

Summary
The mechanistic Target Of Rapamycin (mTOR) integrates numerous stimuli and coordinates the adaptive response of many cellular processes. To accomplish this, mTOR associates with distinct co-factors that determine its signaling output. While multiple such co-factors are known, in many cases their function and regulation remain opaque. The MAPK-interacting kinase (MNK) contributes to rapamycin resistance in cancer cells. Here we demonstrate that MNK sustains mTORC1 activity upon rapamycin treatment and contributes to mTORC1 signaling following T cell activation and growth stimuli in cancer cells. We determined that MNK engages in a complex with mTORC1, promotes mTORC1 association with the Phosphatidyl Inositol 3′ Kinase-related Kinase (PIKK) stabilizer, TELO2, and facilitates mTORC1:substrate binding. Moreover, our data suggest that DEPTOR, the endogenous inhibitor of mTOR, opposes mTORC1:substrate association by preventing TELO2:mTORC1 binding. Thus, MNK orchestrates counterbalancing forces that regulate mTORC1 enzymatic activity.
Keywords: mTOR, MNK, Rapamycin, TELO2, DEPTOR, DDB1, Raptor, T Cell, PIKK, S6 Kinase
Graphical Abstract
Introduction
The atypical Ser-Thr kinase mTOR, which coordinates cellular homeostasis and growth (Laplante and Sabatini, 2012), is controlled by transient protein aggregates that fine-tune signal output to enable multi-faceted adaptations to diverse stimuli. The most thoroughly studied such ensemble is mTOR Complex 1 (C1), which includes Raptor, enabling mTORC1:substrate binding; GβL; PRAS40; DEPTOR, an inhibitor of mTORC1 and C2; and TELO2:TTI1 (Laplante and Sabatini, 2012). It is unknown how DEPTOR suppresses mTOR. TELO2 associates with all PIKKs (Takai et al., 2007), but has not been shown to modulate PIKK enzymatic activity/function.
Cancer cell resistance to the mTORC1 inhibitor rapamycin involves MNK activation via a feedback signaling loop elicited by rapamycin (Carracedo et al., 2008; Wang et al., 2007). MNK1 and 2 are ubiquitously expressed kinases activated by p38α (MNK1) and ERK1/2 (MNK1/2) MAPKs. They are often linked to survival signals in response to a wide variety of stimuli (Adesso et al., 2013; Chrestensen et al., 2007; Ueda et al., 2010). The MNKs are best known for phosphorylating the 5′ 7-methylguanosine (cap)-binding protein, eukaryotic initiation factor (eIF) 4E, by binding to the eIF4E-binding partners eIF4G1/2 (Pyronnet et al., 1999). We recently reported that MNK regulates mTORC1 signaling (Brown et al., 2014a; Brown et al., 2014b). Indeed, a functional relationship of MNK with mTORC1 is well documented. (Eckerdt et al., 2014; Grzmil et al., 2014; Marzec et al., 2011).
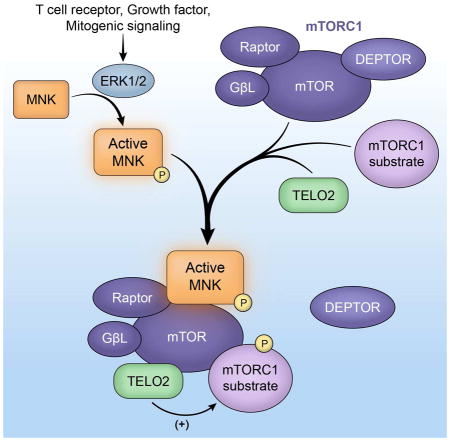
In this work, we show mechanistic evidence that MNK stimulates mTORC1 signaling. We defined a MNK-associated mTORC1 sub-complex and discovered that active MNK facilitates mTORC1 association with its substrates and with TELO2, while discouraging DEPTOR binding. We discovered that MNK-mediated regulation of TELO2:mTORC1 binding critically contributes to T cell activation and mitogenic signaling. Our data suggest physiological roles for TELO2 in supporting mTORC1:substrate association (without altering other mTORC1 associations); and, conversely, for DEPTOR in displacing TELO2 and countering mTORC1:substrate binding. Thus, we uncovered regulation that functionally links DEPTOR and TELO2, and determines mTORC1:substrate proximity in a MNK-responsive manner.
Results
Evidence suggesting a connection between MNK and mTORC1 includes 1) synergy between mTORC1 and MNK inhibitors in restricting cancer cell growth/viability; 2) reduction in mTORC1 activity following MNK inhibition (Brown et al., 2014b; Grzmil et al., 2014; Teo et al., 2015); and 3) activation of MNK via a feedback loop after rapamycin-mediated mTORC1 inhibition (Joubert et al., 2015; Wang et al., 2007). Catalytic mTOR inhibitors do not synergize with MNK inhibition at restricting cancer cell growth, suggesting that MNK acts through regulation of mTOR (Eckerdt et al., 2014; Grzmil et al., 2014). In this study, we sought to elucidate the mechanism(s) underlying MNK-mediated mTORC1 regulation.
MNK inhibition enhances rapamycin-mediated mTORC1 inhibition of growth factor signaling and T cell activation
Due to the preponderance of evidence supporting MNK and mTORC1 inhibitor synergy, we tested whether MNK inhibition alters suppression of mTORC1 signals by rapamycin. To test off-target effects of the prototypical MNK inhibitor, CGP57380 (Knauf et al., 2001), we first compared combination of rapamycin with CGP57380 in either wt or MNK1/2 double knockout (dko) mouse embryo fibroblasts (MEFs) (Fig. 1A) (Ueda et al., 2004). Rapamycin reduced mTORC1 signaling in wt and dko MEFs as shown by the (rapamycin-sensitive) phosphorylation of ribosomal protein S6 kinase (S6K) (Fig. 1A). It also increased p-eIF4E(S209) in wt MEFs, due to feedback to MNK upon rapamycin treatment (Wang et al., 2007). Phosphorylation of rapamycin-insensitive substrates unc51-like autophagy-activating kinase 1 (ULK1) (S757) and 4EBP(S65) was only modestly reduced (Fig. 1A) (Kang et al., 2013). Combining rapamycin with CGP57380 incrementally diminished p-ULK1(S757) and p-4EBP(S65) only in wt MEFs (Fig. 1A), indicating that mTORC1 inhibition in wt MEFs by CGP57380 was due to inhibition of MNK. We conducted similar tests in glioma, breast cancer and melanoma cell lines: in all cases MNK co-inhibition enhanced rapamycin-mediated suppression of p-ULK1(S757) and p-4EBP1(S65) (Fig. 1B). Rapamycin treatment induced MNK-mediated eIF4E(S209) phosphorylation in most cancer cell lines (Fig. 1A, B) (Wang et al., 2007). These data indicate that MNK signaling sustains mTORC1 activity upon rapamycin treatment of cancer cells.
Figure 1. Rapamycin and CGP57380 synergy in inhibiting mTORC1 in MEFs, cancer cells and T cells.

(A) Wt and MNK1/2 dko MEFs or (B) malignant glioma (U87, Du54), breast cancer (MDA-MB231, Sum149) or melanoma (DM440, DM6) cells were treated with DMSO, rapamycin and CGP57380, harvested and analyzed by immunoblot as shown. Quantitations represent the average of two independent series normalized to the rapamycin-only control.
(C, D) Primary mouse splenocytes were stimulated with TPA (100nM) and treated with rapamycin, torin2 and CGP57380 as shown. Cell pellets were lysed for immunoblot analyses (C) and supernatants were collected to test IL-2 and IFN-γ concentrations (ELISA; D). Quantitation of immunoblot and ELISA signals represent the average of two experiments. The quantitative values were normalized between experiments using non-stimulated, mock (DMSO)-treated controls at each time point.
