in order to verify key study characteristics and obtain missing numerical outcome data where possible. We documented correspondence with trialists. We used intention‐to‐treat (ITT) data where possible. We extracted data from per protocol populations and included them if ITT data were not available.
Where we could not obtain standard deviations from trial authors, we imputed them by using standard deviation data from all other trials using the same measure for depression in the review (Furukawa 2006).
Assessment of heterogeneity
We undertook meta‐analysis where treatments, participants and the underlying clinical question were similar enough for pooling to make sense, i.e. where n‐3PUFAs were used as a treatment, where participants had a diagnosis of major/unipolar depressive disorder (or equivalent depressive rating score), and where n‐3PUFAs were implemented as a treatment for major/unipolar depressive disorder. Main analyses include all studies to allow sufficient numbers of studies for analyses to be meaningful, and were conducted using a random‐effects model and Hedges' adjusted g, to allow consideration of the likely heterogeneity between studies (Deeks 2001; Egger 2001; Sterne 2001). We also applied a fixed‐effect model as sensitivity analyses to investigate bias as a result of systematic differences between large and small studies that can be exacerbated by the use of a random‐effects model (Deeks 2001; Egger 2001; Sterne 2001). Large differences between the results of our primary analyses using random‐ and fixed‐effect models would suggest using caution when interpreting results.
We investigated heterogeneity using the I² statistic (Higgins 2002; Higgins 2003). We reported I² statistics and appropriate P values. We grouped the I² statistic into four bands for interpretation, as recommended in theCochrane Handbook (Higgins 2011). These bands were 0% to 40%: might not be important; 30% to 60%: may represent moderate heterogeneity; 50% to 90% may represent substantial heterogeneity; and 75% to 100%: considerable heterogeneity. We identified a priori possible sources of heterogeneity, to include the comparator used, publication bias, the presence or absence of comorbid conditions (physical and psychiatric), use of n‐3PUFAs as a sole or adjunctive therapy, and the risks of bias. We investigated heterogeneity between studies based on the type of participants involved using subgroup analyses, and based on the risks of bias using sensitivity analyses. We also identified additional potential sources of heterogeneity during the review process. These included the use of EPA specifically as a treatment, the inclusion of ALA in placebo capsules, the use of data from per protocol analyses, the use of imputed standard deviations from other studies in analyses, and the consideration of multiple comparison groups from the same trial as individual studies. We explored these potential sources of heterogeneity using sensitivity analyses.
Assessment of reporting biases
We investigated publication bias using funnel plot asymmetry (Sterne 2001). It should be noted that publication bias is one of several possible causes of asymmetry in funnel plots.
Data synthesis
We combined trials reporting mean and standard deviation data using meta‐analysis (Sterne 2001).
For continuous data, we calculated the standardised mean effect for all trials using Hedges' adjusted g (Deeks 2001). Hedges’ adjusted g is a formulation of effect size used in the SMD method that includes an adjustment to correct for small sample bias (Deeks 2001). Studies were weighted using the inverse‐variance method. We used random‐effects models primarily to estimate the SMDs for all analyses (Deeks 2001; Egger 2001; Sterne 2001). The random‐effects model assumes non‐identical effects in different studies, and can be preferable to a fixed‐effect model where heterogeneity between studies is high and unexplained. We also applied a fixed‐effect model as sensitivity analyses. Effect sizes are provided as means and standard deviations, and are related to specific scales to allow understanding by clinicians and practitioners.
For dichotomous data, we used the Mantel‐Haenszel method, and calculated effect sizes as odds ratios.
Subgroup analysis and investigation of heterogeneity
We conducted subgroup analyses investigating effects of n‐3PUFAs on MDD in:
Studies involving individuals with comorbid conditions, studies involving individuals without comorbid conditions, and studies involving a mix of individuals both with and without comorbid conditions. This analysis demonstrates effects due to participant characteristics which may affect treatment recommendations and outcomes. We conducted analyses using the same methods as for the main analyses, using: (i) studies in which participants were clearly identified as having comorbid conditions; (ii) studies in which participants were clearly identified as being without comorbid conditions (based on inclusion and exclusion criteria); and (iii) studies where participants with and without comorbid conditions were mixed, or where the presence or absence of comorbid conditions was not clear.
Studies involving individuals receiving adjunctive therapies, studies involving individuals not receiving adjunctive therapies, and studies involving a mix of individuals both receiving and not receiving adjunctive therapies. This analysis demonstrates effects due to participant characteristics which may affect treatment recommendations and outcomes. Analyses were conducted using the same methods as for the main analyses, using (i) studies in which participants were clearly identified as receiving adjunctive therapies; (ii) studies in which participants were clearly identified as not receiving adjunctive therapies (based on inclusion and exclusion criteria); and (iii) studies where participants receiving and not receiving adjunctive therapies were mixed, or where the presence or absence of adjunctive therapy use was not clear. For the purpose of these analyses, adjunctive therapy included antidepressants, psychotherapy, and any other therapies that may affect mood.
We conducted subgroup analyses only for the n‐3PUFA versus placebo comparison, and only for the primary outcomes.
Sensitivity analysis
We conducted sensitivity analyses to investigate the impact of:
Including all studies versus only studies that we judged to be at low risk of bias. This analysis demonstrates the importance of the use of only those trials at low risk of bias, and the levels of confidence and caution that should be exercised in considering the analyses of all studies. We conducted separate analyses using the same methods as for the main analyses. We defined low risk of bias as in the Cochrane Handbook (Higgins 2011), using (i) selection bias, measured using allocation concealment; (ii) performance bias, using blinding of participants; (iii) attrition bias, using incomplete outcome data. We conducted three separate analyses, one for each risk of bias domain. We chose these domains as the ones most likely to impact on RCTs investigating subjective outcomes (depressive symptomology).
Using a fixed‐effect model as opposed to a random‐effects model. The random‐effects model was used for all main analyses. We conducted fixed‐effect analyses using the same data as for the main analyses.
As a result of differences between studies identified during the review process, we also conducted sensitivity analyses to investigate the impact of:
Including all studies versus only those studies that used a treatment that was solely or predominantly EPA. Recent reviews of n‐3PUFAs in depressive disorders have suggested a benefit from supplementation solely with EPA or predominantly with EPA (Grosso 2014; Martins 2011; Sublette 2011), although the evidence is not conclusive (e.g. Ross 2007). We conducted analyses using the same methods as for the main analyses.
Including all studies versus only those that do not use an oil in the placebo capsules that also contains n‐3PUFAs. We found four studies that used a placebo capsule containing ALA (parent n‐3PUFA of EPA and DHA) and were included in the review due to low conversion rates of ALA to longer chain fatty acids in humans (Ma 1995). We conducted analyses using the same methods as for the main analyses.
Including all studies versus only those studies that provided ITT data for analysis. We conducted analyses using the same methods as for the main analyses.
Including all studies versus only those that did not involve data imputation. Standard deviation data were unavailable for five studies, and we imputed them to allow inclusion of these studies in our main analyses. We conducted analyses using the same methods as for the main analyses.
Including all studies as described versus the inclusion of all trials that were split for analysis as complete trials. Several trials used multiple treatments, and so were split for our primary analyses (as described above) to allow accurate description of all studies as required for subgroup analyses, and to allow consistency between all studies. We combined trials that we had split for the main analyses. We pooled data and conducted analyses using the same methods as for the main analyses.
We conducted sensitivity analyses only for the n‐3PUFA versus placebo comparison. We applied the sensitivity analyses using a fixed‐effect model to all outcomes for completeness, but restricted all other sensitivity analyses to test only our primary outcomes.
'Summary of findings' table
We have provided a 'Summary of findings' table, as recommended in theCochrane Handbook (Higgins 2011). This 'Summary of findings' table is for the comparison of n‐3PUFAs with placebo, and includes all primary and secondary outcomes: depressive symptomology (continuous), adverse events, depressive symptomology (dichotomous remission and response), quality of life, and failure to complete. We assessed the quality of evidence for all outcomes using the GRADE system. This considers within‐study risk of bias (methodological quality), directness of evidence, heterogeneity, precision of effect estimates and risk of publication bias.
Results
Description of studies
Results of the search
This review includes 20 trials with a total of 1458 participants. The searches identified 677 records of potential relevance to our review. Following the removal of duplicates, 575 remained. Initial screening by title and abstract resulted in the removal of a further 422 records, to result in the retrieval of 153 full‐text papers. Of these, 85 records were found to relate to RCTs of relevance to our review, while 68 records were excluded. Records were excluded at this stage because they did not: refer to an RCT, involve individuals or a subgroup of individuals with MDD, involve adults, test n‐3PUFAs, involve a comparator, or they did not include depression outcomes. We only included trials in the review if we were sure that they met the eligibility criteria. Records that related to trials that are currently 'ongoing' and currently 'awaiting classification' remained in the review at this stage, but may be excluded once full details of these trials become available. We provide full details of the search results in the PRISMA flow diagram (Figure 1). We give the primary references to the trials they relate to as references for each study. Of these, the trial by Lucas 2009 involves individuals both with and without MDD (and participants were stratified by diagnosis for randomisation), so we have included only the subgroup of individuals with MDD in our review. The Coryell trial includes tests of two doses of n‐3PUFA (approximately 1 g/d, and approximately 2 g/d); the Da Silva 2005 trial involves individuals who were randomised depending on antidepressant status (antidepressants use/no antidepressant use) at trial entry; the Jazayeri 2008 trial involves two separate comparator groups (placebo/antidepressant); the Mischoulon 2015 trial includes tests of an enriched EPA treatment and an enriched DHA treatment; and the Peet 2002 trial includes tests of three doses of n‐3PUFA (1 g/d, 2 g/d, 4 g/d). In these five trials, all groups were independent, and we have considered each as a separate study. This has resulted in the inclusion in analyses of 26 independent studies (Bot 2010; Carney 2009; Coryell (1g/d); Coryell (2g/d); Da Silva (AD) 2005; Da Silva (nAD) 2005; Gertsik 2012; Gharekhani 2014; Gonzalez 2011; Grenyer 2007; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Lespérance 2011; Lucas 2009; Marangell 2003; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Nemets 2002; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Rondanelli 2010; Silvers 2005; Su 2003).
Figure 1.
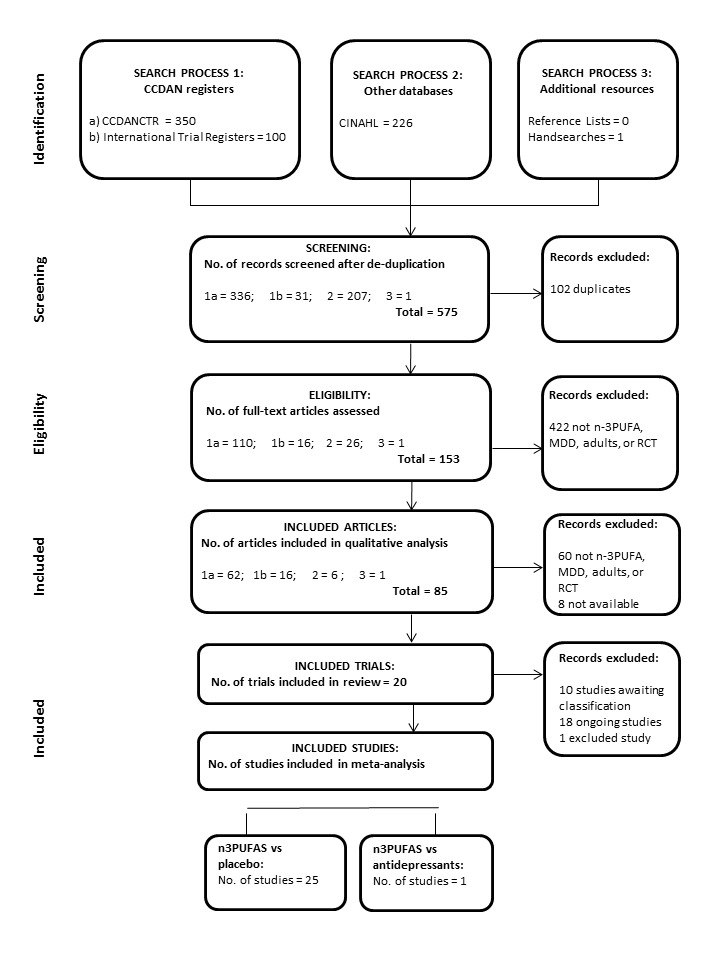
PRISMA Diagram
Published data were available for all 26 independent studies for our primary outcome measure of depressive symptomology. We sought additional data, additional details or clarification from all corresponding authors. Of these, we were unable to contact Alfonso Gonzalez (corresponding author for Gonzalez 2011), and Lauren Marangell (corresponding author for Marangell 2003). The email addresses provided for these individuals did not work, and subsequent web‐based and telephone‐based searches were not fruitful. We received responses, however, from all other corresponding authors. Where additional information was provided by authors, we have detailed this in the Characteristics of included studies tables.
Included studies
We provide full characteristics of the 26 independent studies in the Characteristics of included studies tables. We found considerable differences between studies in all aspects of study methodology. Full detail of the differences in each aspect of study methodology are given below. We used data from all studies in all analyses where possible. Data were missing from analyses due only to insufficient detail, e.g. Da Silva (AD) 2005 and Da Silva (nAD) 2005 report 31 participants and two withdrawals, but fail to provide initial group allocation for the two withdrawals, resulting in these data being unavailable for use in analyses.
Design
All trials included in the review were RCTs involving parallel groups randomised to receive either n‐3PUFAs or a comparator.
Sample sizes
The studies included 1458 participants. Studies varied in sample size, although the majority of studies were small. The number of participants included in each study were as follows: 11 (across both Coryell (1g/d) and Coryell (2g/d)), 20 (Gonzalez 2011; Nemets 2002), 25 (Bot 2010), 28 (Su 2003), 29 (Lucas 2009), 31 (across both Da Silva (AD) 2005 and Da Silva (nAD) 2005), 35 (Park 2015), 36 (Marangell 2003), 41 (Mischoulon 2009), 42 (Gertsik 2012), 46 (Rondanelli 2010), 54 (Gharekhani 2014), 60 (across both Jazayeri (v placebo) 2008 and Jazayeri (v AD) 2008), 70 (across Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002), 77 (Silvers 2005), 83 (Grenyer 2007), 122 (Carney 2009), 196 (across Mischoulon (DHA) 2015; Mischoulon (EPA) 2015) and 432 (Lespérance 2011). In all trials, intervention and comparator groups were composed of approximately equal numbers.
Setting
Participants were recruited from hospitals and clinics (Bot 2010; Carney 2009; Gharekhani 2014; Grenyer 2007; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Park 2015; Su 2003); and community settings (Da Silva (AD) 2005; Da Silva (nAD) 2005; Lucas 2009). Some studies used recruitment methods to capture individuals from both clinical and community settings (Coryell (1g/d); Coryell (2g/d); Gertsik 2012; Lespérance 2011; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Silvers 2005). One study was based in a residential nursing home (Rondanelli 2010). Three studies did not report recruitment setting (Gonzalez 2011, Marangell 2003, Nemets 2002).
Studies were undertaken in the United States (Carney 2009; Coryell (1g/d); Coryell (2g/d); Gertsik 2012; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015), Canada (Lespérance 2011; Lucas 2009), Iran (Gharekhani 2014; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008), Australia (Grenyer 2007), Brazil (Da Silva (AD) 2005; Da Silva (nAD) 2005), Italy (Rondanelli 2010), Korea (Park 2015), the Netherlands (Bot 2010), New Zealand (Silvers 2005), Taiwan (Su 2003), the United Kingdom (Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002) and Venezuela (Gonzalez 2011). Country of study was not reported for the studies by Marangell 2003 or Nemets 2002. These authors are based in the United States and Israel respectively.
Participants
This review relates only to MDD in adults, so all the included studies involved adults. One study uses a local definition of adults (16+ years), and has been included (Gharekhani 2014). Mean ages ranged from a mean of 29 years (across Coryell (1g/d) and Coryell (2g/d)) to a mean of 84 years (Rondanelli 2010). The majority of participants in all studies were women, with the exception of two (Carney 2009; Gharekhani 2014). Percentages of women ranged from 52% (Bot 2010) to 85% (Nemets 2002). Two studies involved only women (Lucas 2009; Rondanelli 2010), and in the studies with a majority of men, the percentages of men were 56% (Gharekhani 2014) and 66% (Carney 2009). Distribution of gender was not reported in four studies (Gertsik 2012; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002).
Five studies included individuals from populations with specific physical comorbidities: diabetes (Bot 2010), coronary heart disease (Carney 2009), end‐stage renal disease (Gharekhani 2014), and Parkinson's disease (Da Silva (AD) 2005; Da Silva (nAD) 2005). The individuals in Da Silva (AD) 2005 and Da Silva (nAD) 2005 may also have had psychiatric comorbidities. Three studies included individuals with no comorbidities (based on exclusion criteria) (Marangell 2003; Mischoulon 2009; Su 2003). Seven studies included individuals with no physical comorbidities, but some/possible psychiatric comorbidities (Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Lucas 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Nemets 2002; Park 2015), while three studies included individuals with no psychiatric comorbidities, but some/possible physical comorbidities (Gertsik 2012; Gonzalez 2011; Rondanelli 2010), and five studies included individuals with some/possible physical and psychiatric comorbidities (Coryell (1g/d); Coryell (2g/d); Grenyer 2007; Lespérance 2011; Silvers 2005). The trial by Peet 2002 (Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002) reports no comorbidities, but also does not report excluding individuals with physical or psychiatric comorbidities.
Studies included individuals who were all receiving adjunctive therapy for depression at the time of the trial (Bot 2010; Carney 2009; Coryell (1g/d); Coryell (2g/d); Da Silva (AD) 2005; Gertsik 2012; Gonzalez 2011; Jazayeri (v placebo) 2008; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002), individuals who were not receiving adjunctive therapy (Da Silva (nAD) 2005; Gharekhani 2014; Jazayeri (v AD) 2008; Lucas 2009; Marangell 2003; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015), and a mix of individuals receiving and not receiving adjunctive therapy (Grenyer 2007; Lespérance 2011; Mischoulon 2009; Nemets 2002; Rondanelli 2010; Silvers 2005; Su 2003). Adjunctive therapy took the form of antidepressant medication in all studies, with the exception of Mischoulon 2009, and included psychotherapy (Lespérance 2011; Mischoulon 2009; Silvers 2005). In Rondanelli 2010, antidepressants were not taken, but participants were permitted to take benzodiazepines, which may have impacted on depressed mood.
Interventions
Studies used either a sole EPA intervention, at doses of 1 g/d (Bot 2010; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Mischoulon 2009; Peet (1g/d) 2002), 2 g/d (Nemets 2002; Peet (2g/d) 2002), 3 g/d (Gonzalez 2011), and 4 g/d (Peet (4g/d) 2002); a sole DHA intervention at a dose of 2 g/d (Marangell 2003); and EPA/DHA combinations, at doses of 1.14 g/d (EPA:DHA ‐ 740:400) (Coryell (1g/d)), 1.2 g/d (EPA:DHA – 720:480) (Da Silva (AD) 2005; Da Silva (nAD) 2005), 1.2 g/d (EPA:DHA –1050:150) (Lespérance 2011; Lucas 2009), 1.8 g/d (EPA:DHA ‐ 1080:720) (Gharekhani 2014), 1.88 g/d (EPA:DHA – 930:750) (Carney 2009), 2.28 g/d (EPA:DHA ‐ 1480:800) (Coryell (2g/d)), 2.76 g/d (EPA:DHA – 0.56:2.2) (Grenyer 2007), 3 g/d (EPA:DHA – 600:2400) (Silvers 2005), 5.22 g/d (EPA:DHA ‐ 3420:1800) (Park 2015) and 6.6 g/d (EPA:DHA – 4400:2200) (Su 2003). Four studies used an intervention consisting of EPA, DHA and other n‐3PUFAs, at doses of 1.224 g/d (EPA:DHA:other ‐ 180:900:144) (Mischoulon (DHA) 2015), 1.436 g/d (EPA:DHA:other ‐ 1060:274:102) (Mischoulon (EPA) 2015), 2.4 g/d (EPA:DHA:other – 1800:400:200) (Gertsik 2012) and 3.13 g/d (EPA:DHA:other – 1670:830:630) (Rondanelli 2010).
All studies used a placebo comparator, with the exception of Jazayeri (v AD) 2008, which compared n‐3PUFAs with antidepressants. Different placebos were used: oil (Coryell (1g/d); Coryell (2g/d)), rapeseed oil (Jazayeri (v placebo) 2008), rapeseed oil plus medium‐chain triglycerides (Bot 2010), corn oil (Carney 2009), olive oil (Gertsik 2012; Grenyer 2007; Silvers 2005; Su 2003), mineral oil (Da Silva (AD) 2005; Da Silva (nAD) 2005), paraffin oil (Gharekhani 2014; Mischoulon 2009; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Rondanelli 2010), safflower oil plus oleic acid (Park 2015), soybean oil (Mischoulon (DHA) 2015; Mischoulon (EPA) 2015), sunflower oil plus 2% fish oil (Lespérance 2011; Lucas 2009). We included studies using rapeseed oil and soybean oil as a comparator, due to likely effects as a result of longer n‐3PUFAs (James 2000; Ruxton 2005) and the reported low conversion rates of ALA to longer n‐3PUFAs (Ma 1995). The oil used in the Coryell studies also contained some ALA (6%). Three studies did not report the placebo used (Gonzalez 2011; Marangell 2003; Nemets 2002). In all cases, the placebo was given in a similar dose to the intervention.
Treatment duration for each trial was as follows: four weeks (Nemets 2002), six weeks (Coryell (1g/d); Coryell (2g/d); Marangell 2003), eight weeks (Gertsik 2012; Gonzalez 2011; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Lespérance 2011; Lucas 2009; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Rondanelli 2010; Su 2003), 10 weeks (Carney 2009), 12 weeks (Bot 2010; Da Silva (AD) 2005; Da Silva (nAD) 2005; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Park 2015; Silvers 2005), and 16 weeks (Gharekhani 2014; Grenyer 2007).
In the trial where n‐3PUFAs were compared with antidepressants (Jazayeri (v AD) 2008), n‐3PUFAs were given using EPA only, at a dose of 1 g/d, and compared with 20 mg/d fluoxetine (antidepressant).
Outcomes
Primary Outcomes
Depressive symptomology (continuous data): Depressive symptomology was reported using continuous data in all studies, at both baseline and study end. Most studies used the Hamilton Depression Rating Scale (HDRS) (Hamilton 1960) (including the HDRS‐short form (Reynolds 1995)), the Montgomery‐Asberg Depression Rating Scale (MADRS) (Montgomery 1979), and/or the Beck Depression Inventory (BDI) (Beck 1987), but the Inventory of Depressive Symptomology Self Report (IDS‐SR) (Trivedi 2004) (Lespérance 2011), the Hopkins Symptom Checklist Depression Scale (HSCL) (Williams 2004) (Lucas 2009), and the Geriatric Depression Scale (GDS) (Yesvage 1983) (Rondanelli 2010) were also used. In almost all studies, depressive symptomology scores were also collected at additional time points between baseline and study end.
Adverse events: Number of individuals experiencing adverse events were reported or provided for 22 studies (Bot 2010; Carney 2009; Coryell (1g/d); Coryell (2g/d); Da Silva (AD) 2005; Da Silva (nAD) 2005; Gertsik 2012; Gharekhani 2014; Grenyer 2007; Lespérance 2011; Lucas 2009; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Nemets 2002; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Rondanelli 2010; Silvers 2005; Su 2003). In some studies only the number of individuals experiencing serious adverse events (Bot 2010; Coryell (1g/d); Coryell (2g/d); Gertsik 2012), clinically relevant adverse events (Nemets 2002) or emerging or worsening adverse events (Mischoulon (DHA) 2015; Mischoulon (EPA) 2015) were reported, and three studies reported only the number of individuals experiencing adverse events reported by at least 5% of participants (Bot 2010; Gertsik 2012; Lespérance 2011). Three studies reported the number of adverse events rather than the number of individuals experiencing them (Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Marangell 2003). Six studies did not report adverse events fully, clearly or in detail (Carney 2009; Da Silva (AD) 2005; Da Silva (nAD) 2005; Gonzalez 2011; Grenyer 2007; Lespérance 2011). Many studies also reported types of adverse event experienced. The majority of adverse events were gastrointestinal, although psychological and other physical events were also reported. We included data on adverse events in analyses, provided the number of individuals reporting adverse events was reported in the n‐3PUFA and placebo group using the same definition of adverse events (serious adverse events, etc.).
Secondary Outcomes
Depressive symptomology (dichotomous data): Depressive symptomology in dichotomous terms was reported in 18 studies (Carney 2009; Coryell (1g/d); Coryell (2g/d); Da Silva (AD) 2005; Da Silva (nAD) 2005; Gertsik 2012; Gonzalez 2011; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Marangell 2003; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Nemets 2002; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Rondanelli 2010). These data were used to provide rates of remission and/or response. As determined by original authors, 'remission' was defined as an end point score within the no/low depression range on the scale utilised (score ≤ 7 on the HDRS (Gertsik 2012; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015), score ≤ 8 on the BDI (Carney 2009), score < 11 on the GDS (Rondanelli 2010)), and 'response' was defined as a 50% improvement in depression scale score.
Quality of life: Quality of life was measured in 13 studies, using a range of validated scales: Clinical Global Impression (CGI) (Guy 1976) (Da Silva (AD) 2005; Da Silva (nAD) 2005; Gertsik 2012; Lucas 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Park 2015), Patient Global Impression (PGI) (Guy 1976) (Gertsik 2012), Global Assessment of Functioning Scale (GAF) (Diguer 1993), (Grenyer 2007; Marangell 2003), Psychological General Well‐being Schedule (PGWB) (Dupuy 1984) (Lucas 2009), the Quality of Life Enjoyment and Satisfaction Questionnaire (QLESQ) (Endicott 1993) (Mischoulon 2009), the Short Form (36) Health Survey (SF‐36) (Ware 1993) (Gharekhani 2014; Rondanelli 2010) and Likert scales (Grenyer 2007). We considered these scales to assess quality of life, although some of them were used as secondary measures of depression in some studies. For the CGI and PGI, higher scores denote poorer quality of life. For the GAF, PGWB, QLESQ and SF‐36, higher scores denote better quality of life.
Failure to complete: All studies reported numbers of individuals who failed to complete, with the exception of Rondanelli 2010, where no details are provided but full data sets are available for all participants, so we presume none failed to complete. For all other studies, figures ranged from 0% (Coryell (1g/d); Coryell (2g/d)) to 55% (Gonzalez 2011). Some studies provided reasons for withdrawal (Bot 2010; Carney 2009; Gharekhani 2014; Grenyer 2007; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Nemets 2002; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Silvers 2005; Su 2003).
Excluded studies
Our searches identified only one trial registration that we have classified as an excluded study (Characteristics of excluded studies). This trial registration (Clayton 2009) details a trial that appears to meet our inclusion criteria, but the study was withdrawn prior to participant enrolment.
Ongoing studies
Sixteen RCTs investigating n‐3PUFAs versus a comparator in adults with MDD are currently ongoing. We provide details of these in the tables of Characteristics of ongoing studies. Details are based on trial registrations (we have had no correspondence with authors of ongoing studies). We have included all potentially relevant studies, to allow subsequent updates of the review to be as inclusive as possible. Some of the studies that are currently included as ongoing studies may be excluded from updates of the review once study details become clearer following completion and publication. Only subgroups of participants in some studies may also be included in subsequent updates, depending on inclusion/exclusion criteria and randomisation procedures. Some trials, for example, focus on adolescents, but include individuals aged up to 25 years (Amminger 2013), and while the majority of respondents in this trial may not be relevant to our review, it may be possible to include a subset of individuals over 18 years, dependent on randomisation procedures.
Studies awaiting classification
Nine trials are currently awaiting classification. Details of these are provided in the tables of Characteristics of studies awaiting classification. These search results comprise two conference abstracts (Kwak 2013; Rees 2005), and seven trial registrations. We cannot yet include the conference abstracts, as we have not so far been able to obtain enough information on these studies to be sure that they are relevant to our review. Neither the first author nor the last author on the abstracts have responded to email requests. The seven trial registrations relate to trials that are now described on trial register websites as 'completed'. We have emailed all contact authors for further information to allow clarification. Corresponding authors for Shinto 2005 and Su 2005 have responded, stating that this trial will be published in due course, and that all details will be available then. We have not received replies relating to the trial registration from the correspondent for Naqvi 2008. Emails for Murck 2004 and Lima 2006 have been returned undelivered and subsequent enquires of study sponsors have not been fruitful. No contact details were available for two registrations (EUCTR2006‐004949‐41‐IT; NCT00816322). The abstract for Rees 2005 has also been linked to a trial registration, but the status of the trial is recorded as 'unknown'. We have again tried to make contact, but have not received replies from study contacts.
Risk of bias in included studies
Details of the risk of bias judgements for each study are given in the tables of Characteristics of included studies, and we present a graphical representation of the overall risk of bias in included studies in Figure 2 and Figure 3. We judged the risks of bias to be very variable between studies.
Figure 2.
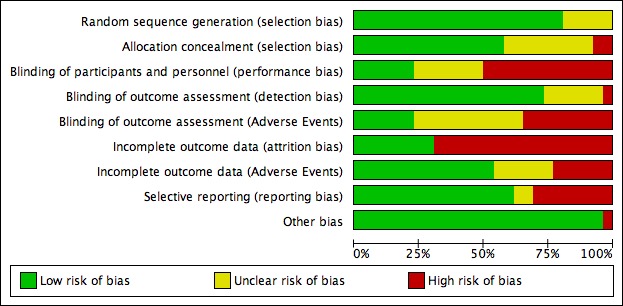
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Figure 3.
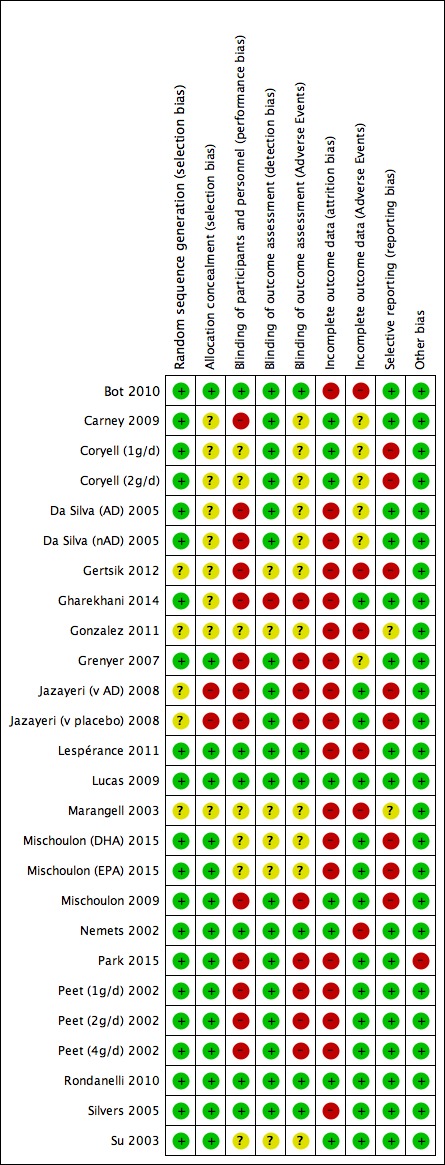
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Random sequence generation
We judged 21 studies to be at low risk of bias for random sequence generation (Bot 2010; Carney 2009; Coryell (1g/d); Coryell (2g/d); Da Silva (AD) 2005; Da Silva (nAD) 2005; Gharekhani 2014; Grenyer 2007; Lespérance 2011; Lucas 2009; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Nemets 2002; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Rondanelli 2010; Silvers 2005; Su 2003). In most of these studies, randomisation was undertaken using a computer‐generated random number generator, but drawing lots (Da Silva (AD) 2005; Da Silva (nAD) 2005) and a random number table (Nemets 2002; Rondanelli 2010) were also used. For all other studies (Gertsik 2012; Gonzalez 2011; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Marangell 2003), insufficient details were provided, resulting in a judgement of unclear risk of bias.
Allocation concealment
We judged 15 studies to be at low risk of bias for allocation concealment (Bot 2010; Grenyer 2007; Lespérance 2011; Lucas 2009; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Nemets 2002; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Rondanelli 2010; Silvers 2005; Su 2003). In these studies, allocation concealment was ensured by individuals outside the main research team conducting allocation, or by using sequential numbering that had been prepared by individuals outside the main research team. We judged two studies to be at high risk of bias (Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008), following comments from the author that the randomisation sequence was not concealed from researchers. For all other studies (Carney 2009; Coryell (1g/d); Coryell (2g/d); Da Silva (AD) 2005; Da Silva (nAD) 2005; Gertsik 2012; Gharekhani 2014; Gonzalez 2011; Marangell 2003), insufficient details were provided, leading to a judgement of unclear risk of bias.
Blinding
Blinding of participants and personnel
We judged six studies to be at low risk of bias for blinding of study participants and personnel to treatment allocation (Bot 2010; Lespérance 2011; Lucas 2009; Nemets 2002; Rondanelli 2010; Silvers 2005). In these studies, blinding was undertaken by adding a small amount of fish oil to the comparator treatment to control for fishy aftertaste and/or adding flavours to both treatments to mask a fishy aftertaste, and following investigation, blinding was found to be successful. We judged 13 studies at high risk of bias (Carney 2009; Da Silva (AD) 2005; Da Silva (nAD) 2005; Gertsik 2012; Gharekhani 2014; Grenyer 2007; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Mischoulon 2009; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002). In these studies, there were no reports of attempting to mask the fishy taste of the intervention, despite good descriptions of the placebo otherwise, and no assessment to check successful concealment. In one study, the majority of participants correctly guessed their allocation (Grenyer 2007). We judged four studies to be at unclear risk of bias (Coryell (1g/d); Coryell (2g/d); Gonzalez 2011; Marangell 2003) due to no report of attempts to mask a fishy taste, but no clear description of other aspects of the placebo. We judged a further three studies to be at unclear risk of bias (Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Su 2003) because flavour was added to the capsules to mask a fishy taste, but there was no assessment to check the success of this precaution.
Blinding of Outcome assessment
We judged the blinding of outcome assessments depending on the individuals making the assessment (participant, researcher, clinician) and the blinding of those persons, as detailed in the blinding of participants and personnel. Thus, we rated participant‐rated measures at a low risk of bias if we considered participants to be successfully blinded to treatment allocation, at unclear risk of bias if blinding was unclear, and at high risk of bias if we considered participants not to be successfully blinded. We treated personnel‐rated measures in a similar fashion. In all cases, we used study reports of the individuals making the assessment if possible, or used standard assessments if details were not specified, e.g. in standard practice, the BDI is a self‐report instrument for completion by patients. Where multiple outcome measures were used and these were given different judgements of risk of bias, we took the key risk of bias judgement to be the one applicable to the outcome measure we used in our analyses.
Mood:
We judged 19 studies to be at low risk of bias, following ratings of adequate blinding of those making the assessments or following adequate blinding of those making the mood assessment used in our analyses (Bot 2010; Carney 2009; Coryell (1g/d); Coryell (2g/d); Da Silva (AD) 2005; Da Silva (nAD) 2005; Grenyer 2007; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Lespérance 2011; Lucas 2009; Mischoulon 2009; Nemets 2002; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Rondanelli 2010; Silvers 2005). We rated six studies at unclear risk of bias, where it was unclear who had made the assessment or whether those individuals were successfully blinded (Gertsik 2012; Gonzalez 2011; Marangell 2003; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Su 2003). We judged one study to be at high risk of bias, where there was a high risk of bias in the blinding of those making the mood assessment (Gharekhani 2014).
