

Abstract
Genetic code expansion (GCE) has become a central topic of synthetic biology. GCE relies on engineered aminoacyl-tRNA synthetases (aaRSs) and a cognate tRNA species to allow codon reassignment by co-translational insertion of non-canonical amino acids (ncAAs) into proteins. Introduction of such amino acids increases the chemical diversity of recombinant proteins endowing them with novel properties. Such proteins serve in sophisticated biochemical and biophysical studies both in vitro and in vivo, they may become unique biomaterials or therapeutic agents, and they afford metabolic dependence of genetically modified organisms for biocontainment purposes. In the Methanosarcinaceae the incorporation of the 22nd genetically encoded amino acid, pyrrolysine (Pyl), is facilitated by pyrrolysyl-tRNA synthetase (PylRS) and the cognate UAG-recognizing tRNAPyl. This unique aaRS•tRNA pair functions as an orthogonal translation system (OTS) in most model organisms. The facile directed evolution of the large PylRS active site to accommodate many ncAAs, and the enzyme’s anticodon-blind specific recognition of the cognate tRNAPyl make this system highly amenable for GCE purposes. The remarkable polyspecificity of PylRS has been exploited to incorporate >100 different ncAAs into proteins. Here we review the Pyl-OT system and selected GCE applications to examine the properties of an effective OTS.
Keywords: genetic code expansion, stop codon suppression, pyrrolysyl-tRNA synthetase, tRNAPyl, synthetic biology, non-canonical amino acid
Introduction
New macromolecular functions often come from the expansion of chemical diversity. In the field of synthetic biology, the increase in chemical complexity originates from a functional demand. Using the genetic code expansion approach, non-canonical amino acids can be incorporated into proteins to study various biological phenomena and allow creation of new biomaterials and entire organisms[1]. Non-canonical amino acids include all amino acids that arose from genetic code expansion either naturally (such as selenocysteine or pyrrolysine[2]) or were added to the genetic code via genetic code expansion (GCE) experiments.
Translation with standard and non-canonical amino acids
Twenty natural amino acids ubiquitously serve as building blocks for protein synthesis. Each amino acid is first covalently attached to its cognate tRNA by a specific aaRS. An elongation factor then binds aminoacyl-tRNA and delivers it to the ribosome, where the amino acid becomes incorporated into a nascent polypeptide chain in response to an assigned codon (Fig 1). Of the 64 triplet codons, 61 sense codons are assigned to a specific standard AA, while 3 stop codons signal for polypeptide chain termination mediated by release factors.
Figure 1.
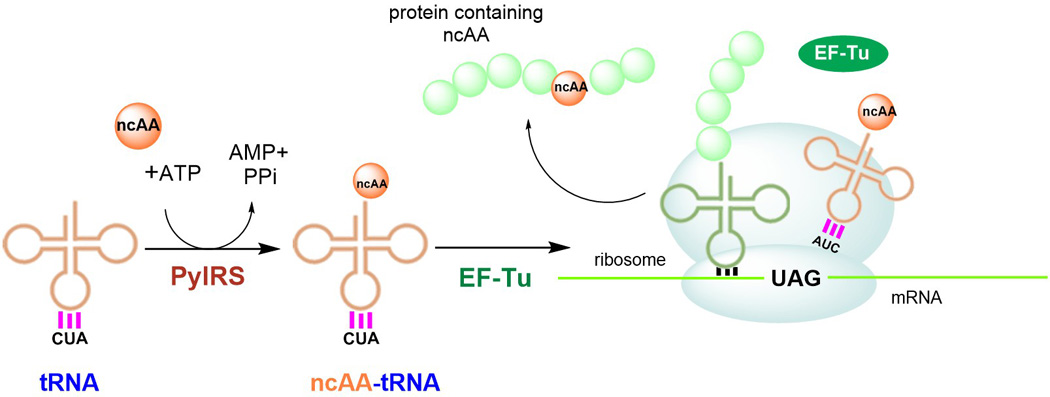
Schematic representation of stop codon suppression strategy. PylRS aminoacylates an orthogonal suppressor tRNA (anticodon CUA) with a non-cognate amino acid (ncAA). NcAA-tRNAPylCUA is delivered to the ribosome by the host elongation factor EF-Tu. At the ribosome, the ncAA-tRNAPylCUA decodes an internal stop codon (UAG) and the ncAA becomes incorporated in the protein of interest.
A genomic code expansion approach to introduce ncAAs in vivo involves site-specific co-translational incorporation through stop codon suppression (SCS, Fig 1). The targeted protein is expressed from an mRNA template containing an internal stop codon reassigned for the ncAA. A ncAA specific aaRS aminoacylates its cognate tRNA devoted to decoding a stop codon (e.g. tRNAPylCUA for recognizing the UAG codon). Such a suppressor tRNA aminoacylated with the ncAA competes with release factors for the binding to the stop codon at the ribosome. If the suppressor tRNA is able to outcompete the release factors, the ncAA is then incorporated in response to the internal stop codon.
Suppressor tRNA and aaRS dedicated to ncAA incorporation should not cross-react with host aaRSs, tRNAs or activate canonical amino acids. This lack of cross-reactivity defines the ncAA-specific aaRS•tRNA as orthogonal (o-pair or orthogonal translation system, OTS).
The best orthogonal translation systems
The success of ncAA incorporation strongly depends on the efficiency of the orthogonal aaRS•tRNA pair. The efficiency of each o-pair is determined by its inherent or engineered orthogonality, the ability of the aaRS to recognize the cognate suppressor tRNA, and the malleability of the tRNA synthetase active site. In addition to the orthogonal aaRS•tRNA pair, factors such as intracellular availability of the ncAA, the sequence context of the targeted codon in the RNA and any effect the incorporated ncAA may have on the recombinant protein’s folding also influence the ncAA incorporation efficiency.
To date, the majority of ncAA incorporation in Escherichia coli and mammalian cells has been achieved by employing the Methanocaldococcus jannaschii TyrRS•tRNATyr or Methanosarcina mazei/Methanosarcina barkeri PylRS•tRNAPyl o-pairs[1a]. The latter is also amenable for use in Saccharomyces cerevisiae, while the E. coli Tyr-OTS provides a better choice than M. jannaschii Tyr-OTS[3] in yeast due to the lack of orthogonality of the archaeal o-pair in S. cerevisiae. Other notable examples for genetic code expansion experiments in E. coli include S. cerevisiae AspRS•tRNAAsp, GlnRS•tRNAGln, TyrRS•tRNATyr and PheRS•tRNAPhe o-pairs. Archaeal O-phosphoseryl-tRNA synthetase (SepRS), a rare synthetase devoted to attaching phosphoserine (Sep) onto tRNACys as a precursor in Cys-tRNACys synthesis has also been exploited for GCE purposes[4]. The small number of OTSs developed that are not based on the Tyr- and Pyl-OTS and the even lower number of ncAAs that these OTSs can incorporate reflects the rigidity of aaRSs with respect to recognizing cognate AAs. This is evident by the relative difficulty of modifying the often used archaeal TyrRS beyond the spectrum of Tyr or Phe analogs[1a].
The PylRS•tRNAPyl system
Pyrrolysyl-tRNA synthetase
PylRS is a homodimeric enzyme that catalyzes the formation of Pyl•tRNAPylCUA. Formation of Pyl-tRNAPyl follows the two step mechanism common to all aaRSs: Pyl is first activated with ATP to form a pyrrolysyl-adenylate (Pyl-AMP) and then transferred to the 3′-hydroxyl group of the tRNAPyl terminal adenosine[5].
Each monomer of PylRS can be roughly divided into its N-terminal and C-terminal portions, which are encoded by a single gene in the Methanosarcinaceae species or by 2 separate genes in Desulfitobacterium hafniense (pylSn and pylSc, respectively[6]). Biochemical analysis of the N-terminal D. hafniense PylRS fragment demonstrate its role in tRNA binding[6]; this is further supported by the lack of in vivo enzyme activity after truncation of the N-terminal PylRS domain[7]. A structural understanding of the role of the N-terminal domain of PylRS is lacking, as it does not have sequence homology with any known protein domain and its low solubility impedes production for crystallization purposes.
While the structure of the N-terminal domain is still unknown, the PylRS C-terminal domain structures have been determined for the archaeal[8] and bacterial[9] enzymes[10]. All structures confirm that this soluble domain forms the catalytic core of the enzyme. The catalytic core is composed of an antiparallel β-sheet surrounded by α-helices, which is characteristic of class II aaRSs[11]. Class-specific motifs are typically located with sequence motif 1 building part of the dimer interface and motifs 2 and 3 recognizing the nucleotide placed at the active site (Fig 2, Fig 3, A).
Figure 2.
