

 An enantioselective and diastereoselective approach toward the synthesis of the polycyclic norditerpenoid ineleganolide is disclosed.
An enantioselective and diastereoselective approach toward the synthesis of the polycyclic norditerpenoid ineleganolide is disclosed.
Abstract
An enantioselective and diastereoselective approach toward the synthesis of the polycyclic norditerpenoid ineleganolide is disclosed. A palladium-catalyzed enantioselective allylic alkylation is employed to stereoselectively construct the requisite chiral tertiary ether and facilitate the synthesis of a 1,3-cis-cyclopentenediol building block. Careful substrate design enabled the convergent assembly of the ineleganolide [6,7,5,5]-tetracyclic scaffold by a diastereoselective cyclopropanation–Cope rearrangement cascade under unusually mild conditions. Computational evaluation of ground state energies of late-stage synthetic intermediates was used to guide synthetic development and aid in the investigation of the conformational rigidity of these highly constrained and compact polycyclic structures. This work represents the first successful synthesis of the core structure of any member of the furanobutenolide-derived polycyclic norcembranoid diterpene family of natural products. Advanced synthetic manipulations generated a series of natural product-like compounds that were shown to possess selective secretory antagonism of either interleukin-5 or interleukin-17. This bioactivity stands in contrast to the known antileukemic activity of ineleganolide and suggests the norcembranoid natural product core may serve as a useful scaffold for the development of diverse therapeutics.
Introduction
Target-directed synthesis provides an irreplaceable platform for the invention of approaches to not only the structures of interest, but also to previously unknown complex molecules of potential biological importance. Our group has been fascinated with the beautifully complex, highly oxygenated, and compact polycyclic norcembranoid ineleganolide (1) since its initial isolation in 1999 (Fig. 1).1 Over the following decades, the subsequent isolation of sinulochmodin C (2)2 and a series of closely related constitutional isomers (3–7)3 as well as the disclosure of the biological activity of this family of norditerpenoids (e.g. antileukemic activity of ineleganolide)1,3a,d,4 has fortified our interest in the synthesis of these molecules. Ineleganolide (1) poses a particularly formidable synthetic challenge. Characterized by a fused [6,7,5]-carbocyclic core, the natural product is constrained within a highly cupped configuration by a bridging dihydrofuranone ring. The periphery of this rigid polycyclic scaffold is decorated with a network of nine stereogenic centers, eight of which are contiguous.
Fig. 1. Isomeric [6,7]- and [7,6]-norcembranoid diterpene natural products (blue and red, respectively).

Owing to intricate structural complexity found in these isomeric norditerpenoids, no de novo synthetic method exists that enables even the construction of the core scaffolding of any member of the family.5 The only laboratory-furnished sample of ineleganolide (1) was produced by Pattenden in 2011 through biomimetic semisynthesis via transannular anionic cyclization cascade (Scheme 1).6 Nicolaou,7 Frontier,8 and Romo9 were unable to construct the carbocyclic core of ineleganolide through the synthetic application of similar cyclization cascades. Moeller sought to forge the ineleganolide skeleton (10) through electrochemical macrocyclization, but could not successfully construct the required cycloheptanone.10 The Vanderwal group planned to construct the same C–C bond in the final stages of the synthetic route by intramolecular cyclization through a latent oxocarbenium (i.e. triflate 11).11 Representing the most advanced synthetic effort toward ineleganolide (1) to date, the prerequisite lactone epimerization could not be accomplished despite exhaustive efforts, thwarting the synthetic strategy.
Scheme 1. Biomimetic semisynthesis and previous synthetic attempts toward ineleganolide (1).

Results and discussion
Exploring the retrosynthetic dissection of ineleganolide (1) with an eye toward the isomeric norcembranoids, the common 1,3-cis-cyclopentanediol moiety became an attractive synthon for the development of a convergent and modifiable synthetic strategy. In contrast to the unsuccessful previous synthetic strategies (cf. Scheme 1), which incorporate the dihydrofuranone ring at an early stage, we believed the late-stage introduction of this heterocycle would facilitate construction of the carbocyclic core. Thus, ineleganolide (1) was envisioned to arise from enone 12 after formation of the dihydrofuranone ring by oxa-Michael addition (Scheme 2). Enone 12 would be synthesized through selective olefin oxidation of cycloheptadiene 13. Cope rearrangement of divinylcyclopropane 14 would forge tetracycle 13.12 Intramolecular cyclopropanation of α-diazoester 15 would construct cyclopropane 14. Cyclization precursor 15 would be assembled by the coupling of carboxylic acid 16 and 1,3-cis-cyclopentenediol 17.
Scheme 2. Retrosynthetic analysis of ineleganolide (1).

Starting with the preparation of diol 17, we adapted a strategy based on our previously reported efficient and highly enantioselective synthesis of 1,3-cis-cyclopentenediol 22 (Scheme 3).13 Using a palladium-catalyzed enantioselective allylic alkylation to form the chiral tertiary ether, silyl enol ether 18 was transformed into ketone 21 in 82% yield with 92% enantiomeric excess (ee). Judicious choice of the chiral ligand, the more readily available and cost-effective (S)-t-BuPHOX ((S)-20) provided chiral tertiary ether 21 in the (S)-configuration. A series of substrate-controlled diastereoselective transformations from ketone 21 then provided diol 22 in five steps, which serves as a building block for norcembranoids in the non-natural enantiomeric series (cf. Fig. 1).14 Methylenation of diol 22 afforded diene 23 in near-quantitative yield. Ultimately, saponification of ester 23 furnished diol coupling partner ent-17 in a combined 63% yield over 9 steps from enol ether 18.