Adverse events:
We rated six studies at low risk of bias following judgements of adequate blinding of those making the assessments (Bot 2010; Lespérance 2011; Lucas 2009; Nemets 2002; Rondanelli 2010; Silvers 2005). We judged nine studies to be at high risk of bias, where assessments were made by those at high risk of performance bias due to inadequate blinding (Gharekhani 2014; Grenyer 2007; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Mischoulon 2009; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002). We rated 11 studies at unclear risk of bias, where it was not apparent who had made the assessment or if those individuals were successfully blinded (Carney 2009; Coryell (1g/d); Coryell (2g/d); Da Silva (AD) 2005; Da Silva (nAD) 2005; Gertsik 2012; Gonzalez 2011; Marangell 2003; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Su 2003).
Incomplete outcome data
Mood:
We rated outcome data for mood as complete if there were no missing outcome data; or if: analyses were conducted using intention‐to‐treat (ITT) data, where ITT was defined as including all those randomised; data were missing for less than 10% of the total randomised population; reasons for missing outcome data were unlikely to be related to true outcome; the difference in missing data between intervention and comparator group was not more than 10% of the total randomised population; and the missing data were not unbalanced between intervention and comparator groups in numbers and reasons.
We rated eight studies at low risk of bias for publication or provision of ITT data (as above) (Carney 2009; Coryell (1g/d); Coryell (2g/d); Lucas 2009; Mischoulon 2009; Nemets 2002; Rondanelli 2010; Su 2003). We judged 18 studies to be at high risk of bias due to the unavailability of ITT data (Bot 2010; Da Silva (AD) 2005; Da Silva (nAD) 2005; Gertsik 2012; Gonzalez 2011; Marangell 2003; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Silvers 2005), or publication or provision of ITT data but a higher than 10% dropout rate (Gharekhani 2014; Grenyer 2007; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Lespérance 2011).
Adverse events:
We judged outcome data for adverse events to be complete if all adverse events were clearly reported, and incomplete if all adverse events were clearly not reported. We rated 14 studies at low risk of bias, due to clear complete reporting of all adverse events (Gharekhani 2014; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Lucas 2009; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Rondanelli 2010; Silvers 2005; Su 2003). We judged six studies to be at high risk of bias due to clear incomplete reporting of all adverse events (Bot 2010; Gertsik 2012; Gonzalez 2011; Lespérance 2011; Marangell 2003; Nemets 2002). We judged six studies at unclear risk of bias where adverse events were not clearly reported (Carney 2009; Coryell (1g/d); Coryell (2g/d); Da Silva (AD) 2005; Da Silva (nAD) 2005; Grenyer 2007).
Selective reporting
We judged 16 studies to be at low risk of bias for selective reporting, where reported outcomes have been checked against protocols (Bot 2010), or where authors have informed us that all planned outcomes have been reported (Carney 2009; Da Silva (AD) 2005; Da Silva (nAD) 2005; Gharekhani 2014; Grenyer 2007; Lespérance 2011; Lucas 2009; Nemets 2002; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Rondanelli 2010; Silvers 2005; Su 2003). Judgements of high risk of reporting bias were given to six studies where all outcomes have not (yet) been reported (Gertsik 2012; Jazayeri (v placebo) 2008; Jazayeri (v AD) 2008; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015), and to two unpublished studies (Coryell (1g/d); Coryell (2g/d)). We judged two studies at unclear risk of bias where protocols were not available and authors have not confirmed complete reporting (Gonzalez 2011; Marangell 2003).
Other potential sources of bias
All studies appeared to be free from other sources of bias, with the exception of Park 2015, where we found a significant imbalance in all measures of mood and quality of life between intervention and comparator groups at baseline.
Effects of interventions
See: Table 1
Comparison 1: n‐3PUFAs versus placebo
Twenty‐five independent studies involving 1438 individuals contribute to this comparison (Bot 2010; Carney 2009; Coryell (1g/d); Coryell (1g/d); Da Silva (AD) 2005; Da Silva (nAD) 2005; Gertsik 2012; Gharekhani 2014; Gonzalez 2011; Grenyer 2007; Jazayeri (v placebo) 2008; Lespérance 2011; Lucas 2009; Marangell 2003; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Nemets 2002; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Rondanelli 2010; Silvers 2005; Su 2003); see also Table 1.
Primary outcomes
1.1 Depressive Symptomology (continuous data)
All 25 studies provided continuous data on depressive symptomology from 1373 individuals, and were included in analyses. Analyses were based on HDRS scores for 17 studies (Carney 2009; Gertsik 2012; Gharekhani 2014; Gonzalez 2011; Grenyer 2007; Jazayeri (v placebo) 2008; Lucas 2009; Marangell 2003; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Nemets 2002; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Silvers 2005; Su 2003), and also on MADRS score for six studies (Bot 2010; Coryell (1g/d); Coryell (2g/d); Da Silva (AD) 2005; Da Silva (nAD) 2005, Lespérance 2011), BDI score for one study (Gharekhani 2014), and GDS score for one study (Rondanelli 2010).
n‐3PUFAs were more effective than placebo: SMD = ‐0.30 (95% CI ‐0.50 to ‐0.10) (see Analysis 1.1, Figure 4, Figure 5), but effect sizes are small to modest, and there was substantial evidence of heterogeneity between studies (I² = 59%). Confidence intervals also range between a very small and a modest effect size, and suggest a possible clinically important effect at their upper end. Using GRADE criteria, we judged the quality of the evidence to be very low. A standardised mean difference of 0.30 represents a difference between groups in scores on the HDRS (17‐item) of approximately 2.1 points (95% CI 0.7 to 3.5).
Analysis 1.1.
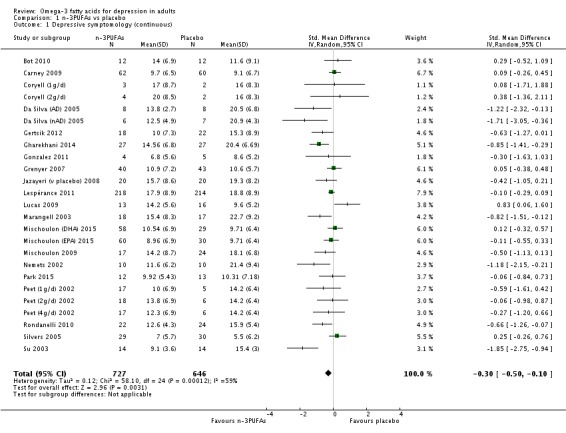
Comparison 1 n‐3PUFAs vs placebo, Outcome 1 Depressive symptomology (continuous).
Figure 4.
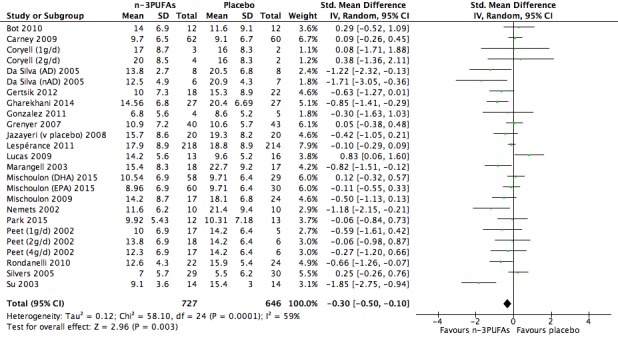
Forest plot of comparison: 1 n‐3PUFAs vs placebo, outcome: 1.1 Depressive symptomology (continuous).
Figure 5.
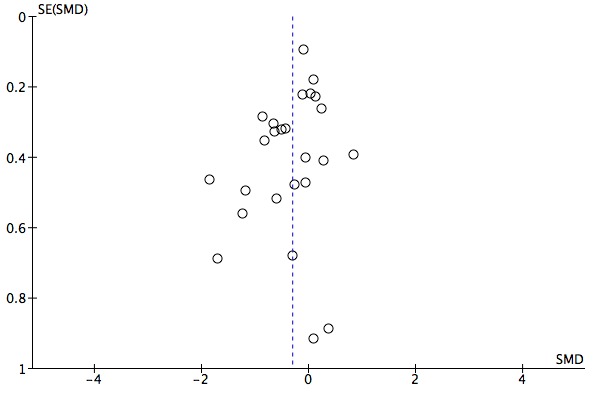
Funnel plot of comparison: 1 n‐3PUFAs vs placebo, outcome: 1.1 Depressive symptomology (continuous).
1.2 Adverse Events
The number of individuals experiencing adverse events was similar in n‐3PUFA and placebo groups: OR = 1.24 (95% CI 0.95 to 1.62), 19 studies, 1207 participants (see Analysis 1.2, Figure 6, Figure 7). Confidence intervals however are wide, and suggest that effects could range from a reduction of 5% to an increase in adverse events in n‐3PUFA groups of 62%, compared with placebo. Using GRADE criteria, we judged the quality of the evidence to be very low. There was no evidence of heterogeneity between groups (I² = 0%).
Analysis 1.2.
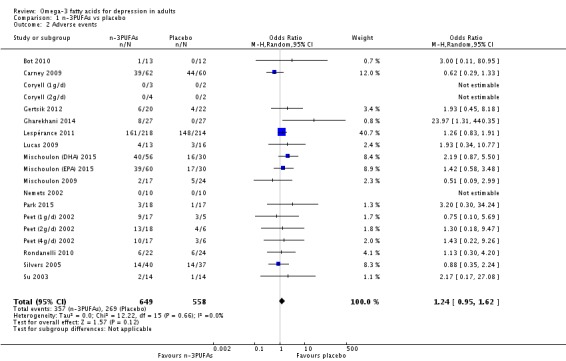
Comparison 1 n‐3PUFAs vs placebo, Outcome 2 Adverse events.
Figure 6.
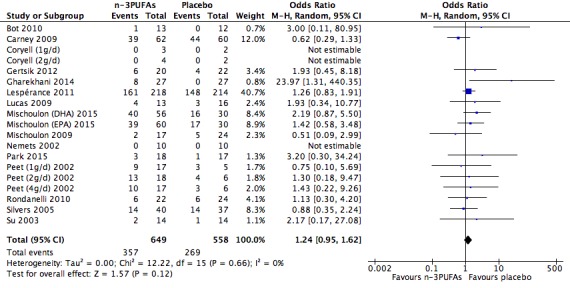
Forest plot of comparison: 1 n‐3PUFAs vs placebo, outcome: 1.2 Adverse events.
Figure 7.
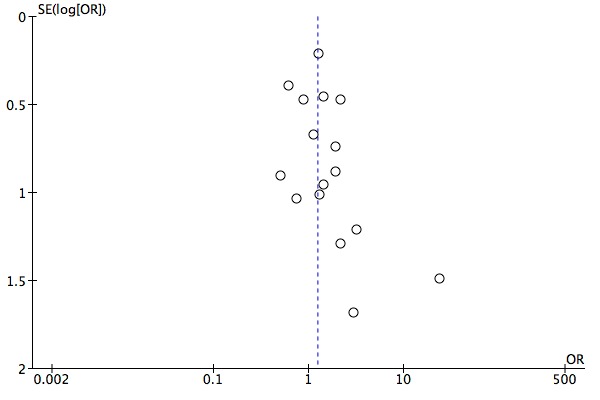
Funnel plot of comparison: 1 n‐3PUFAs vs Placebo, outcome: 1.2 Adverse events.
Secondary outcomes
1.3 Depressive Symptomology (dichotomous data)
There was no evidence of a statistical difference in remission rates following supplementation with n‐3PUFAs compared with placebo: OR = 1.38 (95% CI 0.87 to 2.20), 6 studies, 426 participants (see Analysis 1.3, Figure 8), but confidence intervals are very wide. Confidence intervals suggest a possible effect ranging from a 13% reduction in remission rates with n‐3PUFAs compared with placebo, to a 220% increase in remission rates. Using GRADE criteria, we judged the quality of the evidence to be low. There was little evidence of heterogeneity between groups (I² = 7%).
Analysis 1.3.
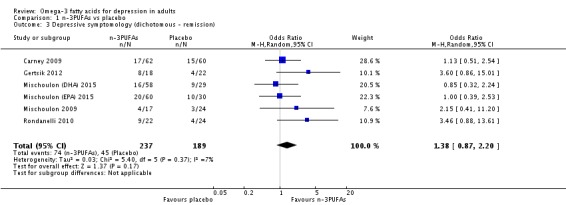
Comparison 1 n‐3PUFAs vs placebo, Outcome 3 Depressive symptomology (dichotomous ‐ remission).
Figure 8.
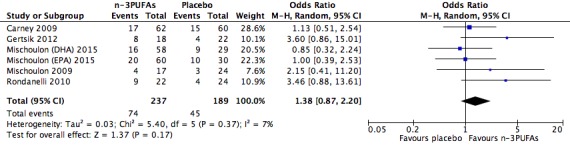
Forest plot of comparison: 1 n‐3PUFAs vs placebo, outcome: 1.3 Depressive symptomology (dichotomous data): remission.
There was no strong evidence of a statistical difference in response rates following supplementation with n‐3PUFAs compared with placebo: OR = 1.39 (95% CI 0.95 to 2.04), 15 studies, 611 participants (see Analysis 1.4, Figure 9), but confidence intervals are again very wide. Confidence intervals suggest a possible effect ranging from a 5% reduction in remission rates with n‐3PUFAs compared with placebo, to a 204% increase in remission rates. Using GRADE criteria, we judged the quality of the evidence to be low. There was little evidence of heterogeneity between groups (I² = 6%).
Analysis 1.4.
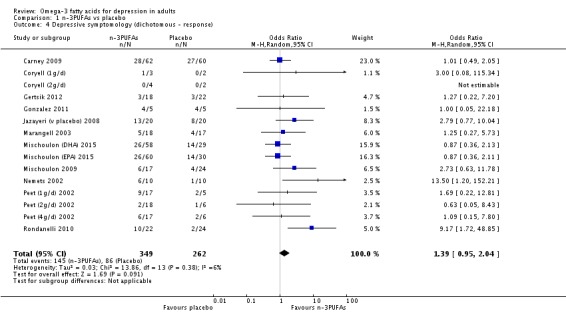
Comparison 1 n‐3PUFAs vs placebo, Outcome 4 Depressive symptomology (dichotomous ‐ response).
Figure 9.
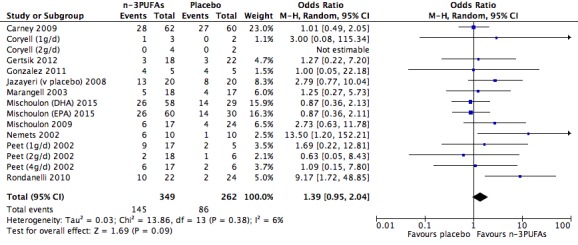
Forest plot of comparison: 1 n‐3PUFAs vs placebo, outcome: 1.4 Depressive symptomology (dichotomous data): response.
1.4 Quality of Life
Continuous data on quality of life were available in 383 participants from nine studies (Da Silva (AD) 2005; Da Silva (nAD) 2005; Gharekhani 2014; Lucas 2009; Marangell 2003; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Park 2015; Rondanelli 2010). We conducted analyses on data from the CGI (Da Silva (AD) 2005; Da Silva (nAD) 2005; Lucas 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Park 2015), the GAF (Marangell 2003) and the SF‐36 (mental health summary scale) (Gharekhani 2014; Rondanelli 2010). We reversed scores for the GAF and SF‐36, so that in all scales a higher score denotes poorer quality of life.
There was no strong evidence of a statistical difference in quality of life between n‐3PUFA and placebo groups: SMD = ‐0.47 (95% CI ‐0.99 to 0.06). Confidence intervals range between a negligible and a large effect size, suggesting both a possible absence of effect at the lower end, and a possible important effect at the upper end. Using GRADE criteria, we judged the quality of the evidence to be very low, and there was considerable evidence of heterogeneity between studies (I² = 82%) (see Analysis 1.5, Figure 10).
Analysis 1.5.
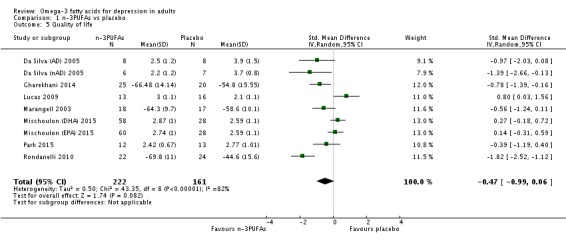
Comparison 1 n‐3PUFAs vs placebo, Outcome 5 Quality of life.
Figure 10.
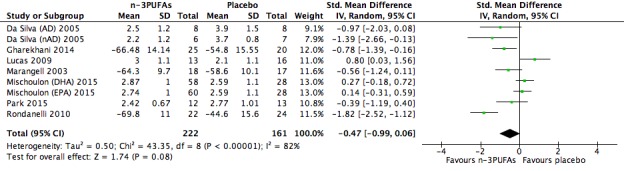
Forest plot of comparison: 1 n‐3PUFAs vs placebo, outcome: 1.5 Quality of life.
1.5 Failure to Complete
Rates for failure to complete were similar in n‐3PUFA and placebo groups: OR = 0.84 (95% CI 0.62 to 1.14), 21 studies, 1344 participants (see Analysis 1.6, Figure 11, Figure 12), but again confidence intervals are wide, and suggest that effects could range from a reduction of 38% to an increase in study withdrawals of 14% in n‐3PUFA groups, compared to placebo. Using GRADE criteria, we judged the quality of the evidence to be very low. There was no evidence of heterogeneity between groups (I² = 0%).
Analysis 1.6.
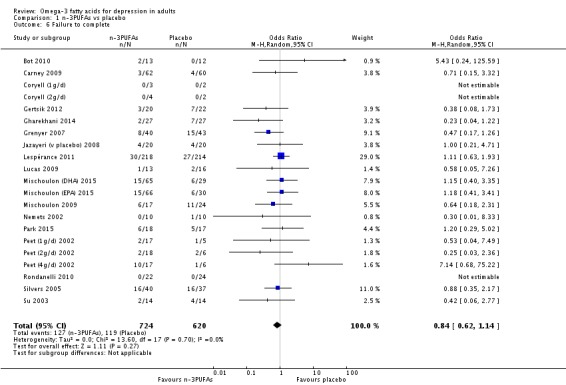
Comparison 1 n‐3PUFAs vs placebo, Outcome 6 Failure to complete.
Figure 11.
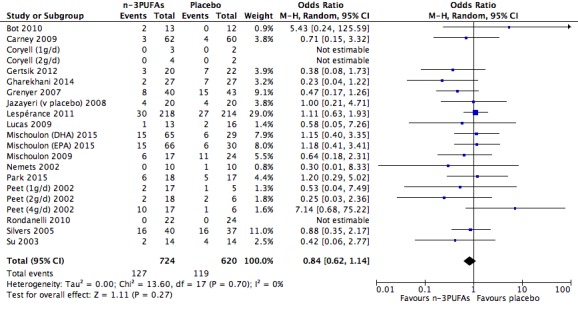
Forest plot of comparison: 1 n‐3PUFAs vs placebo, outcome: 1.6 Failure to complete.
Figure 12.
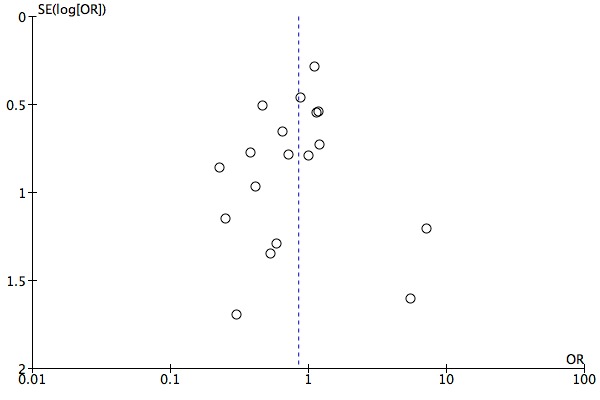
Funnel plot of comparison: 1 n‐3PUFAs vs Placebo, outcome: 1.6 Failure to complete.
Comparison 2: n‐3PUFAs versus antidepressants
Data were only available from one study for this comparison (Jazayeri (v AD) 2008).
Primary outcomes
2.1 Depressive Symptomology (continuous data)
Depressive symptomology based on the HDRS was similar in the n‐3PUFA and antidepressant groups: MD (HDRS(24 item)) = ‐0.70 (95% CI ‐5.88 to 4.48), 1 study, 40 participants (see Analysis 2.1). Confidence intervals however are very wide, and do not rule out a modest benefit or detriment of n‐3PUFAs, compared to antidepressants.
Analysis 2.1.
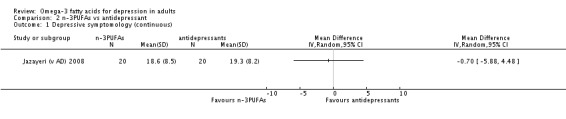
Comparison 2 n‐3PUFAs vs antidepressant, Outcome 1 Depressive symptomology (continuous).
2.2 Adverse Events
Adverse events were only reported in terms of the number of events experienced, as opposed to the number of individuals experiencing at least one event.
Secondary outcomes
2.3 Depressive Symptomology (dichotomous data)
Response rates were similar in n‐3PUFA and antidepressant groups: OR = 1.23 (95% CI 0.35 to 4.31), 1 study, 40 participants (see Analysis 2.2), but confidence intervals are very wide, and do not rule out an important benefit or detriment of n‐3PUFAs, compared to antidepressants. Remission rates were not reported.
Analysis 2.2.
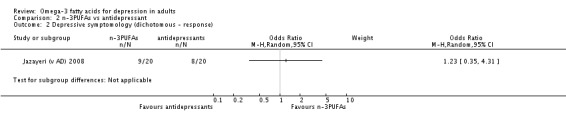
Comparison 2 n‐3PUFAs vs antidepressant, Outcome 2 Depressive symptomology (dichotomous ‐ response).
2.4 Quality of Life
Quality of life was not reported in this study.
2.5 Failure to complete
Rates for failure to complete were similar in n‐3PUFA and antidepressant groups: OR = 1.00 (95% CI 0.21 to 4.71), 1 study, 40 participants (see Analysis 2.3). Confidence intervals however are again very wide, and do not rule out important effects in either direction.
Analysis 2.3.
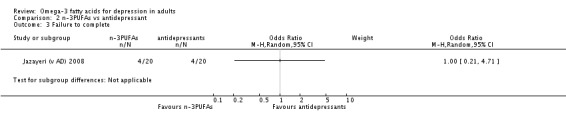
Comparison 2 n‐3PUFAs vs antidepressant, Outcome 3 Failure to complete.
Subgroup analyses
We conducted subgroup analyses only for the n‐3PUFA versus placebo comparison, and only for the primary outcomes, but the number of studies and the number of participants are low. There were insufficient numbers of studies and participants for subgroup analyses to be conducted for other outcomes or for the n‐3PUFA versus antidepressant comparison.
3. Analyses based on comorbidities
There was a suggestion of greater effect sizes (n‐3PUFAs compared to placebo) in depressive symptomology (continuous) in studies including individuals with comorbid conditions: SMD = ‐0.54 (95% CI ‐1.21 to 0.12; 5 studies, 229 participants), and in studies including individuals without comorbid conditions: SMD = ‐0.99 (95% CI ‐1.71 to ‐0.27; 3 studies, 104 participants), compared to studies with a mix of individuals with comorbid and without comorbid conditions: SMD = ‐0.13 (95% CI ‐0.30 to 0.05; 17 studies, 1040 participants) (see Analysis 3.1, Figure 13). The number of studies and the number of individuals in each subgroup however were small, particularly in the subgroup of studies including individuals without comorbid conditions (3 studies, 104 participants), confidence intervals are very wide, suggesting that effects could range from much stronger effects to those that are negligible, and the evidence of heterogeneity within each subgroup was high (I² = 28% to 77%). There was statistical evidence of a difference between subgroups (P = 0.04), but the evidence of heterogeneity between subgroups was high (I² = 68%).
Analysis 3.1.
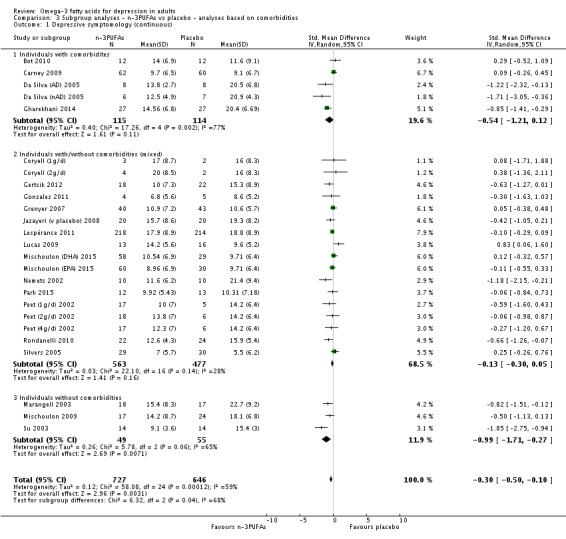
Comparison 3 Subgroup analyses ‐ n‐3PUFAs vs placebo ‐ analyses based on comorbidities, Outcome 1 Depressive symptomology (continuous).
Figure 13.
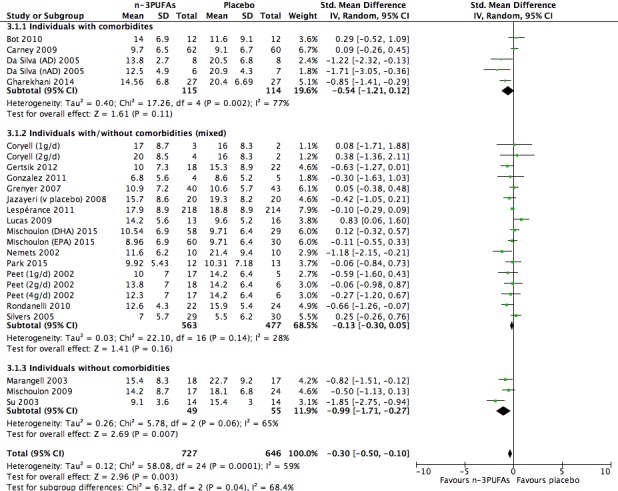
Forest plot of comparison: 1 n‐3PUFAs vs placebo, outcome: 3.1 Depressive symptomology (continuous): Sub‐groups based on presence / absence of comorbidities.
Rates of adverse events were similar across the subgroups. Analysis of studies including individuals with comorbid conditions (OR = 2.66 (95% CI 0.22 to 31.93); 3 studies, 201 participants), studies including individuals without comorbid conditions (OR = 0.82 (95% CI 0.19 to 3.50) 2 studies, 69 participants), and studies with a mix of individuals with and without comorbid conditions (OR = 1.40 (95% CI 1.04 to 1.89) 14 studies, 937 participants) indicated no statistical evidence of a difference between subgroups (P = 0.68), and no evidence of heterogeneity between subgroups (I² = 0%) (see Analysis 3.2). Heterogeneity was high in the subgroup of studies including individuals with comorbid conditions (I² = 71%), but low in the other two subgroups (I² = 0%), and confidence intervals are again very wide, suggesting possible effects that could range between a reduction in events with n‐3PUFAs of 89% to an increase in events of 3193%.
Analysis 3.2.
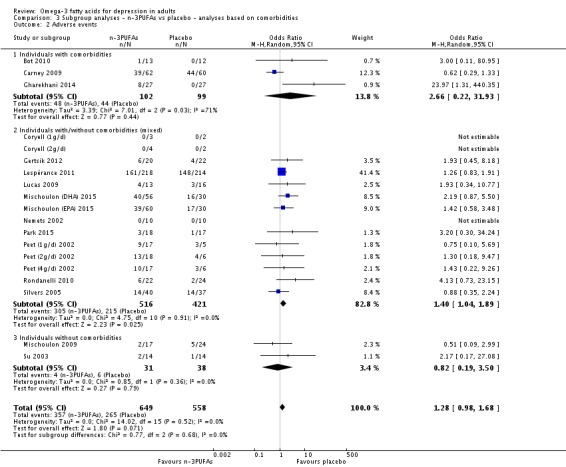
Comparison 3 Subgroup analyses ‐ n‐3PUFAs vs placebo ‐ analyses based on comorbidities, Outcome 2 Adverse events.
4. Analyses based on adjunctive therapy
The effect of n‐3PUFAs compared to placebo in depressive symptomology (continuous) in studies with a mix of individuals receiving and not receiving adjunctive therapy was SMD = ‐0.43 (95% CI ‐0.82 to ‐0.04; 7 studies, 709 participants), in studies with individuals not receiving adjunctive therapy was SMD = ‐0.32 (95% CI ‐0.86 to 0.21; 6 studies, 308 participants), and in studies only including individuals receiving adjunctive therapy was SMD = ‐0.16 (95% CI ‐0.38 to 0.05; 12 studies, 356 participants). There was no statistical evidence of a difference between subgroups (P = 0.48) (see Analysis 4.1, Figure 14). However, the number of studies and the number of individuals in each subgroup were small, and confidence intervals are very wide, suggesting that effects could range from a large beneficial effect of n‐3PUFAs to a small negative effect, compared with placebo. There was no evidence of heterogeneity between subgroups (I² = 0%), and evidence of heterogeneity was low in the subgroup of studies including individuals receiving adjunctive therapy (I² = 0%). However, heterogeneity was high in the other two subgroups (I² = 76% and 77% respectively).
Analysis 4.1.
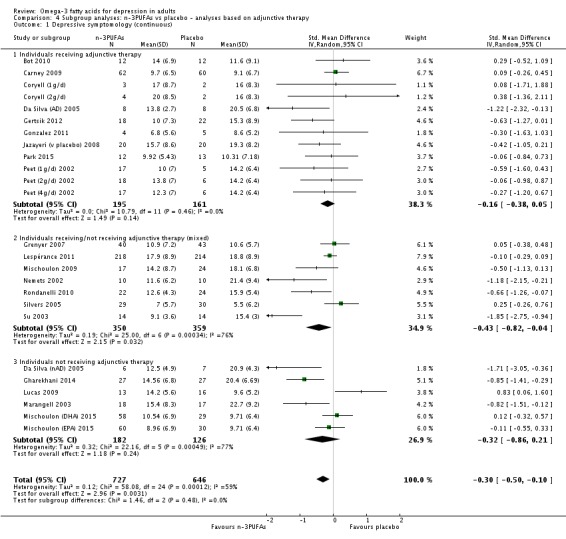
Comparison 4 Subgroup analyses: n‐3PUFAs vs placebo ‐ analyses based on adjunctive therapy, Outcome 1 Depressive symptomology (continuous).
Figure 14.
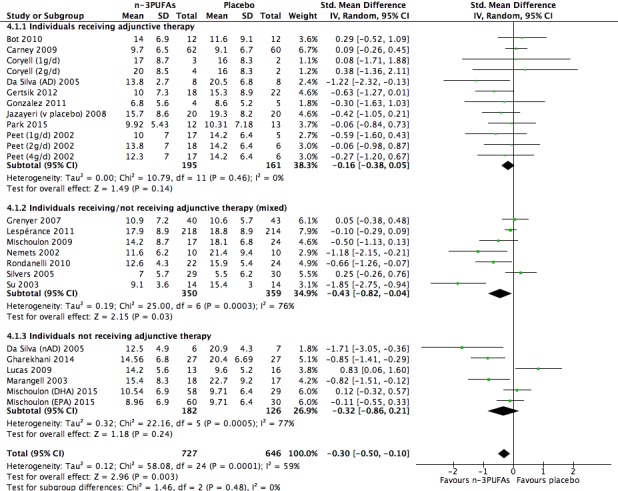
Forest plot of comparison: 1 n‐3PUFAs vs placebo, outcome: 4.1 Depressive symptomology (continuous): Sub‐groups based on use / non‐use of adjunctive therapies.
Rates of adverse events were similar across the subgroups. Analysis of studies with a mix of individuals receiving and not receiving adjunctive therapy: OR = 1.16 (95% CI 0.81 to 1.65; 6 studies, 644 participants), studies including individuals not receiving adjunctive therapy: OR = 2.04 (95% CI 1.03 to 4.03; 4 studies, 259 participants), and studies including individuals receiving adjunctive therapy: OR = 0.97 (95% CI 0.56 to 1.70; 9 studies, 304 participants) indicated that there was no statistical evidence of a difference between subgroups (P = 0.23) and some evidence of heterogeneity between subgroups (I² = 32%), see Analysis 4.2. However, confidence intervals are very wide and suggest possible effects ranging from a reduction in adverse events with n‐3PUFAs to a large increase in adverse events, compared with placebo. Evidence of heterogeneity was low in all subgroups (I² = 0% to 16%).
Analysis 4.2.
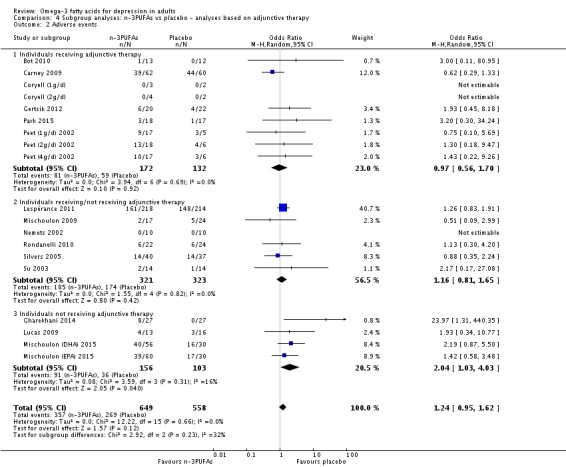
Comparison 4 Subgroup analyses: n‐3PUFAs vs placebo ‐ analyses based on adjunctive therapy, Outcome 2 Adverse events.
Sensitivity analyses
We conducted sensitivity analyses only for the n‐3PUFA versus placebo comparison. The sensitivity analyses using a fixed‐effect model were for all outcomes, while all other sensitivity analyses were conducted only for our primary outcome measures.
5. Low risk of bias
5.1 Selection bias
The results of analyses (random‐effects model) using only the studies that we judged to be at low risk of selection bias based on allocation concealment assessment (Bot 2010; Grenyer 2007; Lespérance 2011; Lucas 2009; Mischoulon 2009; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Nemets 2002; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Rondanelli 2010; Silvers 2005; Su 2003) were:
Depressive symptomology (continuous data): SMD = ‐0.18 (95% CI ‐0.42 to 0.06) (15 studies, 1033 participants), (I² = 60%).
Adverse events: OR = 1.31 (95% CI 0.98 to 1.75) (14 studies, 978 participants), (I² = 0%).
These analyses demonstrate no statistical differences between n‐3PUFA and placebo groups in depressive symptomology or adverse events, but confidence intervals are wide and suggest both a possible clinically important benefit of n‐3PUFAs and a negligible effect in depressive symptomology, and a range of effects in adverse events from a possible reduction of 2% to a possible increase in adverse events of 175%, with n‐3PUFAs compared to placebo.
5.2 Performance bias
The results of all analyses using only the studies that we judged to be at low risk of performance bias based on blinding of participants and personnel assessment (Bot 2010; Lespérance 2011; Lucas 2009; Nemets 2002; Rondanelli 2010; Silvers 2005) were:
Depressive symptomology (continuous data): SMD = ‐0.07 (95% CI ‐0.48 to 0.35) (6 studies, 610 participants), (I² = 70%).
Adverse events: OR = 1.22 (95% CI 0.85 to 1.74) (6 studies, 629 participants), (I² = 0%).
These analyses demonstrate no statistical differences between n‐3PUFA and placebo groups in depressive symptomology or adverse events, but confidence intervals are wide and suggest both a possible clinically important benefit of n3PUFAs and a small detrimental effect of n‐3PUFAs in depressive symptomology, and a range of effects in adverse events from a possible reduction of 15% to a possible increase in adverse events of 174%, with n‐3PUFAs compared to placebo.