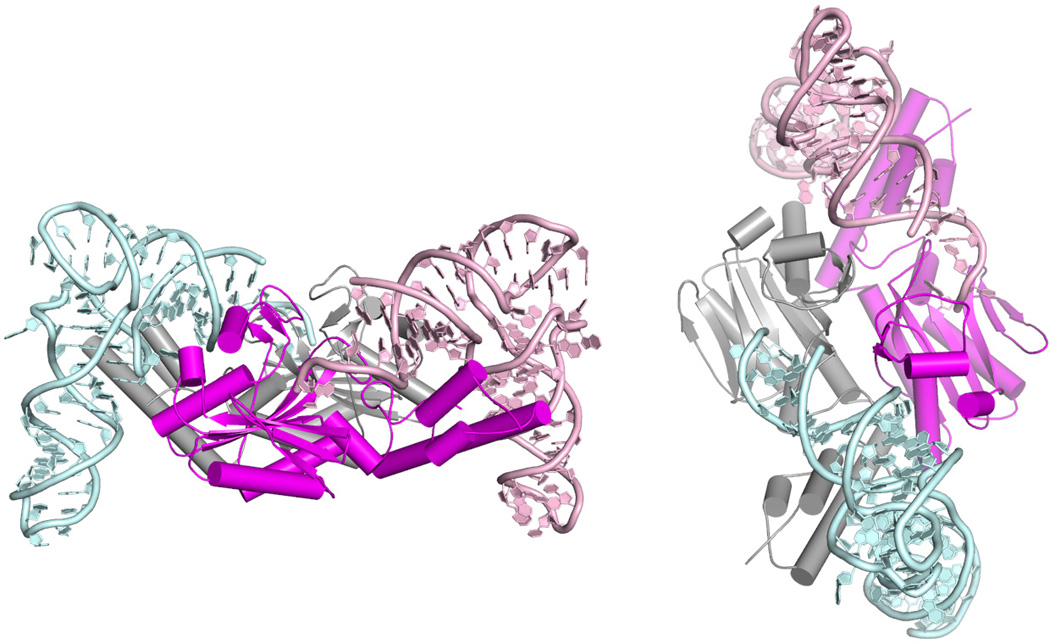
Structure of the PylRS•tRNAPyl complex from D. hafniense (PDB ID 2ZNI) from two perspectives. Individual monomers of the PylRS are shown in grey and magenta, tRNA(I) in light blue and tRNA(II) pink.
Figure 3.
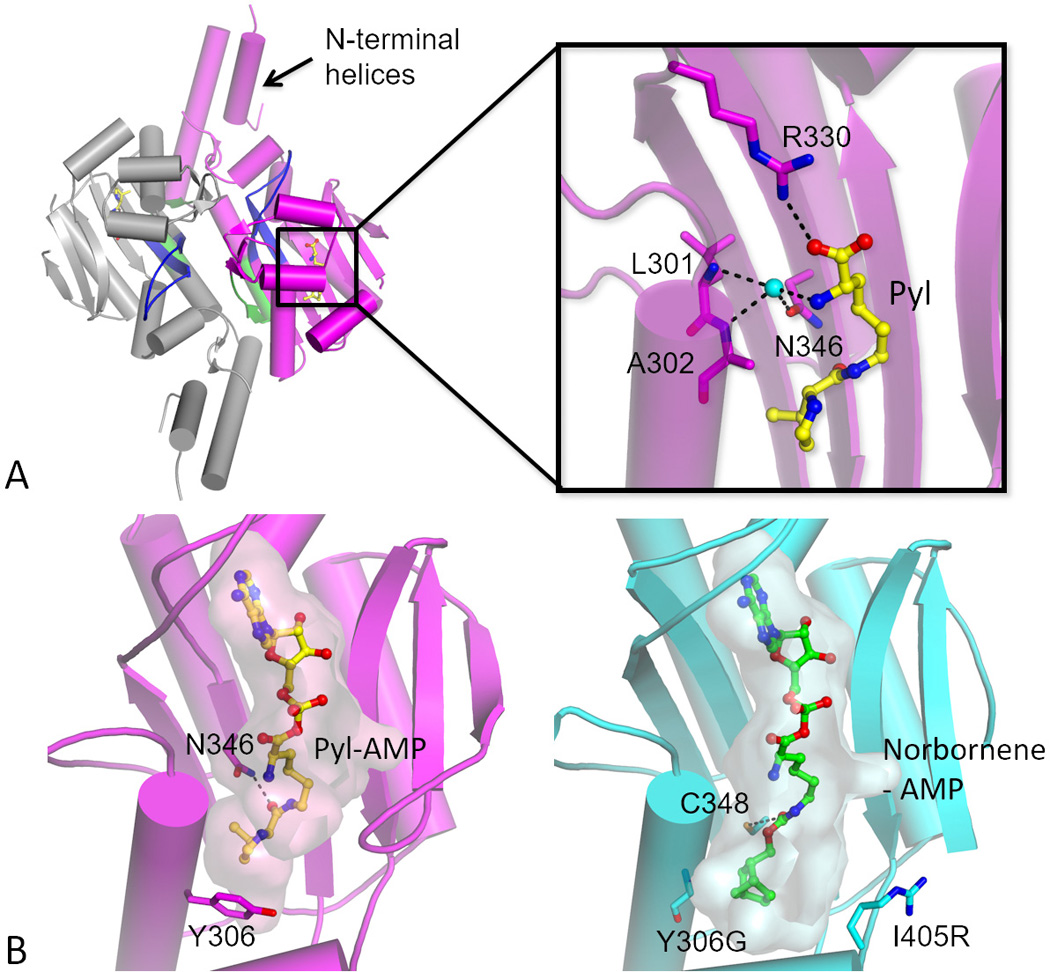
PylRS topology and α-amino group recognition in the active site. The enzyme is composed of two identical monomers (grey and magenta) forming a homodimer. Characteristic class II aaRS motifs are accentuated: motif 1 that forms part of the dimer interface is shown in green and motif 2 loop (involved in tRNA 3′-CCA recognition) in blue. Pyl is bound in the active site and shown in yellow. (Inset) Idiosyncratic recognition of the Pyl α-amino group in M. mazei PylRS (PDB ID 2ZCE). Instead of directly recognizing the α-amino group of the AA substrate PylRS uses a water molecule bound to the Asn-346. Water molecule (cyan) is shown as a ball. Hydrogen bonds are shown as black dashed lines (B) Malleability of the active site as illustrated by comparison of the wild-type enzyme and a norbornene charging mutant (PDB ID 2Q7H and 4BWA, respectively). The wild-type enzyme is shown in magenta and the triple Y306G/Y384F/I405R mutant in cyan. Pyl-AMP and norbornene-AMP (nonhydrolyzable analogs) are given as stick representation in yellow and in green, respectively, with transparent surfaces indicating the substrate-binding pocket. In the triple mutant the Y306G mutation at the base of the AA binding pocket enlarges the binding site thus allowing placement of the norbornene head group (this space is normally occupied by Tyr-306 side chain, see wild-type structure). In the wild-type enzyme Asn-346 forms a hydrogen bond with the carbonyl-amide linkage of Pyl side chain. In contrast, the carbamate linkage of the norbornene does not form a bond with Asn-346 but with Cys-348.
The structure of the bacterial PylRS•tRNAPyl complex shows tRNA binding stoichiometry of two tRNAs per enzyme dimer[9b] (Fig 2). Each tRNA binds along the surface of one of the monomers. Cognate tRNA recognition is ensured through interactions with specific nucleotides (i.e. identity elements) and core binding surface of the enzyme recognizing the characteristic tRNA tertiary structure. The top of the acceptor stem (base pair G1:C72 and R73, Fig 2) is involved in the critical portion of PylRS•tRNAPyl interface. A concave structure on the enzyme surface sterically matches the acceptor helix and directs the tRNAPyl 3′-end into the catalytic site. Residues present within this structure directly communicate with identity elements on tRNAPyl: discriminator base (R73) and the first base pair in the acceptor stem (G1:C72)[9b, 12]. Other directly recognized residues include D-stem base pairs G10:C25 and A11:U24, and G9[9b, 12].
The idiosyncratic core of tRNAPyl is recognized dominantly by the elements of one subunit and fewer contacts are made with the other monomer. PylRS specific tRNA-binding domain 1 makes the majority of the interactions with the tertiary core of tRNAPyl. The PylRS•tRNAPyl structure confirms earlier biochemical data that although bases at position 33 and 37 surrounding the anticodon constitute identity elements[13], PylRS does not use the tRNA anticodon as identity element per se.
