

Abstract
Hirschsprung’s disease (HSCR) is a common cause of neonatal bowel obstruction and the approach to diagnosis and surgical treatment is well defined and accepted. Hirschsprung’s-associated enterocolitis (HAEC) remains a frequent cause of pre-operative and post-operative morbidity and mortality, with unchanged treatment guidelines over multiple decades. Recent advances in our understanding of the genetics underlying HSCR have allowed the development of animal models, some of which recapitulate the HAEC phenotype. These animal models, along with recent translational studies, have implicated multiple facets of mucosal immunity and microbiome dysbiosis in the development of HAEC. Here, we will review the established epidemiology, modes of diagnosis and treatment of HAEC. Furthermore, we will explore emerging concepts in the pathogenesis of this disease; including animal models, alterations in mucosal immunity, dysbiosis of the intestinal microbiome, specific genetic susceptibility, and novel treatment modalities.
Keywords: Hirschsprung disease, Hirschsprung’s disease, enterocolitis, mucosal immunity, microbiome, pathogenesis
Introduction
In 1886, Harald Hirschsprung presented the clinical course and pathology specimens of two unusual patients to the International Congress for Children’s Disease in Berlin[1]. Both patients had profound constipation from birth, demonstrated megacolon without dilation of the rectum, and ultimately expired from a sudden onset of acute abdominal distention, profound diarrhea, and weight loss/dehydration. The presentation of these cases by a world-renowned pediatrician prompted an outpouring of other, similar cases, being presented worldwide. The underlying disease ultimately came to be referred to as “Hirschsprung’s disease” (HSCR, Online Mendelian Inheritance in Man #142623), although we now recognize that the two children Professor Hirschsprung described were, in fact, suffering from Hirschsprung’s-Associated Enterocolitis (HAEC).
Since the time of Harald Hirschsprung, some of the greatest minds in all of medicine engaged in the quest to understand the etiology and pathophysiology of HSCR and to develop treatment strategies[2]. This has led to an understanding of the underlying genetics of HSCR, methods to establish the diagnosis, and a variety of surgical options to resect the aganglionic portion of bowel. However, despite technically adequate surgery and histologic confirmation of removal of the pathologic, aganglionic segment, there is increasing recognition that the majority of Hirschsprung’s patients continue to suffer from bowel dysfunction, soiling or incontinence, HAEC, and decreased quality of life[3]. Of these complications, HAEC has come under increasing scrutiny over the last 10–15 years (Figure 1), likely due to an increasing recognition of its associated morbidity and mortality. Here, we will review aspects of HAEC that are well established (Epidemiology, Diagnosis, Treatment) as well as those that, based on recent translational research, are emerging (Animal Models, Mucosal Immunity, Microbiome, Genetics, Treatment).
Figure 1.
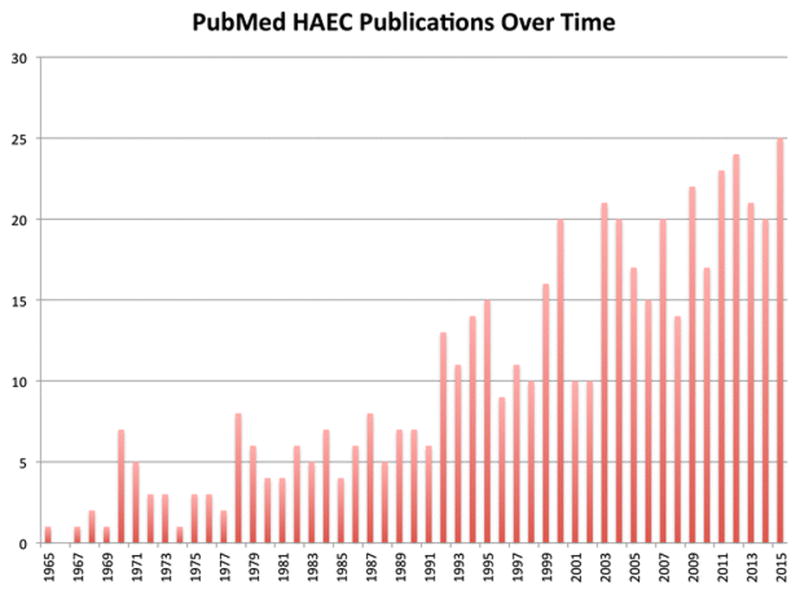
Number of HAEC Publications in PubMed over time. A PubMed search for ((Hirschsprung[Title/Abstract] OR Hirschsprung’s[Title/Abstract]) AND Enterocolitis) was conducted on 31 December 2015 to determine the number of publications related to HAEC over time. The number of publications related to HAEC is increasing, with 25 in 2015.