Scheme 3. Enantioselective synthesis of 1,3-cis-cyclopentenediol coupling partner ent-17.

Construction of the complementary carboxylic acid fragment (ent-16) began with (R)-desmethylcarvone ((R)-24, Scheme 4).15 The 1,2-addition of lithium enolate 25 under cerium-mediated reaction conditions followed by oxidative 1,3-allylic transposition of the intermediate allylic alcohol with oxoammonium salt TEMPO·BF4 (ref. 16) provided cyclohexenone 26. Saponification of ethyl ester 26 followed by coupling with diol fragment ent-17 and diazotransfer using p-ABSA (27) provided the pivotal tandem cyclization cascade precursor, α-diazoester ent-15 in 75% yield over three steps.
Scheme 4. Convergent assembly of the ineleganolide [6,7,5,5]-tetracyclic core.

Pleasingly, initial attempts to accomplish the planned sequential intramolecular cyclopropanation–Cope rearrangement demonstrated that the transformation proceeded efficiently under mild conditions. Exposure of α-diazoester ent-15 to catalytic dirhodium tetraacetate (1 mol%) in dichloromethane at ambient temperature enabled the construction of cycloheptadiene 28, containing the complete carbocyclic core of ent-ineleganolide (ent-1).17 This cyclization cascade is notable given that it is completed under high dilution in less than 20 minutes at ambient temperature and employs an electronically deactivated olefin in the [3,3]-sigmatropic rearrangement. While examples of divinylcyclopropane rearrangements with electron-neutral and electron-rich olefins are abundant,12,18 those employing conjugated olefins are limited and typically require forcing conditions.12c,19
We speculate that the deleterious effect of an electronically deactivated π-bond in the sequential Cope rearrangement is mitigated by substrate design. Cleavage of highly strained bridging cyclopropane ent-14 upon isomerization to the cycloheptadiene product ent-13 is associated with an increased enthalpic gain compared to a prototypical cyclopropane-fused Cope rearrangement that employs an isolated carbocycle.20 Additionally, the unsubstituted vinyl group contributes minimal steric hindrance within the bis-endo configuration (cf. ent-14, Scheme 4) and highly organized boat-like transition state required for the cis-divinylcyclopropane rearrangement to proceed.12a,21
Although the construction of the carbocyclic core of ent-ineleganolide (ent-1) proceeds smoothly from α-diazoester ent-15, no trace of the anticipated diene product (ent-13) was ever detected. Rather, α,β-unsaturated enone 28 was isolated from the reaction mixture as the exclusive product. The 1,3-allylic isomerization is proposed to occur through a base-mediated olefin migration. γ-Deprotonation of α,β-unsaturated ester ent-13 by adventitious acetate and formation of an intermediate conjugated enolate facilitates this process. We hypothesize this isomerization is further aided by a relief of ring strain. These assertions are supported by the observation of an analogous olefin migration under similarly basic reaction conditions.22
Installation of the final requisite atom of ent-ineleganolide was accomplished by a hydroxyl-directed epoxidation of tetracycle 28 to provide epoxide 29 in 85% yield as a crystalline white solid, enabling the confirmation of the relative configuration by single crystal X-ray diffraction (Scheme 5). Henceforth known as ent-isoineleganolide A, epoxide 29 is the first known synthetic isomer of ent-ineleganolide (ent-1) that: (1) contains the full carbocyclic skeleton, (2) possesses all of the required atoms, and (3) has an identical overall oxidation state.
Scheme 5. Synthesis of ent-isoineleganolides A and B.

The direct transformation of ent-isoineleganolide A (29) into enone ent-12 was planned via syn-facial 1,2-hydride migration, but this approach proved unsuccessful despite exhaustive investigation.23 Instead, alternative access to tetracycle ent-12 was developed through the nucleophilic opening of the epoxide moiety within ent-isoineleganolide A (29). This ring opening proceeded with concomitant transannular oxa-Michael addition to furnish bromide 30 in near-quantitative yield. Gratifyingly, direct oxidation of the secondary alkyl bromide could be affected through a Kornblum oxidation manifold,24 affording diketone 31 in 96% yield.
This efficient transformation stands in stark contrast against canonical examples of oxidation under Kornblum conditions. Substrates are usually limited to primary or benzylic halides; only rare and uniformly low yielding examples of the successful Kornblum oxidation of an unactivated secondary halide are found.24b,25 We hypothesize that the bridging furyl oxygen is critical for this transformation, aiding in the abstraction of the secondary bromide and stabilization of the intermediate carbocation (cf. 33, Scheme 5). Indeed, the analog lacking the furyl tether fails to produce any trace of oxidation product under similar conditions.26
Pleased to have achieved access to ketopyran 31, we refocused our attention on advancement toward ent-ineleganolide (ent-1). Chemoselective reduction of ketopyran 31 was observed by tuning the reduction potential of samarium(ii) iodide using lithium chloride as an additive.27 Under these conditions, in situ generated SmCl2 cleaves the α-alkoxyketone bond to provide tetracyclic diol 32. Installation of the hydrogen from the α-face upon protonation of the intermediate Sm-enolate was confirmed by single crystal X-ray diffraction. This stereochemical outcome provides the C(7) configuration found within ent-ineleganolide. Selective dehydration of diol 32 under acidic conditions provided the desired enone (ent-12), another non-natural isomer of ent-ineleganolide (ent-isoineleganolide B).