Properties and engineering of tRNAPyl
Secondary structures of bacterial and archaeal tRNAPyl are remarkably similar, in spite of low sequence similarity[9b, 12]. Most striking features of tRNAPyl include a prolonged anticodon stem (6 bp instead of 5), a tiny D-loop (only 5 bases) and the absence of the almost universally conserved G18G19 and T54ψ55C56 sequences[7, 9b]. It has been proposed that in vivo M. barkeri tRNAPyl is less modified than other cellular tRNAs and harbors only 2 modified nucleotides: a 4-thiouridine and a 1-methyl-pseudouridine[14]. Structure of D. hafniense tRNAPyl shows that deletion of invariant uridine 8 (U8) abolishes the formation of a highly conserved tertiary base pair (U8:A14) and leads to unusual pairing of G14 with T-loop residue C59. Furthermore, U8 deletion allows the base on position 9 to flip away from the tRNA body. These features of tRNAPyl govern its folding into a canonical L-shaped form, but with a very compact core that differs from all known canonical tRNA structures[9b].
Pyrrolysine
In nature, Pyl-OTS and Pyl are confined to a small number of organisms including methanogenic archaea and some bacterial taxa[15]. In these organisms, three consecutive enzymes (PylB, PylC and PylD) form a Pyl biosynthesis cluster responsible for the transformation of two Lys molecules into Pyl[16]. In the first step, PylB, converts Lys to (3R)-methyl-d-ornithine (3MO), which is then ligated to the Nε-amino group of a second Lys molecule by PylC, giving rise to l-lysine-Nε-(3R)-methyl-d-ornithine. The third enzyme, PylD, catalyzes the oxidation of the last intermediate, resulting in the formation of Pyl[16–17]. Pyl is subsequently attached to tRNAPyl by PylRS. In Methanosarcinaceae, tRNAPylCUA is a natural suppressor tRNA that decodes a UAG stop codon present as an internal stop codon in set of methylamine methyltransferases genes[18] and a tRNAHis guanylyltransferase[19]. The incorporation of Pyl in the methyltransferase is critical for enzyme catalysis and enables growth on methylamine as a sole energy source[20]. In contrast, in Methanosarcina acetivorans tRNAHis guanylyltransferase the Pyl residue is not essential and can be exchanged with Trp without a loss of enzymatic activity[19].
PylRS active site architecture
The PylRS active site contains a large amino acid binding pocket where Pyl is mainly recognized through (nonspecific) hydrophobic interactions[8, 21]. Secondary carbonyl group of Pyl forms hydrogen bonds with Asn-346. A comparison of PylRS:Pyl and various PylRS:ncAA complexes[8, 22] reveals that variations in the ncAA side chain are highly tolerated, particularly when they are located in the part of the active site responsible for pyrroline recognition[23]. This property of PylRS contrasts the general features of aaRSs, which are selective against non-cognate substrates. Both structural and in vivo studies demonstrate that PylRS can use substrates that possess a carbamate, carbonyl or amide moiety, which can be positioned to interact with Asn-346 or Cys-348.
The structure of M. mazei PylRS[8, 21] shows that Arg-330 forms a hydrogen bond with the primary carbonyl of Pyl and this interaction is seen in complexes with both Pyl and Pyl analogs. Most interestingly, the primary amino-group is not directly recognized but is bound by a water molecule (Fig 3, A). This water forms hydrogen bonds with amide oxygen of Asp-346 and two backbone amide nitrogen atoms of Leu-301 and Ala-302. Because the α-amino group of Pyl is loosely recognized, Arg-330 appears to be the single residue responsible for the proper orientation of Pyl for the nucleophilic attack of the α-phosphate[8, 22a, 24]. In concordance with this flexible mode of α-amino group recognition come reports showing acylation of tRNAPyl with α-hydroxy acid, d-amino acid and a Nα-methylamino acid[25]. Furthermore, it has been reported that several α-hydroxy acids can be incorporated into proteins by the PylRS•tRNAPyl o-pair[26].
Although the importance of Asn-346 and other Pyl binding residues has been demonstrated through mutational analysis[22a], there is no coherent kinetic data that might disclose the effect of these residues on PylRS aminoacylation catalysis (Table 1). Steady state parameters for Pyl activation have been measured mostly in the absence of tRNAPyl, using a ATP-[32P]PPi exchange assay (Table 1). Conversely, enzyme-kinetic analyses of PylRS were hampered by the lack of synthetically available Pyl, necessitating the use of Pyl analog like Nε-((cyclopentyloxy)carbonyl)-l-lysine more frequently than Pyl.
Table I.
Steady-state kinetic parameters for various ncAAs and PylRS enzymes.
| Organism | ncAA |
Km [mmol.dm−3] |
kcat [1/s] |
Assaya | Enzyme variantb | Structurec | Ref |
|---|---|---|---|---|---|---|---|
| D. hafniense | l-pyrrolysine | 0.044 | 0.325 | 1 | PylSc | - | [6] |
| M. barkeri | Nε-((R)-tetrahydrofuran-2-carbonyl)-l-lysine | 0.39 | 0.062 | 1 | WT | - | [66] |
| Nε-(cyclopentanecarbonyl)-l-lysine | 5.6 | 0.04 | 1 | WT | - | ||
| l-pyrrolysine | 0.055 0.02 |
0.105 0.003 |
1 2 |
WT WT |
- 2Q7H, 2ZIM, 2ZCE |
[66] | |
| Nε-((cyclopentyloxy)carbonyl)-l-lysine | 0.55 | 0.015 | 1 | WT | 2Q7G | [66] | |
| l-pyrrolysine | 0.053 n.d. n.d. |
n.d. 0.1 0.24 |
1 1 1 |
WT WT Δ92PylS |
2Q7H, 2ZIM, 2ZCE 2Q7H, 2ZIM - |
[23] [6] |
|
| Nε-((cyclopentyloxy)carbonyl)-l-lysine | 0.67 | n.d. | 1 | WT | 2Q7G | [23] | |
| Nε-acetyl-l-lysine | 22.3 | 0.0341 | 1 | AcKRS3 | - | [36] | |
| Nε-d-prolyl-l-lysine | 0.5 | n.d. | 1 | WT | - | [23] | |
| M. mazei | 3-iodo-l-phenylalanine | 0.44 | 0.0044 | 2 | IFRS | 4TQD | [22b] |
| l-phenylalanine | 21 14 22.8 |
0.037 0.062 0.075 |
1 1 1 |
FRS1 FRS2 FRS3 |
- - - |
[39a] |
|
| l-pyrrolysine | 0.02 | 0.0083 | 2 | WT | 2Q7H, 2ZIM, 2ZCE | [22b] | |
| Nε-acetyl-l-lysine | 35.3 7.8 |
0.0323 0.00731 |
1 1 |
AcKRS1 AcKRS2 |
4Q6G - |
[36] |
(1) - ATP-[32P]PPi exchange
(2) - aminoacylation
AcKRS1- M. mazei PylRS variant L301M/Y306L/L309A/C348F; AcKRS2- M. mazei PylRS variant L301M/L305M/Y306M/C348S; AcKRS3- M. barkeri PylRS variant L266M/L270I/Y271F/L274A/C313F; FRS1- M. mazei PylRS variant N346A/C348L; FRS2- M. mazei PylRS variant A302L/Y306M/N346S/C348L/Y384L; FRS3- M. mazei PylRS variant A302F/Y306L/N346T/C348F/Y384L; IFRS- M. mazei PylRS variant N346S/C348I
All structures are of M. mazei enzymes.
Steady state kinetic parameters with Pyl for full length M. mazei PylRS and M. barkeri PylRS show that these aaRSs bind the amino acid with a Km of ~50 μmoldm−3 and kcat of 0.1–0.3 s−1 in ATP-[32P]PPi exchange assay. In the presence of tRNAPyl (aminoacylation assay) the affinity for the cognate amino acid is improved (20 μmoldm−3), yet the kcat remains very low (0.008–0.03 s−1). This kcat value is almost 3 orders of magnitude lower than many canonical aaRSs. The lower activity may interrelate with the fact that Pyl is translated 3000 times less often than a sense codon[22b].
Amino acid substrate range
Although low turnover numbers make PylRS a fairly poor catalyst in vitro, to date it has been adapted for incorporation of more than 100 ncAA substrates (full list in [27]). Regardless of the low turnover, in vivo suppression efficiency can amount to almost 80 % with engineered enzymes making this enzyme a powerful tool for GCE experiments[28]. As PylRS can tolerate replacement of the pyrroline ring with a similar substituent, more than 20 lysine derivatives were reported to be suitable substrates for PylRS without active site engineering[23, 27]. In addition to lysine derivatives, Pyl-OTS can be used to incorporate Phe derivatives[27] although a number of Phe derivatives have already been successfully incorporated using M. jannaschii and E. coli TyrRS.