Rapamycin/‘rapalogs’ are standard of care agents for immunosuppression after organ transplantation. Their effects are largely attributable to mTORC1 inhibition in T cells, preventing their proliferation (Kuo et al., 1992). To test whether MNK inhibition exacerbated the immunosuppressive effects of rapamycin, we used primary mouse splenocytes stimulated with the phorbol ester 12-O-Tetradecanoylphorbol-13-acetate (TPA), followed by treatment with DMSO, rapamycin, the catalytic mTOR inhibitor torin2 and CGP57380 as shown (Fig. 1C). TPA caused a modest increase in mTORC1 signaling as evident by p-4EBP1(S65). Similar to the cancer cell lines (Fig. 1B), rapamycin-mediated suppression of p-ULK1(S757) and p-4EBP1(S65) was enhanced by CGP57380 addition (Fig. 1C). This coincided with tempered IL-2 and IFN-γ production, indicators of T cell activation/proliferation (Fig. 1D). A catalytic inhibitor of mTOR (torin2) abolished p-ULK1(S757) and p-4EBP1(S65) (Fig. 1C) along with IL-2 and IFN-y production (Fig. 1D). These findings suggest that MNK modulates mTOR to regulate T cell activity.
MNK knockout/depletion/inhibition reduces basal mTORC1 signaling while increasing DEPTOR abundance
Next we tested the effects of MNK depletion on mTORC1 signaling in HeLa cells (Fig. 2A). Depletion of either MNK1, MNK2, or MNK1/2 broadly attenuated mTORC1 signaling and led to a substantial increase of DEPTOR protein levels (~9-fold in MNK1/2 co-depleted cells; Fig. 2A). DEPTOR is destabilized by mTOR-dependent phosphorylation, possibly as positive feedback to maintain mTOR activation (Gao et al., 2011; Peterson et al., 2009). Thus, elevated DEPTOR levels may reflect reduced mTORC1 activity. We also tested wt, MNK1, 2 and MNK1/2 ko MEFs for changes in basal levels of mTOR-associated proteins (Fig. 2B). The only mTORC1-associated protein affected by MNK ko was DEPTOR, exhibiting >20-fold increased levels in MNK2 ko MEFs (Fig. 2B). It is unclear why maximal DEPTOR induction required MNK1/2 co-depletion in HeLa cells vs. only MNK2 deletion in MEFs. These differences could be due to compensatory adaptations in MEFs (derived of explants from MNK ko mice), which experienced far more cell divisions/adaptation pressure than HeLa cells 72h after siRNA transfection.
Figure 2. MNK regulates mTORC1 signaling and DEPTOR abundance.

(A) HeLa cells were treated with ctrl (−), MNK1, MNK2, or MNK1/2 siRNAs (72h), harvested and analyzed by immunoblot. Quantitation represents the average of three independent series normalized by setting ctrl siRNA values to 1.
(B) Wt, MNK1, MNK2, and MNK1/2 dko MEFs were seeded into 35mm3 dishes (5 × 105 cells per dish) and harvested the next day for analysis of mTORC1-relevant proteins by immunoblot; quantitation represents the average of four independent experiments.
(C) MEFs plated as in (B) were treated with mock or IGF1 as shown, harvested, and analyzed for mTORC1 and AKT activity by immunoblot. Quantitation represents the average of two experiments normalized by setting each untreated control to 1.
HeLa (D) or HEK 293 (E) cells were treated with CGP57380 (10μM) and analyzed at the designated intervals by immunoblot. Quantitations of p-S6(SS240/4), p-4EBP1(S65) and DEPTOR represents the average of three experiments (immunoblots for a representative assay are shown in Figure S1).
DEPTOR overexpression paradoxically activates AKT, due to enhanced mTORC1 inhibition that prevents mTORC1 negative feedback to AKT (Peterson et al., 2009). Consistent with these observations, MNK-depleted MEFs exhibited AKT hyper-activation (compared to wt MEFs) after stimulation with IGF1 (Insulin like growth factor-1), as indicated by p-AKT(T308/S473) and p-PRAS40(T246), an AKT substrate (Fig. 2C). This correlated with DEPTOR levels: MNK2 ko MEFs with the highest DEPTOR expression had the most robust AKT response to IGF1 (Fig. 2B, C). Even with elevated AKT signaling in MNK ko MEFs, downstream mTORC1 phosphorylation of 4EBP1(S65) in response to IGF1 remained similar to wt MEFs (Fig. 2C). This suggests that in MNK ko MEFs, the absence of MNK may temper mTORC1 activity, despite hyper-active AKT.
DEPTOR induction was observed with MNK knockout (MEFs) and short-term >95% MNK depletion (Fig. 2A, B), but we also tested the DEPTOR response to transient MNK inhibition with CGP57380 in HeLa (Fig. 2D) and HEK293 cells (Fig. 2E). The drug rapidly reduced mTORC1 signaling in both cell lines (Fig. 2D, E; see Fig. S1 for corresponding immunoblots). While mTORC1 signals were consistently reduced by CGP57380, changes in DEPTOR stability were less pronounced in HEK293 cells and absent in HeLa cells (Fig. 2D, E). These findings suggest that MNK acts primarily on mTORC1, rather than DEPTOR, and that DEPTOR induction in MNK ko MEFs may be due to chronic mTORC1 suppression. Thus, MNK activity modulates basal mTORC1 signaling.
MNK inhibition reduces mTORC1:substrate association while enhancing mTORC1:DEPTOR binding
To determine how MNK influences mTORC1 interactions, we generated HEK293 cell lines with doxycycline (dox)-inducible expression of Flag-mTOR or HA-Raptor and performed immunoprecipitations (IPs) following CGP57380 vs. mock (DMSO) treatment. We found that MNK inhibition did not alter mTORC1 or C2 integrity, as mTOR associations with Raptor, GβL, PRAS40, DEPTOR, or Sin1 were unchanged (Fig. 3A). However, ULK1 and eukaryotic initiation factor 3f (eIF3f) lost association with mTOR after CGP57380 treatment (Fig. 3A, B). EIF3f serves as the scaffold linking mTORC1 and S6K (Holz et al., 2005). Phosphorylation of ULK1(S757) by mTORC1 inhibits autophagy as part of a broadly anabolic cellular program consistent with phosphorylation of 4EBP1(S65) and S6K(T389) (Kim et al., 2011). We were unable to detect S6K or 4EBP1 association with mTOR by Flag-co-IP. HA-Raptor IP confirmed that MNK inhibition reduced Raptor:ULK1 association (Fig. 3C, D); we could not detect endogenous eIF3f, S6K or 4EBP co-IP with HA-Raptor. Our assays indicate reduced mTORC1:substrate association upon MNK inhibition, rather than potential ULK1 binding with free Raptor. This is because reduced Flag-mTOR:ULK1/eIF3f co-IP was not accompanied by a loss of Raptor binding (Raptor is required for mTORC1:substrate binding; Fig. 3A).
Figure 3. MNK inhibition reduces mTORC1 association with its substrates and increases DEPTOR association with mTORC1.

(A, B) Dox-inducible Flag-mTOR expressing HEK293 cells were treated with mock (DMSO) or CGP57380 as shown. Lysates were subjected to Flag-IP and immunoblot; Raptor, ULK1, and eIF3f binding levels are quantitated in (B).