Established Concepts
Epidemiology
HSCR is a common cause of intestinal obstruction in the newborn, with an incidence that ranges worldwide from 1:2000 to 1:12000 live births, but is most commonly reported as 1:4000[4]. While previously thought to be rare in premature infants, recent analyses indicate that the incidence of HSCR is similar for pre-term and term births[5–7]. The incidence of HAEC reported in the literature is highly variable, with reports as high as 60%, although an incidence of 25–35% is generally quoted in current series[8, 9]. HAEC can occur pre-operatively or post-operatively, with a similar incidence in both settings. It is the presenting symptom of HSCR in 25% of infants and it is these infants that are most likely to experience mortality from the disease[10–13]. Mortality from HAEC ranges from 1–10%, with the majority of deaths occurring in newborns prior to definitive operation[13]. It is unclear if higher mortality in newborns is due to delay in diagnosis, immature immune mechanisms in the newborn, or disease-intrinsic differences from the pre-operative to post-operative period.
Diagnosis
The classic manifestations of HAEC are similar to those originally described by Professor Hirschsprung; abdominal distention, fever, and diarrhea. However, the spectrum of symptoms that children may present with additionally includes vomiting, rectal bleeding, lethargy, loose stools, and obstipation. These symptoms are non-specific, and may lead to alternative diagnoses (e.g. infectious enteritis) or delays in diagnosis. This is reflected the wide range of incidence of HAEC reported in the literature. Ultimately, this has resulted in difficulties in comparing outcomes based on treatment strategies, operative approaches amongst different centers. In an attempt to address the difficulty in establishing the diagnosis of HAEC, a group of 27 gastroenterologists and surgeons participated in a Delphi process[14], starting with 38 features (history, patient characteristics, physical exam signs, laboratory findings, radiology findings, pathology findings) and narrowing this list down to 16 items to develop a HAEC score[15]. Unfortunately, this tool has proven difficulty in routine bedside use, and has not been widely adopted. Currently, overall clinical gestalt based on observation of the classic manifestations remains the mainstay of diagnosis.
Treatment
Current therapy for HAEC is relatively non-specific and consists of systemic antibiotics, rectal irrigations and bowel rest. These measures are directed towards managing the factors that are thought to contribute to pathogenesis and treating the acute symptoms. Systemic antibiotics are used empirically in HAEC, with metronidazole typically chosen to treat anaerobes. This agent has activity against Clostridium difficile, an organism that has been associated with HAEC[16]. Fecal stasis and bacterial overgrowth are treated by rectal irrigations, which are distinct from enemas in which large volumes of fluid may be retained in the already-distended bowel. Additionally, rare patients presenting with severe sepsis or with perforation will benefit from proximal enteric diversion, although cases of persistent and recurrent enterocolitis following diversion have been described[17, 18].
Some authors have advocated for the use of preventive measures in selected patient populations[9]. These measures include routine use of rectal irrigations in the post-operative period, long-term administration of oral metronidazole, use of pro-biotic therapy, and diverting enterostomy. Unfortunately, no definitive evidence to support routine use of these “preventive” measures exists[19].
Development of a targeted therapy for HAEC has the potential to decrease mortality and increase quality of life.