5.3 Attrition bias
The results of all analyses using only the studies that we judged to be at low risk of attrition bias based on assessment of incomplete outcome data (Carney 2009; Coryell (1g/d); Coryell (2g/d); Lucas 2009; Mischoulon 2009; Nemets 2002; Rondanelli 2010; Su 2003) were:
Depressive symptomology (continuous data): SMD = ‐0.39 (95% CI ‐0.96 to 0.17) (8 studies, 297 participants), (I² = 76%).
Adverse events: OR = 0.82 (95% CI 0.46 to 1.45) (8 studies, 297 participants), (I² = 0%).
These analyses also demonstrate no statistical differences between n‐3PUFA and placebo groups in depressive symptomology or adverse events, but confidence intervals are wide and suggest both a possible clinically important benefit of n‐3PUFAs and a negligible effect in depressive symptomology, and a range of effects in adverse events from a possible reduction of 54% to a possible increase in adverse events of 145%, for n‐3PUFAs compared with placebo.
6. Fixed‐effect models
The results of all analyses using a fixed‐effect model were:
Depressive symptomology (continuous data): SMD = ‐0.19 (95% CI ‐0.30 to ‐0.08) (25 studies, 1373 participants).
Adverse events: OR = 1.29 (95% CI 0.99 to 1.67) (19 studies, 1207 participants).
Depressive symptomology (dichotomous data) ‐ remission: OR = 1.38 (95% CI 0.89 to 2.13) (6 studies, 426 participants).
Depressive symptomology (dichotomous data) ‐ response: OR = 1.42 (95% CI 1.00 to 2.01) (15 studies, 611 participants).
Quality of life: SMD = ‐0.35 (95% CI ‐0.56 to ‐0.13) (9 studies, 383 participants).
Failure to complete: OR = 0.85 (95% CI 0.64 to 1.14) (21 studies, 1344 participants).
Results are similar to those achieved using a random‐effects model, although effect sizes are noticeably smaller for measures of depressive symptomology and quality of life. Effect sizes in depressive symptomology are half the size using a fixed‐effect model compared to using a random‐effects model. Differences between n‐3PUFA and placebo groups in quality of life are also statistically significant.
Reporting Bias
The funnel plot for the main analysis of depressive symptomology (continuous) is presented in Figure 5. This figure demonstrates some asymmetry, suggesting possible publication bias in this outcome.
Funnel plots for adverse events and failure to complete also demonstrate some asymmetry, suggesting possible publication bias in these outcomes also (Figure 7 and Figure 12 respectively).
Additional Sensitivity Analyses
7. Use of a treatment that was solely or predominantly eicosapentaenoic acid (EPA)
7.1. Use of a treatment that was solely EPA
Eight studies used an intervention that was solely EPA (Bot 2010; Gonzalez 2011; Jazayeri (v placebo) 2008; Mischoulon 2009; Nemets 2002; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002).
The results of analyses using only these studies were:
Depressive symptomology (continuous data): SMD = ‐0.37 (95% CI ‐0.66 to ‐0.08) (8 studies, 203 participants), (I² = 0%).
Adverse events: OR = 0.99 (95% CI 0.39 to 2.45) (6 studies, 155 participants), (I² = 0%).
These analyses demonstrate a modest benefit for n‐3PUFAs compared to placebo for depressive symptomology and no differences between groups for adverse events, although confidence intervals again suggest an effect size for depressive symptomology that ranges from small to clinically important, and both a possible reduction and an increase in adverse events, for n‐3PUFAs compared to placebo. The overall effect size for depressive symptomology is larger than that for all studies, and the evidence of heterogeneity between studies is lower, but only a small number of studies are included in this analysis.
7.2. Use of a treatment that was predominantly EPA
Thirteen studies used an intervention that was predominantly EPA (Carney 2009; Coryell (1g/d); Coryell (2g/d); Da Silva (AD) 2005; Da Silva (nAD) 2005; Gertsik 2012; Gharekhani 2014; Lespérance 2011; Lucas 2009; Mischoulon (EPA) 2015; Park 2015; Rondanelli 2010; Su 2003).
The results of analyses using only these studies were:
Depressive symptomology (continuous data): SMD = ‐0.40 (95% CI ‐0.72 to ‐0.08) (13 studies, 906 participants), (I² = 71%).
Adverse events: OR = 1.26 (95% CI 0.88 to 1.80) (11 studies, 889 participants), (I² = 9%).
These analyses demonstrate a modest benefit for n‐3PUFAs compared to placebo for depressive symptomology and no differences between groups for adverse events, although confidence intervals again suggest an effect size for depressive symptomology that ranges from small to clinically important, and both a possible reduction and an increase in adverse events, for n‐3PUFAs compared to placebo. The overall effect size for depressive symptomology is larger than that for all studies, but the evidence of heterogeneity between studies is greater.
8. Inclusion of ALA in placebo capsules
Six studies used a placebo containing ALA (an n‐3PUFA) as a comparison (Bot 2010; Coryell (1g/d); Coryell (2g/d); Jazayeri (v placebo) 2008; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015).
The results of analyses without these studies were:
Depressive symptomology (continuous data): SMD = ‐0.40 (95% CI ‐0.65 to ‐0.15) (19 studies, 1121 participants), (I² = 66%).
Adverse events: OR = 1.14 (95% CI 0.85 to 1.54) (14 studies, 995 participants), (I² = 0%).
These analyses demonstrate a modest benefit for n‐3PUFAs compared to placebo for depressive symptomology and no differences between groups for adverse events, although confidence intervals again suggest an effect size for depressive symptomology that ranges from small to clinically important, and both a possible reduction and an increase in adverse events, for n‐3PUFAs compared to placebo. The overall effect size for depressive symptomology is larger than that for all studies, but the evidence of heterogeneity between studies is also higher.
9. Use of data from per protocol analyses
We could not obtain ITT data (either from publications or from authors) for 13 studies (Bot 2010; Da Silva (AD) 2005; Da Silva (nAD) 2005; Gonzalez 2011; Marangell 2003; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Park 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002; Silvers 2005).
The results of analyses without these studies were:
Depressive symptomology (continuous data): SMD = ‐0.36 (95% CI ‐0.66 to ‐0.07) (13 studies, 946 participants), (I² = 70%).
Adverse events: OR = 1.17 (95% CI 0.74 to 1.85) (11 studies, 825 participants), (I² = 21%).
These results are very comparable to those conducted using all studies, although confidence intervals are wider, suggesting less precision in the overall effect sizes.
10. Use of imputed standard deviations from other studies in analyses
We imputed standard deviations for five studies (Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002).
The results of analyses without these studies were:
Depressive symptomology (continuous data): SMD = ‐0.22 (95% CI ‐0.34 to ‐0.10) (20 studies, 1127 participants), (I² = 65%).
Adverse events: OR = 1.15 (95% CI 0.84 to 1.59) (14 studies, 1127 participants), (I² = 2%).
These analyses demonstrate smaller differences between n‐3PUFA and placebo groups for depressive symptomology and adverse events than in our analyses of all studies. Confidence intervals are also narrower, suggesting a greater precision compared to the overall effect sizes, although the evidence of heterogeneity between studies remains high.
11. Consideration of multiple comparison groups from the same study as individual studies
Four trials used multiple treatment groups that we considered in our primary analyses as independent studies (Coryell (1g/d); Coryell (2g/d); Da Silva (AD) 2005; Da Silva (nAD) 2005; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015; Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002). Use of these studies as independent groups may magnify between‐study heterogeneity, although data for all of these studies have been provided for each group separately, so that combining groups results in standard deviations that are estimations based on pooling calculations.
The results of analyses using combined as opposed to split studies were:
Depressive symptomology (continuous data): SMD = ‐0.32 (95% CI ‐0.54 to ‐0.10) (20 studies, 1364 participants), (I² = 68%).
Adverse events: OR = 1.32 (95% CI 0.97 to 1.80) (16 studies, 1183 participants), (I² = 0%).
These results are very comparable to those conducted using all studies as independent studies.
Discussion
Summary of main results
We found trials comparing the impact of n‐3PUFAs on major depressive disorder (MDD) to two different comparators. Twenty‐five studies involving 1438 participants investigated the impact of n‐3PUFAs in MDD compared to placebo, and one study involving 40 participants investigated the impact of n‐3PUFAs in MDD compared to antidepressant treatment.
For the comparison with placebo, we provide a 'Summary of findings' table (Table 1).
Our primary outcomes were depressive symptomology assessed using a continuous measure, and adverse events. Mean depressive symptomology in n‐3PUFA groups was 0.30 (95% CI 0.10 to 0.50) standard deviations lower than placebo following treatment. This small‐to‐modest effect size represents a difference between groups in scores on the HDRS (17‐item) of approximately 2.1 (0.7 to 3.5 respectively). NICE guidelines (NICE 2004) have previously suggested a reduction in HDRS score of 3 or more to be clinically meaningful, thus the clinical significance of our effect size is small. The confidence intervals do not exclude a clinically meaningful effect, but also include a negligible effect at the lower end. Furthermore, the completeness and quality of the evidence was very low (see below).
Numbers of individuals experiencing adverse events were similar between intervention and placebo groups, although assessments of adverse events were suitable for analysis in only 19 of the 25 studies, and our confidence intervals suggest that effects could range from a small reduction to a modest increase in adverse events in n‐3PUFA groups compared with placebo. Furthermore, the completeness and quality of the evidence providing these results was also very low.
Rates of depression remission and response were also similar following n‐3PUFA supplementation compared to placebo, but confidence intervals again suggest a range of possible effects from a small reduction in remission and response rates to a large increase. Quality of life was similar in n‐3PUFA compared with placebo groups, although our confidence intervals again suggest both a possible negligible effect and a possible clinically important benefit of n‐3PUFAs compared to placebo. Rates of failure to complete were also similar between intervention and placebo groups, although our confidence intervals again suggest possible effects that could range from a small reduction to a modest increase in study withdrawals in n‐3PUFA groups compared with placebo.
There was only one study involving 40 participants for the comparison with antidepressants. This study found no differences between treatment with n‐3PUFAs and treatment with antidepressants in depressive symptomology (MD (HDRS(24 item))= ‐0.70 (95% CI: ‐5.88 to 4.48)), rates of response to treatment, or failure to complete. Adverse events were not reported in a manner suitable for analysis, and rates of depression remission and quality of life were not reported.
Overall completeness and applicability of evidence
The evidence for both comparisons and for all outcomes is limited and highly heterogeneous, resulting in findings that are imprecise and potentially biased.
Firstly, for the comparison with placebo, the evidence comes from 25 studies, involving only 1438 participants and with only 1373 participants contributing to the analysis. While data are available from all studies for the analyses on our primary outcome of depressive symptomology, only small numbers of studies contributed to some of our outcomes.
The studies available were highly heterogeneous. All studies were directly relevant to our research question, but we found considerable differences in all aspects of study methodology. Studies differed in the type of participants involved, the interventions used, the comparators used, the duration of supplementation, and the range and measurement of outcomes assessed.
The majority of available studies were also small. Almost half of all participants derive from only three trials: Carney 2009 (122 participants), Lespérance 2011 (432 participants), and the Mischoulon 2015 trial (196 participants). We judged these trials to be at low risk of bias on most measures, but the contribution of these three trials to our overall outcomes was high, even using a random‐effects model (depressive symptomology ‐ 26.9%; adverse events ‐ 82%), and the outcomes of our meta‐analyses reflect the outcomes of these specific trials. All three trials found a negligible mean difference between n‐3PUFA and placebo groups following supplementation. Biases or methodologically specific outcomes in these trials may have contributed to our overall result. All three trials, for example, include participants with comorbidities, use an intervention composed of a combination of EPA and DHA, and assess effects of supplementation after eight to ten weeks. All of these factors may have affected study outcomes, which in turn may have affected our overall outcome.
The funnel plot also suggests an absence of small studies showing null findings. This asymmetry suggests probable publication bias, and suggests that our analyses and overall effect size estimates may be biased towards a positive finding for n‐3PUFAs compared to the true situation. Sensitivity analyses using a fixed‐effect model also demonstrated a smaller standardised mean difference between n‐3PUFA and placebo groups than that found using a random‐effects model, suggesting a positive influence from small positive studies in our main analyses.
Sensitivity analyses using only the studies that we judged to be at low risk of bias also suggest bias in our main analyses towards a positive finding for n‐3PUFAs. Many studies we judged to be at high risk of bias in various domains. Analyses using only studies that we judged to be at low risk of selection bias, performance bias and attrition bias report smaller effect sizes than those found in our main analyses, and confidence intervals include the possibility of no differences between groups. This evidence, alongside that of the funnel plot and the findings using a fixed‐effects model suggest that the true effect of n‐3PUFAs is likely to be smaller than that reported in our main analyses.
Imprecise effect size estimates were found for all outcomes. In all analyses, possible effects range from negligible (and in some analyses from negative) effects to important clinical benefits. While this imprecision does not rule out clinically relevant effects, considerable caution must be used in interpreting all effect size estimates. Further evidence, in the form of adequately‐powered well‐designed trials, is clearly required before firm conclusions can be drawn.
Findings in our primary outcome of depressive symptomology and our secondary outcome of quality of life also demonstrate considerable evidence of heterogeneity. Subgroup analyses investigated possible sources, based on the inclusion of individuals with/without comorbid conditions and the inclusion of individuals using/not using adjunctive therapy, but we found little explanation for this heterogeneity. There is some evidence of different effects depending on the presence or absence of comorbid conditions, and this has also been suggested in individual trials (e.g. Lespérance 2011), but effects are currently far from clear. Limited studies contribute to each subgroup in our analyses, there is considerable evidence of heterogeneity, and there is considerable overlap in effect size estimates for different subgroups. Limited explanation was also gained from the analyses on adjunctive therapy, where few differences between subgroups were found, although findings again are far from conclusive.
Sensitivity analyses also investigated the impact of other aspects of study methodology. Analyses investigating the effects of treatment solely or predominantly with EPA revealed a stronger beneficial effect of n‐3PUFAs compared to placebo than we found in our main analyses, although precision also decreases further, allowing the possibility of negligible effects. Effects size estimates also increase with the removal of studies that use placebos containing ALA. This analysis suggests that ALA may confer some impacts on depressive symptomology similar to those of the longer chain n‐3PUFAs EPA and DHA, but very few studies were available for assessment. The possibility of different effects depending of n‐3PUFA type is interesting, and has been suggested elsewhere in the literature (Martins 2011; Ross 2007; Sublette 2011), although much of this speculation is based on post‐hoc observation, and only one current trial directly compares the impact of EPA and DHA treatments (Mischoulon (DHA) 2015; Mischoulon (EPA) 2015), and found no differences. Mechanisms of action to explain different effects from different n‐3PUFAs are hypothesised (e.g. Ross 2007; Sublette 2011). Proposed mechanisms for action, however, largely focus on the specific actions of EPA and DHA, while offering little suggestion for a potential impact of ALA.
Sensitivity analyses investigating the methods used to conduct analyses reveal few differences in findings from those in our main analyses, although the removal of studies where we imputed standard deviations reduced effect sizes and improved precision.
Only one study was available for the comparison with antidepressant treatment. This study was small, with 20 participants randomised to each treatment arm, and 20% of participants in each arm failed to complete the study. Adverse events were reported by the number of events rather than the number of individuals suffering, remission rates were not reported, but response rates and failure to complete data were supplied.
Quality of the evidence
The quality of the evidence for both comparisons for all outcomes is very low to low. Our judgements of quality according to GRADE are given in the Table 1.
For the placebo comparison, for our primary outcome of depressive symptomology, we considered the quality of evidence to be very low. The body of evidence was composed of limited, predominantly small studies, within which there was substantial evidence of heterogeneity that remains unexplained. Furthermore, the majority of the contributing studies include judgements of high risk of bias in at least one of the domains assessed, and sensitivity analyses reveal different findings in analyses using all studies and analyses using only studies judged to be at low risk of selection bias, performance bias or attrition bias. In all analyses using only studies judged to be at low risk of bias, regardless of the measure of bias used, we found that n‐3PUFAs did not impact on depressive symptomology, compared to placebo. We found similar results from the three large well‐conducted trials mentioned earlier (Carney 2009; Lespérance 2011; Mischoulon (DHA) 2015; Mischoulon (EPA) 2015). As stated earlier, the consideration of risk of bias suggests bias in our main analyses towards a positive finding for n‐3PUFAs, as a result of a high risk of bias in many of the contributing studies. These findings suggest that the positive effect of n‐3PUFAs in our main analyses is a consequence predominantly of the inclusion of studies that may be at high risk of bias. The true effect size estimate is thus likely to be smaller than that provided by our main analyses.
For our second primary outcome of adverse events, the quality of evidence was very low. There was limited evidence of heterogeneity between studies in this analysis, but confidence intervals are wide, suggesting a range of possible effects; data were only available from selected studies, and visual inspection of the funnel plot suggests probable publication bias. Analyses using only studies judged to be at low risk of selection bias, low risk of performance bias, or low risk of attrition bias suggest similar effects as those presented in our main analyses.
For our secondary outcomes of depression remission and response rates, the quality of the evidence was low. Heterogeneity between studies was low, but confidence intervals are very wide, suggesting a broad range of possible effects, selected studies only were available for these analyses, and we judged various elements of these studies to be at high risk of bias.
For our secondary outcome of quality of life, the quality of the evidence was very low. Selected studies only were available for these analyses, confidence intervals are very wide, suggesting a broad range of possible effects, there was high heterogeneity, and we judged various elements of these studies to be at high risk of bias.
For our secondary outcome of failure to complete, the quality of the evidence was low. Most studies were available for this analysis, and heterogeneity between studies was low, but confidence intervals are again wide, suggesting a range of possible effects, and visual inspection of the funnel plot suggests a high probability of publication bias.
For the antidepressant comparison, the quality of the evidence was low. Evidence for this analysis came from only one study. We judged this study to be at high risk of bias for allocation concealment, because the randomisation sequence was not concealed from researchers; for performance bias, because no steps were reported to mask the fishy taste of the intervention or check concealment; for attrition bias, due to a 20% dropout rate; and for reporting bias, because some outcomes have not yet been published. We judged data on depressive symptomology to be at low risk of bias due to good blinding of study personnel, but we judged data on adverse events to be at high risk of detection bias because these were reported by participants, and adequate blinding was unclear. We considered risk of attrition bias for adverse events to be low.
Inconsistency in trial reporting for both comparisons was obvious. Depressive symptomology was frequently reported and analysed using non‐ITT data (assuming a definition of ITT based on number randomised). Adverse events were reported in a variety of ways: by individual and event for all events, by individual for all events, by individual for only serious, likely or frequent events, by event type for all events, by event type for only serious, likely related or frequent events, or a combination of these. Adverse events were included for analysis (based on the number of individuals) in 19 of 25 of the studies. Our secondary outcomes of remission and response in depressive symptomology and quality of life were not well reported. Numbers of participants who failed to complete each trial were well reported.
Potential biases in the review process
The findings of this review are likely to be biased, due to the evidence available to contribute to analyses. Only a limited number of studies were available for assessing all outcomes for both comparisons, only a few studies were available for assessing some outcomes, and there was a high relative weighting in all analyses for the placebo comparison from three large studies.
The review process also may have been biased. Our searches were more likely to detect articles published in English and in mainstream journals. We tried to minimise this bias by including translated articles, but translations were only undertaken for full articles that we selected for inclusion based on titles and abstracts. We were also unable to contact authors of some articles that may have been relevant. We excluded these articles, based on the information available to us, but increased information would have reduced this bias. We made judgements of risk of bias according to predefined rules, but the information required to make these judgements was infrequently published, and our correspondence with authors was again incomplete. Judgements of risk of bias, however, were completed following all data collection, so did not impact on the review process. Reliance on available data (even from authors), also meant that only a few studies could contribute to certain analyses. Remission and response rates were not assessed in all studies, but could have been calculated had raw data been available. Most studies did not assess quality of life. We relied on authors of existing relevant studies or trial registrations for information on unpublished studies. Our searches covered relevant conference‐based publications, but we made no further attempts to find or identify unpublished literature.
Agreements and disagreements with other studies or reviews
Published reviews and meta‐analyses are available investigating a role for n‐3PUFAs for depression compared to placebo. Many of these have focused on randomised controlled trials, but most use a broad definition of depression to consider studies of individuals with a range of depressive diagnoses, including bipolar disorder and postpartum depression, and studies of individuals with depressive symptomology regardless of psychiatric condition. Early reviews tended to use a broader working definition of depressive symptomology, to allow inclusion of adequate studies for analyses, but more recent reviews have used tighter inclusion criteria.
Of recent reviews, the review by Grosso 2014 reports two meta‐analyses, one of studies of individuals with a formal diagnosis, and one of studies of individuals with high levels of depressive symptomology but no formal diagnosis. The analysis of studies of individuals with a formal MDD diagnosis includes nine of the studies included in our analyses, but also includes two additional reports of Rondanelli 2010, and includes both the placebo and the antidepressant comparisons in Jazayeri (v placebo) 2008 and Jazayeri (v AD) 2008), without taking account of the use of the same placebo group. This analysis provides a combined effect size estimate, representing a beneficial effect of n‐3PUFAs, of SMD = 0.56 (95% CI 0.20 to 0.92). The analysis of studies of individuals with high depressive symptomology but no formal diagnosis of MDD includes four of the studies included in our analysis (although individuals in the control group in Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002 are included repeatedly), but also includes seven other studies that we discounted from our review on the basis that individuals with low or moderate depression were also included in intervention groups, and could confound findings. This analysis produced a combined effect size estimate of SMD = 0.22 (95% CI 0.01 to 0.43), and the two analyses together result in a combined effect size estimate of SMD = 0.38 (95% CI 0.18 to 0.59) (I² = 55%). This combined estimate is very similar to that achieved in our analyses, even though these additional studies were included and six of those identified for inclusion in our review did not contribute, including the large trial by Lespérance 2011.
The review by Bloch and Hannestad (Bloch 2012) is a review of 13 studies of individuals with MDD. This analysis includes nine studies that we included in our review, but also includes one study in mild‐moderate depression (Rogers 2008), and three studies in MDD associated with pregnancy. The use of the full study or only the relevant subgroup in Lucas 2009 is also unclear. Ten of the studies in our review, all published in 2008 or later, were not included. The analysis reports no effects of n‐3PUFAs compared to placebo (SMD = 0.11, 95% CI ‐0.04 to 0.26, P = 0.14), but has been criticised elsewhere (Lin 2012) for the inclusion of the trials by Rogers 2008 and Lucas 2009, due to their inclusion of individuals without a formal MDD diagnosis. Reanalysis of the results of the remaining 11 studies demonstrates a small beneficial effect of n‐3PUFAs for MDD compared to placebo, of SMD = 0.29 (95% CI 0.10 to 0.48). This result is very similar to that provided by our analyses.
The review by Martins 2011 includes 35 studies of depressive symptomology, and includes a separate analysis of 14 studies of individuals with MDD. Thirteen of these studies were also included in our review, and their analyses provide a combined effect size estimate, representing a benefit of n‐3PUFAs: SMD = ‐0.45 (95% CI ‐0.75 to ‐0.15). This finding is larger than that found in our analyses, but the confidence intervals are wide and include the effect size estimates from our analyses.
The review by Sublette 2011 covers 19 trials, 13 of which are also included in our review, while two trials were studies of individuals with a primary diagnosis of bipolar disorder, three were studies of individuals with postpartum depression, and one study included individuals with mild‐moderate as opposed to severe depression. This review investigates specifically the percentage of EPA provided (≥ 60% vs < 60%) as a source of heterogeneity, and does not report a combined effect size estimate for all studies.
Similar findings from several meta‐analyses, despite the inclusion and exclusion of different trials, may suggest a consistent effect of n‐3PUFAs versus placebo, but the consistency is more likely in the limited evidence available for investigating these effects. All reviews are based on the same very limited pool of studies, and report wide confidence intervals and so a wide range of possible effects, substantial heterogeneity between studies, and a high probability of publication bias.
Our effect size estimate is also comparable to some degree with that suggested by recent meta‐analyses of the effects of antidepressants for MDD, compared to placebo. Kirsch 2008 reports a weighted mean difference using the HDRS between antidepressant and placebo groups in 35 US Food and Drug Administration (FDA) registered studies of 1.8 HDRS scores, SMD = 0.32 (95% CI 0.25 to 0.40). Turner 2008 reports a mean weighted effect size of 0.37 (95% CI 0.33 to 0.41) from published and 0.15 (95% CI 0.08 to 0.22) from unpublished US FDA‐registered studies. Fountoulakis 2013, in an analysis of recent meta‐analyses, confirms a SMD of 0.32 (95% CI: 0.25 to 0.40), as the result from the most appropriate analysis of the Kirsch 2008 data set. Some evidence is available again suggesting greater effect sizes dependent on depression severity, e.g. Fournier 2010 reports an effect size of d = 0.17 (95% CI 0.04 to 0.30) in a patient‐level analysis of severe MDD cases, and of d = 0.47 (95% CI 0.34 to 0.59) in very severe MDD cases, but limitations in the data available reduce the value of these conclusions. Confidence intervals, however, are much tighter for the analyses of antidepressants than was found in our analyses, suggesting greater precision in these effect size estimates. Analyses of antidepressants suggest effect size estimates that range from small to modest, while our findings on n‐3PUFAs also allow the inclusion of possible negligible effects. The apparent comparability in effect size estimates between our findings and those of reviews on antidepressants should not be taken as evidence in support of n‐3PUFAs. The small size of the overall effect size estimate for both n‐3PUFAs and antidepressants should argue not for a favourable comparison of n‐3PUFAs with antidepressants, but for increased demand for more effective treatments for depressive symptomology from elsewhere.
Authors' conclusions
At present, we do not have sufficient high quality evidence to determine the effects of n‐3PUFAs as a treatment for MDD. Our primary analyses suggest a small‐to‐modest, non‐clinically beneficial effect of n‐3PUFAs on depressive symptomology compared to placebo, although the effect size estimate is imprecise, and the quality of the evidence on which this result is based is low to very low. Sensitivity analyses, funnel plot inspection and comparison of our results with those of large well‐conducted trials also suggest that this effect size estimate is likely to be biased towards a positive finding for n‐3PUFAs, and that the true effect is likely to be smaller. The one study in our review that directly compares n‐3PUFAs and antidepressants finds comparable benefit, but the quality of the evidence here is very low. Our data suggest similar rates of adverse events and numbers failing to complete trials in n‐3PUFA and placebo groups. The data on adverse events and failure to complete are again of low quality, but given the high rates of adverse events associated with some antidepressants, n‐3PUFAs may offer an alternative treatment of possible benefit and reduced side effects. However, whether all possible negative side effects are studied in trials is questionable, and high dropout rates as a result of lack of improvement testify to the negative side effects of false hope. Failure to seek or administer conventional treatment, as a result of treatment with n‐3PUFAs, may also represent an opportunity cost. We need more evidence, and particularly more complete evidence, regarding both the positive and negative effects of n‐3PUFAs for MDD.
More adequately‐powered well‐designed studies are required to increase the evidence base, and explore particularly the heterogeneity found between studies investigating the impact of n‐3PUFAs on depressive symptomology. Many studies are currently underway, but studies that compare n‐3PUFAs with usual antidepressant treatment, and studies to investigate differing effects depending on individual characteristics and study methodology are important. Our review suggests similar effects for n‐3PUFAs and antidepressant treatment for depressive symptomology, but benefits of n‐3PUFAs in terms of adverse events, compliance and patient acceptability are often provided by practitioners. Studies that compare n‐3PUFAs with antidepressant treatment on all possible outcomes are required. Long‐term benefits, long‐term acceptability and long‐term compliance are rarely considered, and neither is cost effectiveness. Studies comparing individuals and different treatments are also needed. Studies do find positive effects, and identification of those who are likely to benefit, or the particular treatments of beneficial impact would be of value. Mechanistic studies are also preferentially required. Hypotheses investigating differential effects depending on participant type or study methodology should be based on proposed mechanisms to increase efficacy, as opposed to post hoc comparisons of individual studies. Future research should target the elucidation of mechanisms both for the development and treatment of MDD, and should identify the possible actions in these pathways for n‐3PUFAs.
Acknowledgements
This work is supported by Bournemouth University, UK, the NIHR Biomedical Research Unit in Nutrition, Diet and Lifestyle, University Hospitals NHS Foudnation Trust and the University of Bristol, UK, and the University of Bristol, UK.
CRG Funding Acknowledgement:
The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Depression, Anxiety and Neurosis Group.
Disclaimer:
The views and opinions expressed herein are those of the authors and do not necessarily reflect those of the NIHR, National Health Service (NHS) or the Department of Health.
Appendices
Appendix 1. How the intervention might work
The positive effects of n‐3PUFAs on depressive illness are thought to occur as a result of integration into the cell membrane phospholipid bilayer, resulting in changes in structure and function (Haag 2003; James 2000; Ruxton 2005). Incorporation into the cell membrane can influence the physical state of the membrane, resulting in increased fluidity and permeability (Ehringer 1990; Hirashima 2004; Tappia 1997), possibly aiding cross‐cell membrane transport and communication (Haag 2003). Secondly, n‐3PUFAs are also thought to have effects on surrounding molecules and cell functions via enzyme activity which results in the release of fatty acids from the phospholipid bilayer to form a number of anti‐inflammatory eicosanoids, prostaglandins, and leukotrienes (Calder 2003; James 2000; Ruxton 2005; Stahl 2008), and via enzyme activity of direct involvement in various neurotransmitter pathways (Haag 2003; James 2000; Ruxton 2005; Stahl 2008). Supplementation with n‐3PUFAs has been found to result in: reduced production of inflammatory cytokines ‐ tumour necrosis factor alpha (TNFa), interleukin 1B, interleukin‐6, C‐reactive protein and serum amyloid A (Calder 2003; Caughey 1996; James 2000; Rallidis 2003); increased serotonergic and dopaminergic activity; and decreased concentrations of noradrenalin (Chalon 2006; De la Presa Owens 1999; Hamazaki 2005; Sawazaki 1999; Yao 2004). Additionally, n‐3PUFA‐deficient diets have been associated with reduced receptor density and disruptions to neurotransmitter activity in serotonergic (De la Presa Owens 1999; Delion 1994; Delion 1996; McNamara 2006), dopaminergic (Chalon 2006; De la Presa Owens 1999; Delion 1994; Delion 1996; McNamara 2006; Takeuchi 2002), and adrenergic systems (Takeuchi 2002) compared to controls. Disruptions to and abnormal cell signalling, inflammatory processes and neurotransmitter system activities have been implicated in MDD (Parker 2006b; Stahl 2008).
Appendix 2. Why it is important to do this review
n‐3PUFAs have been linked to depression in a variety of epidemiological studies (Hibbeln 1998; Noaghiul 2003; Peet 2004; Silvers 2002; Tanskanen 2001); clinical studies (Edwards 1998; Garland 2007; Mamalakis 2002; Mamalakis 2006; Peet 1998); and RCTs (Frangou 2006; Nemets 2002; Stoll 1999; Su 2003).
However, several epidemiological studies have found no association between n‐3PUFA intake and depressive illness (e.g. Appleton 2007; Frangou 2006; Hakkarainen 2004; Miyake 2006; Stoll 1999; Su 2003). Clinical studies have reported no differences in n‐3PUFA levels between individuals diagnosed with MDD and controls (e.g. Browne 2006; Mamalakis 2004) and no clear associations (Appleton 2008a). Several RCTs have also reported no effects of supplementation on MDD (e.g. Grenyer 2007; Silvers 2005), depressive illness (e.g. Keck 2006) or depressed mood (e.g. Rogers 2008).
Reviews in this area clearly demonstrate considerable variability between studies (e.g. Appleton 2006; Appleton 2008b; Appleton 2010; Lin 2007; Parker 2006b; Smith 2011; Stahl 2008). Meta‐analyses also report considerable heterogeneity between studies ( Appleton 2006; Appleton 2010; Lin 2007). Meta‐analyses reveal some small benefit of n‐3PUFAs for depressive disorders (Appleton 2006; Lin 2007), but investigations of the considerable heterogeneity also suggest differential effects of n‐3PUFAs dependent primarily on severity of depressive symptoms at baseline (Appleton 2010). Sensitivity analyses based on severity of depressive symptoms at baseline suggest no benefits of n‐3PUFAs for individuals with mild depressive symptoms or without diagnosis of depression, but also provide some evidence of benefits in individuals with severe depressive symptoms or with depressive diagnoses (Appleton 2010). These findings suggest a possible benefit of n‐3PUFAs for MDD.
Appendix 3. CINAHL search strategy
[Diagnosis]
S1 (MH "Depression") S2 (MH "Depression, Reactive") S3 (MH "Dysthymic Disorder") S4 (MH "Affective Disorders") S5 (MH "Affective Symptoms") S6 (depress* or dysthymi* or “adjustment disorder*” or “affective disorder*” or “affective symptom*” or “mood disorder*”) S7 (S1 or S2 or S3 or S4 or S5 or S6 or S7)
[Intervention]
S8 (MH “FISH OILS”) S9 (MH “FATTY ACIDS, OMEGA‐3”) S10 (MH “DOCOSAHEXAENOIC ACIDS”) S11 (MH “EICOSAPENTAENOIC ACID”) S12 (AB ( (DHA or Docosahex* or Eicosapent* or EPA or “fatty acid*” or fish* or linolenic or omega‐3 or n‐3 or w‐3 or PUFA* or “cod liver oil” or “cod‐liver oil”) ) OR TI ( (DHA or Docosahex* or Eicosapent* or EPA or “fatty acid*” or fish* or linolenic or omega‐3 or n‐3 or w‐3 or PUFA* or “cod liver oil” or “cod‐liver oil”))) S13 (S8 or S9 or S10 or S11 or s12)
[RCT Filter]
S14 (MH "Clinical Trials+") S15 (PT Clinical trial) S16 (TX clini* n‐3 (trial* or study or studies)) S17 (TX ((singl* N1 blind*) or (singl* N1 mask*)) or TX ((doubl* N1 blind*) or (doubl* N1 mask*)) or TX ((tripl* N1 blind*) or (tripl* N1 mask*))) S18 (TX random* n‐3 control*) S19 (MH "Random Assignment") S20 (TX random and (allocat* or assign*)) S21 (TX placebo*) S22 (TX (waitlist* or (wait* and list*)) and (control* or group)) S23 (TX "treatment as usual" or TI TAU or AB TAU) S24 (TX (control* n‐3 (trial* or study or studies or group*))) S25 (MH "Quantitative Studies") S26 (S14 or S15 or S16 or S17 or S18 or S19 or S20 or S21 or S22 or S23 or S24 or S25)
S27 (S7 and S13 and s26)
Appendix 4. Risk of Bias Assessment Tool
n‐3PUFAs and MDD. Risk of Bias Assessment Tool
SEQUENCE GENERATION
|
LOW RISK The investigators describe a random component in the sequence generation process such as: · Referring to a random number table; Using a computer random number generator; Coin tossing; Shuffling cards or envelopes; Throwing dice; Drawing of lots/slips; Minimization*. *Minimization may be implemented without a random element, and this is considered to be equivalent to being random |
|
HIGH RISK The investigators describe a non‐random component in the sequence generation process. Usually, the description would involve some systematic, non‐random approach, for example: · Sequence generated by odd or even date of birth · Sequence generated by some rule based on date (or day) of admission · Sequence generated by some rule based on hospital or clinic record number. Other non‐random approaches happen much less frequently than the systematic approaches mentioned above and tend to be obvious. They usually involve judgement or some method of non‐random categorization of participants, for example: · Allocation by judgement of the clinician · Allocation by preference of the participant · Allocation based on the results of a laboratory test or a series of tests · Allocation by availability of the intervention. |
|
UNCLEAR RISK Insufficient information about the sequence generation process to permit judgement of ‘Yes’ or ‘No’. |
ALLOCATION CONCEALMENT
|
LOW RISK Participants and investigators enrolling participants could not foresee assignment because one of the following, or an equivalent method, was used to conceal allocation: · Central allocation (including telephone, web‐based, and pharmacy‐controlled, randomization) · Sequentially numbered drug containers of identical appearance · Sequentially numbered, opaque, sealed envelopes – all 3 features of the envelopes must be described |
|
HIGH RISK Participants or investigators enrolling participants could possibly foresee assignments and thus introduce selection bias, such as allocation based on: · Using an open random allocation schedule (e.g. a list of random numbers) · Assignment envelopes were used without appropriate safeguards (e.g. if envelopes were unsealed or non‐opaque or not sequentially numbered) · Alternation or rotation · Date of birth · Case record number · Any other explicitly unconcealed procedure. |
|
UNCLEAR RISK Insufficient information to permit judgement of ‘Yes’ or ‘No’. This is usually the case if the method of concealment is not described or not described in sufficient detail to allow a definite judgement – for example if the use of assignment envelopes is described, but it remains unclear whether envelopes were sequentially numbered, opaque and sealed. |
BLINDING OF PARTICIPANTS AND PERSONNEL
|
LOW RISK · Some assessment of blinding at follow‐up, and blinding found to be successful · Flavours of both intervention and control treatments masked by flavour · Small amount of fish oil added to placebo |
|
HIGH RISK · Either participants or study personnel were not blinded. · Participants guessed allocation · No attempts to mask / counter fish oil taste / smell, despite clear description of other aspects of intervention and placebo. |
|
UNCLEAR RISK Insufficient information to permit judgement of ‘Low’ or ‘High Risk’ |
BLINDING OF OUTCOME ASSESSORS
|
LOW RISK · Methods of blinding of outcome assessors described sufficiently and deemed adequate |
|
HIGH RISK Any one of the following: · Methods of blinding of outcome assessors described sufficiently but deemed inadequate · No blinding or incomplete blinding, and the outcome or outcome measurement is likely to be influenced by lack of blinding |
|
UNCLEAR RISK · Insufficient information to permit judgement of ‘Low’ or ‘High Risk’ |
Where more than one outcome measure is used, overall score will be based on the one used in our analyses.