(C) Dox-inducible HA-Raptor expressing HEK293 cells were treated as in (A) and lysates were subjected to HA-IP and immunoblot. Quantitation of DEPTOR, ULK1 and mTOR binding is shown in (D).
(E) Dox-inducible HA-Raptor expressing HEK293 cells were transfected with myc-4EBP1, treated with DMSO or CGP5780, harvested, and the lysates used for HA-IP and immunoblot to detect co-IP of myc-4EBP1 with Raptor. HA-Raptor:myc-4EBP1 co-IP was quantitated (F).
(G) HEK293 cells were transfected with Flag-DEPTOR, treated with DMSO or CGP57380, and subjected to Flag-IP and immunoblot to detect changes in mTORC1 vs. C2 interactions; Flag-DEPTOR:Raptor co-IP was quantitated (H).
(B, D, F, H) Quantitation of immunoblot signals represent the average of 3 (B, D, F) or 2 (H) independent experiments. The values were normalized to the mock (DMSO), dox-induced control.
To test if 4EBP binding to mTORC1 responds to MNK inhibition, we performed HA-Raptor IP in cells with myc-4EBP1 overexpression. Myc-4EBP1:Raptor binding declined with CGP57380 treatment (Fig. 3E, F) similar to ULK1 (Fig. 3C, D). In contrast to Flag-mTOR IP, where DEPTOR co-IP was unchanged by CGP57380 (up to 2h), DEPTOR association with HA-Raptor increased substantially (~5-fold; Fig. 3C, D). Since DEPTOR binds mTOR directly (Peterson et al., 2009), CGP57380 unresponsive Flag-mTOR:DEPTOR co-IP is likely due to DEPTOR binding to mTOR outside of the C1 complex. HA-Raptor:DEPTOR co-IP, however, will only pull down mTORC1-bound DEPTOR (Raptor associates with DEPTOR through mTOR). Flag-DEPTOR IP confirmed that MNK inhibition preferentially induced DEPTOR-association with mTORC1, as DEPTOR binding to Raptor increased relative to Rictor (Fig. 3G, F). Thus, MNK inversely regulates the association of mTORC1 with its substrates and with DEPTOR.
MNK1/2 bind to mTORC1, TELO2, and DDB1 through an eIF4G-independent mechanism
Previous co-IP analyses suggested that MNK2 may interact with mTORC1 (Hu et al., 2012). To define MNK interactions that may explain its effects on mTORC1, we transfected dox-inducible Flag-mTOR expressing cells with HA-tagged MNK variants (Fig. 4A). The variants included wt MNK1 and MNK2, MNK1(D191A) (catalytically inactive), MNK1(Δ4G) (lacking the eIF4G binding domain), and MNK1(T344D) (constitutively active) (Fig. 4A). All HA-tagged MNK variants co-immunoprecipitated with Flag-mTOR (Fig. 4A). Interaction with Flag-mTOR was relatively weak for wt MNK1 and MNK1(D191) and much stronger for MNK1(Δ4G), MNK1(T344D), and MNK2 (Fig. 4A). This suggests that MNK binding to mTOR is independent of MNK:eIF4G binding and is favored by MNK catalytic activity.
Figure 4. MNK1/2 associates with mTORC1 and mTOR-regulatory cofactors TELO2:DDB1.

(A) Top panel: kinase activity and eIF4G-binding characteristics of MNK variants used. *MNK1(Δ4G) kinetic activity [phosphorylation of eIF4E(S209)] is abolished due to lack of eIF4G binding; #MNK2 lacks some auto-inhibitory features of MNK1 and, thus, exhibits high basal eIF4G-binding/eIF4E(S209) phosphorylation relative to MNK1 (Jauch et al., 2006; Shveygert et al., 2010).
Bottom panel: dox-inducible Flag-mTOR expressing HEK293 cells were transfected with HA-tagged wt MNK1, MNK1(D191A), MNK1(Δ4G), MNK1(T344D), or wt MNK2 without (mock) or with dox induction. Lysates were subjected to Flag-IP and immunoblot.
(B) HEK293 cells were transfected with empty vector, Flag-tagged wt MNK1, MNK1(D191A), MNK1(T344D), or wt MNK2. Lysates were subjected to Flag-IP and immunoblot to detect associations with Raptor, mTOR and other mTORC1 co-factors.
(C) HEK293 cells were transfected with wt Flag-tagged MNK1-a, MNK1-a(Δ4G), or wt MNK1-b. Flag-IP was performed from lysates followed by immunoblot analysis to determine mTOR and TELO2 interactions.
(A–C) Experiments were performed in 3 separate series; the results of representative assays are shown. See also Figures S2 and S3.
To complement these studies, we used Flag-MNK variants (wt, D191A, T344D, and MNK2) for Flag co-IP of endogenous mTOR and mTORC1-associated proteins (Fig. 4B). All MNK variants associated with Raptor and mTOR; again, constitutively active MNK2/MNK1(T344D) exhibited stronger binding than wt or kinase dead MNK1 (Fig. 4B). To identify MNK-associated proteins that explain MNK’s effects on mTORC1: substrate binding we also tested for co-IP of known mTOR regulators. This yielded specific, high-affinity co-IP of two mTORC1-associated factors: the DNA Damage-Binding protein 1 (Hussain et al., 2013) (DDB1) and its binding partner DCAF6; and TELO2 (Horejsi et al., 2010) and its binding partners TTI1 and ATM (Fig. 4B). We did not observe MNK co-IP with Rictor, DEPTOR, PRAS40, or several other relevant proteins (Fig. S2). Furthermore, co-IP of other known TELO2 and DDB1 binding partners involved in their PIKK-stabilizing and ubiquitin ligase machinery complexes, respectively, was not detected (Fig. S2). These findings suggest that MNK interactions primarily involve TELO2:DDB1 and occur within the context of mTORC1.
To confirm that MNK:mTORC1 interactions are independent of eIF4G-binding, and favored by MNK catalytic activity, we tested endogenous mTOR and TELO2 co-IP after transient transfection of Flag-wt MNK1-a, MNK1-b, and the MNK1(Δ4G) mutant (Fig. 4C). MNK1 and 2 occur in -a and -b splice variants; MNK1/2-b have higher basal activity than MNK1/2-a because they lack auto-inhibitory features. MNK1-b and MNK(Δ4G) exhibited much stronger mTOR/TELO2 association than MNK1-a, despite low MNK1-b expression after transfection (Fig. 4C). Analogous to MNK2 and MNK1(T344D), strong MNK-1b binding to mTOR/TELO2 may reflect its high basal activity. Increased binding of MNK1-a(Δ4G) with mTOR/TELO2 may be due to a lack of competitive eIF4G-binding or to the loss of inhibitory features in the MNK1-a N-terminus that may interfere with mTOR/TELO2 binding. MNK’s major (known) binding partners, eIF4G1/2, are present in excess over MNK1/2 in HeLa cells (Nagaraj et al., 2011). Thus, MNK association with mTORC1 components may be influenced by eIF4G:MNK binding. Tests in HEK293 cells with dox-inducible eIF4G1 depletion (Dobrikov et al., 2014), however, did not show altered MNK2 association with mTOR, TELO2 or DDB1 (Fig. S3). We were unable to detect Raptor co-IP with any of the MNK1 variants tested in Fig. 4C, corresponding with weak Raptor co-IP with wt Flag-MNK1 in prior assays (Fig. 4B). Our findings suggest that MNK1 associates primarily with TELO2 and/or mTOR within mTORC1, and that MNK association with TELO2 and mTOR is simultaneous.