The lack of specificity towards the functional group at the Nε-atom has allowed for incorporation of Lys analogs bearing reactive chemical groups that can undergo a selective reaction with a desired label. Because the label does not cross-react with cellular (macro)molecules the reaction can occur within a living cell. The earliest reports to introduce a chemical tag via Pyl-OTS included wild-type PylRS. Alkyne (1) and azide (2) derivatives (Fig 4) for Cu(I)-catalyzed alkyne–azide triazole-forming reactions (CuAAC, a type of reaction commonly referred to as “click chemistry”) were introduced into myoglobin as proof-of-principle[29]. For the purposes of monitoring conformational changes of a protein PylRS was used to install a reactive dye for the distance-dependent Förster resonance energy transfer (FRET). While the first member of the FRET pair was generated by alkylation of a Cys residue, alkyne containing Pyl analogs (3 and 4, Fig 4) were introduced into calmodulin via SCS strategy and subsequently labelled with azidocoumarin through a CuAAC reaction[30].
Figure 4.
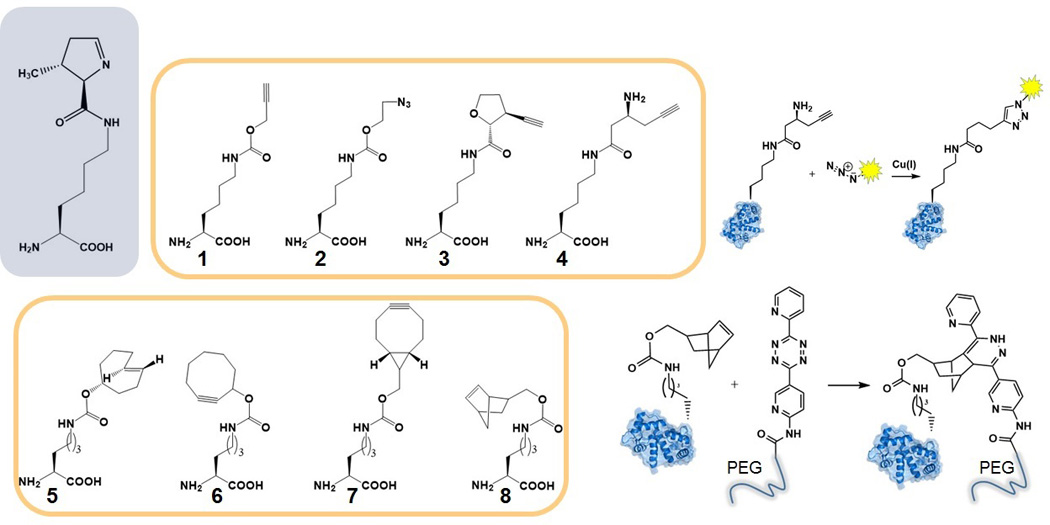
NcAAs that can be co-translationally incorporated into proteins for bioorthogonal labelling through Cu(I) catalyzed cycloaddition (Nε-((prop-2-yn-1-yloxy)carbonyl)-l-lysine (1), Nε-((2-azidoethoxy)carbonyl)-l-lysine (2), Nε-((2R,3S)-3-ethynyltetrahydrofuran-2-carbonyl)-l-lysine (3), Nε-((S)-3-aminohex-5-ynoyl)-l-lysine (4)) or through inverse-electron-demand Diels–Alder reaction (Nε-((((R,E)-cyclooct-4-en-1-yl)oxy)carbonyl)-l-lysine (5), Nε-((cyclooct-2-yn-1-yloxy)carbonyl)-l-lysine (6), Nε-((((1R,8S)-bicyclo[6.1.0]non-4-yn-9-yl)methoxy)carbonyl)-l-lysine (7), Nε-((bicyclo[2.2.1]hept-5-en-2-ylmethoxy)carbonyl)-l-lysine (8)).
To circumvent inherent cytotoxicity of Cu(I) activated cyclooctyne derivatives can also be introduced via Pyl-OTS. Such compounds can react with azide containing labels without additional catalysts. M. mazei PylRS double mutant (Y306A/Y384F) was used to introduce transcyclooctene (5)[31] and cyclooctyne (6 and 7) derivatives into target proteins in E. coli and mammalian cells[32]. Norbornene-bearing ncAA (8) is convenient as it can undergo two different types of conjugation: it can be reacted with azides (Cu(I) catalysis) or with tetrazines (inverse-electron-demand Diels–Alder reaction). Human carbonic anhydrase II was produced containing norbornene derivatized amino acid by using the triple M. mazei PylRS mutant (Y306G/Y384F/I405R). Subsequently, norbornene bearing sites were exploited for polyethylene glycol (PEG) attachment[33]. When comparing active site architectures (Fig 3, B) it is seen that the mutation of Tyr-306 is critical to enlarge the hydrophobic binding pocket and to accommodate bulky head group of norbornene-containing compound.
In addition to chemical groups that can render a protein target susceptible for selective chemistry, many photo-reactive groups act as substrates for both wild-type PylRS and PylRS variants. Photo-reactive ncAAs are attractive; they ensure ‘protected’ co-translational incorporation into target proteins, and subsequent facile deprotection will generate the desired ‘natural’ posttranslational protein modifications. An important modified AA, Nε-methyl-l-lysine, cannot be selectively incorporated via Pyl-OTS as it lacks the side chain carbonyl-oxygen that is recognized by the enzyme. However, protected versions of Nε-methyl-l-lysine contain carbamate and can serve as substrates for PylRS. Upon incorporation they can be deprotected to leave Nε-methyl-l-lysine as the incorporated residue (Fig 5).
Figure 5.
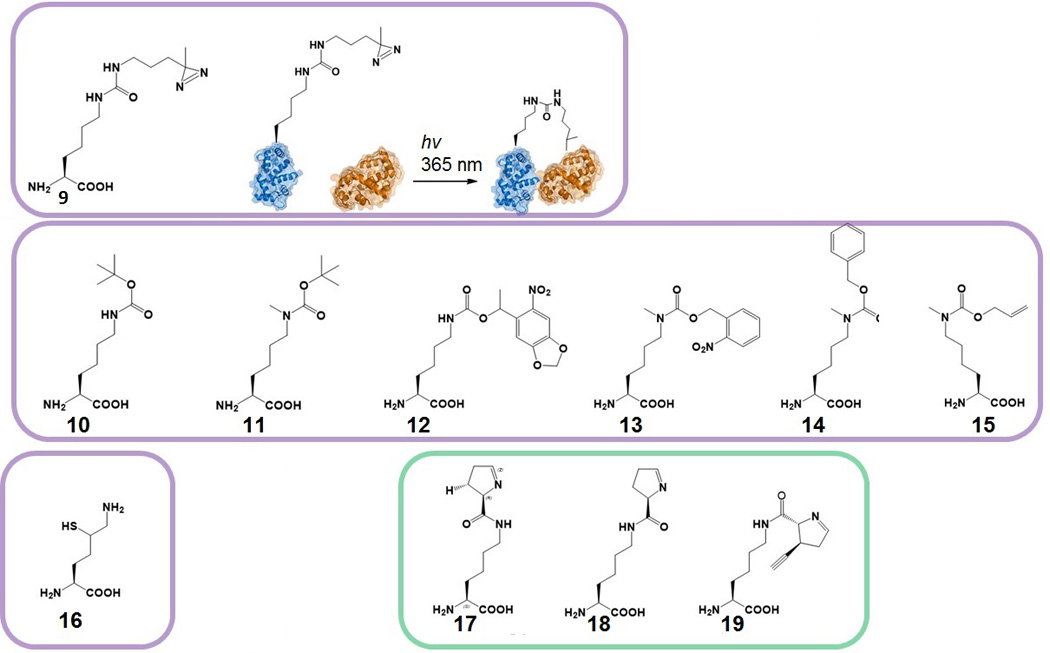
NcAAs with photo-reactive substituents for photo-crosslinking (Nε-((3-(3-methyl-3H-diazirin-3-yl)propyl)carbamoyl)-l-lysine (9)) and photo-caged ncAAs (Nε-(tert-butoxycarbonyl)-l-lysine (10), Nε-(tert-butoxycarbonyl)-Nε-methyl-l-lysine (11), Nε-((1-(6-nitrobenzo[d][1,3]dioxol-5-yl)ethoxy)carbonyl)-l-lysine (12), Nε-methyl-Nε-(((2-nitrobenzyl)oxy)carbonyl)-l-lysine (13), Nε-((benzyloxy)carbonyl)-Nε-methyl-l-lysine (14), Nε-((allyloxy)carbonyl)-Nε-methyl-l-lysine (15)). NcAAs for chemical ligation purposes (2S)-2,6-diamino-5-mercaptohexanoic acid (16). NcAAs that can be synthesized in E. coli using the pylBCD biosynthetic cluster: Nε-((R)-3,4-dihydro-2H-pyrrole-2-carbonyl)-l-lysine (17), Nε-((R)-3,4-dihydro-2H-pyrrole-2-carbonyl)-l-lysine (18) Nε-((2R,3S)-3-ethynyl-3,4-dihydro-2H-pyrrole-2-carbonyl)-l-lysine (19).