Emerging Concepts
Animal models
Because of familial occurrence in 5–20% of patients, association with syndromes, and the existence of spontaneous mutant animals (piebald lethal and lethal spotted mice), a genetic basis for HSCR had long been suspected. The identification of gene defects contributing to HSCR has advanced our ability to study the disease in the laboratory. The Rearranged during transfection (Ret) gene, on the short arm of chromosome 10, was identified in 15 Hirschsprung’s disease pedigrees in the early 1990s[20, 21]. Subsequent to this, multiple genes contributing to Hirschsprung’s disease have been identified (GDNF, NRTN, EDNRB, EDN3, ECE1, ZFHX1B, SOX10, etc.)[22]. Although these gene defects account for less than 30% of sporadic mutations found in humans, animal models in which these genes are mutated have provided valuable insight into the pathogenesis of HSCR and HAEC.
Mutations of RET and of Endothelin receptor B (EdnrB), both of which are involved in ENS formation, are the two most commonly identified HSCR gene defects in humans[22]. Recently, Ret has been shown to be necessary for the development of Peyer’s patches (PP), the primary inductive site for gastrointestinal host defense, which suggests a potential developmental link between the enteric nervous system (ENS) and gastrointestinal mucosal immunity[23]. Unfortunately, Ret−/− animals exhibit a severe phenotype with an ENS absent distal to the stomach and renal agenesis, resulting in death shortly after birth and limiting their utility in studying HAEC[24]. The second most common gene defect identified in patients with HSCR are mutations of EdnrB. EdnrB and its ligand, endothelin 3 (ET-3 or EDN3), regulate enteric neural crest cell proliferation, migration and differentiation[25]. Murine models of EdnrB mutation display aganglionosis of the distal hindgut, mimicking the most common clinical finding in HSCR patients[26, 27]. For this reason, mutation of EdnrB in mice, both in the whole animal (EdnrB−/−)[28–34] and specifically in neural crest cells only (EdnrBNCC−/−)[35, 36], has been used to study the pathogenesis of HAEC, with 9 publications using these models over the last five years.
Mucosal Immunity
The gut-associated lymphoid tissue (GALT) is the largest lymphoid organ in the body and is charged with providing protection against a variety of antigens that may gain access to the host, including food particles, commensal and pathogenic bacteria and their toxins[37]. The GALT can be organized into inductive and effector sites for the ease of discussion and investigation. Peyer’s patches (PP) are the primary inductive site for gut mucosal immunity[38]. PP are distinct collections of immune cells that display follicular architecture similar to lymph nodes and are located along the anti-mesenteric surface of the bowel. Circulating naïve T-lymphocytes and B-lymphocytes, which display α4β7 integrin and L-selectin, migrate from circulation to the PP via binding of MADCAM-1 (mucosal addressin cell adhesion molecule 1), which is expressed in the high endothelial venules associated with PP[39, 40]. The mucosa overlying PP contains specialized epithelial M cells that sample and transport antigen from the lumen of the bowel to the underlying PP, where it is presented to naïve lymphocytes by antigen presenting cells, such as dendritic cells (DC). Activated lymphocytes then enter efferent lymphatic channels to travel to mesenteric lymph nodes (MLN). From the MLN, activated lymphocytes travel through the thoracic duct into the systemic circulation. Homing of activated lymphocytes to the lamina propria (LP), the primary effector site of mucosal immunity, occurs via the coordinated action of a number of cellular adhesion molecules, cytokines and chemokines[41]. Once in the LP, T-lymphocytes produce the pro-inflammatory Th-2 cytokines interleukin-4 (IL-4) and IL-10 to stimulate immunoglobulin A (IgA) production by terminally differentiated B-lymphocytes, termed plasma cells. IgA, the most abundantly produced immunoglobulin isotype in the body, is then actively transported to the mucosal surface in dimeric form by polymeric immunoglobulin receptor (pIgR)[42]. During transport, five of the seven pIgR domains, termed the secretory component (SC), remain attached to the IgA dimer. The secreted complex, referred to as secretory IgA (SIgA), is the principle effector of antigen-specific immune defense along the mucosal surface.
There is abundant evidence in the literature to suggest that multiple aspects of the mucosal immune system detailed above are altered in the setting of HSCR/HAEC. Cheng, etl.al., using the EdnrB−/− mouse, developed a histopathology grading system for quantitative assessment of HAEC[28]. In this system, severity and depth of inflammation are graded separately, and the score summed to arrive at the final number. Similar histologic changes were observed in EdnrBNCC−/− mice during the development of HAEC[35, 36], with mortality in the fourth week of life. Additionally, there was a linear correlation between histology score and bacterial colonization of intraperitoneal organs[28].