MNK regulates TELO2:DDB1 association with mTORC1
To test whether MNK influences TELO2:DDB1 relations with mTORC1, we transfected cells with Flag-TELO2 (Fig. 5A) or Flag-DDB1 (Fig. 5B) and treated them with CGP57380. Interestingly, CGP57380 reduced TELO2:DDB1 association with MNK1 (Fig. 5A, B). It also reduced TELO2 association with Raptor, but not with mTOR or DDB1 (Fig. 5A). Likewise, MNK inhibition discouraged DDB1:Raptor and MNK1 binding, but not TELO2, mTOR or DDB2 (a co-factor of DDB1) associations (Fig. 5B). MNK inhibition did not change TELO2 and DDB1:mTOR co-IP (Fig. 5A, B), despite a reduction in Raptor co-IP, in a manner reminiscent of data in Figure 3A–D. There, CGP57380 enhanced DEPTOR co-IP with Raptor, but not mTOR (Fig. 3A, C). This suggests that, like DEPTOR, MNK regulation of TELO2:DDB1 is specific to mTORC1. Co-IP of mTOR with Flag-TELO2 likely does not respond to MNK inhibition (Fig. 5A, B), because TELO2 interacts with mTOR outside the mTORC1 context. Thus, MNK stimulates TELO2:DDB1 associations with both mTORC1 and itself.
Figure 5. MNK regulates the association of TELO2:DDB1 with a complex containing MNK, mTOR, and Raptor.

(A, B) Flag-TELO2 (A) or Flag-DDB1 (B) were transfected in HEK293 cells pretreated with DMSO or CGP57380 as shown. Lysates were subjected to Flag-IP and immunoblot.
(C) HEK293 cells were transfected with empty vector (PC3), Flag-MNK1, or Flag-MNK2 and treated with DMSO, TPA, or TPA + CGP57380 as shown. Flag-IP of lysates and immunoblot analysis was performed to determine changes in MNK associations defined in Figure 4.
(D) HEK293 cells were transfected with ctrl or TELO2-targeting siRNA followed by transfection with empty vector (PC3) or Flag-MNK2. MNK2 IP and immunoblot analysis were performed as in (C). The assay was performed 3 times; a representative series is shown.
(E) Dox-inducible HA-Raptor expressing HEK293 cells were treated with mock (DMSO) or TPA, lysates were prepared and subjected to HA-IP, and immunoprecipitated proteins were analyzed by immunoblot as shown.
(A–E) Quantitations are averages from 3 (A, C, E) or 2 (B) independent assays, normalizing control values to 1.
Previous reports point to such mTORC1-specific regulation of TELO2, including regulation of TELO2:mTORC1 association during serum starvation (Fernandez-Saiz et al., 2013). Our data suggest that TELO2:DDB1 form a complex that is cooperatively regulated. To further define these interactions, we performed Flag-MNK1 and MNK2 IPs and interrogated TELO2:DDB1:mTOR binding during MNK activation with TPA (Brown et al., 2014a) and inhibition (with CGP57380). In line with Figure 4, where MNK-binding to mTOR and TELO2:DDB1 correlated with MNK catalytic activity, TPA treatment enhanced MNK association with all members of the complex (Fig. 5C). CGP57380, in the presence of TPA, did not affect MNK:mTOR and Raptor associations but markedly reduced MNK binding with TELO2:DDB1, along with the DDB1 binding protein, DCAF6 (Fig. 5C). We found that Flag-MNK1 barely co-immunoprecipitated with these proteins (compared to MNK2; Fig. 5C). Yet, co-IP of endogenous MNK1 consistently occurred in Flag-TELO2 and DDB1 IPs (Fig. 5A, B). It is possible that stoichiometry changes due to ectopic overexpression of Flag-MNK1 may explain this contradiction. Our data further suggest that MNK contacts a TELO2:DDB1:mTORC1 complex and that MNK activity favors TELO2:DDB1 association with mTORC1.
To interrogate MNK’s primary contact within mTORC1, we used Flag-MNK2 IP following TELO2 depletion (Fig. 5D). Intriguingly, TELO2 depletion induced MNK-mediated eIF4E(S209) phosphorylation, likely due to feedback upon mTORC1 suppression (Wang et al., 2007) (Fig. 5D). We speculate that activation of this feedback signal in the presence of overexpressed MNK2 is the reason why mTORC1 signals did not change upon TELO2 depletion (Fig. 5D). Flag-MNK2 IP revealed enhanced mTOR and Raptor co-IP with MNK upon TELO2 depletion (Fig. 5D), indicating that 1) MNK primarily interacts with mTORC1, rather than TELO2:DDB1; 2) MNK activation (e.g. through feedback upon TELO2 depletion) favors MNK:mTORC1 interaction. The latter is supported by evidence that mTOR preferentially interacts with constitutively active MNK1(T344D) or MNK2 (Fig. 4A), and that MNK:mTOR interaction is stimulated by TPA (Fig. 5C). Altogether, these data suggest that catalytically active MNK interacts with mTOR to stimulate mTORC1 interaction with TELO2:DDB1.
TPA induced the association of MNK with TELO2:DBB1 and mTORC1 (Fig. 5C). Thus, if conditions of MNK activation facilitate these interactions, they should affect the broader composition of mTORC1. To address this, we TPA-stimulated HA-Raptor expressing cells followed by HA-Raptor IP (Fig. 5E). TPA induced HA-Raptor:TELO2 binding and, prominently, the association with the mTORC1 substrate ULK1. This was coincident with reduced HA-Raptor:DEPTOR binding (Fig. 5E) as previously reported (Yoon et al., 2015). These data further indicate that active MNK favors mTORC1:TELO2 and substrate association.
TELO2 enhances mTORC1:substrate association while DEPTOR inhibits mTORC1:TELO2 and substrate binding
Our data thus far indicate that MNK activity balances an inverse relationship between mTORC1:DEPTOR and mTORC1:TELO2/substrate binding (Fig. 3, 5). To assess if MNK regulates DEPTOR and substrate binding to mTORC1 through TELO2 and/or DDB1, we tested the impact of DEPTOR, TELO2, or DDB1 overexpression on mTORC1:ULK1 association. Flag-DEPTOR overexpression substantially reduced mTORC1 binding with ULK1 and TELO2 (Fig. 6A). Conversely, Flag-TELO2 overexpression increased HA-Raptor:ULK1 association without affecting Raptor association with DEPTOR (Fig. 6B). Thus, TELO2 does not enhance ULK1:Raptor association by displacing DEPTOR. It follows that the observed reduction of ULK1:Raptor binding upon Flag-DEPTOR overexpression (Fig. 6A) may be explained by displacement of TELO2. Ectopic Flag-DDB1 had no effect on DEPTOR, ULK1, or TELO2 association with Raptor (Fig. 6C). These observations suggest that DEPTOR displaces TELO2 and antagonizes substrate association with mTORC1. Accordingly, TELO2:mTORC1 binding stabilizes mTORC1:substrate binding.
Figure 6. TELO2 controls mTORC1:substrate association and explains MNK regulation of mTORC1.

(A–C) Dox-inducible, HA-Raptor expressing HEK293 cells were transfected with empty vector (PC3), Flag-DEPTOR (A), Flag-TELO2 (B), or Flag-DDB1 (C), lysed, and subjected to Flag-IP followed by immunoblot analysis.
(D) Flag-mTOR IP of HEK293 cells transfected with empty vector (PC3), untagged TELO2, or untagged TELO2 and DEPTOR combined. Co-IP of relevant proteins was assessed by immunoblot.
(E) Dox inducible, HA-Raptor expressing HEK293 cells were transfected with ctrl (-), TELO2, or MNK1-targeting siRNA. HA-Raptor IP was performed and associated proteins were analyzed by immunoblot.