Although we were optimistic this isomer would proceed to ent-ineleganolide (ent-1) spontaneously by tandem olefin isomerization-oxa-Michael addition, this outcome was not observed. Therefore, with enone ent-12 in hand, this transformation was investigated in a stepwise manner. Despite intensive efforts to accomplish the isomerization of tetrasubstituted enone ent-12 to vinylogous diketone 34 (Scheme 6), no trace of either intermediate 34 or ent-ineleganolide (ent-1) was ever detected.28 Surprised by the lack of productive reactivity, density functional theory (DFT) was used to explore the thermodynamics of the desired transformation.29 ent-Isoineleganolide B (ent-12) likely exists in the conformation as shown in Scheme 6 (left).30 In this configuration, the isopropenyl group prefers the pseudoequatorial position and the central cycloheptenone is creased, bisecting the molecule. Relative to the ground state energy of this intermediate, the ground state energy of vinylogous diketone 34 is 13.4 kcal mol–1 higher in its lowest energy product-like conformation, which posits a pseudoaxial isopropenyl moiety and isomerized cycloheptenone. However, the ground state energy of the natural product (ent-1) in its known configuration1 is 14.2 kcal mol–1 lower than vinylogous diketone 34 and overall 0.8 kcal mol–1 lower in energy than ent-isoineleganolide B (ent-12).
Scheme 6. Conformational assessment and relative ground state energies.

The large ground state energy difference between tetra-substituted enone ent-12 and vinylogous diketone 34 in addition to the empirical evidence for the inability to accomplish the desired olefin isomerization in the laboratory suggest the energy barrier for the conversion from enone ent-12 to diketone 34 is experimentally insurmountable. This is likely due to the large conformational shift required for the isomerization event and the reduced enthalpic stability of the conjugated system. The necessary contortion of the central cycloheptenone renders production of the fully conjugated vinylogous diketone moiety unfeasible. The comparative minimum ground state energy of the natural product (ent-1) and the large exergonic difference compared to vinylogous diketone 34 reinforced the use of this intermediate in continued synthetic efforts.
Toward this end, the oxidation state manipulation of carbocyclic diene core 28 was explored (Scheme 7). Although conjugate reduction of enone 28 could not be accomplished using nucleophilic hydride sources, development of samarium(ii) iodide-mediated reaction conditions, including careful control of reaction temperature and sensible selection of additive, enabled diastereoselective conjugate reduction to provide allylic alcohol 35.27 Subsequent hydroxyl-directed epoxidation afforded saturated ketone 36 as a crystalline white solid. The relative configuration of epoxide 36 was established by single crystal X-ray diffraction analysis, confirming the installation of the [6,7]-ring junction in the thermodynamically preferred trans configuration identical to that found within ent-ineleganolide (ent-1). As anticipated, application of the previously developed three step Kornblum oxidation procedure for the conversion of epoxide 36 to ketone 37 failed in the absence of the transannular furyl bridge (vide supra), forcing a strategic revaluation.26
Scheme 7. Redox manipulation of diene 28.

In place of a nucleophilic epoxide opening, methods for a radical-mediated reductive epoxide cleavage were investigated. Employing in situ generated titanocene(iii) chloride using zinc metal as the optimal reductant, epoxide 36 was opened in regioselective fashion to give the 1,3-diol product.31 Elaboration to the cycloheptanone was accomplished by oxidation of the newly revealed secondary alcohol to provide 2H-ent-ineleganolide (37) in 33% yield in only two steps from epoxide 36.
The intermediacy of a tertiary radical at C(7) after the C–O bond scission, in analogous fashion to the reductive opening of ketopyran 31 (see Scheme 5), enabled the installation of the hydrogen on the α-face as desired. Single crystal X-ray diffraction served not only to confirm the assignment of this relative configuration, but also revealed the conformational similarities between 2H-ent-ineleganolide (37) and ent-ineleganolide (cf. ent-1, Scheme 6). 2H-ent-Ineleganolide (37) contains the trans-[6,7]-ring junction, with the isopropenyl substituent in the axial position, and all required stereocenters and functional moieties except the dihydrofuranone bridge. Unfortunately, the conversion of 2H-ent-ineleganolide (37) to ent-ineleganolide (ent-1) proved nontrivial. Formation of the final requisite bond through Suárez reaction,32 selenoxide elimination, palladium-mediated oxidative desaturation33 or by employing either lead(iv) acetate or hypervalent iodine reagents34 failed to produce any traces of oxidation at the apical cycloheptanone methylene.