INCOMPLETE OUTCOME DATA= only relevant to data after randomisation
|
LOW RISK Any one of the following: · No missing outcome data · ITT analysis (includes all those randomized) · Missing outcome data less than 10% of the total randomised population · Missing outcome data for mood less than 5% of the total randomised population · Reasons for missing outcome data unlikely to be related to true outcome (for survival data, censoring unlikely to be introducing bias) · Missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups · Difference in missing data between the groups not greater than 10% · i.e. intervention group of 120 has 6 drop out (5% of trial arm) and control group of 100 has 2 drop out (2% of trial arm): difference in missing data is 3% therefore LOW RISK · Difference in missing data for mood between the groups not greater than 5% |
|
HIGH RISK Any one of the following: · Reason for missing outcome data likely to be related to true outcome, with either imbalance in numbers or reasons for missing data across intervention groups · Difference in missing data between the groups greater than 10% · i.e. intervention group of 120 has 6 drop out (5% of trial arm) and control group of 100 has 26 drop out (26% of trial arm): difference in missing data is 21% therefore HIGH RISK · Difference in missing data for mood between the groups greater than 5% · Overall missing data greater than 10% of the total randomised population · Overall missing data for mood greater than 5% of the total randomised population · Stated as ‘intention‐to‐treat analysis’ but doesn’t use this · Analysed using ‘per protocol’ analyses · ‘As‐treated’ analysis done with substantial departure of the intervention received from that assigned at randomization |
|
UNCLEAR RISK Any one of the following: · Insufficient reporting of attrition/exclusions to permit judgement of ‘Yes’ or ‘No’ (e.g. number randomized not stated, no reasons for missing data provided) · Dropouts not mentioned |
SELECTIVE OUTCOME REPORTING
|
LOW RISK Any of the following: · The study protocol is available and all of the study’s pre‐specified (primary and secondary) outcomes that are of interest in the review have been reported in the pre‐specified way – use trial registration number if available to locate protocol · The study protocol is not available but it is clear that the published reports include all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon). · The study protocol is not available but authors state that all outcomes are reported. |
|
HIGH RISK Any one of the following: · Not all of the study’s pre‐specified primary and secondary outcomes have been reported · Pre‐specified in methods section · Or pre‐specified in protocol · One or more primary or secondary outcomes is reported using measurements, analysis methods or subsets of the data (e.g. subscales) that were not pre‐specified · One or more reported primary or secondary outcomes were not pre‐specified (unless clear justification for their reporting is provided, such as an unexpected adverse effect) · One or more outcomes of interest in the review are reported incompletely so that they cannot be entered in a meta‐analysis – any data excluded from the analysis despite the data being available (i.e. so the reviewers decided not to include it in the meta‐analysis) · The study report fails to include results for a key outcome that would be expected to have been reported for such a study Outcomes refer in all cases to all study outcomes. |
|
UNCLEAR RISK Insufficient information to permit judgement of ‘Yes’ or ‘No’. · No protocol is available, and no contact can be gained with authors. |
OTHER BIAS
|
LOW RISK The study appears to be free of other sources of bias. |
|
HIGH RISK · Stopped early due to some data‐dependent process (including a formal‐stopping rule) · Significant baseline imbalance for mood outcomes |
|
UNCLEAR RISK There may be a risk of bias, but there is either: · Insufficient information to assess whether an important risk of bias exists · Insufficient rationale or evidence that an identified problem will introduce bias. |
Data and analyses
Comparison 1.
n‐3PUFAs vs placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depressive symptomology (continuous) | 25 | 1373 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.50, ‐0.10] |
| 2 Adverse events | 19 | 1207 | Odds Ratio (M‐H, Random, 95% CI) | 1.24 [0.95, 1.62] |
| 3 Depressive symptomology (dichotomous ‐ remission) | 6 | 426 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [0.87, 2.20] |
| 4 Depressive symptomology (dichotomous ‐ response) | 15 | 611 | Odds Ratio (M‐H, Random, 95% CI) | 1.39 [0.95, 2.04] |
| 5 Quality of life | 9 | 383 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.47 [‐0.99, 0.06] |
| 6 Failure to complete | 21 | 1344 | Odds Ratio (M‐H, Random, 95% CI) | 0.84 [0.62, 1.14] |
Comparison 2.
n‐3PUFAs vs antidepressant
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depressive symptomology (continuous) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Depressive symptomology (dichotomous ‐ response) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3 Failure to complete | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only |
Comparison 3.
Subgroup analyses ‐ n‐3PUFAs vs placebo ‐ analyses based on comorbidities
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depressive symptomology (continuous) | 25 | 1373 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.50, ‐0.10] |
| 1.1 Individuals with comorbidites | 5 | 229 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.54 [‐1.21, 0.12] |
| 1.2 Individuals with/without comorbidities (mixed) | 17 | 1040 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.30, 0.05] |
| 1.3 Individuals without comorbidities | 3 | 104 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.99 [‐1.71, ‐0.27] |
| 2 Adverse events | 19 | 1207 | Odds Ratio (M‐H, Random, 95% CI) | 1.28 [0.98, 1.68] |
| 2.1 Individuals with comorbidities | 3 | 201 | Odds Ratio (M‐H, Random, 95% CI) | 2.66 [0.22, 31.93] |
| 2.2 Individuals with/without comorbidities (mixed) | 14 | 937 | Odds Ratio (M‐H, Random, 95% CI) | 1.40 [1.04, 1.89] |
| 2.3 Individuals without comorbidities | 2 | 69 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.19, 3.50] |
Comparison 4.
Subgroup analyses: n‐3PUFAs vs placebo ‐ analyses based on adjunctive therapy
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depressive symptomology (continuous) | 25 | 1373 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.50, ‐0.10] |
| 1.1 Individuals receiving adjunctive therapy | 12 | 356 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.16 [‐0.38, 0.05] |
| 1.2 Individuals receiving/not receiving adjunctive therapy (mixed) | 7 | 709 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐0.82, ‐0.04] |
| 1.3 Individuals not receiving adjunctive therapy | 6 | 308 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.32 [‐0.86, 0.21] |
| 2 Adverse events | 19 | 1207 | Odds Ratio (M‐H, Random, 95% CI) | 1.24 [0.95, 1.62] |
| 2.1 Individuals receiving adjunctive therapy | 9 | 304 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.56, 1.70] |
| 2.2 Individuals receiving/not receiving adjunctive therapy | 6 | 644 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.81, 1.65] |
| 2.3 Individuals not receiving adjunctive therapy | 4 | 259 | Odds Ratio (M‐H, Random, 95% CI) | 2.04 [1.03, 4.03] |
What's new
Last assessed as up‐to‐date: 4 May 2015.
| Date | Event | Description |
|---|---|---|
| 9 December 2016 | Amended | It has recently transpired that the MADRS scores used in our analyses for the trial by Bot et al (2010) were reversed between intervention and placebo groups. These data resulted from correspondence with the authors of this trial and they have recently confirmed an error. Reversal of the data for these groups changes our results minimally, does not change our interpretation of our results and does not change our conclusions. Reversal of these data results in the following changes to the results of the following analyses: 1.1: n‐3PUFAs vs placebo: depressive symptomology (continuous data)(Figures 4,5): original published result – SMD = ‐0.32 (95% CI ‐0.52 to ‐0.12), I2 = 58%; revised result ‐ SMD = ‐0.30 (95% CI ‐0.50 to ‐0.10), I2 = 59%. This result represents a difference between groups in scores on the HDRS (17‐item) of approximately 2.1 points (95% CI 0.7 to 3.5). 3: n‐3PUFAs vs placebo: depressive symptomology (continuous data) subgroup analyses based on presence/absence of comorbidities (Figure 13): Individuals with comorbidities: original published result ‐ SMD = ‐0.65 (95% CI ‐1.28 to ‐0.02), I2 = 74%; revised result ‐ SMD = ‐0.54 (95% CI ‐1.21 to 0.12), I2 = 77%. Statistical evidence of a difference between subgroups remains (p = 0.04), and evidence of heterogeneity between subgroups remains high (I2 = 68%). 4: n‐3PUFAs vs placebo: depressive symptomology (continuous data) subgroup analyses based on presence/absence of adjunctive therapies (Figure 14): Individuals receiving adjunctive therapy: original published result ‐ SMD = ‐0.21 (95% CI ‐0.42 to 0.01), I2 = 0%; revised result ‐ SMD = ‐0.16 (95% CI ‐0.38 to 0.05), I2 = 0%. No statistical evidence of a difference between subgroups remains (p = 0.48), and no evidence of heterogeneity between subgroups remains (I2 = 0%). 5.1: n‐3PUFAs vs placebo: depressive symptomology (continuous data) sensitivity analyses based on selection bias: original published result ‐ SMD = ‐0.21 (95% CI ‐0.45 to 0.03), I2 = 59%; revised result ‐ SMD = ‐0.18 (95% CI ‐0.42 to 0.06), I2 = 60%. 5.2: n‐3PUFAs vs placebo: depressive symptomology (continuous data) sensitivity analyses based on performance bias: original published result ‐ SMD = ‐0.14 (95% CI ‐0.55 to 0.26), I2 = 69%; revised result ‐ SMD = ‐0.07 (95% CI ‐0.48 to 0.35), I2 = 70%. 6: n‐3PUFAs vs placebo: depressive symptomology (continuous data) sensitivity analyses using fixed effects models: original published result ‐ SMD = ‐0.20 (95% CI ‐0.31 to ‐0.09); revised result ‐ SMD = ‐0.19 (95% CI ‐0.30 to ‐0.08). 7.1: n‐3PUFAs vs placebo: depressive symptomology (continuous data) sensitivity analyses based on use of a treatment that was solely EPA: original published result ‐ SMD = ‐0.45 (95% CI ‐0.74 to ‐0.15), I2 = 0%; revised result ‐ SMD = ‐0.37 (95% CI ‐0.66 to ‐0.08), I2 = 0%. 11: n‐3PUFAs vs placebo: depressive symptomology (continuous data) sensitivity analyses based on consideration of multiple comparison groups from the same study as individual studies: original published result ‐ SMD = ‐0.34 (95% CI ‐0.56 to ‐0.12), I2 = 67%; revised result ‐ SMD = ‐0.32 (95% CI ‐0.54 to ‐0.10), I2 = 68%. |
History
Protocol first published: Issue 1, 2004 Review first published: Issue 11, 2015
| Date | Event | Description |
|---|---|---|
| 1 May 2014 | New citation required and major changes | This protocol replaces the withdrawn protocol Silvers 2009 (withdrawn). |
Differences between protocol and review
The following differences between protocol and review have arisen, for the reasons provided:
Protocol: "Only studies involving adults (18 years and over) will be included". Review: One study involving adults (16 years and over) is included (Gharekhani 2014). Age 16 years is the definition of adult in the country in which this study was undertaken.
Protocol: "Studies will be included regardless of participant medication". Review: Studies were included regardless of participant medication and other treatments for depressive symptomology, so we have stated "Studies were included regardless of participant use of adjunctive therapy".
Protocol: "Experimental intervention: Studies will be included regardless of source of n‐3PUFA provided ..., but records of differences will be made". Review: Records of differences based on source of n‐3PUFA provided were made and have been investigated in sensitivity analyses. We conducted sensitivity analyses following the publication of a number of similar comparisons since the conception of this review, and following reviewers' comments.
Protocol: "Where studies use multiple time points, data will be tabulated for all outcomes at all time points where assessments have been made, but only those of longest follow‐up will be included in statistical analyses". Review: Data for all time points have not been tabulated. This has not been done due to the variety of time points used across studies, and the difficulty and low value of comparing across varied time points.
Protocol: "Complementary searches will be conducted .... in BIOSIS Citation Index (1969 to date), and Web of Science (1900 to date)". Review: These searches were not completed. We decided that due to the topic of the review, searches in Biosis and Web of Science would be very unlikely to reveal additional studies.
Protocol: "We will assess the risk of bias according to the following domains. 1. Random sequence generation, 2. Allocation concealment, 3. Blinding of participants and personnel, 4. Blinding of outcome assessment, 5. Incomplete outcome data, 6. Selective outcome reporting, 7. Other bias". Review: We have made assessments of outcome data (blinding of outcome assessment, and incomplete outcome data) separately for each primary outcome. This was done because different judgements could be given to different outcome assessments for some studies, depending on methods of measurement, and it was meaningless to try and combine these.
Protocol: "Data from subgroups of little relevance to the research question, e.g. groups of males and females, will be recorded as reported, and subsequently combined for analysis". Review: Data have not been presented separately for subgroups of little relevance to the research question, because we found none.
Protocol: "Adverse effects and failure to complete data will not be statistically summarised". Review: We have statistically summarised data on adverse effects and failure to complete, where data were available. We did this becuase of the amount of data available and the value of these statistical summaries.
Protocol: Subgroup analyses will be conducted "using only studies in which participants are clearly identified as having comorbid conditions, and using only studies in which participants are clearly identified as being without comorbid conditions. Studies where participants with and without comorbid conditions were mixed, and studies that do not clearly identify whether participants have comorbid conditions or not, will not be included in this analysis". Review: We have conducted subgroup analyses based on comorbidities using all studies. We did this to allow investigation of effects of comorbidities in the whole data set.
Protocol: Subgroup analyses will be conducted "using only studies in which participants are clearly identified as receiving adjunct therapy, and using only studies in which participants are clearly identified as not receiving adjunct therapy. Studies where participants with adjunct therapies are mixed, and studies that do not clearly identify whether participants are receiving or not receiving adjunct therapies will not be included in this analysis". Review: We have conducted subgroup analyses based on adjunctive therapy using all studies. We did this to allow investigation of effects of adjunctive therapy in the whole data set. For these analyses, We have defined adjunctive therapy as including psychotherapy as well as antidepressant medication, and we have limited it to adjunctive therapies for depression.
Protocol: Sensitivity analyses on risk of bias will be conducted where "low risk of bias will be defined as in the Cochrane Handbook (Higgins 2011)". Review: we have further defined low risk of bias as "using (i) selection bias, measured using allocation concealment; (ii) performance bias, using blinding of participants and personnel; (iii) attrition bias, using incomplete outcome data. We conducted three separate analyses, one for each type of bias".
We conducted sensitivity analyses that we had not proposed in the protocol. These analyses investigated possible methodological sources of heterogeneity that became apparent during the review or the write‐up processes, or both. These sensitivity analyses are provided in the review as "additional sensitivity analyses", to distinguish them from our preplanned sensitivity analyses. We applied the sensitivity analyses using a fixed‐effect model to all outcomes for completeness, but restricted all other sensitivity analyses to testing only our primary outcomes.
Planned methods not used in the review
Protocol: Unit of analysis issues: Cross‐over RCTs: We will include only the first study phase of cross‐over RCTs in analyses. We think cross‐over RCTs are unlikely to be used in this field. Cluster‐RCTs: We will include cluster‐RCTs in primary analyses, where cluster will act as the unit of investigation. We think cluster‐RCTs are unlikely to be used in this field. Review: We have not used these methods because we did not find any cross‐over or cluster‐RCTs during our searches. The statements in the protocol will be applied where appropriate in future updates of the review.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Randomised controlled parallel‐arm trial, 12 weeks | |
| Participants |
Participants: 25 participants had a mean age = 54 yrs (SD = 11), 13 women, 12 men, recruited via VU University Medical Centre diabetes outpatient clinic (Amsterdam, NL), advertisements on websites, newspapers and magazines. Participants were recruited between April 2006 and May 2007, the trial was performed between June 2006 and July 2007 Comorbidities: Diabetes Type 1 and 2 in all participants, no other comorbidities Adjunctive therapy: Yes for all participants (usual antidepressants) Inclusion Criteria: aged 18 ‐ 75 years, diagnosed with diabetes (Type 1 or 2, or use of insulin or oral hypoglycaemic agents), on antidepressant medication for at least 2 months, met criteria for MDD using Composite International Diagnositic Interview Exclusion criteria: serious co‐morbid disease, using fish oil supplementation or consuming more than 3 servings of fish/week, alcohol or drug abuse, suicidal ideation, or allergic to fish, fish products or rapeseed oil |
|
| Interventions |
Intervention: E‐EPA (1 g/d, including mixed tocopherols), 2 x 500 mg capsules per day, plus ongoing therapy Comparator: Rapeseed oil + medium chain triglycerides (1 g/d, including mixed tocopherols), 2 x 500 mg capsules per day, plus ongoing therapy Treatment received for 12 weeks |
|
| Outcomes |
Primary: MADRS measured at baseline, 1, 3, 5, 7, 9 and 12 weeks; Adverse Events Secondary: Failure to complete |
|
| Notes |
Funded by Dutch Diabetes Research Foundation and Minami Nutrition, Belgium Supplements provided by Minami Nutrition, Belgium Conflicts of interest: CoI declared by one author Compliance: EPA levels in red blood cell (RBC) phospholipids Depressed mood (continuous): Analysis conducted on MADRS scores at 12 weeks, published per protocol data Adverse events: Adverse events reported in the analyses do not include side effects. 1 individual in the intervention group experienced an allergic reaction. Side effects were not split according to group: no side effects in 8 individuals, prevalent side effects were stomach ache (n = 10), belching (n = 7), nausea (n = 6), diarrhoea (n = 5). Values in the analysis are for adverse events. Failure to complete: Intervention group = 2 (1 allergic reaction, 1 loss to follow‐up), Comparator group = 0. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation occurred with computer‐generated random number performed by an employee of the pharmacy of VU University Medical Centre (P.2, Mocking 2012) |
| Allocation concealment (selection bias) | Low risk | Randomisation performed by a pharmacy employee who was not involved in data collection or analysis (P.2, Mocking 2012) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and researchers were blinded to treatment allocation until completion of data collection. Identical packaging sent out by the pharmacy. Participants instructed not to chew to avoid fishy taste. (P.283 Bot 2010, P.2 Mocking 2012), but no report of masking the fishy taste. "concealment appeared to be successful." ‐ 33% in both groups correctly guessed treatment when questioned. (P.285, Bot 2010) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | MADRS: Research nurse and researchers were blind to treatment allocation until completion of data collection (P.283, Bot 2010, P.2, Mocking 2012) |
| Blinding of outcome assessment (Adverse Events) | Low risk | Adverse events were assessed by nurses based on participant report. Participants did not guess their treatment group (P.285, Bot 2010) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | MADRS: 1 person in intervention group lost to follow‐up, analysis not ITT (although stated as ITT) (P.284, Bot 2010) |
| Incomplete outcome data (Adverse Events) | High risk | Adverse events: AEs reported but only the prevalent side effects (P.285, Bot 2010) |
| Selective reporting (reporting bias) | Low risk | All depression outcomes reported (Protocol included in Mocking 2012) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel‐arm trial, 10 weeks Pre‐randomisation: Paticipants were given a 2½ ‐ 3½ week supply of sertraline (25 mg/ day) plus placebo for 2 weeks then reassessed for depression, compliance and tolerance to medication |
|
| Participants |
Participants: 122 participants with a mean age = 58.3 years, 41 women, recruited from cardiology practices in St Louis, Missouri, US and from cardiac diagnostic labs affiliated with Washington University School of Medicine, USA. They were informed of the study via physicians, study staff or pamphlets. Patients were recruited to the study between May 2005 and December 2008. Comorbidities: CHD in all participants, no psychiatric comorbidities Adjunctive therapy: Yes for all participants ‐ antidepressant sertraline (50 mg/d) Inclusion criteria: score ≥ 16 BDI‐II, DSM‐IV criteria for current MDE (using SCID), CHD as documented by > 50% stenosis in at least 1 major coronary artery, a history of revascularisation or hospitalisation for an acute coronary syndrome; continued to meet DSM criteria, score ≥ 16 BDI‐II, reported no serious adverse events, and took both drugs ≥ 85% of days during pre‐randomisation Exclusion criteria: Cognitive impairment, comorbid psychiatric disorders, psychosis, high risk of suicide or current substance abuse, an acute coronary syndrome within the previous 2 months, a left ventricular ejection fraction of less than 30%, advanced malignancy or physical inability to participate, use of antidepressants, anticonvulsants, lithium, or n‐3PUFA supplements, sensitivity to sertraline or n‐3PUFA or physician/patient refusal |
|
| Interventions |
Intervention: EPA/DHA combination (2 g/d ethyl esters, providing EPA 930 mg, DHA 750 mg), 2 capsules per day, plus 50 mg/d sertraline Comparator: Corn oil (2 g/d), 2 capsules per day, plus 50 mg/d sertraline Treatment received for 10 weeks |
|
| Outcomes |
Primary: BDI‐II (21‐item), HDRS (17‐item) both assessed weekly for 10 weeks, Adverse events Secondary: Response, remission based on BDI, failure to complete |
|
| Notes |
Funded by National Heart Lung and Blood institute, US Supplements provided by GlaxoSmithKline Inc, antidepressants provided by Pfizer Inc. Conflicts of interest: CoIs declared by 2 authors Compliance: RBC membrane levels of EPA and DHA assessed before and after treatment. Capsule counts at each study visit Depressed mood (continuous): Analysis conducted on HDRS (17‐item) scores at 10 weeks, using published ITT data Adverse events: Adverse effects reported as a percentage rather than number of participants reporting 1 new symptom in the 10‐week trial; Intervention group = 63% adverse effects (19% symptoms previously associated with high doses of n‐3PUFAs), Control group = 73% adverse effects (22% symptoms previously associated with high doses of n‐3PUFAs). 14 adverse events, but details do not add up to 14. Values in the analysis are for adverse effects. Failure to complete: Intervention = 3 (1 = health problems related to treatment, 1 = withdrew consent, 1 = wanted other treatment); Comparator = 4 (2 = health problems related to treatment, 1 = withdrew consent, 1 = wanted other treatment) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A SAS permuted‐block randomisation allocation programme (P.1652, Carney 2009) |
| Allocation concealment (selection bias) | Unclear risk | The group assignments were concealed in sealed envelopes and opened at enrolment (P.1652, Carney 2009), not clear if envelopes were opaque |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and nurses were blinded and an identical placebo was used (P.1652, Carney 2009). There was no attempt to mask the fishy taste and no assessment to check concealment |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | BDI (self‐reported scale) ‐ HIGH HDRS professional rating scale ‐ study psychiatrists and nurses were blinded ‐ LOW |
| Blinding of outcome assessment (Adverse Events) | Unclear risk | Adverse events ‐ unclear whether these were reported by clinicians or participants |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT on BDI and HDRS |
| Incomplete outcome data (Adverse Events) | Unclear risk | AEs were not clearly reported |
| Selective reporting (reporting bias) | Low risk | All major outcomes were reported (additional information from authors) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Double‐blind, randomised parallel‐arm trial for 6 weeks, following 4‐week open‐label trial of escitalopram (10 mg/d) to prospectively identify SSRI non‐responders (< 50% improvement) for augmentation | |
| Participants |
Participants: 11 participants with a mean age of 28.8 (SD 9.3, range 18 ‐ 48) years, 9 women and 2 men (split across Coryell (1g/d) and Coryell (2g/d)). Participants recruited via clinician referrals and advertisements, in Iowa City, USA Comorbidities: Possible physical and/or psychiatric comorbidities Adjunctive therapy: Yes for all participants ‐ escitalopram Inclusion: Aged 18 ‐ 55 years; current diagnosis of MDD; meets DSM‐IV criteria Antidepressant for no more than 3 days within the past month or antidepressant for at least the past month with no change in type or dose Exclusion: More than 2 adequate antidepressant trials in the current episode; meets DSM‐IV criteria for substance dependence in the past year; substance abuse within the past month; meets DSM‐IV criteria for an eating disorder in the past year; allergy to fish; bleeding disorder/taking warfarin; omega‐3 supplements for 3 or more days in the past 4‐month period; known to be pregnant; taking medications known to produce affective symptoms; history of non‐response to escitalopram/Lexapro |
|
| Interventions |
Intervention: EPA/DHA combination (740 mg EPA/d + 400 mg DHA/d), 2 capsules, plus 2 placebo capsules Comparator: 4 placebo capsules All participants receive 4 capsules with either 0 or 2 capsules containing EPA. Treatment was received for 6 weeks |
|
| Outcomes |
Primary: MADRS scores, measurements at 6 weeks Secondary: HDRS, adverse events, response based on 50% improvement based on MADRS and HDRS |
|
| Notes |
Supplements provided by Ocean Nutrition Canada Ltd. Conflicts of interest: None Compliance: Capsule counts at each study visit Depressed mood (continuous): Analysis conducted on MADRS scores at 6 weeks, using unpublished ITT data (missing data for HDRS). Placebo group split across 2 intervention groups (1 g/d = 2 participants, 2 g/d = 2 participants). Adverse events: Data on serious and non‐serious adverse events were collected. No serious AEs were reported, but no data on non‐serious adverse events were available. Values in the analysis are for serious adverse events Failure to complete: No withdrawals |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated, simple randomisation (email correspondence from trialist) |
| Allocation concealment (selection bias) | Unclear risk | Researcher was blind to allocation, research nurse was not blind to allocation. Both had contact with participants and unclear who allocated participants (email correspondence from trialist) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Researcher was blinded, research nurse was not blinded, and both had contact with participants. Participants were stated as 'blinded', but no details of blinding of taste. Possible attempts to check blinding, but no data available (email correspondence from trialist) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessments made by researcher, and researcher was blinded (email correspondence from trialist) |
| Blinding of outcome assessment (Adverse Events) | Unclear risk | Outcome assessments made by participants, and unclear if they were blinded (email correspondence from trialist) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Complete MADRS outcome data, and ITT analysis (email correspondence from trialist) |
| Incomplete outcome data (Adverse Events) | Unclear risk | No serious AEs were reported, but data on non‐serious adverse events are not available (email correspondence from trialist) |
| Selective reporting (reporting bias) | High risk | Data not published (email correspondence from trialist) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Double‐blind, randomised parallel‐arm trial for 6 weeks, following 4‐week open‐label trial of escitalopram (10 mg/d) to prospectively identify SSRI non‐responders (< 50% improvement) for augmentation | |
| Participants |
Participants: 11 participants with a mean age of 28.8 (SD 9.3, range 18 ‐ 48) years, 9 women and 2 men (split across Coryell (1g/d) and Coryell (2g/d)). Participants recruited via clinician referrals and advertisements, in Iowa City, USA Comorbidities: Possible physical and/or psychiatric comorbidities Adjunctive therapy: Yes for all participants ‐ escitalopram Inclusion: Aged 18 ‐ 55 years; current diagnosis of MDD; meets DSM‐IV criteria Antidepressant for no more than 3 days within the past month or antidepressant for at least the past month with no change in type or dose Exclusion: More than 2 adequate antidepressant trials in the current episode; meets DSM‐IV criteria for substance dependence in the past year; substance abuse within the past month; meets DSM‐IV criteria for an eating disorder in the past year; allergy to fish; bleeding disorder/taking warfarin; omega‐3 supplements for 3 or more days in the past 4‐month period; known to be pregnant; taking medications known to produce affective symptoms; history of non‐response to escitalopram/Lexapro |
|
| Interventions |
Intervention: EPA/DHA combination (1480 mg EPA/d + 800 mg DHA/d), 4 capsules Comparator: Placebo capsules, 4 capsules All participants receive 4 capsules with either 0 or 4 capsules containing EPA Treatment was received for 6 weeks |
|
| Outcomes |
Primary: MADRS scores, measurements at 6 weeks Secondary: HDRS, adverse events, response based on 50% improvement based on MADRS and HDRS |
|
| Notes |
Supplements provided by Ocean Nutrition Canada Ltd. Conflicts of interest: None Compliance: Capsule counts at each study visit Depressed mood (continuous): Analysis conducted on MADRS scores at 6 weeks, using unpublished ITT data (missing data for HDRS). Placebo group split across 2 intervention groups (1 g/d = 2 participants, 2 g/d = 2 participants) Adverse events: Data on serious and non‐serious adverse events were collected. No serious AEs were reported, but no data on non‐serious adverse events were available. Values in the analysis are for serious adverse events Failure to complete: No withdrawals |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated, simple randomisation (email correspondence from trialist) |
| Allocation concealment (selection bias) | Unclear risk | Researcher was blind to allocation, research nurse was not blind to allocation. Both had contact with participants and unclear who allocated participants (email correspondence from trialist) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Researcher was blinded, research nurse was not blinded, and both had contact with participants. Participants were stated as 'blinded', but no details of blinding of taste. Possible attempts to check blinding, but no data available (email correspondence from trialist) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessments made by researcher, and researcher was blinded (email correspondence from trialist) |
| Blinding of outcome assessment (Adverse Events) | Unclear risk | Outcome assessments made by participants, and unclear if they were blinded (email correspondence from trialist) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Complete MADRS outcome data, and ITT analysis (email correspondence from trialist) |
| Incomplete outcome data (Adverse Events) | Unclear risk | No serious AEs were reported, but data on non‐serious adverse events are not available (email correspondence from trialist) |
| Selective reporting (reporting bias) | High risk | Data not published (email correspondence from trialist) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Pilot randomised controlled parallel‐arm trial, 12 weeks Participants split across Da Silva (AD) 2005 and Da Silva (nAD) 2005, depending on antidepressant use, prior to randomisation |
|
| Participants |
Participants: 31 participants, with a mean age = 64.4 (range 49 ‐ 78) years, 58% women (split across Da Silva (AD) 2005 and Da Silva (nAD) 2005). Participants were selected from Association of Patients with Parkinson's disease of Paraná, Curitiba, Brazil Comorbidities: Parkinson's disease (PD), other possible comorbidities Adjunctive therapy: Yes for all participants: antidepressants Inclusion criteria: Parkinsons disease, DSM‐IV criteria for MDD (MINI plus, and a SCID), score < 2.5 Hoehn & Yahr scale for PD (Hoehn 1967), no signs of dementia (MMSE) (Folstein 1975), UPDRS assessment (Taylor 2005), taking medication for depression for at least 1 yr or refused to take medication Exclusion criteria: initiated antidepressant use after diagnosis, cognitive and memory declines, drug/alcohol dependent. Any participant who presented with an alteration of PD (above 0.5 point on Hoehn and Yahr scale) after 3 months was also excluded |
|
| Interventions |
Intervention: EPA/DHA combination (720 mg/d EPA, 480 mg/d DHA, plus tocopherols), 4 capsules, plus ongoing therapy Comparator: Mineral oil, 4 capsules/d, plus ongoing therapy Treatment received for 3 months |
|
| Outcomes |
Primary: MADRS, BDI assessed at baseline and 12 weeks, Adverse events Secondary: Response based on MADRS, CGI assessed at baseline and 12 weeks, Failure to complete |
|
| Notes |
No funding reported. Supplements provided by Herbarium Foundation for Health and Research Conflicts of interest: None declared Compliance: RBC membrane levels of EPA and DHA assessed before and after treatment Depressed mood (continuous): Analysis conducted on MADRS scores at 12 weeks, per protocol data provided by authors Adverse events: 2 individuals reported adverse events ‐ 1 GI, 1 other physical (split across Da Silva (AD) 2005 and Da Silva (nAD) 2005 ‐ group not reported). Values could not be included in analysis Response (50% improvement in MADRS score) ‐ Intervention group = 42%, comparator group = 6% (split across Da Silva (AD) 2005 and Da Silva (nAD) 2005 ‐ group not reported). Values could not be included in analysis. Quality of Life: Analysis conducted on CGI Failure to complete: 2 individuals withdrew (split across Da Silva (AD) 2005 and Da Silva (nAD) 2005 ‐ group not reported) (1 collateral effects, 1 worsening health status). Values could not be included in analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was done by drawing (additional information from the author) |
| Allocation concealment (selection bias) | Unclear risk | Identification of the groups and separation of the respective capsules were carried out in the lab at University Federal do Parana (P.353) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Neither researcher nor participants knew which substance was given (identical placebo). Not reported if the fishy taste was disguised, and no assessment to check concealment |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | MADRS and BDI (both evaluated by trained psychologist blinded to allocation) (P.353) |
| Blinding of outcome assessment (Adverse Events) | Unclear risk | Adverse events ‐ unclear whether these were reported by clinicians or participants |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Data not ITT |
| Incomplete outcome data (Adverse Events) | Unclear risk | AEs were not clearly reported |