In contrast to Nε-methyl-l-lysine, another important lysine posttranslational modification, Nε-acetyl-l-lysine (AcK), has been successfully incorporated by an evolved PylRS, as the carbonyl oxygen is present in this compound and evolved PylRS does not lose its selectivity. Pyl-OTS mediated incorporation of AcK has been demonstrated in E. coli and mammalian cells. An evolved M. barkeri PylRS•tRNA pair was used to specifically incorporate AcK in myoglobin[34] and histones[35]. M. mazei PylRS enzyme was also evolved for specific incorporation of AcK and this enzyme was later kinetically investigated[22b, 36]. Interestingly, there are several variants of both M. mazei and M. barkeri PylRS that are specific for AcK. It has been proposed that the equivalent efficiency of different PylRS variants originates from the nature of the substrate head group (Table I). Since the acetyl group of AcK is much smaller than pyrroline ring of Pyl, mutations in the Pyl binding site need to occupy the vacant space to compensate for the loss of the bulky pyrroline ring[34].
What is/determines orthogonality?
Feasibility of site-specific incorporation of ncAA depends on the mutual interaction of host translational machinery with the orthogonal aaRS•tRNA pair. Ideally, the o-tRNA would be efficiently recognized by the host elongation factor and ribosome (Fig 1), but not by the host aaRSs. Conversely, o-aaRS should not recognize the host tRNAs or charge standard AAs. The conserved tertiary features of tRNAs and the relatively low complexity of nucleotide determinants often result in engineered tRNAs that interact with the host translation machinery. Attempts to improve orthogonality via tRNA engineering often becomes a complex challenge as adding or removing nucleotide determinants can introduce a determinant for another host aaRS.
In E. coli, decoding of UGA stop codons with tRNAPyl variant leads to high background suppression with Trp[37] due to near cognate UGA suppression by E. coli Trp-tRNATrp [37a]. PylRS and E. coli TrpRS share a number of common identity determinants, which affect the cross-reactivity of tRNAPylUCA. In E. coli, the tRNA anticodon positions C34 and A36, as well as G73 act as a minor determinant for TrpRS, all of which are present in tRNAPylUCA. Mutating the tRNAPylUCA G73 position to U was shown to have increased orthogonality[37a].
While natural amber suppressor tRNAPylCUA is orthogonal in E. coli, in S. cerevisiae the M. barkeri tRNAPylCUA was shown to be the substrate for alanyl-tRNA synthetase (AlaRS)[38] due to the presence of a positive identity determinant, G3:U70. Mutation of this base pair to A3:U70 changed the sequence of the M. barkeri tRNAPyl to that of M. mazei tRNAPyl and orthogonality was obtained[38].
In contrast to the known interaction of tRNAPyl with the host aaRSs, PylRS shows little to no cross-reactivity with the host tRNAs, regardless of the host organism. However, a significant challenge for genetic code expansion with PylRS is in maintaining orthogonality toward the cellular pool of standard AAs. For instance, only 2 active site residues need to be replaced in PylRS to introduce specificity for l-phenylalanine[39]. These active site residues (Asn-346 or Cys-348) are frequently targets of randomization in directed evolution experiments as they bind the carbamate of Pyl or the ncAA substrate[39].
Examples of applications
In mammals and other eukaryotes protein-protein interactions and posttranslational modifications play crucial roles in regulating the function of proteins. Transient protein-protein interactions can be explored in vivo by using photo-active ncAA crosslinkers. Photo-inducible crosslinkers such as p-benzoyl-l-phenylalanine (pBpa) have been used extensively and were mostly incorporated via Tyr-OTS[1a, 40]. Pyl-OTS incorporated (3-(3-methyl-3H-diazirin-3-yl)-propamino-carbonyl-Nε-l-lysine (DiZPK, 9) has the advantage over pBpa because of its flexibility and length and has been incorporated via Pyl-OTS into proteins in bacteria, yeast and mammalian cells[41].
Novel ways of studying posttranslational modifications in mammalian cell lines have been explored by introducing photo-caged AAs into proteins. Incorporation of these AA derivatives with the Pyl-OTS has allowed for a number of investigations that aimed to distinguish how AA modifications influence the behaviour of proteins in vivo. Wild-type PylRS utilizes some of the Boc-protected lysine derivatives without the need for engineering of the enzyme active site. For example, the ncAAs Nε-(tert-butoxycarbonyl)-l-lysine (10) and Nε-(tert-butoxycarbonyl)-Nε-methyl-l-lysine (11) have been incorporated with further deprotection resulting in proteins containing Nε-methylated Lys-residues[29, 42]. To specifically charge the photo-caged lysine derivative (12) a variant of the M. barkeri PylRS has been selected for through directed evolution. This PylRS•tRNAPylCUA pair was used to introduce a critical lysine residue into a kinase MEK1, in a photo-caged state. As the kinase is a part of the signalling pathway, a cellular network could be activated by light irradiation and subsequent release of the caging group[43]. An evolved version of M. barkeri was used to introduce a photo-caged version of Tyr into a STAT1 protein (a part of the interferon mediated signal transduction network) thus enabling studies of Tyr phosphorylation in vivo[44].
Using the SCS strategy for the incorporation of lysine derivatives into histone proteins has allowed for novel approaches to gain insight into the role of histone posttranslational modifications. Using the Pyl-OTS, ncAAs 10 and 11 were introduced into histone H3 and employed to generate Nε, Nε-dimethyl-l-lysine containing H3 proteins. After ncAA insertion and protein isolation, the recombinant histone was subjected to a number of chemical modification steps that enabled methylation on the previously protected site. This approach was undertaken to circumvent the inability of wild-type PylRS to activate Nε, Nε-dimethyl-l-lysine and the lack of specificity of evolved variants of this enzyme[42]. Accordingly, a monomethylated lysine residue (Nε-methyl-l-lysine) was introduced in the photocaged state (13) into myoglobin by a heterologous Pyl o-pair (M. barkeri PylRS and M. mazei tRNA) and the system was shown to be functional in both E. coli and mammalian cells[45]. That and a similar photocaged version of monomethylated lysine (14) were synthesized and introduced into green fluorescence protein (GFP) and Z domain protein. Here, the decaging process included UV photolysis (13) or hydrogenation (14[46]). A similar approach was utilized on a separate occasion, in this case by using the M. mazei enzyme, where a Pyl-OTS was evolved to facilitate Nε-((allyloxy)carbonyl)-Nε-methyl-l-lysine (15) incorporation in bacteria; this AA can be converted to Nε-methyl-l-lysine with a ruthenium catalyst[46].
In addition to introducing posttranslational modifications by decaging translated ncAAs, very large moieties can be incorporated by exploiting native chemical ligation strategy. The strategy involves a thiol group in the first protein that can attack a C-terminal thioester of a second protein. As 1,2-aminothiols can react with thioesters to form an amide bond, ncAA δ-thiol-l-lysine (16) was introduced by Pyl-OTS for protein ubiquitination[47]. The protein containing 16 was then reacted with an ubiquitin thioester and, after desulfurization of the complex, ubiquitin conjugates were isolated.
The challenge of incorporating ncAAs in multicellular organisms was first achieved by using Pyl-OTS in C. elegans[48]. tRNAPyl was used to decode amber stop codon and introduce Nε-(tert-butoxycarbonyl)-l-lysine (10) and Nε-((prop-2-yn-1-yloxy)carbonyl)-l-lysine (1). Since ncAA containing protein was produced from stop codon containing transcripts, nonsense mediated decay pathway that targets transcripts with internal termination codons for destruction had to be circumvented. This was achieved by using a smg2 knockout strain, where product of smg2 gene is a component of the NMD pathway[49].
In D. melanogaster, genetic code expansion was also achieved using Pyl-OTS. Site-specifically labeled proteins were produced containing Nε-((bicyclo[2.2.1]hept-5-en-2-ylmethoxy)carbonyl)-l-lysine (8) reacted with a tetrazine probe[50]. Moreover, same ncAA was stochastically incorporated into proteins in specific tissues and at specific times. This was achieved by crossing flies that expressed Pyl-OTS and a gene of interest with flies expressing Gal4 in specific tissues and at specific times. Transcriptional activator Gal4 is expressed using a tissue-specific promoter and, when present, activates the expression of transgenes under GAL4 upstream activating sequences (Gal4 UAS). Both pylS and the gene of interest with an internal stop codon were placed under Gal4 UAS and in this manner site-specific incorporation of ncAA was enabled only when Gal4 was present, i.e. in a specific tissue and at specific time[50].