Surprised by the difficulty of functionalizing the central cycloheptanone within 2H-ent-ineleganolide (37), we again turned to computational chemistry. The solid-state conformation of 2H-ent-ineleganolide (37AAx, Fig. 2), determined by single crystal X-ray diffraction, revealed the prototypical conformation encountered throughout our synthetic endeavors (e.g. 29, Scheme 5 and ent-12, Scheme 6). Conformational isomerization of the isopropenyl moiety into the equatorial position (37AAx → 37Aequ) increases the ground state energy by 1.0 kcal mol–1. Comparatively, the energy minimized conformations 37Bax and 37Bequ, in which 2H-ent-ineleganolide has adopted the natural product-like configuration within the central cycloheptanone (cf. ent-1, Scheme 6),1 are slightly lower. Although the lowest energy conformations of each state (37Aax vs. 37Bax) are equivalent within the error of the computational method, empirical evidence for the difficulty of functionalizing the apical cycloheptanone methylene suggests that there is a large energy barrier that hinders the interconversion between these conformational isomers. Further computational studies are being pursued in order to more completely understand the energetics of the desired transformation.
Fig. 2. Conformational isomers of 2H-ent-ineleganolide.

Throughout the course of these studies, a reasonably large collection of natural-product like compounds was generated. Owing to the known antileukemic properties of ineleganolide, these “ineleganoloids,” in the non-natural enantiomeric series, were evaluated for their activity in DU145 (human prostate cancer) and A2038 (human melanoma) cell viability assays35 as well as against other oncological targets (EZH2 (ref. 36) and CD73 (ref. 37)). Although no notable activity toward any oncological target was discovered, the concurrent screening of this library for activity against targets in other therapeutic areas (i.e., neurological, cardiological, autoimmune, and endocrine function) revealed activity of various ineleganoloids.38 Diene 28, bromide 30, ketopyran 31, and epoxide 36 were identified as selective interleukin-5 or -17 (IL-5 and IL-17, respectively) secretory inhibitors without significant cytotoxicity (Fig. 3).39 Both IL-5 and IL-17 represent attractive pharmaceutical targets considering their pivotal role in autoimmune response and autoimmune disease (e.g., rheumatoid arthritis).40 The activity of the ineleganoloids stands in stark contrast with the inability of ineleganolide (1) to inhibit cytokine release.3a Further investigation of these preliminary results is ongoing and assessment of the biological activity of the ineleganoloids in the natural enantiomeric series will follow in due course.
Fig. 3. Biological activity of select ineleganoloids.

Conclusions
In conclusion, an efficient enantioselective and diastereoselective synthetic route to the tetracyclic core of ineleganolide has been disclosed. Convergent assembly of the core scaffold was accomplished by the coupling of two enantioenriched fragments, including a 1,3-cis-cyclopentenediol building block common to the polycyclic norcembranoid diterpenes. Tandem intramolecular cyclopropanation–Cope cyclization cascade enabled the diastereoselective construction of the tetracyclic [6,7,5,5]-scaffold of ineleganolide in a single step, providing synthetic access to the core of the polycyclic norcembranoid diterpenes for the first time. Guided by computational data, synthetic advancement facilitated the construction of the first synthetic isomers and analogs of ineleganolide. These natural product-like ineleganoloids advanced the understanding of the conformational restraints influencing chemistry of the highly compact norcembranoid diterpene scaffold and have led to the identification of biologically active ineleganolide analogs.
Acknowledgments
The authors wish to thank the NIH-NIGMS (R01GM080269), Amgen, the Gordon and Betty Moore Foundation, and Caltech for financial support and Eli Lilly & Co. for assistance with biological activity screening. Additionally, the authors gratefully acknowledge Larry Henling and Dr Michael Takase (Caltech) for X-ray crystallographic structural determination, Dr Mona Shahgholi and Naseem Torian (Caltech) for mass spectrometry assistance, and Dr David VanderVelde (Caltech) for NMR experimental assistance and helpful discussions. Additionally, Dr Jeffrey C. Holder, Dr Corey M. Reeves, Prof. Hosea M. Nelson, Dr Jonny R. Gordon, Dr Pamela M. Tadross, and Beau P. Pritchett (Caltech) and Dr Ryan Deluca and Dr Nick Cox (Stanford) are thanked for helpful discussions. R. A. C. gratefully acknowledges the support of this work provided by a fellowship from the National Cancer Institute of the National Institutes of Health (NIH) under Award Number F31A17435. J. L. R. thanks the California Tobacco-Related Disease Research Program of the University of California, Grant Number 14DT-0004 for a predoctoral fellowship. A. C. J. thanks the NIH for the support of this work provided by a postdoctoral fellowship (Award Number F32GM082000).