| Selective reporting (reporting bias) | Low risk | All depression data reported (additional information from author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Pilot randomised controlled parallel‐arm trial, 12 weeks Participants split across Da Silva (AD) 2005 and Da Silva (nAD) 2005, depending on antidepressant use, prior to randomisation |
|
| Participants |
Participants: 31 participants, with a mean age = 64.4 (range 49 ‐ 78) years, 58% women (split across Da Silva (AD) 2005 and Da Silva (nAD) 2005). Participants were selected from Association of Patients with Parkinson's disease of Paraná, Curitiba, Brazil Comorbidities: Parkinson's disease (PD), other possible comorbidities Adjunctive therapy: No for all participants Inclusion criteria: Parkinsons disease, DSM‐IV criteria for MDD (MINI plus, and a SCID), score < 2.5 Hoehn & Yahr scale for PD (Hoehn 1967), no signs of dementia (MMSE) (Folstein 1975), UPDRS assessment (Taylor 2005), taking medication for depression for at least 1 yr or refused to take medication Exclusion criteria: initiated antidepressant use after diagnosis, cognitive and memory declines, drug/alcohol dependent. Any participant who presented with an alteration of PD (above 0.5 point on Hoehn and Yahr scale) after 3 months was also excluded |
|
| Interventions |
Intervention: EPA/DHA combination (720 mg/d EPA, 480 mg/d DHA, plus tocopherols), 4 capsules, plus ongoing therapy Comparator: Mineral oil, 4 capsules/d, plus ongoing therapy Treatment received for 3 months |
|
| Outcomes |
Primary: MADRS, BDI assessed at baseline and 12 weeks, Adverse events Secondary: Response based on MADRS, CGI assessed at baseline and 12 weeks, failure to complete |
|
| Notes |
No funding reported. Supplements provided by Herbarium Foundation for Health and Research Conflicts of interest: None declared Compliance: RBC membrane levels of EPA and DHA assessed before and after treatment Depressed mood (continuous): Analysis conducted on MADRS scores at 12 weeks, per protocol data provided by authors Adverse events: 2 individuals reported adverse events ‐ 1 GI, 1 other physical (split across Da Silva (AD) 2005 and Da Silva (nAD) 2005 ‐ group not reported). Values could not be included in analysis Response (50% improvement in MADRS score) ‐ Intervention group = 42%, comparator group = 6% (split across Da Silva (AD) 2005 and Da Silva (nAD) 2005 ‐ group not reported). Values could not be included in analysis Quality of Life: Analysis conducted on CGI Failure to complete: 2 individuals withdrew (split across Da Silva (AD) 2005 and Da Silva (nAD) 2005 ‐ group not reported) Values could not be included in analysis. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was done by drawing (additional information from the author) |
| Allocation concealment (selection bias) | Unclear risk | Identification of the groups and separation of the respective capsules were carried out in the lab at University Federal do Parana (P.353) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Neither researcher or participants knew which substance was given (identical placebo). Not reported if the fishy taste was disguised, and no assessment to check concealment |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | MADRS and BDI (both evaluated by trained psychologist blinded to allocation) (P.353) |
| Blinding of outcome assessment (Adverse Events) | Unclear risk | Adverse events ‐ unclear whether these were reported by clinicians or participants |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Data not ITT |
| Incomplete outcome data (Adverse Events) | Unclear risk | AEs were not clearly reported |
| Selective reporting (reporting bias) | Low risk | All depression data reported (additional information from author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias. |
| Methods | Randomised controlled parallel‐arm trial, 8 weeks Pre‐randomisation 1 week placebo run‐in phase |
|
| Participants |
Participants: 42 participants, mean age = 40.5 years (SD = 10.2). Distribution of gender was not reported. Recruited via local advertisements or physician referral from the Greater Los Angeles area with preliminary telephone screening Comorbidities: No psychiatric comorbidities, possible physical comorbidities Adjunctive therapy: Yes for all participants, citalopram (20 mg/d) with possible increase in dose after 4 weeks Inclusion criteria: aged 18 ‐ 65 years; DSM IV criteria for MDD via the SCID, score > 17 on HDRS (21‐item), contraception use in women of childbearing age; still qualifying for inclusion after 1 week run‐in Exclusion criteria: psychiatric disorders including psychotic depression and bipolar disorders, current drug/alcohol abuse/dependence or history of such in past 6 months, unstable medical or neurological conditions likely to interfere with treatment, history of allergy to citalopram or n‐3PUFA, finfish or shellfish, history of failure to respond to citalopram, history of seizure disorder, pregnancy, need for concomitant psychotropic medication including other antidepressants, active suicidal ideation or safety concerns, exposure to fluoxetine or MAOIs in previous 2 months, anticoagulant therapy, dietary intake of > 3 g n‐3PUFAs/day at baseline |
|
| Interventions |
Intervention: EPA/DHA combination (EPA = 1800 mg/d, DHA= 400 mg/d, other n‐3PUFAs = 200mg/d), 2 capsules, twice daily with meals, plus citalopram (20 mg/d) Comparator: Olive oil (4 g/d), 2 capsules, twice daily with meals, plus citalopram (20 mg/d) Treatment received for 8 weeks |
|
| Outcomes |
Primary: HDRS (21‐item), BDI, MADRS ‐ all assessed at baseline, randomisation, 2, 4, 6 and 8 weeks; Adverse events Seconday: Remission and response based on HDRS; CGI, PGI; failure to complete |
|
| Notes |
Funded by NIH National Centre for Complementary and Alternative Medicine & National Centre for Research Resources, USA Supplements provided by Nordic Naturals Conflicts of Interest: CoIs declared by 2 authors Compliance: Capsule counts and assessment of citalopram blood levels Depressed mood (continuous): Analysis conducted on HDRS (21‐item) scores at 8 weeks, ITT data taken from published graph Adverse events: Only reported for completers. No significant adverse events. Only frequently‐reported adverse effects were reported: Intervention group = 6 (all GI), comparator group = 4 (all GI). Less than 5% of participants in either group reported other adverse events, e.g. headache, sedation or sexual dysfunction. Numbers reported in the analysis relate to frequently‐reported adverse effects Quality of life: CGI, PGI, data not reported. Failure to complete: Intervention group = 3 (2 undisclosed exclusion criteria, 1 lost to follow‐up), comparator group = 7 (2 lack of efficacy, 5 lost to follow‐up) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Block randomised by sex to receive citalopram. Half of the subjects also received omega‐3 and the other half received placebo" but method of sequence generation not reported (P.62) |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study was described as "masked" but not clear who was blinded (P.61). It was unclear if the fishy taste was disguised and no assessment to check concealment |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | HDRS, MADRS and BDI ‐ all unclear ‐ The study was described as "masked" but it was unclear who was blinded (P.61). |
| Blinding of outcome assessment (Adverse Events) | Unclear risk | AEs ‐ measured by Treatment Emergent Symptoms Scale (Guy 1976). The study was described as "masked" but not clear who was blinded (P.61) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | HDRS, MADRS, BDI ‐ Analysis not ITT and > 10% missing data |
| Incomplete outcome data (Adverse Events) | High risk | AEs ‐ only reported for completers |
| Selective reporting (reporting bias) | High risk | No protocol to check for additional outcome measures. CGI, PGI not reported |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel‐arm trial, 4 months | |
| Participants |
Participants: 54 participants. Details reported only for completers ‐ mean age by group (Intervention = 56.8 (SD = 13.09) years; Comparator = 57.2 (SD = 15.19) years), 20 women, 25 mens. Participants recruited from haemodialysis (HD) units of 2 teaching hospitals affiliated with Tehran University of Medical Sciences, Iran Comorbidities: end‐stage renal disease, no psychiatric comorbidities Adjunctive treatment: no, for all participants Inclusion criteria: Adult patients who had been treated with HD for at least 3 months Exclusion criteria: BDI < 16, pregnancy, malabsorption syndrome, malignancy, inflammatory or infectious diseases, hypothyroidism, medical or surgical illness in recent 3 months, haemoglobinopathies, asthma, chronic obstructive pulmonary disease, coagulopathies, known psychiatric disorders, lack of tolerance or hypersensitivity to fish products as well as those who were receiving corticosteroid, non‐steroidal anti‐inflammatory drugs, omega‐3 fatty acids in the previous 3 months, anticoagulants including warfarin, immunomodulator or immunosuppressive were excluded |
|
| Interventions |
Intervention: EPA/DHA combination (1080 mg/d EPA: 720 mg/d DHA). 2 capsules, 3 x daily with meals for 4 months Comparator: Placebo (paraffin oil), 2 capsules, 3 x daily with meals for 4 months |
|
| Outcomes |
Primary: BDI, Adverse events Secondary: SF‐36 (mental health component summary), failure to complete BDI and SF‐36 assessed at baseline and 4 months whilst undergoing haemodialysis |
|
| Notes |
Funded: Tehran University of Medical Sciences (grant No: 17020) Conflicts of Interest: None reported Compliance: Pill counts Depressed mood (continuous): Analysis conducted on BDI at 4 months, ITT data provided by authors Adverse events: All adverse events reported (side effects). Intervention group = 8 (all GI), comparator group = 0 Quality of life: SF‐36 means and SDs, data provided by authors. Mental health summary scale used in analyses. Failure to complete: Intervention group = 2 (1 non‐compliance, 1 surgery), comparator group = 7 (1 hospitalisation, 1 undergoing renal transplantation, 2 discomfort from taking large capsule, 1 death due to CHD, 2 changing HD centre) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The study used a 9‐block permuted randomisation procedure to allocate participants randomly into 2 groups. Each block contained an equal number of omega‐3 and control group selections, with the order of the blocks permuted. Random numbers to allocate blocks and randomise group selection were generated using Microsoft Office Excel software, P.3 |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No masking of fishy taste and no assessment to check concealment |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | BDI (self report) |
| Blinding of outcome assessment (Adverse Events) | High risk | AEs (self report) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | BDI ‐ ITT data provided by authors, but > 10% dropout |
| Incomplete outcome data (Adverse Events) | Low risk | All AEs reported (correspondence from authors) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported (correspondence from authors) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel‐arm trial, 8 weeks | |
| Participants |
Participants: 20 participants. 10 completing participants were a mean age = 38.77 years (SD = 10.74, range 30 ‐ 54), 8 women. Comorbidities: none reported, possible physical comorbidities Adjunctive treatment: Yes for all participants, fluoxetine (20 mg/d) Inclusion criteria: aged 18 ‐ 60 years; diagnosis of MDD (assessed by SCID‐ID), single or recurrent episode according to DSM‐IV‐TR criteria Exclusion criteria: not on antidepressants for at least 1 month prior to first blood sample collection, other psychiatric conditions; fish allergy; coagulopathies or taking aspirin |
|
| Interventions |
Intervention: EPA (3 g/d), 3 capsules, plus fluoxetine (20 mg/d) Comparator: Placebo, 3 capsules, plus fluoxetine (20 mg/d) |
|
| Outcomes |
Primary: HDRS assessed at baseline, 2, 4, 6 and 8 weeks Secondary: Response based on HDRS, failure to complete Treatment for 8 weeks |
|
| Notes |
No funding reported. Conflicts of Interest: not reported. Compliance: not reported Depressed mood (continuous): Analysis conducted on HDRS (17‐item) scores at 8 weeks, calculated from published per protocol data Adverse events: AEs not reported fully or clearly. Values could not be included in analyses Failure to complete: 10 people withdrew (due to collateral effects, development of medical diseases, and stopped attending the psychiatric clinic), but group allocation unclear. Data is not reported for 1 additional individual |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomised on a double‐blind basis" but method of sequence generation not reported (abstract) |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Double‐blind" (abstract) not reported who is blind to treatment. It was unclear if the fishy taste was disguised, and no assessment to check concealment |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | HDRS ‐ unclear whether outcome assessor was blind to treatment |
| Blinding of outcome assessment (Adverse Events) | Unclear risk | AEs ‐ unclear whether outcome assessor was blind to treatment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | HDRS ‐ not ITT, > 10% dropout (50%) |
| Incomplete outcome data (Adverse Events) | High risk | AEs ‐ "10 did not continue the study for several reasons. Among these, treatment withdrawal was due to collateral effects, development of medical diseases..." (translation) (P.74) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available to check for additional outcome measures |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel‐arm trial, 16 weeks | |
| Participants |
Participants: 83 outpatients from Northfields Clinics, University of Wollongong, Australia, mean age 45.3 (range 18 ‐ 70) years, 51 women Comorbidities: Yes in some participants: anxiety (54%), personality disorder (57%) Adjunctive therapy: Yes in some participants: 74% currently taking therapeutic doses of antidepressants Inclusion criteria: aged 18 ‐ 75 years, SCID DSM‐IV primary diagnosis of MDD, HDRS > 16 Exclusion criteria: serious medical condition, non‐consent for venipuncture, comorbid substance abuse, psychotic, bipolar, OCD or eating disorder |
|
| Interventions |
Intervention: EPA/DHA combination (tuna fish oil providing 2.2 g/d DHA, 0.56 g/d EPA, plus 80 mg vit E), 8 x 1 g capsules, plus ongoing therapy Comparator: Olive oil, 8 g/d, 8 x 1 g capsules per day, plus ongoing therapy Treatment received for 16 weeks |
|
| Outcomes |
Primary: HDRS, BDI ‐ baseline, 3 week intervals until 16 weeks, Adverse events Secondary: GAF, Likert scales of aches/pains, energy, fatigue, sleep, appetite; failure to complete |
|
| Notes |
Funded by Clover Corporation Plc, Australia, University of Wollongong, Australia, and the Australian Research Council Supplements provided by Clover Corporation Plc, Australia Conflict of Interest: Not reported Compliance: Fortnightly capsule counts, EPA and DHA in RBC membranes, plasma cholesterol and alpha‐tocopherol at baseline, 6 weeks and 16 weeks Depressed mood (continuous): Analysis conducted on HDRS (17‐item) scores at 16 weeks, ITT data provided by authors Adverse events: Only prespecified adverse events are reported. ˜⅓ of sample noticed changes in stools due to capsules across both groups. Only significant differences between groups also reported (belching, noticeable aftertaste in the mouth and breath), but no values. Study could not be included in analyses Quality of life: GAF measured, but no data available Failure to complete: Interventon group = 8, comparator group = 15. Reasons ‐ 8 time/commitment, 4 moved away, 3 hospitalised, 2 time constraints, 6 lost to follow‐up (reasons not split by group) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "urn" randomisation balanced by prognostic factors of age, sex, therapy, HDRS score (P.1394) Randomisation was undertaken by a person unconnected with the study in a different location, who used a computer randomisation programme. Researchers gave them the blocking variables and the allocation was emailed back (correspondence with author) |
| Allocation concealment (selection bias) | Low risk | Randomisation and capsule packing performed externally (P.1394) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants, clinicians and researchers were blind to allocation. Identical placebo and capsules odourless, however when checked the majority (90% fish oil group, 64% placebo group) of participants correctly guessed their group (P.1395) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | HDRS ‐ clinician‐rated: physicians blinded to allocation (LOW) BDI ‐ self report (HIGH) |
| Blinding of outcome assessment (Adverse Events) | High risk | AEs ‐ participant‐rated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ITT analysis conducted but 28% dropped out |
| Incomplete outcome data (Adverse Events) | Unclear risk | AEs not clearly reported |
| Selective reporting (reporting bias) | Low risk | All relevant outcomes reported (correspondence with author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel 3‐arm trial, 8 weeks | |
| Participants |
Participants: 60 outpatients from the Roozbeh Psychiatry Hospital, Tehran, Iran (split across Jazayeri (v placebo) 2008 and Jazayeri (v AD) 2008)). 48 participants completing the study had a mean age = 34.8 years, 33 women Comorbidities: No physical comorbidities, possible psychiatric comorbidities Adjunctive therapy: No for all participants Inclusion criteria: Aged 20 ‐ 59 years, DSM‐IV criteria for MDD (SCID), no psychotic features, scoring > 15 HDRS (24‐item), medication‐free for at least 6 weeks Exclusion criteria: comorbid psychiatric diagnosis (other than dysthymia and anxiety), significant medical illness established by medical history, physical examination or laboratory tests, suicidal thoughts, substance abuse, history of hypomanic/manic/mixed episode, pregnancy and lactation, consumption of n‐3PUFAs in the previous year and dietary intake of > 1 serving of fish per week, use of non‐steroid anti‐inflammatory drugs and other drugs 2 weeks before or during the intervention. |
|
| Interventions |
Intervention: E‐EPA (1.1 g/d providing 1 g/d pure EPA, plus 11 mg vitamin E), 2 x 550 mg capsules, plus 20 mg/d fluoxetine placebo (starch and avicel) (EPA group) Comparator: Rapeseed oil (1.1 g/d, plus 11 mg vitamin E), 2 x 550 mg capsules, plus 20 mg/d fluoxetine (Fluoxetine group) Treatment received for 8 weeks |
|
| Outcomes |
Primary: HDRS (24‐item) ‐ baseline, 2, 4, 6, 8 weeks; Adverse events Secondary: Response based on HDRS; Failure to complete |
|
| Notes |
Supported by Vice Chancellor for Research, Tehran University of Medical Sciences, Iran Supplements provided by Minami Nutrition, Belgium Conflicts of Interest: not reported Compliance: Capsule counts Depressed mood (continuous): Analysis conducted on HDRS (24‐item) scores at 8 weeks, ITT data provided by authors Adverse events: Number of events reported rather than number of participants experiencing events. Intervention group = 5 adverse events (3 GI, 1 psychological, 1 other physical), Comparator group = 28 adverse events (6 GI, 10 psychological, 12 other physical). Study could not be included in analyses. Failure to complete: Intervention group = 4 (1 developing suicidal ideation, 1 non‐compliance, 2 lost to follow‐up), Comparator group = 4 (1 drowsiness, 1 non‐compliance, 2 lost to follow‐up) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Prearranged block randomisation" (P.193) but unclear how sequence was generated Permuted‐block randomisation (correspondence with authors) |
| Allocation concealment (selection bias) | High risk | The randomisation sequence was not concealed from researchers (correspondence with authors) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Double dummy" placebo technique used to blind participants; however, no steps taken to mask fish taste and no assessment to check concealment (P.194 ‐ 5) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | HDRS ‐ physicians blind to treatment allocation |
| Blinding of outcome assessment (Adverse Events) | High risk | AEs ‐ participant‐reported. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | HDRS ‐ ITT data provided by authors, > 10% dropout from each group |
| Incomplete outcome data (Adverse Events) | Low risk | All AEs reported (Table 4, p.196) |
| Selective reporting (reporting bias) | High risk | Some outcomes not yet published (correspondence with authors) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel 3‐arm trial, 8 weeks | |
| Participants |
Participants: 60 outpatients from the Roozbeh Psychiatry Hospital, Tehran, Iran (split across Jazayeri (v placebo) 2008 and Jazayeri (v AD) 2008)). 48 participants completing the study had a mean age = 34.8 years, 33 women Comorbidities: No physical comorbidities, possible psychiatric comorbidities Adjunctive therapy: No for all participants Inclusion criteria: Aged 20 ‐ 59 years, DSM‐IV criteria for MDD (SCID), no psychotic features, scoring > 15 HDRS (24‐item), medication‐free for at least 6 weeks Exclusion criteria: comorbid psychiatric diagnosis (other than dysthymia and anxiety), significant medical illness established by medical history, physical examination or laboratory tests, suicidal thoughts, substance abuse, history of hypomanic/manic/mixed episode, pregnancy and lactation, consumption of n‐3PUFAs in the previous year and dietary intake of > 1 serving of fish per week, use of non‐steroid anti‐inflammatory drugs and other drugs 2 weeks before or during the intervention. |
|
| Interventions |
Intervention: E‐EPA (1.1 g/d providing 1 g/d pure EPA, plus 11 mg vitamin E), 2 x 550 mg capsules, plus 20 mg/d fluoxetine (Fluoxetine + EPA combination group) Comparator: Rapeseed oil (1.1 g/d, plus 11 mg vit E), 2 x 550 mg capsules, plus 20 mg/d fluoxetine (Fluoxetine group) Treatment received for 8 weeks |
|
| Outcomes |
Primary: HDRS (24‐item) ‐ baseline, 2, 4, 6, 8 weeks; Adverse events Secondary: Response based on HDRS; Failure to complete |
|
| Notes |
Supported by Vice Chancellor for Research, Tehran University of Medical Sciences, Iran Supplements provided by Minami Nutrition, Belgium Conflicts of Interest: not reported Compliance: Capsule counts Depressed mood (continuous): Analysis conducted on HDRS (24‐item) scores at 8 weeks, ITT data provided by authors Adverse events: Number of events reported rather than number of participants experiencing events. Intervention group = 20 adverse events (6 GI, 4 psychological, 10 other physical), Comparator group = 28 adverse events (6 GI, 10 psychological, 12 other physical). Study could not be included in analyses. Failure to complete: Intervention group = 4 (1 steatorrhoea, 1 physical conditions, 1 non‐compliance, 1 lost to follow‐up), Comparator group = 4 (1 drowsiness, 1 non‐compliance, 2 lost to follow‐up) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Prearranged block randomisation" (P.193) but unclear how sequence was generated Permuted‐block randomisation (correspondence with authors) |
| Allocation concealment (selection bias) | High risk | The randomisation sequence was not concealed from researchers (correspondence with authors) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Double dummy" placebo technique used to blind participants, however no steps taken to mask fish taste and no assessment to check concealment (P.194 ‐ 5) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | HDRS ‐ physicians blind to treatment allocation |
| Blinding of outcome assessment (Adverse Events) | High risk | AEs ‐ participant‐reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | HDRS ‐ ITT data provided by authors, > 10% dropout from each group |
| Incomplete outcome data (Adverse Events) | Low risk | All AEs reported (Table 4, p.196) |
| Selective reporting (reporting bias) | High risk | Some outcomes not yet published (correspondence with authors). |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel‐arm trial, 8 weeks | |
| Participants |
Participants: 432 outpatients with mean age = 46.0 (SD = 12.4) years, 68.5% women, were recruited via adverts, physician referrals and caseloads of study investigators from 8 academic and psychiatric clinics in Canada. The study ran from Oct 2005 to Jan 2009 Comorbidities: Yes in some participants: anxiety disorders (52.8%), possible physical comorbidities Adjunctive therapy: Yes for some participants: 40.3% antidepressants at baseline, 14.8% undergoing psychotherapy, 27.1% regularly used at least 1 other psychotropic medication Inclusion criteria: aged 18 years and over, met diagnostic criteria for MDE (MINI 5), score ≥ 27 IDS‐SR, clinically significant depressive symptoms for ≥ 4 weeks, if taking antidepressants ‐ to have been at maximum dosage for > 4 weeks, or if not on antidepressants to have been intolerant for ≥ 2 previous antidepressants or refused to take them despite medical advice Exclusion criteria: known allergy or intolerance to fish/sunflower oil, taken > 14 g of n‐3PUFA supplements during past 4 weeks, diagnosis of alcohol/drug abuse/dependency during past 12 months or bipolar disorder (MINI), significant suicidal risk based on clinical judgement, history of MI, pancreatic insufficiency or coagulation diseases, regularly taking drugs or herbs with antiplatelet or anticoagulant properties, non‐menopausal pregnant women or those not taking contraception |
|
| Interventions |
Intervention: EPA/DHA combination (EPA = 1050 mg/d, DHA = 150 mg/d), 3 x capsules daily, plus ongoing therapy Comparator: Sunflower oil + 2% fish oil (to help blind), 3 x capsules daily, plus ongoing therapy Treatment received for 8 weeks |
|
| Outcomes |
Primary: IDS‐SR, MADRS at baseline, 1, 2, 4 and 8 weeks, Adverse events Secondary: Failure to complete |
|
| Notes |
Funded by Isodis Natura and Foundation Du Centre Hospitalier de l'Universite de Montreal and the CRCHUM Supplements provided by Isodis Natura Conflicts of Interest: CoIs declared by 3 authors Compliance: Reported in results, but method of assessment not reported Depressed mood (continuous): Analysis conducted on MADRS scores at 8 weeks, unadjusted ITT data provided by authors. Adverse events: Adverse events only gained from completers, only includes events reported by ≥ 5% population. Serious adverse events reported by event not by individual. Serious adverse events reported: Intervention group = 7 (3 physical, 4 psychological), Comparator group = 4 (4 physical). Number of participants with non‐serious adverse events: Intervention group = 322 events in 161 participants (215 GI, 107 other), Comparator group = 294 events in 148 participants (181 GI, 113 other). Data in the analysis are for non‐serious adverse events Failure to complete: Intervention = 30 (reasons not reported), Comparator = 27 (reasons not reported) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated, randomly permuted blocks of 2 and 4, stratified by site and baseline antidepressant use/non‐use. (P.1056 and correspondence from author) |
| Allocation concealment (selection bias) | Low risk | Group assignment using sequentially‐numbered containers, generated by co‐ordinating centre. Only technician preparing containers had access to randomisation codes (P.1056) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study research personnel and participants were blinded. 2% fish oil was added to placebo to control for fishy aftertaste. James' blinding index used to check blinding of treatment allocation (P. 1056) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | IDS‐SR and MADRS both low ‐ study psychiatrists, personnel were blinded |
| Blinding of outcome assessment (Adverse Events) | Low risk | AEs ‐ participant‐assessed |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ITT analysis and although similar dropouts in each group, > 10% dropout |
| Incomplete outcome data (Adverse Events) | High risk | AEs ‐ only reported AEs reported by > 5% of participants |
| Selective reporting (reporting bias) | Low risk | All outcomes reported (correspondence with author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias. |
| Methods | Randomised controlled parallel‐arm trial, 8 weeks. Subgroup analysis of 29 women with MDD (randomisation stratified according to MDD diagnosis) | |
| Participants |
Participants: 29 postmenopausal women with mean age = 49.6 years were recruited from the general population in Quebec, Canada through newspaper, radio and television advertisements, and flyers posted in clinics and by clinicians. Participants were recruited from March 2005 to November 2006, study ran until February 2007 Comorbidities: No physical comorbidities, possible psychiatric comorbidities Adjunctive therapy: No for all participants Inclusion criteria: aged 40 ‐ 55 years, postmenopausal, score ≤ 72 on the PGWB and score < 26 on the HDRS (21‐item) Exclusion criteria: score ≥ 26 on the 21‐item HDRS, physical conditions known to affect mental health, substance abuse/dependence, high consumption of fish (> 3 serving per week), fish allergies, past or current schizophrenia or bipolar disorder, risk of suicide or homicide, postmenopausal for more than 5 years, use of St John's Wort, antidepressants, hormone replacement therapy or fish oil supplements in previous 3 months, use of anticoagulants |
|
| Interventions |
Intervention: EPA/DHA combination (1.5 g/d ethyl esters, providing 1050 mg/d EPA, 150 mg/d DHA), 3 capsules daily Comparator: Sunflower oil (1.5 g/d, plus 0.2% regular fish oil [18% EPA/12% DHA]), 3 capsules daily Treatment received for 8 weeks |
|
| Outcomes |
Primary: 21‐item HDRS, 20‐item HSCL (Williams 2004) measured at baseline, 4 and 8 weeks. Adverse events Secondary: PGWB, CGI, Failure to complete |
|
| Notes |
Supported by Laval University, Canada Supplements provided by Isodus Natura, Belgium Conflicts of Interest: CoIs declared by one author Compliance: Capsule counts, and RBC membrane analysis Depressed mood (continuous): Analysis conducted on HDRS (21‐item) scores at 8 weeks, ITT data provided by authors Adverse events: Adverse events reported by event, not by individuals. Only includes events reported by ≥ 5% population. Adverse events are not published separately for the subgroup. Adverse events (number of individuals) in the analysis were provided by the authors Quality of life: CGI data used in the analysis Failure to complete: Intervention group = 1 (lack of efficacy), comparator group = 2 (adverse events) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified by history of major depressive episode. Computer‐generated stratified randomisation lists prepared by a statistician. (P.642) |
| Allocation concealment (selection bias) | Low risk | Researchers responsible for seeing participants allocated next available entry number. Statistician gave randomisation list to pharmacy who packaged capsules. (P.642) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants, investigators and staff were blind to treatment assignment until the last participants completed study (P.642) Capsules were obtained directly from the pharmacist Matching placebo with added fish for aftertaste There was no difference in the number of people guessing their allocation correctly. (P.645) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | HDRS/CGS/HSCL/PGWB ‐ all low. Participants, investigators and staff were blind to treatment assignment until the last participants completed study |
| Blinding of outcome assessment (Adverse Events) | Low risk | AEs ‐ reported by participants who were blinded to treatment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis ‐ additional information from authors |
| Incomplete outcome data (Adverse Events) | Low risk | All AEs reported ‐ information provided by the authors |
| Selective reporting (reporting bias) | Low risk | All outcome measures reported (correspondence with author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel‐arm trial, 6 weeks | |
| Participants |
Participants: 36 participants. 35 participants completing the study had a mean age = 47.3 years, 28 women Comorbidities: No Adjunctive therapy: No Inclusion criteria: aged 18 ‐ 65 years, met DSM‐IV criteria for MDD without psychotic features (assessed by SCID), score ≥ 12 on the MADRS and score ≥ 17 on the 28‐item HDRS, medication‐free for ≥ 2 weeks prior to enrolment, dietary intake of ≤ 1 serving of fish per week Exclusion criteria: physical conditions or psychiatric comorbidities, treatment resistance |
|
| Interventions |
Intervention: DHA (2 g/d) Comparator: placebo (2 g/d) Treatment received for 6 weeks |
|
| Outcomes |
Primary: MADRS, HDRS (28‐item) measured at baseline, 2 and 6 weeks; Adverse events Secondary: Response based on MADRS; GAF; Failure to complete |
|
| Notes |
Funded by Martek Biosciences Corporation, USA Conflicts of Interest: not reported Compliance: RBC DHA levels Depressed mood (continuous): Analysis conducted on HDRS (28‐item) scores at 6 weeks, per protocol data as published Adverse events: Number of events reported rather than number of participants with at least 1 adverse event. Intervention group = 25 events (19 GI, 6 other physical), comparator group = 5 (1 GI, 4 other physical). Failure to complete: 1 participant withdrew (group allocation unclear) (reason not reported). Study could not be included in analyses |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not reported other than "double blind" specified in title. It was unclear if the fishy taste was disguised and no assessment to check concealment. It was unclear whether or not the placebo was identical |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | MADRS/HDRS ‐ unclear whether assessor was blinded to treatment |
| Blinding of outcome assessment (Adverse Events) | Unclear risk | AEs ‐ unclear whether assessor was blinded to treatment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | MADRS/HDRS ‐ not ITT analysis |
| Incomplete outcome data (Adverse Events) | High risk | Not all AEs reported ‐ "AEs included ..." P.997. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available to check prespecified outcome measures |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Multi‐centre parallel design randomised controlled trial, 8 weeks | |
| Participants |
Participants: 196 participants (split across Mischoulon (DHA) 2015 and Mischoulon (EPA) 2015): 177 participants considered evaluable (provided 1 post‐baseline assessment); Mean age 45.8 (SD 12.5) years, 59.3% women (n = 105), 40.7% men (n = 72). Participants recruited at Massachusetts General Hospital and Cedars‐Sinai Medical Center through advertisements and referrals from outpatient programmes, from May 2006 to June 2011 Comorbidities: anxiety disorders/dysthymia in some participants, no serious/unstable physical comorbidities Adjunctive therapy: no, for all participants Inclusion criteria: A diagnosis of MDD per the SCID‐I/P), a CGI‐S score ≥ 3, and a baseline 17‐item HDRS‐17 score ≥ 15 Exclusion criteria: pregnancy or women of childbearing potential who were not using a medically‐accepted means of contraception; suicidality or homicidality; serious or unstable medical illness; current or past history of organic mental disorders, substance use disorders, any psychotic disorders, and bipolar disorder; history of multiple adverse drug reactions or allergy to the study compounds; concurrent use of psychotropic medications, systematic corticosteroid or steroid antagonists, anticoagulants, or immunosuppressant agents; electroconvulsive therapy during the current episode; any trial of ≥ 6 weeks with citalopram 40 mg/d or equivalent antidepressant during the current episode (to select a less refractory sample that would be more likely to respond to treatment); history of use of 1 g/d of n‐3 supplements; history of a bleeding disorder; psychotherapy; smoking 10 cigarettes per day; vitamin E supplementation > 400 IU; menstruating individuals unable to have baseline and post‐treatment blood drawn during the follicular phase; and individuals unable to refrain from nonsteroidal anti‐inflammatory use for > 72 hours prior to blood work. People with a CGI‐I score of 1 or 2 (i.e. “much improved” or “very much improved”) during the baseline visit (1 week after the screen visit) were excluded from the study |
|
| Interventions |