While the incorporation of ncAAs into proteins with the Pyl-OTS has been extensively reported in eukaryotes, one notable exception has been yeast, where there have been only two reports of Pyl-OTS use in S. cerevisiae for proof-of-principle and optimization purposes. To date Pyl-OTS has not been used in any yeast species other than S. cerevisiae. The first reported use of the Pyl-OTS in yeast utilized the M. mazei PylRS and M. mazei tRNAPyl pair[51]. The authors used a M. mazei PylRS variant that had been selected for in E. coli to utilize Nε-(tert-butyloxycarbonyl)-l-lysine (10). Using a screen where a variant of the transcription activator Gal4 containing two stop codons in its coding sequence is used to drive expression of reporter genes, resulting in phenotypes that allow for both positive and negative selection[3a]. A weak phenotype dependent on the presence of the ncAA was observed, demonstrating both the orthogonality and function of the M. mazei PylRS•tRNAPyl pair in S. cerevisiae. Although a weak phenotype was observed, a reporter protein was not isolated and the ncAA incorporation was not confirmed through MS, making it difficult to assess the efficiency of incorporation.
Further work to characterize and optimize the Pyl-OTS in S. cerevisiae successfully utilized the M. barkeri PylRS•M. mazei tRNAPyl pair and derivatives for the incorporation of several lysine analogs[38]. Initially, the authors used the M. barkeri PylRS•M. barkeri tRNAPyl pair, which had successfully been used in other organisms[40]; however, due to a lack of orthogonality the M. barkeri tRNAPyl was mutated to that of M. mazei tRNAPyl. Expression of the tRNAPyl was a challenge and the use of external RNA polymerase III promoters that have been effective in heterologous tRNA expression in yeast did not produce sufficient amounts of functional tRNAPyl for effective suppression. This was overcome through engineering of the yeast dicistronic tRNA operon encoding tRNAArg and tRNAAsp, where the two mature tRNAs are produced from a dimeric precursor RNA[52]. In this instance the tRNAAsp was replaced by the gene for tRNAPyl and both tRNA genes transcribed from the internal promoter of the tRNAArg gene. Using the M. mazei tRNAPyl with variants of M. barkeri PylRS resulted Gal4 read-through based phenotypes dependent on the required ncAA. While incorporation was confirmed for 3 of the 5 analogs utilized, the yields of reporter protein obtained (30–100 µg/L) were nearly two orders of magnitude below what can be obtained with other ncAAs and orthogonal pairs in yeast[53].
While only two studies have been reported, it is clear that both expression and orthogonality tRNAPyl is different in yeast when compared to other eukaryotic cells. The distinct challenges of using this OTS in yeast in conjunction with the known low solubility of PylRS in other organisms and the low activity of PylRS and variants toward ncAAs combined likely account for the low incorporation efficiency observed with this orthogonal pair in yeast.
Outlook
New routes to Pyl
A complete understanding of kinetic features of the Pyl-OTS is still lacking due to the difficult nature of Pyl chemical synthesis. The first reported chemical synthesis of Pyl involved coupling of (4R,5R)-4-methylpyrroline-5-carboxylic acid to lysine[54]. During synthesis, the sensitive carboxypyrroline ring is exposed to strong acidic conditions, which may be responsible for pyrroline ring degradation and low Pyl yields. To avoid this, an alternative synthetic route was developed where Pyl was synthesized with the generation of the pyrroline ring occurring in the penultimate step of the synthesis which resulted in a 2 times higher yield of Pyl[55].
An appealing alternative to chemical synthesis is the transplantation of the Pyl biosynthetic cluster to E. coli for Pyl producing purposes. Because Pyl is synthesized from Lys, no additional amino acid is needed to be supplemented in the culture media. This strategy was used to produce Pyl containing proteins in E. coli, as Pyl-OTS can to be co-transformed in the same strain[56]. Interestingly, supplementing culture medium with d-ornithine results with the production of pyrroline-carboxy-lysine (Pcl, 17) instead of Pyl. Furthermore, different 3′-substituted d-ornithine derivatives (3MO analogs) can be added to create Pyl analogs with the help of incomplete Pyl biosynthetic cluster[57]. Only genes for PylC and PylD are needed to produce Pyl, if 3MO or 3MO analog is added. For this reason, adding d-ornithine or (3S)-ethynyl-d-ornithine to the media results with desmethyl-pyrrolysine (dmPyl, 18) and ethynyl-pyrrolysine production (ePyl, 19). Although the production of Pyl analogs was streamlined for amber suppression and incorporation[57], manipulating Pyl biosynthetic cluster opens new perspectives on intracellular ncAA production.
Stop codon suppression vs. sense codon reassignment
In-frame stop codons targeted for ncAA incorporation in a gene of interest are not the only such stop codons in a transcriptome of a model organism (with the exception of recoded E. coli strains[1b, 58]) as the same codons occur as natural stop codons in a variety of host’s cellular proteins. Decoding such sites with the aid of introduced o-pair may have deleterious consequences as illustrated by the Sep-OTS in E. coli[59] or Pyl-OTS in certain mammalian cell lines[60]. In E. coli, natural stop codons are not equally susceptible to o-tRNA mediated decoding and this susceptibility depends upon the presence of the appropriate release factor and/or introduction of ribosomal proteins with lower affinity for the same release factor[61]. In mammalian cell lines, transcriptional response to amber suppression varies greatly between different cell lines with the transcription of over a 1000 genes dysregulated after introducing Pyl-OTS (or a derivative AcKRS-OTS) into mouse embryonic stem cells and only 11 in mouse embryonic fibroblast[60].
Apart from the toxic effect that a suppressor tRNA may exert on cellular fitness, protein production via SCS suffers from variable yields as a consequence of the competition between the suppressor tRNA and the cellular release factors for binding to the same stop codon. This problem is partially alleviated by using strains with release factor deletions[58]. An alternative to using a release factor deletion strain is to target sense codons. As PylRS does not recognize the tRNA anticodon, any sense codon can be targeted for ncAA incorporation by Pyl-OTS. Sense codon reassignment was previously attempted in Mycoplasma capricolum. As this organism’s genome contains only 6 instances of the CGG (arginine) codon and lacks the corresponding tRNAArgCCG, this codon was considered to be an “open” sense codon. Unfortunately, engineered tRNAPylCCG was shown not to be orthogonal in this organism as it was recognized by M. capricolum ArgRS[62]. This problem was circumvented in E. coli by introducing serine and leucine anticodons into tRNAPyl [28]. As neither the E. coli LeuRS or SerRS use the tRNA anticodon as a recognition element, these enzymes do not cross-react with tRNAPylCAG or tRNAPylACU, resulting in orthogonal tRNAPyl variants that are able to decode sense codons. Using the M. mazei PylRS IFRS variant, which aminoacylates 3-iodo-l-phenylalanine (3-I-Phe), it was possible to quantify incorporation at targeted Ser AGU codons. The extent of 3-I-Phe incorporation at the targeted site in a GFP reporter protein was ~65%[28]. The fact that the efficient IFRS can, in part, accomplish reassigning a high-frequency serine codon[28] opens new opportunities for using this enzyme as an integral part of E. coli translation machinery.
Another strategy to utilize sense codons for GCE purposes consists in targeting the degenerate codons that occur in the genome at a low frequency (rare codons) and are decoded by a low abundance tRNA isoacceptor. The approach requires elimination of the host’s dedicated tRNA and the synonymous replacement of targeted codons within the genome (at least in essential genes). Recently, a critical step toward successful emancipation of the rare AUA (Ile) codon was achieved by introducing Mycoplasma mobile tRNAIleUAU into E. coli. M. mobile tRNAIleUAU is able to translate AUA codons without modification of its anticodon while E. coli tRNAIle2CAU depends on tRNAIle-lysidine synthetase (encoded by tilS) to modify its C34 to 2-lysyl-cytidine for proper decoding of rare AUA codons. As M. mobile tRNA can rescue the lethal tilS deletion through direct AUA codon reading it is now possible to introduce an orthogonal IleRS and reassign the AUA codon with a desired ncAA[63]. Successful reassignment of a rare Arg codon (AGG) was also achieved in E. coli with M. mazei PylRS•tRNAPylCCU[64]. In opposition to rare AUA codons, AGG codons are recognized by 2 native tRNAs, tRNAArg4UCU (translates both AGA and AGG codons) and the low abundance tRNAArg5CCU (devoted only to AGG decoding). Although tRNAArg5CCU is dispensable in E. coli, tRNAArg4UCU is required to decode AGA codons; however, the levels of tRNAArg4UCU needed to be reduced to avoid wobble decoding of AGG codons and enable preferential reading by the orthogonal tRNAPylCCU. By using a modified version of tRNAArg4UCU placed under a weak promotor, the authors succeeded in reassigning the rare AGG codon from arginine to l-homoarginine which was introduced using an engineered version of M. mazei PylRS and tRNAPylCCU[64b]. Sense codon reassignment offers the exciting possibility introducing artificial amino acids throughout the organism’s proteome.