Footnotes
References
- Duh C.-Y., Wang S.-K., Chia M.-C., Chiang M. Y. Tetrahedron Lett. 1999;40:6033. [Google Scholar]
- Tseng Y.-J., Ahmed A. F., Dai C.-F., Chiang M. Y., Sheu J.-H. Org. Lett. 2005;7:3813. doi: 10.1021/ol051513j. [DOI] [PubMed] [Google Scholar]
- (a) Lillsunde K.-E., Festa C., Adel H., de Marino S., Lombardi V., Tilvi S., Nawrot D., Zampella A., D'Souza L., D'Auria M., Tammela P. Mar. Drugs. 2014;12:4045. doi: 10.3390/md12074045. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Radhika P., Subba Rao P. V., Anjaneyulu V., Asolkar R. N., Laatsch H. J. Nat. Prod. 2002;65:737. doi: 10.1021/np010528x. [DOI] [PubMed] [Google Scholar]; (c) Sheu J.-H., Ahmed A. F., Shiue R.-T., Dai C.-F., Kuo Y.-H. J. Nat. Prod. 2002;65:1904. doi: 10.1021/np020280r. [DOI] [PubMed] [Google Scholar]; (d) Iguchi K., Kajiyama K., Yamada Y. Tetrahedron Lett. 1995;36:8807. [Google Scholar]
- (a) Huang C.-Y., Tseng Y.-J., Chokkalingam U., Hwang T.-L., Hsu C.-H., Dai C.-F., Sung P.-J., Sheu J.-H. J. Nat. Prod. 2016;79:1339. doi: 10.1021/acs.jnatprod.5b01142. [DOI] [PubMed] [Google Scholar]; (b) Tseng Y.-J., Wang S.-K., Duh C.-Y. Mar. Drugs. 2013;11:3288. doi: 10.3390/md11093288. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huang K.-J., Chen Y.-C., El-Shazly M., Du Y.-C., Su J.-H., Tsao C.-W., Yen W.-H., Chang W.-B., Su Y.-D., Yeh Y.-T., Lu M.-C. Molecules. 2013;18:2924. doi: 10.3390/molecules18032924. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kamel H. N., Slattery M. Pharm. Biol. 2005;43:253. [Google Scholar]
- While most synthetic efforts toward the polycyclic norcembranoids have been focused on ineleganolide (1), one attempt was made to synthesize the core of yonarolide (5), see: Ueda Y., Abe H., Iguchi K., Ito H., Tetrahedron Lett., 2011, 52 , 3379 . [Google Scholar]
- (a) Li Y., Pattenden G. Tetrahedron. 2011;67:10045. [Google Scholar]; (b) Li Y., Pattenden G. Nat. Prod. Rep. 2011;28:1269. doi: 10.1039/c1np00023c. [DOI] [PubMed] [Google Scholar]; (c) Li Y., Pattenden G. Nat. Prod. Rep. 2011;28:429. doi: 10.1039/c0np00029a. [DOI] [PubMed] [Google Scholar]
- Pratt B. A., Ph.D. dissertation, Scripps Research Institute, La Jolla, California, 2008. [Google Scholar]
- (a) O'Connell C., Ph.D. dissertation, Queen's Universtiy of Belfast, Belfast, Northern Ireland, U.K., 2006. [Google Scholar]; (b) O'Connell C. E. and Frontier A. J., Abstracts of Papers, 32nd Northeast Regional Meeting of the American Chemical Society, American Chemical Society, Rochester, NY, October 31−November 3 2004, GEN-088. [Google Scholar]
- (a) Liu G., Ph.D. dissertation, Texas A&M University, College Station, Texas, 2011. [Google Scholar]; (b) Liu G., and Romo D., Abstracts of Papers, 237th American Chemical Society National Meeting, American Chemical Society, Salt Lake City, UT, March 22−26 2009, ORGN-083. [Google Scholar]
- (a) Tang F., Ph.D. dissertation, Washington University in St. Louis, St. Louis, Missouri, 2009. [Google Scholar]; (b) Tang F., Moeller K. D. Tetrahedron. 2009;65:10863. [Google Scholar]; (c) Tang F., Moeller K. D. J. Am. Chem. Soc. 2007;129:12414. doi: 10.1021/ja076172e. [DOI] [PubMed] [Google Scholar]
- (a) Horn E. J., Silverston J. S., Vanderwal C. D. J. Org. Chem. 2016;81:1819. doi: 10.1021/acs.joc.5b02550. [DOI] [PubMed] [Google Scholar]; (b) Horn E. J., Ph.D. dissertation, University of California at Irvine, Irvine, CA, 2014. [Google Scholar]
- (a) Krüger S., Gaich T. Beilstein J. Org. Chem. 2014;10:183. doi: 10.3762/bjoc.10.14. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davies H. M. L., Stafford D. G., Doan B. D., Houser J. H. J. Am. Chem. Soc. 1998;120:3326. [Google Scholar]; (c) Davies H. M. L. Tetrahedron. 1993;49:5203. [Google Scholar]; (d) Vogel E. Angew. Chem., Int. Ed. 1963;2:1. [Google Scholar]