Intervention: 1000 mg DHA enriched mix (consisting of 45 mg EPA / 225 mg DHA [EPA:DHA 1:5], plus 10% docosapentaenoic acid [DPA, n‐3], 2% heneicosapentaenoic acid [HPA, n‐3], 1% stearidonic acid [SDA, n‐3], 1% eicosatetraenoic acid [ETA, n‐3], 0.4% α‐linolenic acid [ALA, n‐3], 1% arachidonic acid [AA, n‐6], 0.5% linoleic acid [LA, n‐6], and 20% unspecified fatty acids) per soft‐gel capsule. 4 DHA enriched capsules (plus EPA arm placebo capsules) every morning for 8 weeks Comparator: 980 mg soybean oil per capsule (formed of 53.6% LA, 7.1% ALA, 0.1% myristic acid, 11% palmitic acid, 4% stearic acid, 0.2% palmitoleic acid, and 24% oleic acid), 4 capsules every morning (plus EPA arm placebo capsules) for 8 weeks |
|
| Outcomes |
Primary: HDRS (17‐item), QIDS‐SR16, every 2 weeks for 8 weeks, Adverse events (PRISE scale) Secondary: Depression remission and response; CGI (Scale), CGI (Improvement), WBS (Ryff 1995), QLESQ, every 2 weeks for 8 weeks Failure to complete |
|
| Notes |
Supported by NIH Grant Supplements provided by Nordic Naturals Conflicts of Interest: CoIs reported for several authors Compliance: NR Depressed mood (continuous): Analysis conducted on HDRS (17‐item) scores at 8 weeks, published modified ITT (at least 1 post‐baseline assessment) data used for analyses, end outcome scores calculated from change data, SDs imputed from other studies using the HDRS (17‐item). Placebo group split across 2 intervention groups (DHA = 29 participants, EPA = 30 participants) Adverse events: Adverse events reported by individuals, 20 ‐ 30% of participants endorsed some baseline PRISE physical or depressive symptoms. The following participants experienced emerging or worsening adverse events: Intervention = 40 of 56, Comparator = 33 of 60 (correspondence from author). Values included in the analysis are for emerging or worsening AEs. Depression remission defined as final HDRS (17‐item) score ≤ 7; Depression response defined as improvement ≥ 50% in HDRS (17‐item). Quality of life: CGI scale data used in analyses Failure to complete: Intervention group = 15 (2 insufficient time/energy, 5 lost to follow‐up, 3 violated protocol, 2 family emergency, 3 NR), comparator group = 12 (1 health problems related to treatment, 1 scheduling issues, 3 lost to follow‐up, 3 violated protocol, 4 NR) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A fixed‐block size of 30 participants (MGH) or a randomly‐permuted block size between 6 and 15 participants (CSMC). P55 |
| Allocation concealment (selection bias) | Low risk | Only blind treatment codes, co‐ordinated between both site pharmacies, were noted on randomisation lists provided to study staff. P55 |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Flavours added to mask taste but no check to assess blinding (correspondence from authors) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Self‐reported for mood scales, P55 |
| Blinding of outcome assessment (Adverse Events) | Unclear risk | AEs rated by participants, P55 |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Mood scales ‐ not ITT and > 10% dropout ‐ P55, and correspondence from authors |
| Incomplete outcome data (Adverse Events) | Low risk | All reported (correspondence from authors) |
| Selective reporting (reporting bias) | High risk | Well being scale and n‐3PUFA blood levels still to be reported (correspondence from authors) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Multi‐centre parallel design randomised controlled trial, 8 weeks | |
| Participants |
Participants: 196 participants (split across Mischoulon (DHA) 2015 and Mischoulon (EPA) 2015): 177 participants considered evaluable (provided 1 post‐baseline assessment); Mean age 45.8 (SD 12.5) years, 59.3% women (n = 105), 40.7% men (n = 72). Participants recruited at Massachusetts General Hospital and Cedars‐Sinai Medical Center through advertisements and referrals from outpatient programmes, from May 2006 to June 2011 Comorbidities: anxiety disorders/dysthymia in some participants, no serious/unstable physical comorbidities Adjunctive therapy: no, for all participants Inclusion criteria: A diagnosis of MDD per the SCID‐I/P), a CGI‐S score ≥ 3, and a baseline 17‐item HDRS‐17 score ≥ 15 Exclusion criteria: pregnancy or women of childbearing potential who were not using a medically‐accepted means of contraception; suicidality or homicidality; serious or unstable medical illness; current or past history of organic mental disorders, substance use disorders, any psychotic disorders, and bipolar disorder; history of multiple adverse drug reactions or allergy to the study compounds; concurrent use of psychotropic medications, systematic corticosteroid or steroid antagonists, anticoagulants, or immunosuppressant agents; electroconvulsive therapy during the current episode; any trial of ≥ 6 weeks with citalopram 40 mg/d or equivalent antidepressant during the current episode (to select a less refractory sample that would be more likely to respond to treatment); history of use of 1 g/d of n‐3 supplements; history of a bleeding disorder; psychotherapy; smoking 10 cigarettes per day; vitamin E supplementation > 400 IU; menstruating individuals unable to have baseline and post‐treatment blood drawn during the follicular phase; and individuals unable to refrain from nonsteroidal anti‐inflammatory use for > 72 hours prior to blood work. People with a CGI‐I score of 1 or 2 (i.e. “much improved” or “very much improved”) during the baseline visit (1 week after the screen visit) were excluded from the study |
|
| Interventions |
Intervention: 1000 mg EPA enriched mix (consisting of 530 mg EPA / 137 mg DHA per soft gel [EPA:DHA 4:1], plus 7% stearidonic acid [SDA, n‐3], 1% heneicosapentaenoic acid [HPA, n‐3], 1% docosapentaenoic acid [DPA, n‐3], 1% eicosatetraenoic acid [ETA, n‐3], 0.2% α‐linolenic acid [ALA, n‐3], 3% arachidonic acid [AA, n‐6], 0.2% linoleic acid [LA, n‐6], and 10% – 11% unspecified fatty acids) per soft‐gel capsule. 2 EPA enriched capsules (plus DHA arm placebo capsules) every morning for 8 weeks Comparator: 980 mg soybean oil per capsule (formed of 53.6% LA, 7.1% ALA, 0.1% myristic acid, 11% palmitic acid, 4% stearic acid, 0.2% palmitoleic acid, and 24% oleic acid), 2 capsules every morning (plus DHA arm placebo capsules) for 8 weeks |
|
| Outcomes |
Primary: HDRS (17‐item), QIDS‐SR16, every 2 weeks for 8 weeks, Adverse events (PRISE) Secondary: Depression remission and response; CGI‐S, CGI‐I, WBS (Ryff 1995), QLESQ, every 2 weeks for 8 weeks. Failure to complete |
|
| Notes |
Supported by NIH Grant Supplements provided by Nordic Naturals Conflicts of Interest: CoIs reported for several authors Compliance: NR Depressed mood (continuous): Analysis conducted on HDRS (17‐item) scores at 8 weeks, published modified ITT (at least 1 post‐baseline assessment) data used for analyses, end outcome scores calculated from change data, SDs imputed from other studies using the HDRS (17‐item). Placebo group split across 2 intervention groups (DHA = 29 participants, EPA = 30 participants) Adverse events: Adverse events reported by individuals, 20 ‐ 30% of participants endorsed some baseline PRISE physical or depressive symptoms. The following participants experienced emerging or worsening adverse events: Intervention = 40 of 56, Comparator = 33 of 60 (correspondence from author). Values included in the analysis are for emerging or worsening AEs. Depression remission defined as final HDRS (17‐item) score ≤ 7; Depression response defined as improvement ≥ 50% in HDRS (17‐item). Quality of life: CGI scale data used in analyses Failure to complete: Intervention group = 15 (2 insufficient time/energy, 5 lost to follow‐up, 3 violated protocol, 2 family emergency, 3 NR), comparator group = 12 (1 health problems related to treatment, 1 scheduling issues, 3 lost to follow‐up, 3 violated protocol, 4 NR) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A fixed‐block size of 30 participants (MGH) or a randomly‐permuted block size between 6 and 15 participants (CSMC). P55 |
| Allocation concealment (selection bias) | Low risk | Only blind treatment codes, co‐ordinated between both site pharmacies, were noted on randomisation lists provided to study staff. P55 |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Flavours added to mask taste but no check to assess blinding (author correspondence) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Self‐reported for mood scales, P55 |
| Blinding of outcome assessment (Adverse Events) | Unclear risk | AEs rated by participants, P55 |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Mood scales ‐ not ITT and > 10% dropout ‐ P55, and correspondence from author |
| Incomplete outcome data (Adverse Events) | Low risk | All reported (correspondence from author) |
| Selective reporting (reporting bias) | High risk | Well being scale and n‐3PUFA blood levels still to be reported (correspondence from author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel‐arm trial, 8 weeks | |
| Participants |
Participants: After 57 participants were randomised at a screening visit, 41 completed a baseline visit and entered into the study; mean age 43 years (SD = 13), 63% women. These participants were recruited via advertisements and referrals to the Massachussets General Hospital Depression Clinical and Research Programme, from Jan 2003 to June 2006. Comorbidities: No Adjunctive therapy: Yes in some participants: concurrent psychotherapy if receiving therapy prior to enrolment Inclusion criteria: aged 18 ‐ 80 years, DSM‐IV diagnosis of MDD (using SCID‐IP), score ≥ 18 on the 17‐item HDRS and ≥ 3 on the CGI‐SI scale, ability to provide informed written consent, free from antidepressant, antipsychotic or mood‐stabilisation medication Exclusion criteria: unstable medical conditions, psychiatric or psychotic comorbidities, current serious suicide or homicidal risk, substance abuse, currently taking n‐3PUFA supplements, history of adverse drug reactions or allergy to study drugs, pregnancy or no use of medically‐approved contraception among women of child‐bearing potential, breastfeeding, failure to respond to ≥ 1 antidepressant trial, history of unstable seizure disorder, history of electroconvulsive therapy in previous 6 months, anticoagulant use |
|
| Interventions |
Intervention: E‐EPA (1 g/d, plus 0.2% alpha tocopherol), 2 x 500 mg capsules twice daily or both at once Comparator: Paraffin oil (1 g/d, plus 0.2% alpha tocopherol), 2 x 500 mg capsules twice daily or both at once Treatment received for 8 weeks |
|
| Outcomes |
Primary: HDRS (17‐item) measured every 2 weeks for 8 weeks; Adverse events Secondary: Remission and response based on HDRS; QLESQ; Failure to complete |
|
| Notes |
Funded by the National Center for Complementary and Alternative Medicine, NIH, USA Supplements provided by Amarin Neuroscience Ltd, UK Conflicts of Interest: CoIs declared from many authors. Compliance: Capsule counts at each visit; Plasma n‐3PUFA levels measured Depressed mood (continuous): Analysis conducted on HDRS (17‐item) scores at 8 weeks, ITT data provided by authors Adverse events: Adverse events were reported in 7 individuals ‐ Intervention group = 2 (2 GI), comparator group = 5 (5 GI) Quality of life: QLESQ ‐ data not reported. Failure to complete: Intervention group = 6 (1 non‐response, 1 commuting, 4 lost to follow‐up), comparator group = 11 (2 non‐response, 1 feeling better, 8 lost to follow‐up) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation performed by research pharmacy using www.randomization.com (P.1637) |
| Allocation concealment (selection bias) | Low risk | Assigned medications were coded and sent to treatment team by research pharmacy (P.1637) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Study clinicians and participants remained blind to assignment for duration of study (P.1637). It was unclear if the fishy taste was disguised and no assessment to check concealment |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | HDRS ‐ Study clinicians remained blind to assignment for duration of study |
| Blinding of outcome assessment (Adverse Events) | High risk | AEs rated by participants |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | HDRS ‐ ITT numbers obtained through correspondence with author |
| Incomplete outcome data (Adverse Events) | Low risk | All AEs reported |
| Selective reporting (reporting bias) | High risk | All primary outcome measures reported (correspondence with author). QLESQ was a planned outcome and measured but not analysed or reported (correspondence with author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel‐arm trial, 4 weeks | |
| Participants |
Participants: 20 participants with mean age = 53.4 (SD = 11.7, range 28 ‐ 73) years, 17 women Comorbidities: Yes in some participants: 1 participant had comorbid OCD Adjunctive therapy: Yes in some participants: all with the exception of 1 participant Inclusion criteria: recurrent MDD (according to DSM‐IV criteria) from ≥ 2 clinical interviews with ≥ 2 specialist psychiatrists spaced at least 1 week apart, aged 18 ‐ 75 years, no unstable medical disease, no psychotic or psychiatric comorbidities other than panic disorder, dysthymic disorder or OCD, no substance abuse |
|
| Interventions |
Intervention: E‐EPA (2 g/d), 2 x 500 mg capsules, twice daily, plus ongoing therapy Placebo: placebo, 2 x 500 mg capsules, twice daily, plus ongoing therapy Treatment received for 4 weeks |
|
| Outcomes |
Primary: HDRS (24‐item) measured at baseline and weekly for 4 weeks; Adverse events Secondary: Response based on HDRS, Failure to complete |
|
| Notes |
Funding: not reported Supplements provided by Laxdale Ltd., UK. Conflicts of Interest: not reported Compliance: not reported Depressed mood (continuous): Analysis conducted on HDRS (24‐item) scores at 4 weeks, ITT data calculated from publication Adverse events: Only clinically relevant adverse events were investigated, none found. Values in the analysis are for clinically relevant AEs Failure to complete: Intervention group = 0, comparator group = 1 (symptoms worsened) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised according to a random‐number table (correspondence with author) |
| Allocation concealment (selection bias) | Low risk | Senior investigator generated random‐number table and was in a different building to senior clinician. Senior clinician was not aware of the randomisation sequence. (Correspondence with author) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. Intervention and placebo capsules were matching, although no attempt to match taste. No participants reported fishy sensations when asked specifically, and debriefing recorded a completely random guess rate by participant and clinician (P.477, 478) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | HDRS ‐ assessors blind to treatment assignment |
| Blinding of outcome assessment (Adverse Events) | Low risk | AEs ‐ participant‐rated, participants blind to treatment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | HDRS ‐ 1 participant dropped out, but possible to conduct ITT analysis using LOCF |
| Incomplete outcome data (Adverse Events) | High risk | Only clinically relevant AEs reported |
| Selective reporting (reporting bias) | Low risk | All outcomes reported (correspondence with author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel‐arm trial, 12 weeks | |
| Participants |
Participants: 35 participants, mean age only reported by group (Intervention = 43.5 (SD = 3.72) years; comparator = 39.41 (SD = 3.58) years); 27 women, 8 men. Participants recruited from Hanyang University Hospital, Korea, from 2010 to 2013 Comorbidities: None reported, possible psychiatric comorbidities Adjunctive therapy: Yes, usual care and antidepressant medications in all participants Inclusion criteria: CES‐D‐K (Cho 1998) score > 24, confirmed by psychiatrist according to DMS‐IV Exclusion criteria: pregnant, lactating, < 18 / > 65 years old, taking supplements containing n‐3PUFAs, medical comorbidity (CV disease, dementia), chronic depression lasting > 2 years or treatment‐resistant depression, other primary psychiatric disorders (bipolar or schizophrenia) |
|
| Interventions |
Intervention: E‐EPA/DHA combination (EPA = 3420 mg/d, DHA = 1800 mg/d), 3 capsules daily for 12 weeks Comparator: safflower oil and oleic acid (3g), 3 capsules daily for 12 weeks |
|
| Outcomes |
Primary: HDRS (17‐item), CES‐D‐K measured at baseline, 4, 8, 12 weeks; Adverse events Secondary: CGI, CGI‐IS, dietary data, blood samples, failure to complete |
|
| Notes |
Funded by the Korean Research Foundation Supplements provided by DSM Nutritional Products, Switzerland Conflicts of Interest: None declared, however Dr Y Park is a founder of Omega Quant Asia (a laboratory specialising in fatty acid analysis) Compliance: Plasma n‐3PUFA levels measured Depressed mood (continuous): Analysis conducted on HDRS (17‐item) scores at 12 weeks, data using modified ITT (at least 1 post‐baseline visit) provided by authors Adverse events: Adverse events were reported in 4 individuals: Intervention group = 3 (3 fishy eructation), comparator group = 1 (1 fishy eructation) Quality of life: Analysis conducted on CGI (scale) Failure to complete: Intervention group = 6 (1 rejected blood sampling, 5 participant decision), comparator group = 5 (1 rejected blood sampling, 4 participant decision) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Independent statistician, computer‐generated randomisation scheme allowing for randomisation blocks, P143 |
| Allocation concealment (selection bias) | Low risk | Sequentially‐numbered containers with either n‐3PUFAs or placebo; randomly assigned to participants. Identity codes were concealed in sequentially‐numbered opaque envelopes managed by the study investigators, P143 |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No attempt to mask flavour or check blinding, P142 |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | HDRS scores were measured by psychiatrist who was blinded to treatment groups |
| Blinding of outcome assessment (Adverse Events) | High risk | AEs ‐ participant‐rated (participants not blinded effectively) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | HDRS ‐ > 10% missing in the overall sample and not ITT analysis |
| Incomplete outcome data (Adverse Events) | Low risk | All AEs reported, P144 |
| Selective reporting (reporting bias) | Low risk | All outcomes reported (correspondence from authors) |
| Other bias | High risk | Significant baseline imbalance for mood disorders, P144 |
| Methods | Randomised controlled multicentre parallel‐arm trial, 12 weeks | |
| Participants |
Participants: 70 participants with a mean age of 44.7 years were recruited by family physicians in the UK who had an interest in depression and experience in conducting clinical trials (split across Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002) Comorbidities: none reported, but possible physical and/or psychiatric comorbidities Adjunctive therapy: Yes in all participants: antidepressants Inclusion criteria: aged 18 ‐ 70 years, score ≥ 15 on the 17‐item HDRS despite ongoing treatment with a standard antidepressant at an adequate dose |
|
| Interventions |
Intervention: E‐EPA (1 g/d + 3 g/d placebo), 4 x 500 mg capsules, twice daily Comparator: liquid paraffin (4 g/d), 4 x 500 mg capsules, twice daily Treatment received for 12 weeks |
|
| Outcomes |
Primary: HDRS (17‐item), MADRS, BDI were all measured at baseline, 4, 8 and 12 weeks; Adverse events Secondary: Response based on HDRS, MADRS and BDI; failure to complete |
|
| Notes |
Funding: not reported Conflicts of Interest: CoIs declared by one author. Other author works for Laxdale Ltd., UK. Compliance: Capsule counts Depressed mood (continuous): Analysis conducted on HDRS (17‐item) scores at 12 weeks, published ITT data (although 1 participant from the placebo group is missing from these data). Placebo group split across all 3 intervention groups (1 g/d = 5 participants, 2 g/d = 6 participants, 4 g/d = 6 participants), SDs calculated from all other studies also using the HDRS (17‐item) Adverse events: Intervention group: 18 events experienced by 9 participants (7 GI, 4 psychological, 7 other physical), comparator group: 23 events experienced by 10 participants (4 GI, 2 psychological, 17 other physical) Failure to complete: Intervention groups (2 per group, reasons not separated by group 1 g/d, 2g/d, 4 g/d) = 6 (3 withdrew consent, 1 lack of efficacy, 1 violated protocol, 1 adverse event), comparator group = 4 (1 withdrew consent, 1 violated protocol, 1 adverse event, 1 lost to follow‐up) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomly allocated by PCI clinical services computer" (P.914) |
| Allocation concealment (selection bias) | Low risk | Capsules were packed and coded by PCI clinical services. Participants were randomly allocated on entry to study, PCI Clinical Services had no involvement with the rest of the trial. (P.914) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants took the same number of capsules, placebo and intervention capsules were identical in appearance. Participants, researchers and assessors blind to treatment allocation. (P.914) It was unclear if they disguised the fishy taste and no assessment to check concealment |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | HDRS/MADRS ‐ assessors blind to treatment allocation (LOW) BDI ‐ participant‐rated (HIGH) |
| Blinding of outcome assessment (Adverse Events) | High risk | AEs assessed by participants |
| Incomplete outcome data (attrition bias) All outcomes | High risk | HDRS/MADRS/BDI ‐ Not ITT analysis (only 17 participants used in the analysis of placebo group) |
| Incomplete outcome data (Adverse Events) | Low risk | All AEs reported |
| Selective reporting (reporting bias) | Low risk | All outcomes reported (correspondence from author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled multicentre parallel‐arm trial, 12 weeks | |
| Participants |
Participants: 70 participants with a mean age of 44.7 years were recruited by family physicians in the UK who had an interest in depression and experience in conducting clinical trials (split across Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002) Comorbidities: none reported, but possible physical and/or psychiatric comorbidities Adjunctive therapy: Yes in all participants: antidepressants Inclusion criteria: aged 18 ‐ 70 years, score ≥ 15 on the 17‐item HDRS despite ongoing treatment with a standard antidepressant at an adequate dose |
|
| Interventions |
Intervention: E‐EPA (2 g/d + 2 g/d placebo), 4 x 500 mg capsules, twice daily Comparator: liquid paraffin (4 g/d), 4 x 500 mg capsules, twice daily Treatment received for 12 weeks |
|
| Outcomes |
Primary: HDRS (17‐item), MADRS, BDI were all measured at baseline, 4, 8 and 12 weeks; Adverse events Secondary: Response based on HDRS, MADRS and BDI; Failure to complete |
|
| Notes |
Funding: not reported Conflicts of Interest: CoIs declared by one author. Other author works for Laxdale Ltd., UK. Compliance: Capsule counts Depressed mood (continuous): Analysis conducted on HDRS (17‐item) scores at 12 weeks, published ITT data (although 1 participant from the placebo group is missing from these data). Placebo group split across all 3 intervention groups (1 g/d = 5 participants, 2 g/d = 6 participants, 4 g/d = 6 participants), SDs calculated from all other studies also using the HDRS (17‐item) Adverse events: Intervention group: 18 events experienced by 9 participants (7 GI, 4 psychological, 7 other physical), comparator group: 23 events experienced by 10 participants (4 GI, 2 psychological, 17 other physical) Failure to complete: Intervention groups (2 per group, reasons not separated by group 1 g/d, 2g/d, 4 g/d) = 6 (3 withdrew consent, 1 lack of efficacy, 1 violated protocol, 1 adverse event), comparator group = 4 (1 withdrew consent, 1 violated protocol, 1 adverse event, 1 lost to follow‐up) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomly allocated by PCI clinical services computer" (P.914) |
| Allocation concealment (selection bias) | Low risk | Capsules were packed and coded by PCI clinical services. Participants were randomly allocated on entry to study, PCI Clinical Services had no involvement with the rest of the trial. (P.914). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants took the same number of capsules, placebo and intervention capsules were identical in appearance. Participants, researchers and assessors blind to treatment allocation. (P.914) It was unclear if they disguised the fishy taste and no assessment to check concealment. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | HDRS/MADRS ‐ assessors blind to treatment allocation (LOW) BDI ‐ participant‐rated (HIGH) |
| Blinding of outcome assessment (Adverse Events) | High risk | AEs assessed by participants |
| Incomplete outcome data (attrition bias) All outcomes | High risk | HDRS/MADRS/BDI ‐ Not ITT analysis (only 17 participants used in the analysis of placebo group) |
| Incomplete outcome data (Adverse Events) | Low risk | All AEs reported |
| Selective reporting (reporting bias) | Low risk | All outcomes reported (correspondence from author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled multicentre parallel‐arm trial, 12 weeks | |
| Participants |
Participants: 70 participants with a mean age of 44.7 years were recruited by family physicians in the UK who had an interest in depression and experience in conducting clinical trials (split across Peet (1g/d) 2002; Peet (2g/d) 2002; Peet (4g/d) 2002) Comorbidities: none reported, but possible physical and/or psychiatric comorbidities Adjunctive therapy: Yes in all participants: antidepressants Inclusion criteria: aged 18 ‐ 70 years, score ≥ 15 on the 17‐item HDRS despite ongoing treatment with a standard antidepressant at an adequate dose |
|
| Interventions |
Intervention: E‐EPA (4 g/d), 4 x 500 mg capsules, twice daily Comparator: liquid paraffin (4 g/d), 4 x 500 mg capsules, twice daily Treatment received for 12 weeks |
|
| Outcomes |
Primary: HDRS (17‐item), MADRS, BDI were all measured at baseline, 4, 8 and 12 weeks; Adverse events Secondary: Response based on HDRS, MADRS and BDI; Failure to complete |
|
| Notes |
Funding: not reported Conflicts of Interest: CoIs declared by one author. Other author works for Laxdale Ltd., UK. Compliance: Capsule counts Depressed mood (continuous): Analysis conducted on HDRS (17‐item) scores at 12 weeks, published ITT data (although 1 participant from the placebo group is missing from these data). Placebo group split across all 3 intervention groups (1 g/d = 5 participants, 2 g/d = 6 participants, 4 g/d = 6 participants), SDs calculated from all other studies also using the HDRS (17‐item) Adverse events: Intervention group: 18 events experienced by 9 participants (7 GI, 4 psychological, 7 other physical), comparator group: 23 events experienced by 10 participants (4 GI, 2 psychological, 17 other physical) Failure to complete: Intervention groups (2 per group, reasons not separated by group 1 g/d, 2g/d, 4 g/d) = 6 (3 withdrew consent, 1 lack of efficacy, 1 violated protocol, 1 adverse event), comparator group = 4 (1 withdrew consent, 1 violated protocol, 1 adverse event, 1 lost to follow‐up) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomly allocated by PCI clinical services computer" (P.914) |
| Allocation concealment (selection bias) | Low risk | Capsules were packed and coded by PCI clinical services. Participants were randomly allocated on entry to study, PCI Clinical Services had no involvement with the rest of the trial. (P.914) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants took the same number of capsules, placebo and intervention capsules were identical in appearance. Participants, researchers and assessors blind to treatment allocation. (P.914) It was unclear if they disguised the fishy taste and no assessment to check concealment. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | HDRS/MADRS ‐ assessors blind to treatment allocation (LOW) BDI ‐ participant‐rated (HIGH) |
| Blinding of outcome assessment (Adverse Events) | High risk | AEs assessed by participants |
| Incomplete outcome data (attrition bias) All outcomes | High risk | HDRS/MADRS/BDI ‐ Not ITT analysis (only 17 participants used in the analysis of placebo group) |
| Incomplete outcome data (Adverse Events) | Low risk | All AEs reported |
| Selective reporting (reporting bias) | Low risk | All outcomes reported (correspondence from author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel‐arm trial, 8 weeks | |
| Participants |
Participants: 46 women with a mean age of 83.9 years, resident in a nursing home in Pavia, Italy for ≥ 3 months. Data were gathered between January 2006 and December 2007 Comorbidities: No psychiatric comorbidities, arthritis in some individuals Adjunctive therapy: No antidepressants, possible use of other therapies Inclusion criteria: aged 65 ‐ 95 years, BMI of 19 ‐ 30 kg/m2, score > 10 on the GDS, MMSE score > 24, met DSM‐IV criteria for MDD or dysthymia, as assessed by senior psychiatrist Exclusion criteria: presence of clinically uncontrolled organic disease or clinically relevant lab abnormalities, any psychotic or psychiatric comorbidities, including suicidal ideation, current use of psychotropic drugs other than benzodiazepines. Ongoing pharmacological treatment for physical conditions, at the time of enrolment, was maintained during the study |
|
| Interventions |
Intervention: EPA/DHA combination (3.13 g/d ‐ EPA = 1.67 g/d, DHA = 0.83 g/d, other n‐3PUFAs = 0.63 g/d) Comparator: Paraffin oil (2.5 g/d) Treatment received for 8 weeks |
|
| Outcomes |
Primary: GDS was measured before and after treatment at week 0 and week 8. Adverse events Secondary: Remission and response based on GDS; SF‐36 (mental health summary score); Failure to complete |
|
| Notes |
Funded by Regione Lomdardia, Italy Intervention provided by Also SpA Div. Also‐Enervit, Zelbio (Co), Italy. Conflicts of Interest: None declared. Compliance: EPA and DHA levels in RBC membranes Depressed mood (continuous): Analysis conducted on GDS scores at 8 weeks, published ITT data Adverse events: No serious adverse events reported. Minor adverse events: Intervention group = 6 (6 GI), comparator group = 6 (5 GI, 1 other physical). Values in the analysis are for minor events Failure to complete: Not mentioned, but full data sets provided for all participants |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Bottles for each treatment group were assigned a participant number according to a coded (AB) block randomisation table prepared by an independent statistician. (P.57) |
| Allocation concealment (selection bias) | Low risk | As participants were enrolled they were assigned a progressive participant number. Investigators were blinded to the randomisation table, the code assignments and the procedure. (P.58) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Investigators were blinded to the randomisation table, the code assignments and the procedure. Bottles of oily preparation were identical for each treatment group and lemon flavour was added to both oils. No participants complained about a fish smell or eructation or made any comment about the contents of the supplement or perception of being in 1 of the 2 groups. (P.58, 60) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | GDS ‐ Investigators and participants blind to treatment |
| Blinding of outcome assessment (Adverse Events) | Low risk | AEs ‐ investigators and participants blind to treatment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | GDS ‐ ITT analysis |
| Incomplete outcome data (Adverse Events) | Low risk | All AEs are reported |
| Selective reporting (reporting bias) | Low risk | All outcome measures reported (correspondence with author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel‐arm trial, 12 weeks | |
| Participants |
Participants: 77 participants with a mean age = 38.8 years, 41 women, recruited through a Community Mental Health Service, general practices and advertisements in community newspapers in New Zealand. Participants were recruited between July 2000 and September 2001 Comorbidities: possible physical and psychiatric comorbidities Adjunctive therapy: Yes for some participants: 61 participants taking antidepressants, 21 participants receiving psychotherapy Inclusion criteria: current depressive episode, aged 18 ‐ 65 years, stable medication for ≥ 2 months prior to enrolment, willing to provide blood samples and, if female, premenopausal with a normal menstrual cycle, available for the length of the study Exclusion criteria: any psychotic or psychiatric comorbidities other than anxiety disorders, currently taking n‐3PUFA supplements, allergy to seafood or objection to taking fish‐/olive oil‐based products, blood clotting disorders or use of anticoagulants, any unstable medical conditions or conditions likely to affect gastrointestinal absorption |
|
| Interventions |
Intervention: EPA/DHA combination (8 g/d DHA enriched tuna oil providing 0.6 g/d EPA, 2.4 g/d DHA, 80 mg vitamin E), 4 x 1 g capsules, twice daily, plus ongoing therapy Comparator: Olive oil (8 g/d) 4 x 1 g capsules, twice daily, plus ongoing therapy Treatment received for 12 weeks |
|
| Outcomes |
Primary: HDRS Short Form (9‐item) (score of > 10 represents severe depression) and BDI‐II were measured at baseline and weeks 2, 4, 8 and 12. Adverse events Secondary: Failure to complete |
|
| Notes |
Funded by Foundation for Research, Science and Technology, New Zealand. Supplements provided by Clover Corporation Plc, Australia Conflicts of Interest: No CoIs declared. Compliance: RBC membrane EPA and DHA levels measured, participants completing exit interview asked about compliance Depressed mood (continuous): Analysis conducted on HDRS (9‐item) scores at 12 weeks, per protocol data obtained from authors Adverse events: Intervention group ‐ 20 events in 14 participants (11 GI, 7 other physical, 2 not reported); comparator group ‐ 16 events in 14 participants (8 GI, 2 psychological, 5 other physical, 1 not reported) Failure to complete: Intervention group: 16 (2 withdrew before baseline, 9 discontinued intervention, 1 head trauma, 1 physical disorder, 2 scored < 6 on HDRS at week 0, 1 not reported); comparator group 16 (2 withdrew before baseline, 5 discontinued intervention, 1 head trauma, 3 personality disorders, 1 bipolar disorder, 4 scored < 6 on HDRS at week 0) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation according to a prearranged computer‐generated code (P.212) |
| Allocation concealment (selection bias) | Low risk | Randomisation sequence generated by statistician not directly involved in the study (P.212). Allocation sequence was concealed from both participants and the research psychologists (P.213) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Capsules looked identical, fish smell and taste were minimal. Participants were told only that both oils were natural and aftertaste might be experienced. No evidence that participants guessed their treatment allocation (P = 0.804) (P.215) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | HDRS/BDI ‐ both researchers and participants blind to treatment allocation |
| Blinding of outcome assessment (Adverse Events) | Low risk | AEs ‐ participant‐rated, participants blind to treatment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | HDRS/BDI ‐ analysis conducted on only those providing 1 follow‐up (not ITT), and > 10% dropout |
| Incomplete outcome data (Adverse Events) | Low risk | All AEs reported |
| Selective reporting (reporting bias) | Low risk | All relevant outcome measures reported (correspondence with author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
| Methods | Randomised controlled parallel‐arm trial, 8 weeks Pre‐randomisation: all participants received single‐blind placebo capsules for 1 week, those with a ≥ 20% decrease in HDRS score (placebo responders) were excluded |
|
| Participants |
Participants: 28 outpatients referred by Taipei Medical University‐Wan Fang Hospital. 22 participants completing the trial had a mean age = 38.4 years, 18 women Comorbidities: No Adjunctive therapy: Yes in some, if participants on stable medication at enrolment Inclusion criteria: aged 18 ‐ 60 years, diagnosis with DSM‐IV MDD and no other comorbid Axis I or Axis II psychiatric disorder, rated > 18 on the HDRS (21‐item), stable medication or psychotherapy for 4 weeks before enrolment, physically healthy under evaluations of medical history, physical examinations, and laboratory tests and competent to understand the study and give written informed consent Exclusion criteria: Participants receiving antipsychotics or mood stabilizers, ≥ 20% decrease in HDRS score (placebo responders) following pre‐randomisation |
|
| Interventions |