(F) HEK293 cells were transfected with ctrl (-) or MNK1 and 2-targeting siRNA, transfected with empty vector or flag-TELO2. Cell lysates were assessed by Flag-TELO2 IP and immunoblot.
(G) HeLa cells were transfected with ctrl (-) or MNK1-targeting siRNA followed by transfection with empty vector (PC3), Flag-TELO2, or Flag-MNK1 (T344D). Lysates were analyzed by immunoblot for markers of mTORC1/MNK activation.
Quantitation represents the average of two (A, C, D) or three (B, E, F, G) independent experiments normalized by setting empty vector (PC3) control to 1 (A–C), setting TELO2 + PC3 control to 1 (D), or by setting ctrl siRNA samples to 1 (E–G).
To confirm these data with Flag-mTOR IPs, cells with dox-inducible Flag-mTOR expression were transfected with empty vector (PC3) or untagged TELO2 with and without simultaneous untagged DEPTOR overexpression (Fig. 6D). Corroborating the results of the HA-Raptor IPs (Fig. 6A, B), TELO2 overexpression alone enhanced mTOR binding with ULK1. Adding simultaneous DEPTOR overexpression suppressed TELO2 and ULK1 association with mTOR (Fig. 6D). Thus, TELO2 favors mTORC1:substrate binding and is antagonized by DEPTOR. Therefore, MNK-mediated stabilization of TELO2:mTORC1 may explain MNK’s effects of enhancing mTORC1:substrate association. It is noteworthy that DEPTOR did not change mTORC1 signaling with simultaneous Raptor overexpression (Fig. 6A); in the absence of ectopic Raptor, however, DEPTOR overexpression reduced mTORC1 signals in the presence of ectopic Flag-mTOR (Fig. 6D). This suggests that Raptor overexpression in some way negates or dilutes DEPTOR regulation of mTORC1 signaling.
MNK controls mTORC1:substrate binding and activity through TELO2
To confirm our observations, we depleted TELO2 or MNK1 in dox-induced, HA-Raptor expressing cells (Fig. 6E) and probed for TELO2 and ULK1 interactions with mTORC1. TELO2 depletion led to an intriguing reduction in DEPTOR levels, presumably an adaptation mechanism (Fig. 3E; input panel), and suppressed HA-Raptor:ULK1 association, consistent with Figure 6B (Fig. 6E). MNK1 depletion enhanced DEPTOR expression (consistent with Fig. 2) and DEPTOR co-IP with HA-Raptor (as shown in Fig. 3C) (Fig. 6E). MNK1 depletion also reduced HA-Raptor:TELO2 co-IP (Fig. 6E), confirming previous evidence that MNK favors TELO2:mTORC1 association (Fig. 5). However, there was no decrease in HA-Raptor:ULK1 co-IP following MNK1 depletion (Fig. 6E). This may be explained by the limited decrease of TELO2:mTORC1 binding in cells with MNK1 depletion where MNK2 is still active. Also, increased mTORC1:DEPTOR binding, without reducing mTORC1:ULK1 association, suggests that DEPTOR’s primary effect is displacing TELO2 from mTORC1 rather than blocking mTORC1:substrate binding (Fig. 6E). The modest effect of siMNK1 on HA-Raptor:TELO2 co-IP, compared to DEPTOR overexpression (Fig. 6A), may explain the relative lack of an effect on ULK1 binding with mTORC1 and suggests that MNK primarily functions to regulate DEPTOR and TELO2 interactions with mTORC1.
To further delineate MNK’s role in regulating TELO2:mTORC1 binding, we co-depleted MNK1 and 2 in Flag-TELO2 expressing cells, followed by Flag-TELO2 IP (Fig. 6F). MNK1/2 depletion reduced (endogenous) Raptor:TELO2 binding but not TELO2 binding to mTOR (Fig. 6F), confirming data in Figure 5. This indicates that MNK specifically affects TELO2 interactions with mTORC1, but not with mTOR outside the C1 complex. If MNK regulation of TELO2:mTORC1 binding explains its effects on mTORC1 signaling, then over-expression of TELO2 or constitutively active MNK1(T344D) should compensate for the effects of MNK1 depletion (Fig. 6G). As in Figure 2, MNK1 depletion reduced 4EBP1(S65) phosphorylation (Fig. 6G). However, overexpression of either TELO2 or MNK1(T344D) almost completely reversed this effect (Fig. 6G). Thus, MNK acts through TELO2 to determine mTORC1:substrate associations and mTORC1 activity. TELO2 overexpression/depletion had no significant impact on mTORC1 signaling (Fig. 5A; Fig. 6A, D–G). This is also true for Raptor overexpression (Fig. 5E; Fig. 6A–C). This is expected, as our assays did not involve stimuli or inhibitors of mTORC1, and because altering mTORC1 cofactor levels alone is unlikely to shift mTORC1 catalytic output controlled by a vast, integrated regulatory network (Laplante and Sabatini, 2012). Therefore, we assessed MNK’s control over TELO2:mTORC1 association and mTORC1:substrate binding in physiological systems with endogenous, unaltered mTORC1 components.
MNK controls TELO2/substrate binding to mTORC1 upon T cell activation and growth factor signaling
To ascertain the biological impact of MNK on endogenous TELO2:mTORC1 binding, we tested MNK’s role in T cell activation and mitogenic stimulation, conditions that activate MNK. Hence, combined CD3/CD28-mediated activation of Jurkat T cells induced MNK [(p-eIF4E(S209)] and mTORC1 [p-S6K(T389)] signaling (Fig. 7A). Similar to wt MEFs, human cancer cell lines or mouse splenocytes (Fig. 1A–C), MNK inhibition reduced mTORC1 activity [p-ULK1(S757), pS6(SS240/4) and p-4EBP1(S65); Fig. 7B]. Endogenous Raptor IP revealed CD3/CD28-induced TELO2:mTORC1 and ULK1 binding that was antagonized by inhibiting MNK, while mTOR:Raptor association was unchanged (Fig. 7B). Endogenous TELO2 IP from the same samples revealed MNK1:TELO2 binding upon CD3/CD28-induced mTORC1/MNK activation, which was suppressed by CGP57380 (Fig. 7B). Primary human T cells responded in a similar manner in this assay: CD3/CD28-activation of mTORC1 [phosphorylation of S6K(T389)] was nearly abolished by CGP57380 (Fig. 7C). Accordingly, inhibiting MNK abrogated CD3/CD28-activated IFN-y induction (Fig. 7C). These findings corroborate our mechanistic studies in a system of physiologic mTORC1 activation and delineate MNK’s influence on endogenous mTORC1.
Figure 7. MNK regulation of TELO2:mTORC1 binding facilitates T cell activation and growth factor signaling.

(A) Jurkat T cell leukemia cells were untreated or treated with anti-CD3 and anti-CD28 antibodies as shown, harvested to produce lysates, and tested for MNK and mTORC1 activation.
(B) Jurkat T cells were untreated or treated as in (A) in the presence of DMSO (-) or CGP57380. Lysates were subjected to IP of endogenous proteins using control IgG, anti-Raptor IgG, or anti-TELO2 IgG followed by immunoblot analysis. The quantitation shown is for TELO2:(endogenous) Raptor co-IP (top panel).
(C) Primary human T cells were treated as in (B), harvested, and analyzed for MNK and mTORC1 activation and IFNγ release (ELISA).
(D) Sum149 cells were treated with insulin and or CGP57380 as shown. Cells were harvested, lysed, and analyzed by immunoblot. Statistics compare CGP57380 vs. DMSO control at each time point (ANOVA protected, Tukey’s HSD).