- Craig II R. A., Roizen J. L., Smith R. C., Jones A. C., Stoltz B. M. Org. Lett. 2012;14:5716. doi: 10.1021/ol3027297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The naturally occurring enantiomers of ineleganolide (1) and the related norcembranoid diterpenes (2–7) were originally established by analogy to the known absolute stereochemistry of sinulochmodin C (2, see ref. 2). The natural enantiomers of norditerpenoids 1–7 could be accessed using (R)-t-BuPHOX ((R)-20), which has been synthesized and used by our group in the enantioselective total synthesis of (+)-liphagal, see: Day J. J., McFadden R. M., Virgil S. C., Kolding H., Alleva J. L., Stoltz B. M., Angew. Chem., Int. Ed., 2011, 50 , 6814 , . Alternatively, more cost-effective PHOX ligands derived (R)-valine have been shown to be comparably effective to (R)-t-BuPHOX for the enantioselective formation of ketone 21 from enol ether 18, see: Craig II R. A., Stoltz B. M., Tetrahedron Lett., 2015, 56 , 4670 . [Google Scholar]
- (R)-Desmethylcarvone ((R)-24) is available in 6 steps from (R)-carvone, see: ; (a) González M. A., Ghosh S., Rivas F., Fischer D., Theodorakis E. A. Tetrahedron Lett. 2004;45:5039. [Google Scholar]; (b) Chen J., Marx J. N. Tetrahedron Lett. 1997;38:1889. [Google Scholar]; (c) Lavallée J.-F., Spino C., Ruel R., Hogan K. T., Deslongchamps P. Can. J. Chem. 1991;70:1406. [Google Scholar]
- Shibuya M., Tomizawa M., Iwabuchi Y. J. Org. Chem. 2008;73:4750. doi: 10.1021/jo800634r. [DOI] [PubMed] [Google Scholar]
-
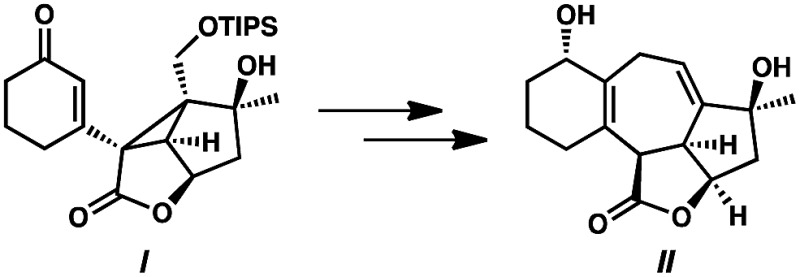
Although no trace of cyclopropane ent-14 has been detected during the conversion of α-diazoester ent-15 to cycloheptadiene 28, the isolation of cyclopropane I and the subsequent step-wise advancement to cycloheptadiene II supports the assertion that a tandem cyclopropanation–Cope rearrangement cascade is operative. Without isolated intermediates, however, we are unable to conclusively rule out the formation of cycloheptadiene product 28 through a direct (4 + 3) cycloaddition from α-diazoester ent-15, see: Guzmán P. E., Lian Y., Davies H. M. L., Angew. Chem., Int. Ed., 2014, 53
, 13083
,
 . [DOI] [PMC free article] [PubMed] [Google Scholar]
. [DOI] [PMC free article] [PubMed] [Google Scholar] - (a) Gaydou M., Miller R. E., Delpont N., Ceccon J., Echavarren A. M. Angew. Chem., Int. Ed. 2013;52:6396. doi: 10.1002/anie.201302411. [DOI] [PubMed] [Google Scholar]; (b) Vaswani R. G., Day J. J., Wood J. L. Org. Lett. 2009;11:4532. doi: 10.1021/ol901746c. [DOI] [PubMed] [Google Scholar]; (c) Davies H. M. L., Calvo R. L., Townsend R. J., Ren P., Churchill R. M. J. Org. Chem. 2000;65:4261. doi: 10.1021/jo991959b. [DOI] [PubMed] [Google Scholar]; (d) Kende A. S., Smalley Jr T. L., Huang H. J. Am. Chem. Soc. 1999;121:7431. [Google Scholar]
- (a) Osler J. D., Unsworth W. P., Taylor R. J. K. Org. Biomol. Chem. 2013;11:7587. doi: 10.1039/c3ob41617h. [DOI] [PubMed] [Google Scholar]; (b) Ito H., Takeguchi S., Kawagishi T., Iguchi K. Org. Lett. 2006;8:4883. doi: 10.1021/ol061947u. [DOI] [PubMed] [Google Scholar]; (c) Fukuyama T., Liu G. J. Am. Chem. Soc. 1996;118:7426. [Google Scholar]; (d) Wender P. A., Eissenstat M. A., Filosa M. P. J. Am. Chem. Soc. 1979;101:2196. [Google Scholar]
- For comparisons of the strain energy of isolated cyclopropanes with fused cyclopropane ring systems, see: ; (a) Borst M. L. G., Ehlers A. W., Lammertsma K. J. Org. Chem. 2005;70:8110. doi: 10.1021/jo0513010. [DOI] [PubMed] [Google Scholar]; (b) Wiberg K. B., Kass S. R., de Meijere A., Bishop III K. C. J. Am. Chem. Soc. 1985;107:1003. [Google Scholar]
- (a) Zora M. J. Mol. Struct.: THEOCHEM. 2004;681:113. [Google Scholar]; (b) Zora M. J. Org. Chem. 2003;68:9635. doi: 10.1021/jo035173w. [DOI] [PubMed] [Google Scholar]; (c) Zora M., Özkan I., Danişman M. F. J. Mol. Struct.: THEOCHEM. 2003;636:9. [Google Scholar]; (d) Doering W. V. E., Roth W. R. Angew. Chem., Int. Ed. 1963;2:115. [Google Scholar]
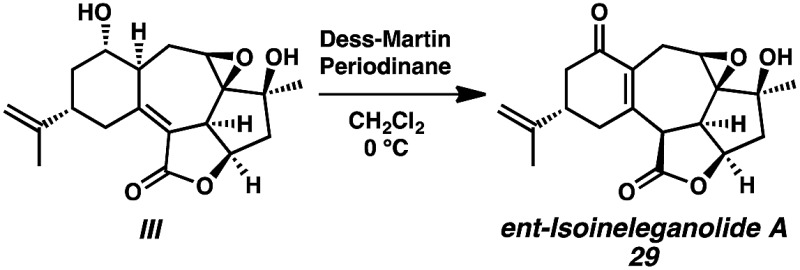