Intervention: EPA/DHA combination (6.6 g/d ‐ 4.4 g/d EPA and 2.2 g/d DHA, plus tocopherols and tertiary‐butylhydroquinone), 5 capsules, twice daily Comparator: Olive oil ethyl esters (plus tocopherols and tertiary‐butylhydroquinone), 5 capsules, twice daily Treatment received for 8 weeks |
|
| Outcomes |
Primary: HDRS (21‐item) measured at ‐1, 0, 2, 4, 6 and 8 weeks. Adverse events Secondary: Failure to complete |
|
| Notes |
Funded by National Science Council, and China Chemical and Pharmaceutical Company, Taiwan Supplements provided by China Chemical and Pharmaceutical Company, Taiwan Conflicts of Interest: not reported Compliance: EPA and DHA levels from RBCs Depressed mood (continuous): Analysis conducted on HDRS (21‐item) scores at 8 weeks, ITT data provided by authors Adverse events: Intervention group: 1 GI, 1 psychological; comparator group = 1 other physical Failure to complete: Intervention group = 2 (1 non‐compliance, 1 lost to follow‐up), comparator group = 4 (1 non‐compliance, 3 lost to follow‐up) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random‐number sheet generated in Excel (correspondence with author) |
| Allocation concealment (selection bias) | Low risk | Packages were consecutively numbered according to randomisation schedule by an independent nutritionist (correspondence with author) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Orange flavour was added to the capsules, which were identical to blind the participants (P.268). However there was no assessment to check concealment |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | HDRS ‐ unclear whether assessors were blind to treatment allocation |
| Blinding of outcome assessment (Adverse Events) | Unclear risk | AEs ‐ unclear whether participants were blind to treatment allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | HDRS ‐ ITT analysis obtained from author |
| Incomplete outcome data (Adverse Events) | Low risk | All AEs reported |
| Selective reporting (reporting bias) | Low risk | All outcomes reported (correspondence with author) |
| Other bias | Low risk | Study appeared to be free from other sources of bias |
BDI: Beck depression inventory CES‐D‐K: Center for Epidemiological Studies depression scale Korean version CGI: clinical global impression CHD: coronary heart disease DHA: docosahexaenoic acid DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, Fourth edition EPA: eicosapentaenoic acid GAF: global assessment of functioning GDS: geriatric depression scale GI: gastrointestinal HSCL: Hopkins symptom checklist depression scale ITT: intention‐to‐treat HDRS: Hamilton depression rating scale LOCF: last observation carried forward MADRS: Montgomery‐Asberg depression rating scale MAOI: monoamine oxidase inhibitor MDD: major depressive disorder MDE: major depressive episode MMSE: mini mental state examination OCD: Obsessive‐compulsive disorder PGWB: psychological general well being RBC: red blood cell QLESQ: quality of life enjoyment and satisfaction questionnaire SCID: structured clinical interview (depression) SD: standard deviation SSRI: selective serotonin reuptake inhibiting UPDRS: Unified Parkinson disease rating scale WBS: well‐being scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Clayton 2009 | Study record indicates study withdrawn prior to enrolment |
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | Randomised, placebo‐controlled, double‐blind study |
| Participants | Adults aged between 18 and 65, affected by MDD or recurrent depressive disorder according to DSM‐IV‐TR and the HDRS |
| Interventions | Intervention: fish oil 30 EPA/DHA plus SSRI Comparator: placebo, plus SSRI |
| Outcomes | Primary: Improvement in HDRS and CGI score |
| Notes |
| Methods | 12‐week, parallel‐group, double‐blind addition of choline alfoscerate or E‐EPA to ongoing antidepressant therapy |
| Participants | Adults aged over 60 years with depression |
| Interventions | Intervention: E‐EPA 2 g/d plus usual treatment Comparator: Choline alfoscerate 800 mg/d plus usual treatment |
| Outcomes | Executive function: Controlled Oral Word Association Test; Korean Stroop Color‐Word Test; Trail Making Test part B Depressive symptoms: Korean Geriatric depression scale (K‐GDS); Quick Inventory of Depressive Symptomology‐Self Report (QIDSSR) |
| Notes | Unsure if an RCT and unsure of MDD diagnosis ‐ no correspondence from author |
| Methods | Randomised controlled trial |
| Participants | Adults age 18 ‐ 60 years with major depressive episode, according to DSM‐IV criteria |
| Interventions | Intervention: Fluoxetine (oral) 20 mg/day plus omega‐3 (oral) 900 mg/day Comparator: Fluoxetine (oral) 20 mg/day plus placebo |
| Outcomes | Primary: 1. Response to differential treatment at 2, 4 and 6 weeks 2. Magnitude of the response at 2, 4 and 6 weeks 3. Biochemical analyses on blood samples at 0 and 6 weeks: 3.1. Neurotransmitters in plasma 3.2. Isolation of lymphocytes 3.3. Neurotransmitters in lymphocytes 3.4. Detection of tryptophan hydroxylase 3.5. Folate levels 3.6. Homocysteine levels 3.7. Vitamin B12 levels 4. In participants who took omega‐3, brain‐derived neurotrophic factor (BDNF) in serum and lymphocytes will be determined Secondary: Correlation between response to antidepressant and biochemical measurements |
| Notes |
| Methods | Multicentre, double‐blind, randomised, parallel‐group, placebo‐controlled trial |
| Participants | Adults aged 18 ‐ 75 with: 1. Score of ≥ 16 on the HDRS 2.Treatment for ≥ 8 weeks with 1 or more standard antidepressants, at stable dose for ≥ 3 weeks 3. Currently receiving at least the minimum therapeutic dose of 1 or more standard antidepressants, as defined in the BNF 4. Diagnosis of major depressive disorder (DSM‐IV) |
| Interventions | Intervention: 1 g/d ethyl EPA Comparator: Placebo |
| Outcomes | Not reported |
| Notes |
| Methods | Allocation: Randomised Endpoint Classification: Efficacy Study Intervention Model: Parallel Assignment Masking: Double‐blind (participant, caregiver, investigator, outcomes assessor) Primary Purpose: treatment |
| Participants | Adolescents between the ages of 13 and 21 currently under standard care treatment at the Child Division of the Department of Psychiatry at Cedars‐Sinai Medical Center Diagnosed with MDD using the DSM‐IV diagnostic criteria |
| Interventions | Intervention: Cognitive behaviour therapy in combination with omega‐3 fatty acid supplements Comparator: Cognitive behaviour therapy in combination with placebo |
| Outcomes | Primary: CDI, HDRS, both 8 times for an average of 8 weeks |
| Notes |
| Methods | Allocation: Randomised Endpoint classification: Safety/efficacy study Intervention Model: Parallel Assignment Masking: Double‐blind (participant, caregiver, investigator, outcomes assessor) Primary Purpose: treatment |
| Participants | Adults aged between 18 and 65 years meeting DSM‐IV criteria for MDD |
| Interventions | Intervention: Omega‐3 fatty acids Comparator: placebo |
| Outcomes | Primary: HDRS Secondary: BDI; adverse effects; recurrence rate |
| Notes |
| Methods | Allocation: Randomised Endpoint classification: Safety/efficacy study Intervention model: Parallel assignment Masking: Double‐blind Primary Purpose: treatment 2 studies reported in abstract, depending on therapy at time of entry: Adjunctive study and monotherapy study |
| Participants | Adults aged: 21 ‐ 65 years Inclusion Criteria:
Adjunctive study: Participants with a first or new episode of MDD Monotherapy Study: participants who have MDD but are not currently on an antidepressant |
| Interventions | Adjunctive study: Intervention: 6 g/d fish oil plus standard treatment; comparator: placebo plus standard treatment; treatment received for 4 weeks Monotherapy study: Intervention: Fish oil; comparator: placebo; treatment received for 6 weeks |
| Outcomes | Primary: Change from pretreatment score on Depression Rating Scale at 6 weeks Secondary: Weekly measure of depressive symptoms; Weekly measure of anxiety symptoms; Weekly measure of functional status; Blood levels of n‐3PUFAs pre‐ and post‐treatment |
| Notes |
| Methods | Allocation: Randomised Endpoint classification: Safety/efficacy study Intervention Model: Parallel assignment Masking: Double‐blind (participant, investigator, outcomes assessor) Primary Purpose: treatment |
| Participants | Adults aged between 18 and 85 years with: Diagnosis of relapsing‐remitting MS Diagnosis of depressive disorder Score between 11 and 30 on the MADRS Score of 25 or greater on the MMSE |
| Interventions | Intervention: Fish oil concentrate (triglyceride form) at a dose of 6 g/d (1.95 g EPA and 1.45 g DHA) Comparator: Placebo oil |
| Outcomes | Primary: MADRS Secondary: Quality of life (SF‐36) |
| Notes |
| Methods | Allocation: Randomised Endpoint classification: Efficacy study Intervention model: Parallel assignment Masking: Double‐blind (participant, caregiver, investigator, outcomes assessor) Primary Purpose: treatment |
| Participants | Adults aged 18 ‐ 65 years meeting DSM‐IV criteria for MDD |
| Interventions | Intervention: DHA/EPA (1.6 ˜ 2.8 g/d (5 capsules)) Comparator: placebo (5 g/d (5 capsules)) |
| Outcomes | Primary: HDRS Secondary: BDI; Adverse events |
| Notes |
BDI: Beck depression inventory BNF: British National Formulary CDI: Children's depression inventory CGI: Clinical global impression DHA: docosahexaenoic acid DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, Fourth edition EPA: eicosapentaenoic acid HDRS: Hamilton depression rating scale MADRS: Montgomery‐Asberg Depression Rating Scale MDD: major depressive disorder MMSE: Mini‐Mental State Examination SSRI: selective serotonin reuptake inhibiting
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Youth Depression Alleviation: A randomised controlled trial of omega‐3 fatty acids (fish oil) for major depressive disorder in young people (YoDA‐F) |
| Methods | Randomised placebo‐controlled trial |
| Participants | Participants aged 15 ‐ 25 years, seeking help for psychological distress A score between 11 and 20 on the QIDS‐A17‐C at first contact with the service AND after 1 week (plus 1 ‐ 5 days if the client is unable to attend earlier) at the second assessment, or at 2 subsequent (weekly) follow‐up assessments A diagnosis of MDD using the SCID‐I/P |
| Interventions | Intervention: Cognitive behavioural case management plus 4 capsules of marine fish oil per day (providing approximately 840 mg of EPA, approximately 560 mg of DHA, and approximately 5 mg of Vitamin E) Comparator: Cognitive behavioural case management plus 4 capsules of placebo per day (approximately 700 mg paraffin oil) |
| Outcomes | Primary: Change in depressive symptoms as assessed by QIDS‐A17‐C between baseline and 12 weeks Secondary: Change in depressive symptoms as assessed by QIDS‐A17‐C between baseline and 26 weeks, Remission rate at 12 and 26 week follow‐up, Changes to symptomology and psychosocial functioning assessed across a range of domains assessed at baseline and weeks 4, 8, 12, and 26 |
| Starting date | February 2014 |
| Contact information | G Paul Amminger, Orygen Youth Health Research Centre |
| Notes | ACTRN12613001352796 |
| Trial name or title | Omega‐3 for depression and other cardiac risk factors ‐ 2 |
| Methods | Allocation: Randomised Endpoint classification: Safety/efficacy Study Intervention model: Parallel assignment Masking: Double‐blind (participant, caregiver, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Adults aged 30 ‐ 75 years with: Documented coronary heart disease Diagnosis of MDD based on structured interview |
| Interventions | Intervention: 2 g/d EPA, plus 50 mg/d sertraline for 10 weeks Comparator: 2 g/d corn oil, plus 50 mg/d sertraline for 10 weeks |
| Outcomes | Primary: BDI‐II Secondary: HDRS (17‐item); heart rate variability; interleukin‐6 Measurements taken at baseline and 10 weeks |
| Starting date | April 2014 |
| Contact information | Patricia Herzing, Washington University School of Medcine |
| Notes |
| Trial name or title | The role Of omega‐3 fatty acids in adolescent depression |
| Methods | Allocation: randomised Endpoint classification: Efficacy study Intervention model: Parallel assignment Masking: Double‐blind (participant, caregiver, investigator, outcomes assessor) Primary Purpose: treatment |
| Participants | Adolescents aged between 12 and 19 meeting DSM‐IV‐TR criteria for MDD MDD duration of at least 8 weeks and a severity score of at least 40 on the CDRS‐R Age at first onset MDD of at least 12 years |
| Interventions | Intervention: n‐3PUFAs: the initial dose will be 1.2 g/d. This will be increased gradually by 0.6 g/d per 2 weeks to a possible maximum daily dose of 3.6 g/d Comparator: Corn oil; the dosage will correspond to the titration schedule of the omega‐3 fatty acid experimental treatment |
| Outcomes | Primary: CDRS‐R Secondary: CGI |
| Starting date | December 2005 |
| Contact information | Vilma Gabbay, Mount Sinai School of Medicine |
| Notes |
| Trial name or title | Omega‐3 fatty acid supplementation for symptoms of depression in patients with cardiovascular disease |
| Methods | Randomised controlled trial, parallel, blinded |
| Participants | Adults aged between 18 ‐ 75 years with: (a) angiographically‐documented coronary artery disease, defined as > 50% stenosis in an epicardial coronary artery on selective coronary angiography (b) comorbid depression as determined by a score of ≥ 16 on the CES‐D scale |
| Interventions | Intervention: 4 x 1 g/d capsules of EPA‐rich fish oil for 6 months (each capsule will contain 500 mg EPA and 25 mg DHA) Comparator: 4 x 1 g/d capsules of soybean/corn oil for 6 months (each capsule will contain 500 mg soybean oil and 500 mg corn oil) |
| Outcomes | Primary: HDRS Secondary: SF‐36; SAQ; flow mediated dilatation in the brachial artery; Changes in cerebral blood flow measured by transcranial Doppler ultrasound Measurements taken at baseline, 3 months (HDRS, SF‐36, SAQ) and 6 months |
| Starting date | November 2008 |
| Contact information | Professor Peter Howe, Nutritional Physiology Research Centre, University of South Australia |
| Notes |
| Trial name or title | Omega 3 for treatment of depression in patients with heart failure (OCEAN) |
| Methods | Allocation: Randomized Endpoint classification: Safety/efficacy study Intervention model: Parallel assignment Masking: Double‐blind (participant, caregiver, investigator, outcomes assessor) Primary Purpose: treatment |
| Participants | Adults aged 21 years and over with: Diagnosis of MDD determined by the DSM‐IV‐TR criteria with a HDRS score ≥ 18 New York Heart Association Class ≥ II |
| Interventions | Arm 1: 400/200 EPA/DHA fish oil 2 g/d Arm 2: Almost pure EPA 2 g/d Arm 3: Matched placebo corn oil capsules Treatment given for 12 weeks |
| Outcomes | Primary: Change in HDRS score; change in RBC/Plasma EPA |
| Starting date | May 2014 |
| Contact information | Wei Jiang, Duke University |
| Notes |
| Trial name or title | Omega 3 FA supplements as augmentation in the treatment of depression |
| Methods | Allocation: Randomised Endpoint classification: Safety/efficacy study Intervention model: Parallel assignment Masking: Double‐blind (participant, caregiver, investigator, outcomes assessor) Primary Purpose: treatment |
| Participants | Adults aged 18 years and over with: A diagnosis of depression Cardiovascular disease, diabetes or cancer |
| Interventions | Intervention: Desvenlafaxine (50 mg/d) and omega 3 FA supplement (range 2.4 g/d ‐ 4.8 g/d) over a 12‐week period Comparator: Desvenlafaxine (50 mg/day) and placebo (for omega 3 FA supplement) over a 12‐week period |
| Outcomes | Primary: HADS Secondary: MADRS; SF‐12; visual analogue scale for energy; visual analogue scale for pain; LSEQ |
| Starting date | February 2013 |
| Contact information | Jayesh Kamath, University of Connecticut Health Center |
| Notes |
| Trial name or title | Comparing efficacy of omega‐3 and placebo in reducing Beck Depression Score in HIV/AIDS patients |
| Methods | Randomised, double‐blind clinical trial |
| Participants | HIV‐positive patients aged between 18 ‐ 65 years old, receiving antiretroviral therapy for at least 1 year, CD4 count ≥ 350 and Beck Depression Score ≥16 |
| Interventions | Intervention: Soft gelatin cap of omega‐3 (Cap 1000 mg, Zahravi Pharmaceutical Company, Tabriz, Iran), 1 cap orally twice daily, for 8 weeks Comparator: Soft gelatin cap of placebo (cap 1000 mg, Zahravi Pharmaceutical Company, Tabriz, Iran), 1 cap orally twice daily, for 8 weeks |
| Outcomes | Primary: BDI at baseline, week 4 and 8 |
| Starting date | October 2014 |
| Contact information | Hossein Khalili, Tehran University of Medical Sciences |
| Notes |
| Trial name or title | Treating depression in coronary artery disease with omega‐3 fatty acids |
| Methods | Allocation: Randomised Endpoint classification: Efficacy study Intervention model: Parallel assignment Masking: Double‐blind (participant, caregiver, investigator, outcomes assessor) Primary Purpose: treatment |
| Participants | Adults aged 45 ‐ 80 years with: DSM‐IV criteria for MDE or minor depression as assessed by the SCID‐I depression module Stable coronary artery disease (based on no hospitalisation for cardiac events for at least 7 weeks prior) Angiographic documentation of presence and extent of coronary artery disease |
| Interventions | Intervention: 3 capsules (3 x 1 g) fish oil‐derived concentrated ethyl esters, providing 1.9 g omega‐3 fatty acids (1.2 g EPA, 0.6 g DHA, 0.1 g other omega‐3 fatty acids) Comparator: 3 capsules (3 x 1 g) of 50/50 soybean/corn oil blend containing less than 0.12 g of omega‐3 fatty acids with negligible EPA and DHA |
| Outcomes | Primary: HDRS Secondary: SF‐36; BDI‐II Measurements at baseline, 4, 8 and 12 weeks |
| Starting date | June 2010 |
| Contact information | Abby Li, Sunnybrook Health Sciences Centre |
| Notes |
| Trial name or title | Evaluating effects of omega‐3 supplementation on weight and depression among overweight or obese women with depression compared to placebo |
| Methods | Randomised, double‐blind, 12‐week, placebo‐controlled study |
| Participants | Females aged 18 to 50 years with: BMI > 25 Mild (minor) depression based on semi‐structured diagnostic interview by a psychiatrist |
| Interventions | Intervention: 3 gm omega‐3 (2 capsules with each meal) for 12 weeks Comparator: 2 placebo capsules with each meal for 12 weeks |
| Outcomes | Primary: weight, BMI and HDRS at baseline and weeks 2, 4, 8 and 12 Secondary: Central fat mass at baseline and weeks 2, 4, 8, and 12 |
| Starting date | June 2014 |
| Contact information | Seyed‐Ali Mostafavi, Psychiatric Research Center, Roozbeh Hospital, South Kargar St., Tehran, Iran |
| Notes |
| Trial name or title | Augmentation of omega‐3 fatty acid with antidepressants for major depressive disorder: a double‐blind, randomised controlled trial |
| Methods | Randomised, double‐blind, parallel‐groups, controlled trial |
| Participants | Adults aged between 20 ‐ 65 years old with a MDE, where: the person did not receive any antidepressant drugs for major depression, has a HDRS (17‐item) score, the major depressive episode is the focus of the treatment and the treating physician has judged escitalopram to be the appropriate first‐line drug, and is a native Japanese speaker |
| Interventions | Intervention: Omega‐3 polyunsaturated fatty acid Comparator: placebo |
| Outcomes | Primary: HDRS, at 12 weeks Secondary: MADRS; BDI; QIDS‐J; CGI‐S; RS‐14; Serum BDNF, proBDNF, MMP‐9, fatty acid level; Plasma IL‐6 |
| Starting date | April 2014 |
| Contact information | Wakako Nakano, University of Occupational and Environmental Health Department of Psychiatry |
| Notes |
| Trial name or title | A study of omega‐3 as an augmentor of antidepressant treatment for major depression |
| Methods | Allocation: randomised Endpoint classification: Safety/efficacy study Intervention model: Parallel assignment Masking: Double‐blind Primary purpose: treatment |
| Participants | Adults aged between 18 and 65 years presenting with a first or new episode of DSM‐IV non‐psychotic MDD warranting treatment with antidepressant mediation |
| Interventions | Intervention: Omega‐3 (fish oil) Comparator: placebo (paraffin oil) |
| Outcomes | Primary: Change from pretreatment score on Depression Rating scale at 4 weeks Secondary: Daily mood rating; weekly measure of depression; weekly measure of anxiety; weekly measure of functional status |
| Starting date | February 2006 |
| Contact information | Catherine Owen, University of New South Wales |
| Notes |
| Trial name or title | Effects of a Mediterranean‐style diet and fish oil supplements on mood and health |
| Methods | Randomised controlled trial (participants non‐blinded) |
| Participants | Adults aged 18 ‐ 65 with: Poor diet indicated by poor diet quality score Self‐reported depressive symptoms |
| Interventions | Intervention: Fortnightly food hampers (containing extra virgin olive oil, seasonal fruit/vegetables ‐ approx 2 fruits and 5 vegetables ‐ and nuts) 2‐hour fortnightly cooking workshops (using selected simple tasty affordable recipes based on Mediterranean diet principles) for 3 months Fish oil capsules (2/day containing a total of 1 g DHA+EPA) for 6 months, commencing at baseline 2‐hour group nutrition education session following baseline assessments Comparator: Fortnightly social groups for 3 months |
| Outcomes | Primary: DASS 21; Apolipoprotein B/A1 ratio in serum; AQoL‐8d Quality of life questionnaire Secondary: Sodium/potassium ratio in urine; blood pressure using an automatic sphygmomanometer, seated after 5 minutes rest; 20‐item PANAS; 14‐item Mediterranean diet questionnaire; SDQ; 3‐day food diaries to measure dietary intake at food group level; erythrocyte fatty acid analysis in red blood cells; fasting glucose and insulin in serum; carotenoids in plasma; inflammatory markers IL1b, IL6, IL8, IL10, TNF, IL18, MIC‐1 and oxidative stress markers reduced glutathione and oxidised glutathione ‐ all in serum; anthropometric measures (weight, height, waist and hip circumference) using ISAK protocols as per the International Standards for Anthropometric Assesment protocols. All outcomes measured at baseline, 3 months, 6 months |
| Starting date | April 2014 |
| Contact information | Natalie Parletta, School of Population Health, University of South Australia |
| Notes |
| Trial name or title | Adjunctive natural low dose docosahexaenoic acid (DHA) omega‐3 in a 16 week random double‐blind placebo controlled (RDBPC) cross‐over withdrawal study in a group of chronic, psychiatric out‐patients with anxiety and mood disorders |
| Methods | Randomised controlled, double‐blind, cross‐over trial Following the open‐label phase (first 4 weeks of the study) there will be 2 double‐blind cross‐over phases, each of 8 weeks duration, where the participant will first take DHA omega‐3 then look‐alike placebo capsule containing safflower oil, or placebo then DHA omega‐3. In the final 4 weeks phase all participants receive DHA omega‐3 |
| Participants | Adults aged 20 ‐ 70 who are: 1. Outpatients with chronic anxiety and/or depressive symptoms 2. Patients currently taking DHA (NeuroSpark) capsules for at least 3 months prior to study entry |
| Interventions | Intervention: Natural low‐dose docosahexaenoic acid (DHA) omega‐3 (NeuroSpark) 130 ‐ 390 mg per day in addition to standard psychiatric treatments Comparator: safflower oil capsules Treatment given for 16 weeks |
| Outcomes | Primary: HAM‐A, HDRS, LSEQ, Fatigue questionnaire Secondary: Change from baseline in cognitive function; levels of metabolites of Arachidonic acid (AA); cytokines (e.g. TNF‐alpha and others), inflammatory markers (CRP), RBC membrane PUFA analyses to measure PUFA levels Measurements taken at weeks 0, 4, 12, 20 and 24 (various measures at each time point) |
| Starting date | May 2014 |
| Contact information | Michael Piperoglou, University of Melbourne |
| Notes |
| Trial name or title | An 8‐week randomised, double‐blind, placebo controlled trial investigating the role of adjunctive bioactive lipids specifically; docosahexaenoic acid (DHA) versus eicosapentaenoic acid (EPA) in Major Depressive Disorder ‐ with a 6 week open label extension of DHA in patients aged 18‐65 years. |
| Methods | 8 week randomised, double‐blind, placebo‐controlled trial |
| Participants | Adults aged between 18 ‐ 65 years diagnosed with a MDE |
| Interventions | Arm 1: DHA (2 tablets (260 mg/day)) Arm 2: EPA (2 tablets or 360 mg/day) Arm 3: Sunflower oil (2 tablets or 2000 mg/day) In addition and where possible patient’s background antidepressant medication will remain as a fixed dose for the 8 week study period |
| Outcomes | Primary: HDRS, change from baseline at 8 weeks Secondary: BDNF levels, change from baseline at 8 weeks |
| Starting date | October 2010 |
| Contact information | Deirdre Smith, The Professorial Research Unit, University of Melbourne |
| Notes |
| Trial name or title | Omega‐3 polyunsaturated fatty acids and psychological interventions for workers with mild to moderate depression: a randomised controlled trial |
| Methods | Parallel, randomised, double‐blind, placebo‐controlled trial |
| Participants | Japanese workers aged between 20 ‐ 65 years |
| Interventions | Intervention: Psychological interventions + n‐3PUFAs Comparator: Psychological interventions |
| Outcomes | Primary: BDI‐II Secondary: Kessler K6, CES‐D |
| Starting date | September 2014 |
| Contact information | Jun Tayama, Nagasaki University |
| Notes |
| Trial name or title | Decreasing risk of coronary artery disease in schizophrenia by omega‐3 fatty acid supplementation (CAD) |
| Methods | Allocation: Randomised Endpoint classification: Efficacy study Intervention model: Parallel assignment Masking: Double‐blind (participant, caregiver, investigator, outcomes assessor) Primary Purpose: treatment |
| Participants | Adults aged 18 or over meeting: DSM‐IV criteria for schizophrenia (or schizoaffective disorder), major depression, or bipolar (depressed phase) disorder who are treated with antipsychotic, antidepressant or antimanic drugs and a lipid‐lowering drug (statin) for 2 months or longer |
| Interventions | Intervention: EPA (2 g in 4 x 500 mg soft gels daily) + antipsychotic drug (doctor's choice) treatment for baseline, 1 month, 2 months and 4 months duration Comparator: Placebo (soy bean oil, 2 g in 4 x 500 mg soft gels daily) + antipsychotic drug (doctor's choice) treatment for baseline, 1 month, 2 months and 4 months duration |
| Outcomes | Primary: To assess whether EPA supplementation can lead to improvement in further reducing CAD risk profile Secondary: To test whether EPA supplementation can simultaneously improve the psychiatric status of patients with schizophrenia |
| Starting date | September 2005 |
| Contact information | Jeffrey Yao, University of Pittsburgh and VA Pittsburgh Healthcare System |
| Notes |
BDI: Beck depression inventory BDNF: Brain‐derived neurotropic factor CAD: coronary artery disease CES‐D: Center for Epidemiologic Studies – Depression CGI: Clinical global impression DASS: Depression anxiety stress scale CDRS‐R: children's depression rating scale ‐ revised DHA: docosahexaenoic acid DSM‐IV: DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, Fourth edition EPA: eicosapentaenoic acid HADS: Hospital anxiety and depression scale HAM‐A: Hamilton Anxiety Scale HDRS: Hamilton depression rating scale LSEQ: Leeds sleep evaluation questionnaire MDD: major depressive disorder MDE: major depressive episode PANAS: Positive And Negative Affect Scale QIDS‐A17‐C: Quick inventory for depressive symptomatology ‐ adolescent version SAQ: Seattle Angina Questionnaire SCID‐IP: Structured Clinical Interview for DSM‐IV Axis I Disorders, patient version SDQ: Simple dietary questionnaire
Contributions of authors
KA wrote the protocol. All authors checked and subsequently revised this draft.
For the review, HS and RP screened all articles identified by searches, and extracted data from all eligible studies. KA also extracted data from all eligible studies. KA, HS and RP collectively resolved disagreements. HS and RP entered all data into Review Manager 5. KA checked all entered data, conducted all analyses, and wrote up the review. All authors checked and subsequently revised this draft.
Sources of support
Internal sources
-
Bournemouth University, UK.
Researcher time
-
University of Bristol, UK.
Researcher time
External sources
No sources of support supplied
Declarations of interest
KA: None known HS: None known RP: None known AN: None known RC: None known
Edited (no change to conclusions)
References
References to studies included in this review
- Bot M, Pouwer F, Assies J, Jansen E, Beekman ATF, Jonge P. Supplementation with eicosapentaenoic omega‐3 fatty acid does not influence serum brain‐derived neurotrophic factor in diabetes mellitus patients with major depression: A randomized controlled pilot study. Neuropsychobiology 2011;63(4):219‐23. [] [DOI] [PubMed] [Google Scholar]; Bot M, Pouwer F, Assies J, Jansen E, Diamant M, Snoek FJ, et al. Eicosapentaenoic acid as an add‐on to antidepressant medication for co‐morbid major depression in patients with diabetes mellitus: A randomized, double‐blind placebo‐controlled study [ISRCTN30877831; NTR624]. Journal of Affective Disorders 2010;126(1‐2):282‐6. [] [DOI] [PubMed] [Google Scholar]; Mocking RJ, Assies J, Bot M, Jansen E, Schene AH, Pouwer F. Biological effects of add‐on eicosapentaenoic acid supplementation in diabetes mellitus and co‐morbid depression: a randomized controlled trial. PloS One 2012;7(11):e49431. [] [DOI] [PMC free article] [PubMed] [Google Scholar]; Pouwer F. Addition of eicosapentaenoic acid to maintenance anti‐depressant therapy in diabetes patients with major depressive disorder: a double‐blind, placebo‐controlled pilot study. www.controlled‐trials.com/ISRCTN30877831 (Accessed 21 August 2014). [] ; 3000967
- Anonymous. News breaks. Nutrition Today 2009;44(6):234‐5. [] [Google Scholar]; Anonymous. Use of omega‐3 with treatment for depression in heart disease patients may not provide benefit. Nurse Prescribing 2009;7(11):523‐4. [] [Google Scholar]; Bot M, Carney RM, Freedland KE, Rubin EH, Rich MW, Steinmeyer BC, et al. Inflammation and treatment response to sertraline in patients with coronary heart disease and comorbid major depression. Journal of Psychosomatic Research 2011;71(1):13‐7. [] [DOI] [PMC free article] [PubMed] [Google Scholar]; Carney RM. Omega‐3 fatty acids to improve depression and reduce cardiovascular risk factors. ClinicalTrials.gov/show/NCT00116857 (Accessed 21 August 2014). [] ; Carney RM, Freedland KE, Rubin EH, Rich MW, Steinmeyer MS, Harris WS. Omega‐3 augmentation of sertraline in treatment of depression in patients with coronary heart disease: A randomized controlled trial [NCT00116857]. JAMA 2009;302(15):1651‐7. [] [DOI] [PMC free article] [PubMed] [Google Scholar]; Carney RM, Freedland KE, Steinmeyer BC. 'Omega‐3 fatty acids for CHD with depression': Reply to comment. JAMA 2010;303(9):836. [] [DOI] [PubMed] [Google Scholar]; D'Anci KE. Nutrition updates. Nutrition Reviews 2010;68(1):71‐3. [] [DOI] [PubMed] [Google Scholar]; Roest AM, Carney RM, Stein PK, Freedland KE, Meyer H, Steinmeyer BC, et al. Obstructive sleep apnea/hypopnea syndrome and poor response to sertraline in patients with coronary heart disease. Journal of Clinical Psychiatry 2012;73(1):31‐6. [] [DOI] [PMC free article] [PubMed] [Google Scholar]; 3000972
- Coryell WH. Essential fatty acids for major depression. ClinicalTrials.gov/show/NCT00256412 (Accessed 22/07/2014). [] ; Fiedorowicz JG, Hale N, Spector AA, Coryell WH. Neuroticism but not omega‐3 fatty acid levels correlate with early responsiveness to escitalopram. Annals of Clinical Psychiatry 2010;22(3):157‐63. [] [PMC free article] [PubMed] [Google Scholar]; 3000981
- Coryell WH. Essential fatty acids for major depression. ClinicalTrials.gov/show/NCT00256412 (Accessed 22/07/2014). [] ; Fiedorowicz JG, Hale N, Spector AA, Coryell WH. Neuroticism but not omega‐3 fatty acid levels correlate with early responsiveness to escitalopram. Annals of Clinical Psychiatry 2010;22(3):157‐63. [] [PMC free article] [PubMed] [Google Scholar]; 3000984
- Silva TM, Munhoz RP, Alvarez C, Naliwaiko K, Kiss A, Andreatini R, et al. Depression in Parkinson's disease: a double‐blind, randomized, placebo‐controlled pilot study of omega‐3 fatty‐acid supplementation. Journal of Affective Disorders 2008;111(2‐3):351‐9. [] [DOI] [PubMed] [Google Scholar]; 3000987
- Silva TM, Munhoz RP, Alvarez C, Naliwaiko K, Kiss A, Andreatini R, et al. Depression in Parkinson's disease: a double‐blind, randomized, placebo‐controlled pilot study of omega‐3 fatty‐acid supplementation. Journal of Affective Disorders 2008;111(2‐3):351‐9. [] [DOI] [PubMed] [Google Scholar]; 3000989
- Gertsik L. PUFA augmentation in treatment of major depression. ClinicalTrials.gov/show/NCT00067301 (Accessed 21 August 2014). [] ; Gertsik L, Poland RE, Bresee C, Rapaport MH. Omega‐3 fatty acid augmentation of citalopram treatment for patients with major depressive disorder. Journal of Clinical Psychopharmacology 2012;32(1):61‐4. [] [DOI] [PMC free article] [PubMed] [Google Scholar]; 3000991
- Dashti‐Khavidaki S, Gharekhani A, Khatami M‐R, Miri E‐S, Khalili H, Razeghi E, et al. Effects of omega‐3 fatty acids on depression and quality of life in maintenance hemodialysis patients. American Journal of Therapeutics 2014;21(4):275‐87. [] [DOI] [PubMed] [Google Scholar]; Gharekhani A, Khatami M‐R, Dashti‐Khavidaki S, Razeghi E, Noorbala A‐A, Hashemi‐Nazari S‐S, et al. The effect of omega‐3 fatty acids on depressive symptoms and inflammatory markers in maintenance hemodialysis patients: A randomized, placebo‐controlled clinical trial. European Journal of Clinical Pharmacology 2014;70(6):655‐65. [] [DOI] [PubMed] [Google Scholar]; Gharekhani A, Khatami MR, Dashti‐Khavidaki S, Razeghi E, Abdollahi A, Hashemi‐Nazari SS, et al. Potential effects of omega‐3 fatty acids on anemia and inflammatory markers in maintenance hemodialysis patients. DARU 2014;22(1):11. [] [DOI] [PMC free article] [PubMed] [Google Scholar]; Khatami MR. Evaluating therapeutic effects of omega‐3 fatty acids on anemia and nutritional status in hemodialysis patients with depression. www.irct.ir/searchresult.php?id=3043&number=5 (Accessed 25th June 2015). [] ; 3000994
- Gonzalez A, Mata S, Sanchez P, Gonzalez D, Urbina M, Fazzino F, et al. Omega‐3 fatty acids as adjunctive of antidepressant therapy and its effects on brain‐derived neurotrophic factor in serum, monocytes and lymphocytes. Archivos Venezolanos de Farmacologia y Terapeutica 2011;30(4):72‐8. [] [Google Scholar]; 3000999
- Grenyer BF, Crowe T, Meyer B, Owen AJ, Grigonis‐Deane EM, Caputi P, et al. Fish oil supplementation in the treatment of major depression: a randomised double‐blind placebo‐controlled trial. Progress in Neuro‐psychopharmacology & Biological Psychiatry 2007;31(7):1393‐6. [] [DOI] [PubMed] [Google Scholar]; Meyer BJ, Grenyer BFS, Crowe T, Owen AJ, Grigonis‐Deane EM, Howe PR. Improvement of major depression is associated with increased erythrocyte DHA. Lipids 2013;48(9):863‐868. [] [DOI] [PubMed] [Google Scholar]; Rotblatt M, Grenyer BFS. Fish oil not effective for chronic refractory depression. Focus on Alternative & Complementary Therapies 2008;13(1):30‐1. [] [Google Scholar]; 3001001
- Jazayeri S, Keshavarz SA, Tehrani‐Doost M, Djalali M, Hosseini M, Amini H, et al. Effects of eicosapentaenoic acid and fluoxetine on plasma cortisol, serum interleukin‐1beta and interleukin‐6 concentrations in patients with major depressive disorder. Psychiatry Research 2010;178(1):112‐5. [] [DOI] [PubMed] [Google Scholar]; Jazayeri S, Tehrani‐Doost M, Keshavarz SA, Hosseini M, Djazayery A, Amini H, et al. Comparison of therapeutic effects of omega‐3 fatty acid eicosapentaenoic acid and fluoxetine, separately and in combination, in major depressive disorder. Australian and New Zealand Journal of Psychiatry 2008;42(3):192‐8. [] [DOI] [PubMed] [Google Scholar]; 3001005
- Jazayeri S, Keshavarz SA, Tehrani‐Doost M, Djalali M, Hosseini M, Amini H, et al. Effects of eicosapentaenoic acid and fluoxetine on plasma cortisol, serum interleukin‐1beta and interleukin‐6 concentrations in patients with major depressive disorder. Psychiatry Research 2010;178(1):112‐5. [] [DOI] [PubMed] [Google Scholar]; Jazayeri S, Tehrani‐Doost M, Keshavarz SA, Hosseini M, Djazayery A, Amini H, et al. Comparison of therapeutic effects of omega‐3 fatty acid eicosapentaenoic acid and fluoxetine, separately and in combination, in major depressive disorder. Australian and New Zealand Journal of Psychiatry 2008;42(3):192‐8. [] [DOI] [PubMed] [Google Scholar]; 3001008