Is orthogonality impairing functionality?
Although unusual tRNAPyl structure helps maintain orthogonality to host aaRSs and tRNAs, ncAA-tRNAPyl still has to be efficiently recognized by the host elongation factor and the ribosome. It was recently recognized that tRNAPyl might not be an optimal substrate for the bacterial EF-Tu. Improved tRNAPyl binding was achieved through mutagenesis of nucleotides in the acceptor and T-stems (which are expected to interact with elongation factor)[65]. When using the optimized version of tRNAPyl (tRNAPyl-opt) the authors were able to show increased multiple AcK incorporation into three different proteins by an engineered PylRS variant. In vitro kinetic measurements with PylRS variants showed that tRNAPyl-opt is basically undistinguishable from wild-type tRNAPyl thus inferring that the improved suppression efficiency of tRNAPyl-opt originates from its improved interaction with EF-Tu.
Conclusion
The Pyl-OTS is a powerful tool for genetic code expansion. To a large extent this rests on the astonishing polyspecificity of the enzyme toward ncAAs and the orthogonality of Pyl-OTS in the majority of common host organisms. While biochemical and structural studies have aided in our understanding of Pyl and ncAA binding, a thorough kinetic analysis is still lacking due to the limited availability (through chemical synthesis) of Pyl and the unexplored possibility of large scale preparation of Pyl from in vivo sources. As shown above, the Pyl-OTS offers a robust methodology for ncAA incorporation, and it has been widely applied. However, much still remains to be discovered to really make the Pyl-OTS a superior synthetic biology tool for GCE.
Acknowledgments
We thank Chenguang Fan, Sergey Melnikov and Oscar Vargas-Rodriguez for discussions. Work in the authors’ laboratory was supported by grants from the National Institute of General Medical Sciences (GM22854), and from the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the Department of Energy (DE-FG02-98ER20311).
Footnotes
Dedication
Dedicated to the memory of Ivana Weygand-Đurašević, an outstanding scientist, remarkable and caring mentor, and inspiring collaborator. She imparted the pursuit of rigorous science and the love of tRNA in many of us.
References
- 1.a) Liu CC, Schultz PG. Annu. Rev. Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]; b) Rovner AJ, Haimovich AD, Katz SR, Li Z, Grome MW, Gassaway BM, Amiram M, Patel JR, Gallagher RR, Rinehart J, Isaacs FJ. Nature. 2015;518:89–93. doi: 10.1038/nature14095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ambrogelly A, Palioura S, Söll D. Nat. Chem. Biol. 2007;3:29–35. doi: 10.1038/nchembio847. [DOI] [PubMed] [Google Scholar]
- 3.a) Chin JW, Cropp TA, Chu S, Meggers E, Schultz PG. Chem. Biol. 2003;10:511–519. doi: 10.1016/s1074-5521(03)00123-6. [DOI] [PubMed] [Google Scholar]; b) Fechter P, Rudinger-Thirion J, Tukalo M, Giege R. Eur. J. Biochem. 2001;268:761–767. doi: 10.1046/j.1432-1327.2001.01931.x. [DOI] [PubMed] [Google Scholar]
- 4.a) Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart J, Söll D. Science. 2011;333:1151–1154. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rogerson DT, Sachdeva A, Wang K, Haq T, Kazlauskaite A, Hancock SM, Huguenin-Dezot N, Muqit MM, Fry AM, Bayliss R, Chin JW. Nat. Chem. Biol. 2015;11:496–503. doi: 10.1038/nchembio.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Englert M, Moses S, Hohn M, Ling J, O'Donoghue P, Söll D. FEBS Lett. 2013;587:3360–3364. doi: 10.1016/j.febslet.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang R, Krzycki JA. J. Biol. Chem. 2012;287:32738–32746. doi: 10.1074/jbc.M112.396754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herring S, Ambrogelly A, Gundllapalli S, O'Donoghue P, Polycarpo CR, Söll D. FEBS Lett. 2007;581:3197–3203. doi: 10.1016/j.febslet.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kavran JM, Gundllapalli S, O'Donoghue P, Englert M, Söll D, Steitz TA. Proc. Natl. Acad. Sci. U.S.A. 2007;104:11268–11273. doi: 10.1073/pnas.0704769104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Lee MM, Jiang R, Jain R, Larue RC, Krzycki J, Chan MK. Biochemical and Biophysical Research Communications. 2008;374:470–474. doi: 10.1016/j.bbrc.2008.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nozawa K, O'Donoghue P, Gundllapalli S, Araiso Y, Ishitani R, Umehara T, Söll D, Nureki O. Nature. 2009;457:1163–1167. doi: 10.1038/nature07611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yanagisawa T, Umehara T, Sakamoto K, Yokoyama S. Chembiochem. 2014;15:2181–2187. doi: 10.1002/cbic.201402266. [DOI] [PubMed] [Google Scholar]
- 11.Moras D. Trends Biochem. Sci. 1992;17:159–164. doi: 10.1016/0968-0004(92)90326-5. [DOI] [PubMed] [Google Scholar]
- 12.Herring S, Ambrogelly A, Polycarpo CR, Söll D. Nucleic Acids Res. 2007;35:1270–1278. doi: 10.1093/nar/gkl1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ambrogelly A, Gundllapalli S, Herring S, Polycarpo C, Frauer C, Söll D. Proc. Natl. Acad. Sci. U.S.A. 2007;104:3141–3146. doi: 10.1073/pnas.0611634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polycarpo C, Ambrogelly A, Berube A, Winbush SM, McCloskey JA, Crain PF, Wood JL, Söll D. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12450–12454. doi: 10.1073/pnas.0405362101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan J, O'Donoghue P, Ambrogelly A, Gundllapalli S, Sherrer RL, Palioura S, Simonovic M, Söll D. FEBS Lett. 2010;584:342–349. doi: 10.1016/j.febslet.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krzycki JA. Curr. Opin. Chem. Biol. 2013;17:619–625. doi: 10.1016/j.cbpa.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 17.a) Hertweck C. Angew. Chem. Int. Ed. Engl. 2011;50:9540–9541. doi: 10.1002/anie.201103769. [DOI] [PubMed] [Google Scholar]; b) Gaston MA, Zhang L, Green-Church KB, Krzycki JA. Nature. 2011;471:647–650. doi: 10.1038/nature09918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paul L, Ferguson DJ, Krzycki JA. J. Bacteriol. 2000;182:2520–2529. doi: 10.1128/jb.182.9.2520-2529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinemann IU, O'Donoghue P, Madinger C, Benner J, Randau L, Noren CJ, Söll D. Proc. Natl. Acad. Sci. U.S.A. 2009;106:21103–21108. doi: 10.1073/pnas.0912072106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krzycki JA. Curr. Opin. Microbiol. 2005;8:706–712. doi: 10.1016/j.mib.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 21.Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S. J. Mol. Biol. 2008;378:634–652. doi: 10.1016/j.jmb.2008.02.045. [DOI] [PubMed] [Google Scholar]
- 22.a) Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S. Chem. Biol. 2008;15:1187–1197. doi: 10.1016/j.chembiol.2008.10.004. [DOI] [PubMed] [Google Scholar]; b) Guo LT, Wang YS, Nakamura A, Eiler D, Kavran JM, Wong M, Kiessling LL, Steitz TA, O'Donoghue P, Söll D. Proc. Natl. Acad. Sci. U.S.A. 2014;111:16724–16729. doi: 10.1073/pnas.1419737111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Polycarpo CR, Herring S, Berube A, Wood JL, Söll D, Ambrogelly A. FEBS Lett. 2006;580:6695–6700. doi: 10.1016/j.febslet.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flügel V, Vrabel M, Schneider S. PLoS One. 2014;9:e96198. doi: 10.1371/journal.pone.0096198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi T, Yanagisawa T, Sakamoto K, Yokoyama S. J. Mol. Biol. 2009;385:1352–1360. doi: 10.1016/j.jmb.2008.11.059. [DOI] [PubMed] [Google Scholar]