- Oxidation of diol III resulted in reformation of isoineleganolide A (29) after olefin migration in the presence of the adventitious acetate byproduct derived from Dess-Martin periodinane
- A characteristic list of the conditions attempted to induce the desired epoxide isomerization: H2SO4/CHCl3, SiO2/CHCl3, DBU/CHCl3, KOt-Bu/THF, Pd(CH3CN)2Cl2/benzene, BF3·Et2O/CHCl3, In(OTf)3/CHCl3, Al(Oi-Pr)3/CHCl3, Ti(Oi-Pr)4/THF, Zn(OTf)2/toluene, Mg(OTf)2/toluene, MgCl2/toluene, MgBr2/toluene
- (a) Ly T. W., Liao J.-H., Shia K.-S., Liu H.-J. Synthesis. 2004:271. [Google Scholar]; (b) Ganem B., Boeckman Jr R. K. Tetrahedron Lett. 1974;15:917. [Google Scholar]; (c) Kornblum N., Jones W. J., Anderson G. J. J. Am. Chem. Soc. 1959;81:4113. [Google Scholar]
- (a) Liu H.-J., Sun D. Tetrahedron Lett. 1997;38:6159. [Google Scholar]; (b) Dave P., Byun H.-S., Engel R. Synth. Commun. 1986;16:1343. [Google Scholar]
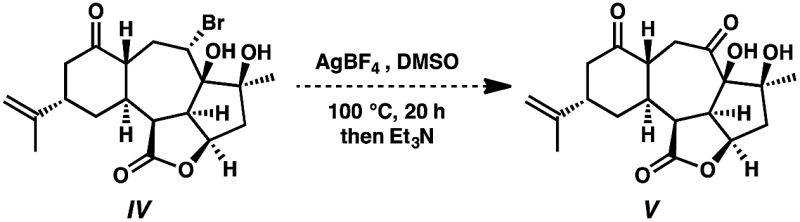
- Under analogous Kornblum oxidation conditions, bromide IV fails to undergo any oxidative transformation. In fact, bromide IV does not react until heated above 120 °C, at which point only dehydration of the substrate is observed
- (a) Szostak M., Spain M., Procter D. J. J. Org. Chem. 2014;79:2522. doi: 10.1021/jo4028243. [DOI] [PubMed] [Google Scholar]; (b) Reisman S. E., Ready J. M., Weiss M. M., Hasuoka A., Hirata M., Tamaki K., Ovaska T. V., Smith C. J., Wood J. L. J. Am. Chem. Soc. 2006;128:1448. doi: 10.1021/ja057640s. [DOI] [PubMed] [Google Scholar]; (c) Miller R. S., Sealy J. M., Shabangi M., Kuhlman M. L., Fuchs J. R., Flowers R. A. J. Am. Chem. Soc. 2000;122:7718. [Google Scholar]
- A characteristic list of the conditions attempted to induce the desired olefin isomerization: DBU/CH2Cl2, PdCl2(CH3CN)2/CH2Cl2, Grubbs Gen. II/MeOH, RhCl3·H2O/EtOH, LHMDS/THF
- Calculations were performed with Spartan '10 (Wavefunction, Inc., Irvine, CA). The in vacuo equilibrium geometry for each structure was calculated by a series of sequential calculations as follows: Hartree–Fock computation (equilibrium geometry, 3-21G basis set), DFT (equilibrium geometry, B3LYP/6-31G basis set), DFT (energy, B3LYP/6-311+G** basis set), DFT (equilibrium geometry, B3LYP/6-311+G** basis set). The error from these calculations is ±0.23 kcal mol–1, thus all energy differences larger than 0.46 kcal mol–1 were considered significant. Except for molecular mechanics and semi-empirical models, the calculation methods used in Spartan have been documented in: Shao Y., Molnar L. F., Jung Y., Kussmann J., Ochsenfeld C., Brown S. T., Gilbert A. T. B., Slipchenko L. V., Levchenko S. V., O'Neill D. P., DiStasio Jr R. A., Lochan R. C., Wang T., Beran G. J. O., Besley N. A., Herbert J. M., Lin C. Y., Van Voorhis T., Chien S. H., Sodt A., Steele R. P., Rassolov V. A., Maslen P. E., Korambath P. P., Adamson R. D., Austin B., Baker J., Byrd E. F. C., Dachsel H., Doerksen R. J., Dreuw A., Dunietz B. D., Dutoi A. D., Furlani T. R., Gwaltney S. R., Heyden A., Hirata S., Hsu C.-P., Kedziora G., Khalliulin R. Z., Klunzinger P., Lee A. M., Lee M. S., Liang W. Z., Lotan I., Nair N., Peters B., Proynov E. I., Pieniazek P. A., Rhee Y. M., Ritchie J., Rosta E., Sherrill C. D., Simmonett A. C., Subotnik J. E., Woodcock III H. L., Zhang W., Bell A. T., Chakraborty A. K., Chipman D. M., Keil F. J., Warshel A., Hehre W. J., Schaefer III H. F., Kong J., Krylov A. I., Gill P. M. W., Head-Gordon M., Phys. Chem. Chem. Phys., 2006, 8 , 3172 .16902710 [Google Scholar]
- The conformation of ent-isoineleganolide B (ent-12) was established by two-dimensional NOE studies and by analogy to the conformation of precursors 28–32, as established by single crystal X-ray diffraction. The ground state energies of the other conformations of ent-isoineleganolide B (ent-12) were additionally assessed by our computational method and are all within ±1.8 kcal mol–1 of one another. For full details, see ESI.