- Lespérance F. Double‐blind, placebo‐controlled, randomised trial of eicosapentaenoic acid (EPA) for major depression. www.controlled‐trials.com/ISRCTN47431149/ (Accessed 21 August 2014). [] ; Lespérance F, Frasure‐Smith N, St‐André E, Turecki G, Lesperance P, Wisniewski SR. The efficacy of omega‐3 supplementation for major depression: A randomized controlled trial [ISRCTN47431149]. Journal of Clinical Psychiatry 2011;72(8):1054‐62. [] [DOI] [PubMed] [Google Scholar]; 3001011
- Lucas M. Double‐blind, placebo‐controlled, randomised, clinical trial of eicosapentaenoic acid in the treatment of mood disorders among middle‐aged women. www.controlled‐trials.com/ISRCTN69617477 (Accessed 21 August 2014). [] ; Lucas M, Asselin G, Mérette C, Poulin MJ, Dodin S. Ethyl‐eicosapentaenoic acid for the treatment of psychological distress and depressive symptoms in middle‐aged women: a double‐blind, placebo‐controlled, randomized clinical trial. American Journal of Clinical Nutrition 2009;89(2):641‐51. [] [DOI] [PubMed] [Google Scholar]; 3001014
- Marangell LB, Martinez JM, Zboyan HA, Kertz B, Kim HF, Puryear LJ. A double‐blind, placebo‐controlled study of the omega‐3 fatty acid docosahexaenoic acid in the treatment of major depression. American Journal of Psychiatry 2003;160(5):996‐8. [] [DOI] [PubMed] [Google Scholar]; Marangell LB, Zboyan HA, Cress KK, Benisek D, Arterburn L. A double‐blind, placebo‐controlled study of docosahexaenoic acid (DHA) in the treatment of depression. 4th Congress of the International Society for the Study of Lipids and Fatty Acids, Tsukuba, Japan. 2000:105. [] [Google Scholar]; 3001017
- Mischoulon D. Omega‐3 fatty acids for treatment of major depression: differential effects of EPA and DHA, and associated biochemical and immune parameters. ClinicalTrials.gov/ct2/show/NCT00361374 (Accessed 25th June 2015). [] ; Mischoulon D, Nierenberg AA, Schettler PJ, Kinkead B, Fehling K, Martinson M, et al. Clinical predictors of response to omega‐3 fatty acids in major depressive disorder [abstract]. Biological Psychiatry [abstracts of the 70th Annual Scientific Convention and Meeting of the Society of Biological Psychiatry, SOBP; 2015 May 14 ‐ 16;Toronto, ON Canada]. 2015:147S. [] [Google Scholar]; Mischoulon D, Nierenberg AA, Schettler PJ, Kinkead BL, Fehling K, Martinson MA, et al. A double‐blind, randomized controlled clinical trial comparing eicosapentaenoic acid versus docosahexaenoic acid for depression. Journal of Clinical Psychiatry 2015;76(1):54‐61. [] [DOI] [PMC free article] [PubMed] [Google Scholar]; Rapaport MH. Inflammation as a predictive biomarker for response to omega‐3 fatty acids in major depressive disorder [abstract]. Biological Psychiatry [abstracts of the 70th Annual Scientific Convention and Meeting of the Society of Biological Psychiatry, SOBP; 2015 May 14 ‐ 16; Toronto, ON Canada]. 2015:146S‐7S. [] [Google Scholar]; Rapaport MH, Mischoulon D. Omega‐3 fatty acids for treatment of major depression: differential effects of EPA and DHA, and associated biochemical and immune parameters. ClinicalTrials.gov/ct2/show/NCT00517036 (Accessed 25th June 2015). [] ; Rapaport MH, Nierenberg AA, Schettler PJ, Kinkead B, Cardoos A, Walker R, et al. Inflammation as a predictive biomarker for response to omega‐3 fatty acids in major depressive disorder: a proof‐of‐concept study. Molecular Psychiatry 2015 Mar 24 [Epub ahead of print]. [] [DOI] [PMC free article] [PubMed]; Rapaport MH, Schettler P, Pace TW, Kinkead B, Nierenberg AA, Mischoulon D. Inflammation, depression and N‐3 fatty acids: A case of personalized medicine abstract. Neuropsychopharmacology [abstracts of the 52nd Annual Meeting of the American College of Neuropsychopharmacology, ACNP; 2013 Dec 8 ‐ 12; Hollywood, FL United States]. 2013:S353‐S4. [] [Google Scholar]; 3001020
- Mischoulon D. Omega‐3 fatty acids for treatment of major depression: differential effects of EPA and DHA, and associated biochemical and immune parameters. ClinicalTrials.gov/ct2/show/NCT00361374 (Accessed 25th June 2015). [] ; Mischoulon D, Nierenberg AA, Schettler PJ, Kinkead B, Fehling K, Martinson M, et al. Clinical predictors of response to omega‐3 fatty acids in major depressive disorder [abstract]. Biological Psychiatry [abstracts of the 70th Annual Scientific Convention and Meeting of the Society of Biological Psychiatry, SOBP; 2015 May 14 ‐ 16;Toronto, ON Canada]. 2015:147S. [] [Google Scholar]; Mischoulon D, Nierenberg AA, Schettler PJ, Kinkead BL, Fehling K, Martinson MA, et al. A double‐blind, randomized controlled clinical trial comparing eicosapentaenoic acid versus docosahexaenoic acid for depression. Journal of Clinical Psychiatry 2015;76(1):54‐61. [] [DOI] [PMC free article] [PubMed] [Google Scholar]; Rapaport MH. Inflammation as a predictive biomarker for response to omega‐3 fatty acids in major depressive disorder [abstract]. Biological Psychiatry [abstracts of the 70th Annual Scientific Convention and Meeting of the Society of Biological Psychiatry, SOBP; 2015 May 14 ‐ 16; Toronto, ON Canada]. 2015:146S‐7S. [] [Google Scholar]; Rapaport MH, Mischoulon D. Omega‐3 fatty acids for treatment of major depression: differential effects of EPA and DHA, and associated biochemical and immune parameters. ClinicalTrials.gov/ct2/show/NCT00517036 (Accessed 25th June 2015). [] ; Rapaport MH, Nierenberg AA, Schettler PJ, Kinkead B, Cardoos A, Walker R, et al. Inflammation as a predictive biomarker for response to omega‐3 fatty acids in major depressive disorder: a proof‐of‐concept study. Molecular Psychiatry 2015 Mar 24 [Epub ahead of print]. [] [DOI] [PMC free article] [PubMed]; Rapaport MH, Schettler P, Pace TW, Kinkead B, Nierenberg AA, Mischoulon D. Inflammation, depression and N‐3 fatty acids: a case of personalized medicine abstract. Neuropsychopharmacology [abstracts of the 52nd Annual Meeting of the American College of Neuropsychopharmacology, ACNP; 2013 Dec 8 ‐ 12; Hollywood, FL United States]. 2013:S353‐S4. [] [Google Scholar]; 3001028
- Anonymous. Omega‐3 fatty acid effective as monotherapy for MDD. Brown University Psychopharmacology Update 2009;20(12):3‐4. [] [Google Scholar]; Mischoulon D. Ethyl eicosapentanoic acid (Ethyl‐EPA) for treating major depression. ClinicalTrials.gov/show/NCT00096798 (Accessed 22nd July 2014). [] ; Mischoulon D, Papakostas GI, Dording CM, Farabaugh AH, Sonawalla SB, Agoston AM, et al. A double‐blind, randomized controlled trial of ethyl‐eicosapentaenoate for major depressive disorder [NCT00096798]. Journal of Clinical Psychiatry 2009;70(12):1636‐44. [] [DOI] [PMC free article] [PubMed] [Google Scholar]; 3001036
- Nemets B, Stahl Z, Belmaker RH. Addition of omega‐3 fatty acid to maintenance medication treatment for recurrent unipolar depressive disorder. American Journal of Psychiatry 2002;159(3):477‐9. [] [DOI] [PubMed] [Google Scholar]; Nemets B, Stahl Z, Belmaker RH. Omega‐3 fatty acid treatment of depressive breakthrough during unipolar maintenance. International Journal of Neuropsychopharmacology 2002;5(Suppl 1):149. [] [Google Scholar]; Stahl Z, Nemets B, Belmaker RH. Omega‐3 fatty acid treatment of depressive breakthrough during unipolar maintenance. European Neuropsychopharmacology 2002;12(Suppl 3):S178. [] [Google Scholar]; 3001040
- Park Y. Omega‐3 index as a risk factor for depression in Korean and effect of Omega‐3 fatty acids on Korean patients with depression [KCT0001206]. cris.nih.go.kr/cris/en/search/search_result_st01.jsp?seq=4315 (Accessed 25th June 2015). [] ; Park Y, Park Y‐S, Kim SH, Oh DH, Park Y‐C. Supplementation of n‐3 polyunsaturated fatty acids for major depressive disorder: A randomized, double‐blind, 12‐week, placebo‐controlled trial in Korea. Annals of Nutrition & Metabolism 2015;66(2‐3):141‐148. [] [DOI] [PubMed] [Google Scholar]; 3001044
- Peet M, Horrobin DF. A dose‐ranging study of the effects of ethyl‐eicosapentaenoate in patients with ongoing depression despite apparently adequate treatment with standard drugs. Archives of General Psychiatry 2002;59(10):913‐9. [] [DOI] [PubMed] [Google Scholar]; 3001047
- Peet M, Horrobin DF. A dose‐ranging study of the effects of ethyl‐eicosapentaenoate in patients with ongoing depression despite apparently adequate treatment with standard drugs.. Archives of General Psychiatry 2002;59(10):913‐9. [] [DOI] [PubMed] [Google Scholar]; 3001049
- Peet M, Horrobin DF. A dose‐ranging study of the effects of ethyl‐eicosapentaenoate in patients with ongoing depression despite apparently adequate treatment with standard drugs.. Archives of General Psychiatry 2002;59(10):913‐9. [] [DOI] [PubMed] [Google Scholar]; 3001051
- Rizzo AM, Corsetto PA, Montorfano G, Opizzi A, Faliva M, Giacosa A, et al. Comparison between the AA/EPA ratio in depressed and non depressed elderly females: Omega‐3 fatty acid supplementation correlates with improved symptoms but does not change immunological parameters. Nutrition Journal 2012;11:82. [] [DOI] [PMC free article] [PubMed] [Google Scholar]; Rondanelli M, Giacosa A, Opizzi A, Pelucchi C, Vecchia C, Montorfano G, et al. Effect of omega‐3 fatty acids supplementation on depressive symptoms and on health‐related quality of life in the treatment of elderly women with depression: A double‐blind, placebo‐controlled, randomized clinical trial. Journal of the American College of Nutrition 2010;29(1):55‐64. [] [DOI] [PubMed] [Google Scholar]; Rondanelli M, Giacosa A, Opizzi A, Pelucchi C, Vecchia C, Montorfano G, et al. Long chain omega 3 polyunsaturated fatty acids supplementation in the treatment of elderly depression: effects on depressive symptoms, on phospholipids fatty acids profile and on health‐related quality of life. Journal of Nutrition, Health & Aging 2011;15(1):37‐44. [] [DOI] [PubMed] [Google Scholar]; 3001053
- Anonymous. Adjunctive fish oil for depression. Nurses' Drug Alert 2005;29(3):22‐3. [] [Google Scholar]; Silvers KM, Woolley CC, Hamilton FC, Watts PM, Watson RA. Randomised double‐blind placebo‐controlled trial of fish oil in the treatment of depression. Prostaglandins, Leukotrienes, and Essential Fatty Acids 2005;72(3):211‐8. [] [DOI] [PubMed] [Google Scholar]; 3001057
- Su KP, Huang SY, Chiu CC, Shen WW. Corrigendum to "Omega‐3 fatty acids in major depressive disorder A preliminary double‐blind, placebo‐controlled trial" [Eur Neuropsychopharmacol. 13 (2003) 267‐271]. European Neuropsychopharmacology 2004;14(2):173. [] [DOI] [PubMed] [Google Scholar]; Su KP, Huang SY, Chiu CC, Shen WW. Omega‐3 fatty acids in major depressive disorder. A preliminary double‐blind, placebo‐controlled trial. European Neuropsychopharmacology 2003;13(4):267‐71. [] [DOI] [PubMed] [Google Scholar]; 3001060
References to studies excluded from this review
- Clayton A. Study of supplementation of antidepressants with fish oil to improve time to clinical response (SADFAT). ClinicalTrials.gov/show/NCT00963196 (Accessed 11 September 2014). [] ; 3001063
References to studies awaiting assessment
- EUCTR2006‐004949‐41‐IT. Randomized double‐blind study to evaluate the adjuvant effect of polyunsaturated fatty acids omega‐3 in therapy with SSRI paroxetine mesylate in unipolar mood depression and recurrent depression. www.ClinicalTrialsregister.eu/ctr‐search/trial/2006‐004949‐41/IT (Accessed 22nd July 2014). [] ; 3001065
- Kwak KP, Lee KH. A comparative study of addition therapy of choline alfoscerate and omega 3 fatty acid in older depressed patients with or without executive dysfunction. International Psychogeriatrics 2014;25:S75‐192. [] [Google Scholar]; 3001067
- Lima L. A randomised study on the antidepressive effect of fluoxetine and folic acid, as possible augmenter, and the SYNThesis of serotonin (5‐HT) in lymphocytes prior and after treatment. isrctn.org/ISRCTN06123818 (Accessed 26th June 2015). [] ; 3001069
- Murck H. A multicentre, double‐blind, randomised, parallel group, placebo‐controlled trial of Lax‐101 (ethyl eicosapentaenoate) as adjunct therapy in patients who remain depressed following treatment with standard antidepressant therapy. National Research Register2004. [] ; 3001071
- Naqvi S. Treating adolescent depression with fish oils. ClinicalTrials.gov/show/NCT00658476 (Accessed 26th June 2015). [] ; 3001073
- NCT00816322. The effect of fish oil in major depressive disorder. ClinicalTrials.gov/show/NCT00816322 (Accessed 22nd July 2014). [] ; 3001075
- Rees AM, Parker GB, Owen C. A study of omega‐3 as a treatment for major depression. ClinicalTrials.gov/show/NCT00238758 (Accessed 26th June 2015). [] ; Rees AM, Parker GB, Owen CA. Omega‐3 polyunsaturated fatty acids as a treatment for major depression. Australian and New Zealand Journal of Psychiatry 2005;39 Suppl 2:A52. [] [Google Scholar]; 3001077
- Shinto L. Fish oil for the treatment of depression in patients with multiple sclerosis. ClinicalTrials.gov/show/NCT00122954 (Accessed 25th June 2015). [] ; 3001080
- Su KP. The effect of fish oil in major depressive disorder. ClinicalTrials.gov/show/NCT01371383 (Accessed 26th June 2015). [] ; 3001082
References to ongoing studies
- Amminger GP, Rice S. Youth Depression Alleviation: a randomised controlled trial of omega‐3 fatty acids (fish oil) for major depressive disorder in young people (YoDA‐F). www.anzctr.org.au/ACTRN12613001352796.aspx (Accessed 26th June 2015). [] ; Rice SM, Hickie IB, Yung AR, Mackinnon A, Berk M, Davey C, et al. Youth depression alleviation: the Fish Oil Youth Depression Study (YoDA‐F): A randomized, double‐blind, placebo‐controlled treatment trial. Early Intervention in Psychiatry 13 Aug 2014 [Epub ahead of print]. [] [DOI] [PubMed]; 3001084
- Carney RM. Omega‐3 for depression and other cardiac risk factors ‐ 2. ClinicalTrials.gov/show/NCT02021669 (Accessed 26th June 2015). [] ; 3001087
- Gabbay V. Omega‐3 fatty acids in adolescent depression. ClinicalTrials.gov/show/NCT00312897 (Accessed 26th June 2015). [] ; Gabbay V. The role of omega‐3 fatty acids In adolescent depression. ClinicalTrials.gov/show/NCT00962598 (Accessed 26th June 2015). [] ; 3001089
- Howe P. Fish oil treatment for depression in cardiovascular disease. www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=82950 ([Accessed 26th June 2015). [] ; 3001092
- Jiang W. Omega 3 for treatment of depression in patients with heart failure (OCEAN). ClinicalTrials.gov/show/NCT02057406 (Accessed 26th June 2015). [] ; 3001094
- Kamath J. Omega 3 FA supplements as augmentation in the treatment of depression. ClinicalTrials.gov/show/NCT01803711 (Accessed 26th June 2015). [] ; 3001096
- Khalili H. Comparing efficacy of omega‐3 and placebo in reducing Beck depression score in HIV/AIDS patients. www.irct.ir/searchresult.php?id=3449&number=17 (Accessed 25th June 2015). [; IRCT201410073449N17] ; 3001098
- Lanctôt K, Hermann N. Treating depression in coronary artery disease with omega‐3 fatty acids. ClinicalTrials.gov/show/NCT00981383 (Accessed 26th June 2015). [] ; 3001100
- Mostafavi S‐A, Akhondzadeh S. Evaluating effects of omega‐3 supplementation on weight and depression among overweight or obese women with depression compared to placebo. www.irct.ir/searchresult.php?id=16465&number=3 (Accessed 25th June 2015). [; IRCT2014053016465N3] ; 3001102
- Nakano W. Augmentation of omega‐3 fatty acid with antidepressants for major depressive disorder: a double‐blind, randomized controlled trial. apps.who.int/trialsearch/Trial.aspx?TrialID=JPRN‐UMIN000013525 (Accessed 26th June 2015). [] ; 3001104
- Parker GB. A study of omega‐3 as an augmentor of antidepressant treatment for major depression. ClinicalTrials.gov/show/NCT00289484 (Accessed 26th June 2015). [] ; 3001106
- Parletta N. Effects of a Mediterranean‐style diet and fish oil supplements on mood and health. www.anzctr.org.au/ACTRN12614000438651.aspx (Accessed 26th June 2015). [] ; 3001108
- Piperoglou M. Adjunctive natural low dose docosahexaenoic acid (DHA) omega‐3 in a 16 week random double‐blind placebo controlled (RDBPC) cross‐over withdrawal study in a group of chronic, psychiatric out‐patients with anxiety and mood disorders. www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=365804 (Accessed 26th June 2015). [] ; 3001110
- Smith D. An 8 week randomised, double‐blind, placebo controlled trial investigating the role of adjunctive bioactive lipids specifically; docosahexaenoic acid (DHA) versus eicosapentaenoic acid (EPA) in major depressive disorder ‐ with a 6 week open label extension of DHA in patients aged 18 ‐ 65 years. www.anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12610000710022 (Accessed 26th June 2015). [] ; 3001112
- Tayama J. Omega‐3 polyunsaturated fatty acids and psychological interventions for workers with mild to moderate depression: a randomized controlled trial. www.umin.ac.jp/ctr/index.htm (Accessed 26th June 2015). [] [DOI] [PubMed]; 3001114
- Yao JK. Coronary artery disease (CAD) risk in schizophrenia: effect of omega‐3 fatty acid supplementation. ClinicalTrials.gov/show/NCT00167310 (Accessed 26th June 2015). [] ; 3001116
Additional references
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th Edition. Washington, DC: American Psychiatric Association, 2013. [Google Scholar]
- Appleton KM, Hayward RC, Gunnell D, Peters TJ, Rogers PJ, Kessler, D, et al. Effects of n3 long chain polyunsaturated fatty acids on depressed mood: systematic review of published trials. American Journal of Clinical Nutrition 2006;84(6):1308‐16. [DOI] [PubMed] [Google Scholar]
- Appleton KM, Peters TJ, Hayward RC, Heatherley SV, McNaughton SA, Rogers PJ, et al. Depressed mood and n‐3 polyunsaturated fatty acid intake from fish: non‐linear or confounded association?. Social Psychiatry and Psychiatric Epidemiology 2007;42(2):100‐4. [DOI] [PubMed] [Google Scholar]
- Appleton KM, Gunnell D, Peters TJ, Ness AR, Kessler D, Rogers PJ. No clear evidence of an associations between plasma concentrations of n‐3 long chain polyunsaturated fatty acids and depressed mood in a non‐clinical population. Prostaglandins, Leukotrienes and Essential Fatty Acids 2008;78:337‐42. [DOI] [PubMed] [Google Scholar]
- Appleton KM, Rogers PJ, Ness AR. Is there a role for n‐3 long‐chain polyunsaturated fatty acids in the regulation of mood and behaviour? A review of the evidence to date from epidemiological studies, clinical studies and intervention trials. Nutrition Research Reviews 2008;21(1):13‐41. [DOI] [PubMed] [Google Scholar]
- Appleton KM, Rogers PJ, Ness AR. Updated systematic review and meta‐analysis of the effects of n‐3 long‐chain polyunsaturated fatty acids on depressed mood. American Journal of Clinical Nutrition 2010;91(3):757‐70. [DOI] [PubMed] [Google Scholar]
- Beck AT, Steer RA. Beck Depression Inventory Manual. San Antonio, Texas: Psychological Corporation, 1987. [Google Scholar]
- Bloch MH, Hannestad J. Omega‐3 fatty acids for the treatment of depression: Systematic review and meta‐analysis. Molecular Psychiatry 2012;17(12):1272‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- British Nutrition Foundation. BNF Briefing paper: n‐3 fatty acids and health. London: British Nutrition Foundation, 1999. [Google Scholar]
- Browne JC, Scott KM, Silvers KM. Fish consumption in pregnancy and omega‐3 status after birth are not associated with postnatal depression. Journal of Affective Disorders 2006;90(2‐3):131‐9. [DOI] [PubMed] [Google Scholar]
- Calder PC. n‐3 polyunsaturated fatty acids and inflammation: from molecular biology to the clinic. Lipids 2003;38(4):342‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey GE, Mantzioris E, Gibson RA, Cleland LG, James MJ. The effect on human tumor necrosis factor alpha and interleukin 1B production of diets enriched in n‐3 fatty acids from vegetable oil or fish oil. American Journal of Clinical Nutrition 1996;63:116‐22. [DOI] [PubMed] [Google Scholar]
- Chalon S. Omega‐3 fatty acids and monoamine neurotransmission. Prostaglandins, Leukotrienes, and Essential Fatty Acids 2006;75(4‐5):259‐69. [DOI] [PubMed] [Google Scholar]
- Cho MJ, Kim MH. Use of the Center for Epidemiological Studies Depression (CES‐D) scale in Korea. J Nerv Ment Dis 1998;186:304‐10. [DOI] [PubMed] [Google Scholar]
- Presa Owens S, Innis SM. Docosahexaenoic and arachidonic acid prevent a decrease in dopaminergic and serotoninergic neurotransmitters in frontal cortex caused by a linoleic and alpha‐linolenic acid deficient diet in formula fed piglets. Journal of Nutrition 1999;129(11):2088‐93. [DOI] [PubMed] [Google Scholar]
- Deeks JJ, Altman DG, Bradbrun MJ. Statistical methods for examining heterogeneity and combining results from several studies in meta‐analysis. In: Egger M, Davey Smith G, Altman DG editor(s). Systematic Reviews in Health Care: Meta‐analysis in Context. London: BMJ Publishing Group, 2001:285‐312. [Google Scholar]
- Delion S, Chalon S, Hérault J, Guilloteau D, Besnard JC, Durand G. Chronic dietary alpha‐linolenic acid deficiency alters dopaminergic and serotonergic neurotransmission in rats. Journal of Nutrition 1994;124(12):2466‐76. [DOI] [PubMed] [Google Scholar]
- Delion S, Chalon S, Guilloteau D, Besnard JC, Durand G. Alpha‐linolenic acid dietary deficiency alters age related changes in dopaminergic and serotonergic neurotransmission in the rat frontal cortex. Journal of Neurochemistry 1996;66(4):1582‐91. [DOI] [PubMed] [Google Scholar]
- Dennis CL, Dowswell T. Interventions (other than pharmacological, psychosocial or psychological) for treating antenatal depression. Cochrane Database of Systematic Reviews 2013, Issue 7. [DOI: 10.1002/14651858.CD006795.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diguer L, Barber J, Luborsky L. Three concommitants: personality disorders, psychiatric severity, and outcome of dynamic psychotherapy of major depression. American Journal of Psychiatry 1993;150(8):1246‐8. [DOI] [PubMed] [Google Scholar]
- Dupuy H. The psychological general well‐being (PGWB) index. In: Wenger NK, Mattson ME, Furber CD, et al. editor(s). Assessment of Quality of Life in Clinical Trials of Cardiovascular Therapies. New York, US: Le Jaq Publishing Inc., 1984:170‐83. [Google Scholar]
- Edwards R, Peet M, Shay J, Horrobin D. Depletion of docosahexaenoic acid in red blood cell membranes of depressive patients. Biochemical Society Transactions 1998;26:s142. [DOI] [PubMed] [Google Scholar]
- Egger M, Davey Smith G. Principles of and procedures for systematic reviews. In: Egger M, Davey Smith G, Altman DG editor(s). Systematic Reviews in Health Care: Meta‐analysis in Context. London: BMJ Publishing Group, 2001:23‐42. [Google Scholar]
- Ehringer W, Belcher D, Wassall SR, Stillwell W. A comparison of the effects of linolenic acid (18:3 omega 3) and docosahexaenoic (22:6 omega 3) acids on phospholipid bilayers. Chemistry and Physics of Lipids 1990;54(2):79‐88. [DOI] [PubMed] [Google Scholar]
- Endicott J, Nee J, Harrison W, Blumenthal R. Quality of Life Enjoyment and Satisfaction Questionnaire: a new measure. Psychopharmacology Bulletin 1993;29(2):321‐6. [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. "Min‐mental state": a practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research 1975;12(3):189‐98. [DOI] [PubMed] [Google Scholar]
- Fountoulakis KN, Veroniki AA, Siamouli M, et al. No role for initial severity on the efficacy of antidepressants: results of a multi‐meta‐analysis. Annals of General Psychiatry 2013;12(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier JC, DeRubeis RJ, Hollon SD, Dimidjian S, Amsterdam JD, Shelton RC, et al. Antidepressant drug effects and depression severity: a patient‐level meta‐analysis. JAMA 2010;303(1):47‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangou S, Lewis M, McCrone P. Efficacy of ethyl‐eicosapentaenoic acid in bipolar depression: randomised double‐blind placebo‐controlled study. British Journal of Psychiatry 2006;188:46‐50. [DOI] [PubMed] [Google Scholar]
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(1):7‐10. [DOI] [PubMed] [Google Scholar]
- Garland MR, Hallahan B, McNamara M, Carney PA, Grimes H, Hibbeln JR, et al. Lipids and essential fatty acids in patients presenting with self‐harm. British Journal of Psychiatry 2007;190:112‐7. [DOI] [PubMed] [Google Scholar]
- Gregory J, Foster K, Tyler H, Wiseman M. National Diet and Nutritional Survey of British Adults. London: Her Majesty's Stationary Office, 2000. [Google Scholar]
- Grosso G, Pajak A, Marventano S, Castellano S, Galvano F, Bucolo C, et al. Role of omega‐3 fatty acids in the treatment of depressive disorders: a comprehensive meta‐analysis of randomized clinical trials. PloS One 2014;9(5):e96905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy W. Clinical Global Impressions. ECDEU Assessment Manual for Psychopharmacology, revised. Rockville, MD: National Institute of Mental Health, 1976. [Google Scholar]
- Haag M. Essential fatty acids and the brain. Canadian Journal of Psychiatry 2003;48(3):195‐203. [DOI] [PubMed] [Google Scholar]
- Hakkarainen R, Partonen T, Haukka J, Virtamo J, Albanes D, Lönnqvist J. Is dietary intake of omega‐3 fatty acids associated with depression?. American Journal of Psychiatry 2004;161(3):567‐9. [DOI] [PubMed] [Google Scholar]
- Hamazaki K, Itomura M, Huan M, Nishizawa H. Effects of w‐3 fatty acid‐containing phospholipids on blood catecholamine concentrations in healthy volunteers: a randomized, placebo controlled, double blind trial. Nutrition 2005;21:705‐10. [DOI] [PubMed] [Google Scholar]
- Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbeln JR. Fish consumption and major depression. Lancet 1998;351(9110):1213. [DOI] [PubMed] [Google Scholar]
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21(11):1539‐58. [DOI] [PubMed] [Google Scholar]
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. 5.1. Chichester, UK: The Cochrane Collaboration & John Wiley & Sons, Ltd.
- Hirashima F, Parow AM, Stoll AL, Demopulos CM, Damico KE, Rohan ML, et al. Omega‐3 fatty acid treatment and T2 whole brain relaxation times in bipolar disorder. American Journal of Psychiatry 2004;161(10):1922‐4. [DOI] [PubMed] [Google Scholar]
- Hoehn M, Yahr M. Parkinsonism: onset, progression and mortality. Neurology 1967;17(5):427‐42. [DOI] [PubMed] [Google Scholar]
- James MJ, Gibson RA, Cleland LG. Dietary polyunsaturated fatty acids and inflammatory mediator production. American Journal of Clinical Nutrition 2000;71(1 Suppl):343s‐8s. [DOI] [PubMed] [Google Scholar]
- Keck PE Jr, Mintz J, McElroy SL, Freeman MP, Suppes T, Frye MA, et al. Double‐blind, randomized, placebo‐controlled trials of ethyl‐eicosapentanoate in the treatment of bipolar depression and rapid cycling bipolar disorder. Biological Psychiatry 2006;60(9):1020‐2. [DOI] [PubMed] [Google Scholar]
- Kirsch I, Deacon BJ, Huedo‐Medina TB, Scoboria A, Moore TJ, Johnson BT. Initial severity and antidepressant benefits: a meta‐analysis of data submitted to the Food and Drug Administration. PLoS Medicine 2008;5(2):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PY, Su KP. A meta‐analytic review of double‐blind, placebo‐controlled trials of anti‐depressant efficacy of omega‐3 fatty acids. Journal of Clinical Psychiatry 2007;68(7):1056‐61. [DOI] [PubMed] [Google Scholar]
- Lin P‐Y, Mischoulon D, Freeman MP, et al. Are omega‐3 fatty acids anti‐depressants or just mood‐improving agents?. Mol Psychiatry 2012;17:1161‐1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde K, Berner MM, Kriston L. St John's wort for major depression. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD000448.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Folsom AR, Eckfeldt JH, Lewis L, Chambless LE. Short‐ and long‐term repeatability of fatty acid composition of human plasma phospholipids and cholesterol esters: The Atherosclerosis Risk in Communities (ARIC) Study Investigators. American Journal of Clinical Nutrition 1995;62(3):572‐8. [DOI] [PubMed] [Google Scholar]
- Mamalakis G, Tornaritis M, Kafatos A. Depression and adipose essential polyunsaturated fatty acids. Prostaglandins, Leukotrienes and Essential Fatty Acids 2002;67(5):311‐8. [DOI] [PubMed] [Google Scholar]
- Mamalakis G, Kiriakakis M, Tsibinos G, Kafatos A. Depression and adipose polyunsaturated fatty acids in an adolescent group. Prostaglandins, Leukotriens and Essential Fatty Acids 2004;71(5):289‐94. [DOI] [PubMed] [Google Scholar]
- Mamalakis G, Kiriakakis M, Tsibinos G, Hatzis C, Flouri S, Mantzoros C, et al. Depression and serum adiponectin and adipose omega‐3 and omega‐6 fatty acids in adolescents. Pharmacology, Biochemistry and Behavior 2006;85(2):474‐9. [DOI] [PubMed] [Google Scholar]
- Martins JG. EPA but not DHA appears to be responsible for the efficacy of omega‐3 LC‐PUFA supplementation in depression: Evidence from an updated meta‐analysis of randomized controlled trials. OCL ‐ Oleagineux Corps Gras Lipides 2011;18:188‐98. [DOI] [PubMed] [Google Scholar]
- McNamara RK, Richtand NM, Levant B. Omega‐3 fatty acid deficiency decreases dopamine D2 receptor binding and increases serotonin 5‐HT2A receptor binding in the adult rat prefrontal cortex. Biological Psychiatry 2006;59:146S. [Google Scholar]
- Miller BJ, Murray L, Beckmann MM, Kent T, Macfarlane B. Dietary supplements for preventing postnatal depression. Cochrane Database of Systematic Reviews 2013, Issue 10. [DOI: 10.1002/14651858.CD009104.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Y, Sasaki A, Yokoyama T, Tanaka K, Ohya Y, Fukushima W. Risk of postpartum depression in relation to dietary fish and fat intake in Japan: the Osaka Maternal and Child Health Study. Psychological Medicine 2006;36(12):1727‐37. [DOI] [PubMed] [Google Scholar]
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382‐9. [DOI] [PubMed] [Google Scholar]
- National Institute for Clinical Excellence. Depression: Management of Depression in Primary and Secondary Care. London, England: National Institute for Clinical Excellence, 2004. [Google Scholar]
- Noaghiul S, Hibbeln JR. Cross‐national comparisons of seafood consumption and rates of bipolar disorders. American Journal of Psychiatry 2003;160(12):2222‐7. [DOI] [PubMed] [Google Scholar]
- Parker G, Gibson NA, Brotchie H, Heruc G, Rees AM, Hadzi‐Pavlovic D. Omega‐3 fatty acids and mood disorders. American Journal of Psychiatry 2006;163(6):969‐78. [DOI] [PubMed] [Google Scholar]
- Peet M, Murphy B, Shay J, Horrobin D. Depletion of omega‐3 fatty acid levels in red blood cell membranes of depressive patients. Biological Psychiatry 1998;43(5):315‐9. [DOI] [PubMed] [Google Scholar]
- Peet M. International variations in the outcome of schizophrenia and the prevalence of depression in relation to national dietary practices: an ecological analysis. British Journal of Psychiatry 2004;184:404‐8. [DOI] [PubMed] [Google Scholar]
- Rallidis LS, Paschos G, Liakos GK, Velissaridou AH, Anastasiadis G, Zampelas A. Dietary alpha‐linolenic acid decreases C‐reactive protein, serum amyloid A and interleukin‐6 in dyslipidaemic patients. Atherosclerosis 2003;167(2):237‐42. [DOI] [PubMed] [Google Scholar]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
- Reynolds WM, Kobak KA. Hamilton Depression Inventory. Psychological Assessment Resources. Odessa, FL: Psychological Assessment Resources, 1995. [Google Scholar]
- Rogers PJ, Appleton KM, Kessler D, Peters TJ, Gunnell D, Hayward RC, et al. No effect of n‐3 long chain polyunsaturated fatty acid (EPA and DHA) supplementation on depressed mood and cognitive function: a randomized controlled trial. British Journal of Nutrition 2008;99(2):421‐31. [DOI] [PubMed] [Google Scholar]
- Ross BM, Seguin J, Sieswerda LE. Omega‐3 fatty acids as treatments for mental illness: which disorder and which fatty acid?. Lipids in Health and Disease 2007;6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruxton CHS, Calder PC, Reed SC, Simpson MJA. The impact of long chain n‐3 polyunsaturated fatty acids on human health. Nutrition Research Reviews 2005;18(1):113‐29. [DOI] [PubMed] [Google Scholar]
- Ryff CD, Keyes CL. The structure of psychological well‐being revisited. J Pers Soc Psychol 1995;69:719‐27. [DOI] [PubMed] [Google Scholar]
- Sawazaki S, Hamazaki T, Yazawa K, Kobayashi M. The effect of docosahexaenoic acid on plasma catecholamine concentrations and glucose tolerance during long‐lasting psychological stress: a double blind placebo controlled study. Journal of Nutritional Science and Vitaminology 1999;45(5):655‐65. [DOI] [PubMed] [Google Scholar]
- Silvers KM, Scott KM. Fish consumption and self‐reported physical and mental health status. Public Health Nutrition 2002;5(3):427‐31. [DOI] [PubMed] [Google Scholar]
- Simopoulos AP. Evoluntionary aspects of omega‐3 fatty acids in the food supply. Prostaglandins, Leukotrienes and Essential Fatty Acids 1999;60(5‐6):421‐9. [DOI] [PubMed] [Google Scholar]
- Smith MA, Beilin LJ, Mori TA, Oddy WH. Essential fatty acids and mood: a systematic review of observational studies. American Journal of Food and Nutrition 2011;1:14‐27. [Google Scholar]
- Stahl LA, Begg DP, Weisinger RS, Sinclair AJ. The role of omega‐3 fatty acids in mood disorders. Current Opinion in Investigational Drugs 2008;9(1):57‐64. [PubMed] [Google Scholar]
- Sterne JAC, Egger M, Davey Smith G. Investigating and dealing with publication and other biases. In: Egger M, Davey Smith G, Altman DG, eds editor(s). Systematic Reviews in Health Care: Meta‐analysis in Context. London: BMJ Publishing Group, 2001:189‐208. [Google Scholar]
- Stoll AL, Severus WE, Freeman MP, Rueter S, Zboyan HA, Diamond E, et al. Omega‐3 fatty acids in bipolar disorder – a preliminary double‐blind, placebo‐controlled trial. Archives of General Psychiatry 1999;56(5):407‐12. [DOI] [PubMed] [Google Scholar]
- Sublette ME, Ellis SP, Geant AL, Mann JJ. Meta‐analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. Journal of Clinical Psychiatry Diseases of the Nervous System 2011;72(12):1577‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Fukumoto Y, Harada E. Influence of a dietary n‐3 fatty acid deficiency on the cerebral catecholamine contents, EEG and learning ability in rat. Behavioral Brain Research 2002;131(1‐2):193‐203. [DOI] [PubMed] [Google Scholar]
- Tanskanen A, Hibblen JR, Hintikka J, Haatainen K, Honkalampi K, Viinamäki K. Fish consumption, depression, and suicidality in a general population. Archives of General Psychiatry 2001;58(5):512‐3. [DOI] [PubMed] [Google Scholar]
- Tappia PS, Ladha S, Clark DC, Grimble RF. The influence of membrane fluidity, TNF receptor binding, cAMP production and GTPase activity on macrophage cytokine production in rats fed a variety of fat diets. Molecular and Cellular Biochemistry 1997;166(1‐2):135‐43. [DOI] [PubMed] [Google Scholar]
- Taylor T, Jefferis GS, Koop M, Hill BC, Hastie T, Heit G, et al. Quantitative measurements of alternating finger tapping in Parkinson's disease correlate with UPDRS motor disability and reveal the improvement in fine motor control from medication and deep brain stimulation. Movement Disorders 2005;20:01286‐98. [DOI] [PubMed] [Google Scholar]
- Trivedi MH, Rush AJ, Ibrahim HM, Carmody TJ, Biggs MM, Suppes T, et al. The Inventory of Depressive Symptomology, clinician rating (IDS‐C) and self report (IDS‐SR), and the Quick Inventory of Depressive Symptomology, clinician rating (QIDS‐C) and self‐report (QIDS‐SR) in public sector patients with mood disorders; a psychometric evaluation.. Psychological Medical 2004;34(1):73‐82. [DOI] [PubMed] [Google Scholar]
- Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its influence on apparent efficacy. New England Journal of Medicine 2008;358(3):252‐60. [DOI] [PubMed] [Google Scholar]
- Ware JE, Snow KK, Kosinski M, Gandek B. SF‐36 Health Survey: Manual and Interpretation Guide. Boston MA: The Health Institute, 1993. [Google Scholar]
- World Health Organization. Global Burden of Disease. www.who.int/healthinfo/global_burden_disease/gbd/en/index.html (Accessed 1st February 2014).
- Williams JW Jr, Stellato CP, Cornell J, Barrett JE. The 13‐ and 20‐item Hopkins Symptom Checklist Depression Scale: psychometric properties in primary care patients with minor depression or dysthymia. International Journal of Psychiatry in Medicine 2004;34(1):37‐50. [DOI] [PubMed] [Google Scholar]
- Yao JK, Magan S, Sonel AF, Gurklis JA, Sanders R, Reddy RD. Effects of omega‐3 fatty acid on platelet serotonin responsivity in patients with schizophrenia. Prostaglandins, Leukotrienes and Essential Fatty Acids 2004;71(3):171‐6. [DOI] [PubMed] [Google Scholar]
- Yesavage JA, Brink TL, Rose TL, Lum O, Huang V, Adey M, Leirer VP. Development and validation of a geriatric depression screening scale: a preliminary report. Journal of Psychiatric Research 1983;17(1):37‐49. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
- Silvers KM, Hackett ML, Scott KM. Omega 3 fatty acids for depression. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD004692.pub2] [DOI] [Google Scholar]