(E) Sum149 cells were serum-starved (12h) followed by stimulation with fetal bovine serum or insulin as shown; cell lysates were subjected to endogenous Raptor IP followed by immunoblot as in (B).
(A–E) Quantitations represent the average of 3 independent experiments, normalizing control values to 1.
We confirmed these findings using growth factor stimulation of Sum149 breast cancer cells, which depend on MNK signals to maintain mTORC1 activity in the presence of rapamycin (Fig. 1B). Insulin spurred a robust increase in p-AKT(S473) and p-S6K(T389), but also activated MNK (Fig. 7D). Pretreatment with CGP57380 abolished eIF4E(S209) phosphorylation and substantially decreased mTORC1-mediated S6K(T389) phosphorylation (Fig. 7D). Endogenous Raptor co-IP revealed that serum stimulation or insulin induced TELO2:mTORC1 binding, correlating with MNK [p-eIF4E(S209)] and mTORC1 activation [p-4EBP1(S65), p-S6(SS240/4)] (Fig. 7E). TELO2:mTORC1 binding was stimulated to a greater extent than Raptor:mTOR, suggesting that mitogenic signals increase TELO2 binding with mTORC1 in a manner that is not explained by enhanced Raptor:mTOR association (Fig. 7E). This suggests mTORC1-specific regulation of TELO2:mTOR binding in response to growth stimuli, an effect that our mechanistic investigations demonstrate to depend on MNK.
Discussion
mTOR’s complex functions at the crossroads of vital homeostatic signaling networks are controlled by dynamic, variable multi-protein aggregates and the functional relations of their components. In this work, we identified MNK as an important regulator of mTORC1. Our studies define inverse relations between DEPTOR and TELO2 that determine mTORC1:substrate binding and signaling activity and coordinate mTORC1’s role in T cell activation and mitogenic signaling. MNK binding with an mTOR:TELO2: DDB1 module supports TELO2:mTORC1 association to stimulate mTORC1:substrate binding and signaling. Further studies are needed to determine the precise composition of such MNK-anchored ensembles and the regulation of their assembly.
MNK’s effects on mTORC1 involve the critical mTORC1-associated factors TELO2, DDB1 and DEPTOR. TELO2, first recognized in s. cerevisiae for telomere-binding and controlling telomere length (Lustig and Petes, 1986), associates with and stabilizes all 6 members of the PIKK family (Takai et al., 2007). Our data suggest that TELO2 may also influence the enzymatic activity of PIKKs, i.e. mTORC1 signaling. Beyond its role in regulating mTORC1 assembly (Kaizuka et al., 2010), TELO2-binding determines mTORC1:substrate binding possibly by holding mTOR in an ‘active’ confirmation with Raptor. MNK’s relationship with TELO2:DDB1 may impinge on other PIKKs, and influence signaling networks other than those coordinated by mTORC1, e.g. in the DNA damage response (Hurov et al., 2010; Wakasugi et al., 2002). Indeed, MNK inhibition was proposed to enhance DNA-damaging chemotherapy (Grzmil et al., 2016). Exploring this possibility is beyond the scope of this work.
MNK:TELO2 association closely correlated with DDB1 co-IP, suggesting coordinated regulation of mTOR:TELO2:DDB1 interactions. DDB1 is part of a ubiquitin E3 ligase complex that interacts with Raptor, facilitates Raptor ubiquitination and, thus, participates in control over mTORC1 (Hussain et al., 2013). mTORC1:TELO2 binding may influence DDB1 recruitment, and subsequent DDB1-mediated Raptor ubiquitination (Hussain et al., 2013).
DEPTOR inhibits both mTOR complexes but shows unique specificity for mTORC1 (Peterson et al., 2009). Our data suggest that active MNK, through interaction with mTORC1, displaces DEPTOR from mTORC1. This may be a prerequisite for induced TELO2 binding. The primary biological consequence of these events is enhanced mTORC1:TELO2 binding (determining mTORC1 proximity to its substrates), because TELO2 overexpression prevented the effects of MNK depletion on mTORC1 and mirrored MNK’s effects on mTORC1:substrate binding.
We propose that DEPTOR inhibits mTORC1:TELO2 association to regulate mTORC1:substrate proximity, and thus, activity. Most importantly, our work to unravel the effects of the MAPK effector MNK on mTORC1 signaling revealed roles for TELO2 beyond its known stabilizing functions for PIKKs, and implicates TELO2 in control over PIKK signaling output.
Experimental Procedures
Cell lines, inhibitors, stimulants, mice
MNK wt and dko MEFs were a gift from Dr. R. Fukunaga (Ueda et al., 2004); DM6 and DM443 cell lines were a gift of Dr. D. Tyler (UTMB, TX, USA); U87, Du54, MDA-MB231, and Sum149 lines were a gift of Drs. D. Bigner and S. Nair (Duke Univ., NC, USA); Jurkat T cells were obtained from American Type Culture Collection and grown in 10% FBS containing RPMI (Invitrogen). Pre-sorted CD3+ T cells were purchased from Astarte Biologics. Murine splenocytes were obtained from naïve C57Bl6 mice (6–8 week old) and were purified as previously described (Kruisbeek, 2001); the mice were surplus animals used under an IACUC-approved protocol. All cells were grown in 10% FBS-containing DMEM (Invitrogen) or, for Sum149, DMEM F-12 (Lonza). Doxycycline (dox; Sigma-Aldrich) was dissolved in water and used at a concentration of 1μg/mL. Dox-inducible eIF4G1 shRNA HEK293s were previously described (Dobrikov et al., 2014); dox-inducible Flag-mTOR and HA-Raptor lines were generated using the Flp-In TRX system (Invitrogen). Dox-inducible cell lines were grown in 10% FBS-containing DMEM, hygromycin (200μg/mL) and blasticidin (15μg/mL, Sigma-Aldrich); dox was added to deplete eIF4G1 (96h) or for Ha-Raptor/Flag-mTOR expression (12h). IGF1 (Sigma-Aldrich) was dissolved in water; TPA and CGP57380 (both Tocris) were dissolved in DMSO. To induce T cell activation, 1 μg/mL of anti-CD3/CD28 (BioLegend) and goat anti mouse IgG (Jackson Immunoresearch) were added to cell cultures for 30 min; CGP57380 or DMSO was added 30 min prior to antibody addition when indicated. For primary T cell activation, half of the cells were harvested 1h post antibody addition for immunoblot or 4h post treatment for IFN-γ analysis.
DNA constructs
HA-tagged MNK1/2 have been described (Shveygert et al., 2010). Flag-tagged wt MNK1-a, MNK1-b, MNK1(D191A), MNK1(Δ4G), and MNK1(T334D) were generated (by replacing the HA-tags) using primers (1) 5′ Phos-TTAAGATGGACTACAAAGACGATGACGACAAGG-3′ and (2) 5′ Phos-GATCCCTTGT CGTCATCGTCTTTGTAGTCCATC. The primers were annealed and inserted into AflII and BamHI-digested HA-tagged MNK constructs. pcDNA3-Flag-mTOR [a gift of Dr. J. Chen (#26603) (Vilella-Bach et al., 1999)]; pRK5-HA Raptor [a gift from Dr. D. Sabatini (#8513) (Kim et al., 2002)]; pRK5-Flag-DEPTOR [a gift from Dr. D. Sabatini (#21334) (Peterson et al., 2009)]; pcDNA3-Flag-DDB1 [a gift from Dr. Y. Xiong (#19918) (Hu et al., 2008)]; p3x-Flag-CMV10-hTel2 [a gift from Dr. N. Mizushima (#30214) (Kaizuka et al., 2010)] were obtained from Addgene (Cambridge, MA, USA). To sub-clone mTOR into pcDNA5, the pcDNA3 Flag-mTOR construct was digested with NotI and the resulting mTOR fragment was inserted into NotI-digested pcDNA5. pcDNA5-Raptor was produced by digesting pRK-Raptor with SalI, treating with Klenow fragment (NEB), cleaved with NotI and inserted into EcoRV/NotI-digested pcDNA5. pRK5-Flag-DEPTOR was treated in the same way for insertion into pcDNA3.1 to produce untagged DEPTOR. For untagged TELO2, p3x-Flag-CMV10-hTel2 was cleaved with BglII, treated with Klenow fragment, and inserted into EcoRV-digested pcDNA3.1.