- (a) Jiménez T., Campaña A. G., Bazdi B., Paradas M., Arráez-Román D., Segura-Carretero A., Fernández-Gutiérrez A., Oltra J. E., Robles R., Justicia J., Cuerva J. M. Eur. J. Org. Chem. 2010:4288. [Google Scholar]; (b) Gansäuer A., Pierobon M., Bluhm H. Angew. Chem., Int. Ed. 1998;37:101. doi: 10.1002/1521-3773(20020902)41:17<3206::AID-ANIE3206>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]; (c) RajanBabu T. V., Nugent W. A. J. Am. Chem. Soc. 1994;116:986. [Google Scholar]; (d) RajanBabu T. V., Nugent W. A., Beattie M. S. J. Am. Chem. Soc. 1990;112:6408. [Google Scholar]
- (a) Betancor C., Freire R., Pérez-Martín I., Prangé T., Suárez E. Tetrahedron. 2005;61:2803. [Google Scholar]; (b) de Armas P., Concepción J. I., Francisco C. G., Hernández R., Salazar J. A., Suárez E. J. Chem. Soc., Perkin Trans. 1. 1989:405. [Google Scholar]; (c) Concepción J. I., Francisco C. G., Hernández R., Salazar J. A., Suárez E. Tetrahedron Lett. 1984;25:1953. [Google Scholar]
- A wide variety of conditions were screened for palladium-meditated oxidative desaturation both directly from ketone 37 as well as stepwise through the intermediate silyl enol ether without success. For select examples, see: ; (a) Diao T., Stahl S. S. J. Am. Chem. Soc. 2011;133:14566. doi: 10.1021/ja206575j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ohmori N. J. Chem. Soc., Perkin Trans. 1. 2002:755. [Google Scholar]; (c) Degrado S. J., Mizutani H., Hoveyda A. H. J. Am. Chem. Soc. 2001;123:755. doi: 10.1021/ja003698p. [DOI] [PubMed] [Google Scholar]
- Efforts were focused on the application of 2-iodoxybenzoic acid (IBX) and PhI(OH)(OTs), see: ; (a) Nicolaou K. C., Montagnon T., Baran P. S., Zhong Y. L. J. Am. Chem. Soc. 2002;124:2245. doi: 10.1021/ja012127+. [DOI] [PubMed] [Google Scholar]; (b) Moriarty R. M., Vaid R. K., Hopkins T. E., Vaid B. K., Prakash O. Tetrahedron Lett. 1990;31:201. [Google Scholar]
- Compounds 28, 29, 30, 31, and 32 were screened against DU145 and A2038 cell viability assays in triplicate, revealing no significant activity. Special, personal thanks to Prof. David Horne and Prof. Sangkil Nam (City of Hope, Duarte, CA) for their assistance in performing these cell viability assays
- Chase A., Cross N. C. P. Clin. Cancer Res. 2011;17:2613. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- Young A., Mittal D., Stagg J., Smyth M. J. Cancer Discovery. 2014;4:879. doi: 10.1158/2159-8290.CD-14-0341. [DOI] [PubMed] [Google Scholar]
- Biological activity data generated through the Open Innovation Drug Discovery Program (OIDD) and screening data were supplied courtesy of Eli Lilly and Company—used with Eli Lilly's permission. To learn more about the Lilly Open Innovation Drug Discovery program, please visit the program website at https://openinnovation.lilly.com, last accessed on 06-15-2016.
- Biological activity data was generated employing a high throughput screen based on primary blood mononuclear cells (PMBCs) and using 10 μM of compound. The results are reported as follows: compound number (% secretory inhibition IL-5, % secretory inhibition IL-17, % PMBC cell death). Diene 28 (7.8, 38.1, 3.9), bromide 30 (47.2, 3.8, 4.9), ketopyran 31 (40.2, <2, 6.7), epoxide 36 (40.2, <2, 5.9). See ESI and Table S1 for full details
- (a) Chang S. H., Dong C. Cell. Signalling. 2011;23:1069. doi: 10.1016/j.cellsig.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kouro T., Takatsu K. Int. Immunol. 2009;21:1303. doi: 10.1093/intimm/dxp102. [DOI] [PubMed] [Google Scholar]; (c) Simmons D. L. Drug Discovery Today. 2006;11:210. doi: 10.1016/S1359-6446(05)03721-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.