- 26.Li YM, Yang MY, Huang YC, Li YT, Chen PR, Liu L. ACS Chem. Biol. 2012;7:1015–1022. doi: 10.1021/cb300020s. [DOI] [PubMed] [Google Scholar]
- 27.Wan W, Tharp JM, Liu WR. Biochim. Biophys. Acta. 2014;1844:1059–1070. doi: 10.1016/j.bbapap.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho JM, Reynolds NM, Rivera K, Connolly M, Guo LT, Ling J, Pappin DJ, Church GM, Söll D. ACS Synth. Biol. 2015 doi: 10.1021/acssynbio.5b00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen DP, Lusic H, Neumann H, Kapadnis PB, Deiters A, Chin JW. J. Am. Chem. Soc. 2009;131:8720–8721. doi: 10.1021/ja900553w. [DOI] [PubMed] [Google Scholar]
- 30.Fekner T, Li X, Lee MM, Chan MK. Angew. Chem. Int. Ed. Engl. 2009;48:1633–1635. doi: 10.1002/anie.200805420. [DOI] [PubMed] [Google Scholar]
- 31.a) Plass T, Milles S, Koehler C, Schultz C, Lemke EA. Angew. Chem. Int. Ed. Engl. 2011;50:3878–3881. doi: 10.1002/anie.201008178. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Plass T, Milles S, Koehler C, Szymanski J, Mueller R, Wiessler M, Schultz C, Lemke EA. Angew. Chem. Int. Ed. Engl. 2012;51:4166–4170. doi: 10.1002/anie.201108231. [DOI] [PubMed] [Google Scholar]
- 32.Borrmann A, Milles S, Plass T, Dommerholt J, Verkade JM, Wiessler M, Schultz C, van Hest JC, van Delft FL, Lemke EA. ChemBioChem. 2012;13:2094–2099. doi: 10.1002/cbic.201200407. [DOI] [PubMed] [Google Scholar]
- 33.Schneider S, Gattner MJ, Vrabel M, Flügel V, López-Carrillo V, Prill S, Carell T. Chembiochem. 2013;14:2114–2118. doi: 10.1002/cbic.201300435. [DOI] [PubMed] [Google Scholar]
- 34.Neumann H, Peak-Chew SY, Chin JW. Nat. Chem. Biol. 2008;4:232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- 35.Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. Mol. Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Umehara T, Kim J, Lee S, Guo LT, Söll D, Park HS. FEBS Lett. 2012;586:729–733. doi: 10.1016/j.febslet.2012.01.029. [DOI] [PubMed] [Google Scholar]
- 37.a) Odoi KA, Huang Y, Rezenom YH, Liu WR. PLoS One. 2013;8:e57035. doi: 10.1371/journal.pone.0057035. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) O'Donoghue P, Prat L, Heinemann IU, Ling J, Odoi K, Liu WR, Söll D. FEBS Lett. 2012;586:3931–3937. doi: 10.1016/j.febslet.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hancock SM, Uprety R, Deiters A, Chin JW. J. Am. Chem. Soc. 2010;132:14819–14824. doi: 10.1021/ja104609m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.a) Ko JH, Wang YS, Nakamura A, Guo LT, Söll D, Umehara T. FEBS Lett. 2013;587:3243–3248. doi: 10.1016/j.febslet.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang YS, Russell WK, Wang Z, Wan W, Dodd LE, Pai PJ, Russell DH, Liu WR. Mol. Biosyst. 2011;7:714–717. doi: 10.1039/c0mb00217h. [DOI] [PubMed] [Google Scholar]
- 40.Dumas A, Lercher L, Spicer CD, Davis BG. Chem.Sci. 2015;6:50–69. doi: 10.1039/c4sc01534g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.a) Chou CJ, Uprety R, Davis L, Chin JW, Deiters A. Chem.Sci. 2011;2:480–483. [Google Scholar]; b) Zhang M, Lin S, Song X, Liu J, Fu Y, Ge X, Fu X, Chang Z, Chen PR. Nat. Chem. Biol. 2011;7:671–677. doi: 10.1038/nchembio.644. [DOI] [PubMed] [Google Scholar]
- 42.Nguyen DP, Garcia Alai MM, Virdee S, Chin JW. Chem. Biol. 2010;17:1072–1076. doi: 10.1016/j.chembiol.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 43.Gautier A, Deiters A, Chin JW. J. Am. Chem. Soc. 2011;133:2124–2127. doi: 10.1021/ja1109979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arbely E, Torres-Kolbus J, Deiters A, Chin JW. J. Am. Chem. Soc. 2012;134:11912–11915. doi: 10.1021/ja3046958. [DOI] [PubMed] [Google Scholar]
- 45.Groff D, Chen PR, Peters FB, Schultz PG. ChemBioChem. 2010;11:1066–1068. doi: 10.1002/cbic.200900690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang YS, Wu B, Wang Z, Huang Y, Wan W, Russell WK, Pai PJ, Moe YN, Russell DH, Liu WR. Mol. Biosyst. 2010;6:1557–1560. doi: 10.1039/c002155e. [DOI] [PubMed] [Google Scholar]
- 47.Virdee S, Kapadnis PB, Elliott T, Lang K, Madrzak J, Nguyen DP, Riechmann L, Chin JW. J. Am. Chem. Soc. 2011;133:10708–10711. doi: 10.1021/ja202799r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greiss S, Chin JW. J. Am. Chem. Soc. 2011;133:14196–14199. doi: 10.1021/ja2054034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gatfield D, Unterholzner L, Ciccarelli FD, Bork P, Izaurralde E. EMBO J. 2003;22:3960–3970. doi: 10.1093/emboj/cdg371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bianco A, Townsley FM, Greiss S, Lang K, Chin JW. Nat. Chem. Biol. 2012;8:748–750. doi: 10.1038/nchembio.1043. [DOI] [PubMed] [Google Scholar]
- 51.Mukai T, Kobayashi T, Hino N, Yanagisawa T, Sakamoto K, Yokoyama S. Biochem. Biophys. Res. Commun. 2008;371:818–822. doi: 10.1016/j.bbrc.2008.04.164. [DOI] [PubMed] [Google Scholar]
- 52.Schmidt O, Mao J, Ogden R, Beckmann J, Sakano H, Abelson J, Söll D. Nature. 1980;287:750–752. doi: 10.1038/287750a0. [DOI] [PubMed] [Google Scholar]
- 53.Wang Q, Wang L. J. Am. Chem. Soc. 2008;130:6066–6067. doi: 10.1021/ja800894n. [DOI] [PubMed] [Google Scholar]
- 54.Hao B, Zhao G, Kang PT, Soares JA, Ferguson TK, Gallucci J, Krzycki JA, Chan MK. Chem. Biol. 2004;11:1317–1324. doi: 10.1016/j.chembiol.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 55.Wong ML, Guzei IA, Kiessling LL. Org. Lett. 2012;14:1378–1381. doi: 10.1021/ol300045c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Longstaff DG, Larue RC, Faust JE, Mahapatra A, Zhang L, Green-Church KB, Krzycki JA. Proc. Natl. Acad. Sci. U.S.A. 2007;104:1021–1026. doi: 10.1073/pnas.0610294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ehrlich M, Gattner MJ, Viverge B, Bretzler J, Eisen D, Stadlmeier M, Vrabel M, Carell T. Chemistry. 2015;21:7701–7704. doi: 10.1002/chem.201500971. [DOI] [PubMed] [Google Scholar]
- 58.Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA, Rohland N, Schultz PG, Jacobson JM, Rinehart J, Church GM, Isaacs FJ. Science. 2013;342:357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heinemann IU, Rovner AJ, Aerni HR, Rogulina S, Cheng L, Olds W, Fischer JT, Söll D, Isaacs FJ, Rinehart J. FEBS Lett. 2012;586:3716–3722. doi: 10.1016/j.febslet.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elsässer SJ, Ernst RJ, Walker OS, Chin JW. Nature Methods. 2016;13 doi: 10.1038/nmeth.3701. 158-+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aerni HR, Shifman MA, Rogulina S, O'Donoghue P, Rinehart J. Nucleic Acids Res. 2015;43 doi: 10.1093/nar/gku1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krishnakumar R, Prat L, Aerni HR, Ling J, Merryman C, Glass JI, Rinehart J, Söll D. Chembiochem. 2013;14:1967–1972. doi: 10.1002/cbic.201300444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bohlke N, Budiša N. FEMS Microbiol. Lett. 2014;351:133–144. doi: 10.1111/1574-6968.12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.a) Zeng Y, Wang W, Liu WR. ChemBioChem. 2014;15:1750–1754. doi: 10.1002/cbic.201400075. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mukai T, Yamaguchi A, Ohtake K, Takahashi M, Hayashi A, Iraha F, Kira S, Yanagisawa T, Yokoyama S, Hoshi H, Kobayashi T, Sakamoto K. Nucleic Acids Research. 2015;43:8111–8122. doi: 10.1093/nar/gkv787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fan C, Xiong H, Reynolds NM, Söll D. Nucleic Acids Res. 2015;43:e156. doi: 10.1093/nar/gkv800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li WT, Mahapatra A, Longstaff DG, Bechtel J, Zhao G, Kang PT, Chan MK, Krzycki JA. J. Mol. Biol. 2009;385:1156–1164. doi: 10.1016/j.jmb.2008.11.032. [DOI] [PubMed] [Google Scholar]