Antibodies, Immunoblots, and ELISA
Antibodies against p-ULK1(S757), ULK1, p-4EBP1(S65)/(T37), 4EBP1, p-S6K(T389), S6K, p-eIF4E(S209), eIF4E, p-S6(SS240/4), S6, DEPTOR, p-AKT(S473)/(T308), AKT, MNK1, p-MNK1(TT197/202), mTOR, Raptor, p-PRAS40(T246), PRAS40, p-ERK1/2(T202/Y204), ERK1/2, GβL, eIF4G1, DDB1, ATM, HSP90, βTRC, Cul4A, RBX1, DDB2, Rictor, Sin1 (Cell Signaling Technologies); tubulin, HA-tag, Flag-tag (Sigma-Aldrich); TELO2 (Proteintech); TTI1 (Santa-Cruz Biotechnology); DCAF6, WDR26, eIF3f (Bethyl Labs); and Cul4B (Genetex). Immunoblots were performed as previously described (Dobrikov et al., 2011). Immunoblot signals were obtained on and quantified using the Li-COR Odyssey FC2 imaging system and Image Studio software. IL-2 (R&D systems) and IFN-γ (Invitrogen) ELISAs were performed using the manufacturer’s instructions.
Immunoprecipitations (IP), transfections, and siRNA
For IPs, ~70% confluent 150mm dishes of cells treated as described in the figure legends were lysed with CHAPs buffer (Kim et al., 2002) that was modified to contain: 40mM HEPES (pH 7.5), 120mM NaCl, 10mM glycerophosphate, 0.3% CHAPs detergent, 15mM MgCl2, 1mM DTT (all Sigma Aldrich), and EDTA-free proteinase & phosphatase inhibitor (Thermo-Fisher). IPs were performed as previously described (Brown et al., 2014a). After overnight incubation, for HA-IPs, beads were washed 5 times in CHAPs lysis buffer, HA-bead associated proteins were eluted with LDS buffer (Invitrogen) at 95°C (10min). For Flag-IPs, beads (Sigma) were washed 3 times in CHAPs lysis buffer, followed by 2 washes in low (0.03%)-CHAPs buffer, and flag-peptide (Sigma-Aldrich) elution in low-CHAPs buffer at 4°C (1h) following the manufacturer’s instructions. Flag-affinity beads were removed using 30μM chromatography columns (Thermo-Fisher), and eluted proteins were concentrated using a 10kDa cutoff protein concentration column (Millipore). For endogenous IPs, 3 μg of either rabbit anti-Raptor (Millipore) or -TELO2 (Proteintech) antibody was added to lysates and rotated overnight. Pre-blocked protein A (Thermo-Fisher) bound agarose beads were added for one hour to pull down antibody bound complexes and beads were washed and eluted the same as for HA-IP. For immunoblot detection of proteins following endogenous IP, a conformation-specific mouse anti rabbit secondary antibody (Cell Signaling Technologies) was used prior to an anti-mouse secondary antibody. Transfections of plasmid DNA (48h) or siRNA (72h) were performed using Lipofectamine 2000 or RNAiMax Lipofectamine, respectively, (Invitrogen) following the manufacturer’s instructions. SiRNAs used were specific to MNK1, MNK2, TelO2, Raptor or All-stars control siRNA (Qiagen). In experiments with double transfections of expression vectors or siRNA, equal total amounts of DNA and/or siRNA were transfected within each experimental sample using empty vector or control siRNA to supplement controls and single expression vector/siRNA transfections.
Statistics
Statistical analysis was performed using JMP 13 software (SAS). All error bars represent Standard Error of the Mean (SEM). For multiple group comparisons all groups from each experimental repeat were compared using ANOVA. If the ANOVA test was significant (p<0.05), post-hoc Tukey’s HSD was performed. Paired t-tests were used for data comparing only 2 groups. All asterisks denote a significant p-value defined by p<0.05.
Supplementary Material
Acknowledgments
This work was supported by PHS grants CA124756 and CA190991 (M.G.). Graphical abstract and slider image by Lauren Halligan, with permission Duke University 2016.
Footnotes
AUTHOR CONTRIBUTIONS
M.C.B. and M.G. conceived of the project and empirical strategy. M.C.B. performed all experiments. M.C.B. and M.G. interpreted data and composed the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adesso L, Calabretta S, Barbagallo F, Capurso G, Pilozzi E, Geremia R, Delle Fave G, Sette C. Gemcitabine triggers a pro-survival response in pancreatic cancer cells through activation of the MNK2/eIF4E pathway. Oncogene. 2013;32:2848–2857. doi: 10.1038/onc.2012.306. [DOI] [PubMed] [Google Scholar]
- Brown MC, Bryant JD, Dobrikova EY, Shveygert M, Bradrick SS, Chandramohan V, Bigner DD, Gromeier M. Induction of viral, 7-methyl-guanosine cap-independent translation and oncolysis by mitogen-activated protein kinase-interacting kinase-mediated effects on the serine/arginine-rich protein kinase. J Virol. 2014a;88:13135–13148. doi: 10.1128/JVI.01883-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Dobrikov MI, Gromeier M. Mitogen-activated protein kinase-interacting kinase regulates mTOR/AKT signaling and controls the serine/arginine-rich protein kinase-responsive type 1 internal ribosome entry site-mediated translation and viral oncolysis. J Virol. 2014b;88:13149–13160. doi: 10.1128/JVI.01884-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrestensen CA, Eschenroeder A, Ross WG, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Sturgill TW. Loss of MNK function sensitizes fibroblasts to serum-withdrawal induced apoptosis. Genes to cells : devoted to molecular & cellular mechanisms. 2007;12:1133–1140. doi: 10.1111/j.1365-2443.2007.01122.x. [DOI] [PubMed] [Google Scholar]
- Dobrikov M, Dobrikova E, Shveygert M, Gromeier M. Phosphorylation of eukaryotic translation initiation factor 4G1 (eIF4G1) by protein kinase C{alpha} regulates eIF4G1 binding to Mnk1. Mol Cell Biol. 2011;31:2947–2959. doi: 10.1128/MCB.05589-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrikov MI, Shveygert M, Brown MC, Gromeier M. Mitotic phosphorylation of eukaryotic initiation factor 4G1 (eIF4G1) at Ser1232 by Cdk1:cyclin B inhibits eIF4A helicase complex binding with RNA. Mol Cell Biol. 2014;34:439–451. doi: 10.1128/MCB.01046-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerdt F, Beauchamp E, Bell J, Iqbal A, Su B, Fukunaga R, Lulla RR, Goldman S, Platanias LC. Regulatory effects of a Mnk2-eIF4E feedback loop during mTORC1 targeting of human medulloblastoma cells. Oncotarget. 2014;5:8442–8451. doi: 10.18632/oncotarget.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Saiz V, Targosz BS, Lemeer S, Eichner R, Langer C, Bullinger L, Reiter C, Slotta-Huspenina J, Schroeder S, Knorn AM, et al. SCFFbxo9 and CK2 direct the cellular response to growth factor withdrawal via Tel2/Tti1 degradation and promote survival in multiple myeloma. Nature cell biology. 2013;15:72–81. doi: 10.1038/ncb2651. [DOI] [PubMed] [Google Scholar]
- Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P, Wan L, Zhai B, Chin YR, Shaik S, et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell. 2011;44:290–303. doi: 10.1016/j.molcel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzmil M, Huber RM, Hess D, Frank S, Hynx D, Moncayo G, Klein D, Merlo A, Hemmings BA. MNK1 pathway activity maintains protein synthesis in rapalog-treated gliomas. J Clin Invest. 2014;124:742–754. doi: 10.1172/JCI70198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzmil M, Seebacher J, Hess D, Behe M, Schibli R, Moncayo G, Frank S, Hemmings BA. Inhibition of MNK pathways enhances cancer cell response to chemotherapy with temozolomide and targeted radionuclide therapy. Cell Signal. 2016 doi: 10.1016/j.cellsig.2016.06.005. [DOI] [PubMed] [Google Scholar]
- Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Horejsi Z, Takai H, Adelman CA, Collis SJ, Flynn H, Maslen S, Skehel JM, de Lange T, Boulton SJ. CK2 phospho-dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Mol Cell. 2010;39:839–850. doi: 10.1016/j.molcel.2010.08.037. [DOI] [PubMed] [Google Scholar]
- Hu J, Zacharek S, He YJ, Lee H, Shumway S, Duronio RJ, Xiong Y. WD40 protein FBW5 promotes ubiquitination of tumor suppressor TSC2 by DDB1-CUL4-ROC1 ligase. Genes & development. 2008;22:866–871. doi: 10.1101/gad.1624008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu SI, Katz M, Chin S, Qi X, Cruz J, Ibebunjo C, Zhao S, Chen A, Glass DJ. MNK2 inhibits eIF4G activation through a pathway involving serine-arginine-rich protein kinase in skeletal muscle. Sci Signal. 2012;5:ra14. doi: 10.1126/scisignal.2002466. [DOI] [PubMed] [Google Scholar]
- Hurov KE, Cotta-Ramusino C, Elledge SJ. A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev. 2010;24:1939–1950. doi: 10.1101/gad.1934210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S, Feldman AL, Das C, Ziesmer SC, Ansell SM, Galardy PJ. Ubiquitin hydrolase UCH-L1 destabilizes mTOR complex 1 by antagonizing DDB1-CUL4-mediated ubiquitination of raptor. Mol Cell Biol. 2013;33:1188–1197. doi: 10.1128/MCB.01389-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauch R, Cho MK, Jakel S, Netter C, Schreiter K, Aicher B, Zweckstetter M, Jackle H, Wahl MC. Mitogen-activated protein kinases interacting kinases are autoinhibited by a reprogrammed activation segment. EMBO J. 2006;25:4020–4032. doi: 10.1038/sj.emboj.7601285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert PE, Stapleford K, Guivel-Benhassine F, Vignuzzi M, Schwartz O, Albert ML. Inhibition of mTORC1 Enhances the Translation of Chikungunya Proteins via the Activation of the MnK/eIF4E Pathway. PLoS Pathog. 2015;11:e1005091. doi: 10.1371/journal.ppat.1005091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaizuka T, Hara T, Oshiro N, Kikkawa U, Yonezawa K, Takehana K, Iemura S, Natsume T, Mizushima N. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem. 2010;285:20109–20116. doi: 10.1074/jbc.M110.121699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB, Sabatini DM. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science. 2013;341:1236566. doi: 10.1126/science.1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf U, Tschopp C, Gram H. Negative regulation of protein translation by mitogenactivated protein kinase-interacting kinases 1 and 2. Mol Cell Biol. 2001;21:5500–5511. doi: 10.1128/MCB.21.16.5500-5511.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruisbeek AM. Isolation of mouse mononuclear cells. Curr Protoc Immunol. 2001;Chapter 3(Unit 3):1. doi: 10.1002/0471142735.im0301s39. [DOI] [PubMed] [Google Scholar]
- Kuo CJ, Chung J, Fiorentino DF, Flanagan WM, Blenis J, Crabtree GR. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature. 1992;358:70–73. doi: 10.1038/358070a0. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig AJ, Petes TD. Identification of yeast mutants with altered telomere structure. Proc Natl Acad Sci U S A. 1986;83:1398–1402. doi: 10.1073/pnas.83.5.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzec M, Liu X, Wysocka M, Rook AH, Odum N, Wasik MA. Simultaneous inhibition of mTOR-containing complex 1 (mTORC1) and MNK induces apoptosis of cutaneous T-cell lymphoma (CTCL) cells. PLoS One. 2011;6:e24849. doi: 10.1371/journal.pone.0024849. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol. 2011;7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyronnet S, Imataka H, Gingras AC, Fukunaga R, Hunter T, Sonenberg N. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J. 1999;18:270–279. doi: 10.1093/emboj/18.1.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shveygert M, Kaiser C, Bradrick SS, Gromeier M. Regulation of eukaryotic initiation factor 4E (eIF4E) phosphorylation by mitogen-activated protein kinase occurs through modulation of Mnk1-eIF4G interaction. Mol Cell Biol. 2010;30:5160–5167. doi: 10.1128/MCB.00448-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Wang RC, Takai KK, Yang H, de Lange T. Tel2 regulates the stability of PI3Krelated protein kinases. Cell. 2007;131:1248–1259. doi: 10.1016/j.cell.2007.10.052. [DOI] [PubMed] [Google Scholar]
- Teo T, Yu M, Yang Y, Gillam T, Lam F, Sykes MJ, Wang S. Pharmacologic co-inhibition of Mnks and mTORC1 synergistically suppresses proliferation and perturbs cell cycle progression in blast crisis-chronic myeloid leukemia cells. Cancer letters. 2015;357:612–623. doi: 10.1016/j.canlet.2014.12.029. [DOI] [PubMed] [Google Scholar]
- Ueda T, Sasaki M, Elia AJ, Chio II, Hamada K, Fukunaga R, Mak TW. Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and Mnk2) delays tumor development. Proc Natl Acad Sci U S A. 2010;107:13984–13990. doi: 10.1073/pnas.1008136107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol. 2004;24:6539–6549. doi: 10.1128/MCB.24.15.6539-6549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella-Bach M, Nuzzi P, Fang Y, Chen J. The FKBP12-rapamycin-binding domain is required for FKBP12-rapamycin-associated protein kinase activity and G1 progression. The Journal of biological chemistry. 1999;274:4266–4272. doi: 10.1074/jbc.274.7.4266. [DOI] [PubMed] [Google Scholar]
- Wakasugi M, Kawashima A, Morioka H, Linn S, Sancar A, Mori T, Nikaido O, Matsunaga T. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J Biol Chem. 2002;277:1637–1640. doi: 10.1074/jbc.C100610200. [DOI] [PubMed] [Google Scholar]
- Wang X, Yue P, Chan CB, Ye K, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Fu H, Khuri FR, Sun SY. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinasedependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Mol Cell Biol. 2007;27:7405–7413. doi: 10.1128/MCB.00760-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MS, Rosenberger CL, Wu C, Truong N, Sweedler JV, Chen J. Rapid mitogenic regulation of the mTORC1 inhibitor, DEPTOR, by phosphatidic acid. Mol Cell. 2015;58:549–556. doi: 10.1016/j.molcel.2015.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.