A role for IgA in HAEC pathogenesis was demonstrated by Imamura, et.al, who found an increase in IgA containing plasma cells along the length of resected aganglionic bowel from patients with HAEC as compared to HSCR patients without HAEC[43]. They also found decreased luminal IgA (SIgA) in the same patients, suggesting decreased production or impaired transport to the lumen. Recently, a similar observation, decreased luminal SIgA, has been made in EdnrBNCC−/− mice[44]. Interestingly, this finding was specific to the gut, with normal levels of bronchial and nasal IgA seen in these animals. In the mouse, a specific subset of B-lymphocytes (IgM+IgDhigh), termed mature B-lymphocytes, from the PP are responsible for the majority of intestinal IgA production[45]. Unlike the Ret−/− animals, which lack PP, the EdnrBNCC−/− animals display a normal number of PP, but demonstrate decreased B220+IgM+IgDhigh (mature) B-lymphocytes in PP[44]. In the mouse, mature B-lymphocytes in PP are primarily of splenic origin[45]. Small splenic size and splenic lymphopenia was first noted in the EdnrB−/− animal[30]. They noted a relative reduction of B-versus T-lymphocytes as well as a negative correlation between splenic lymphocyte counts and intestinal inflammation on histologic analysis. These findings were extended in the EdnrBNCC−/−, in which B-cell lymphopenia was also observed[36]. Furthermore, they observed a decrease in marginal zone B-lymphocytes, suggesting impaired B-lymphocyte development or trafficking. Both groups have noted that similar lymphocyte population alterations may be observed in the setting of stress (e.g. HAEC). In an attempt to delineate the contribution of the EdnrB−/− genotype to the HAEC phenotype, Frykman, etl.al., performed bone marrow transplants from EdnrB−/− animals to Rag2−/− recipients and, separately, induced bowel obstruction in WT animals[33]. They concluded that stress from obstruction resulted in lymphocyte alterations similar to those seen in the HAEC models. However, they have additionally shown that, in the EdnrB−/− model, animals that undergo surgical relief of obstruction continue to carry a 40% risk of developing HAEC[46]. Additionally, they noted small splenic size in the EdnrB−/− bone marrow transplant recipients, further underscoring a role for EdnrB in lymphocyte development and/or trafficking.
Another critical facet of mucosal immunity is barrier defense. In the steady state, bacterial invasion across the colonic epithelium is prevented mucus, composed of glycosylated proteins of the MUC family and secreted by goblet cells[47]. This prevents the adherence of pathologic organisms to enterocytes, reducing susceptibility to infection. Multiple studies have demonstrated impaired intestinal barrier function in HAEC patients, specifically decreased secretion of mucin and increased adherence of bacteria to enterocytes[48–50]. Recently, Thiagarajah, etl.al., used human HSCR colonic biopsies and the EdnrB−/− mouse to investigate goblet cell alterations in the setting of HSCR and HAEC[32]. They found increased goblet cell numbers in HSCR patients and distal EdnrB−/− colons, along with up-regulation of goblet cell differentiation genes. Finally, they noted decreased amounts of membrane-bound Mucin 4 (Muc4), but not secreted Muc2, in the distal colons of EdnrB−/− animals. Further, functional studies have been performed in the same animal model[34]. In these studies, reduced rates of transport of microparticles and bacteria, both active and passive, were observed in the aganglionic and ganglionated bowel. Of note, increased goblet cell size was also noted in the EdnrBNCC−/− model, further suggesting alterations in goblet cell structure and function in the setting of HAEC[35]. In this study, the authors focused on the proximal, ganglionated bowel, and investigated Paneth cells, which are the primary source of antimicrobial compounds for epithelial protection[51]. They found reduced Paneth cell sPLA2 expression in ileal tissue and decreased sPLA2 activity in the lumen, along with a blunted sPLA2 response to bacterial challenge[35].
Together, these studies have delineated a series of mucosal immune defects in HSCR/HAEC that involve both the aganglionic, as well as ganglionated bowel, and extend beyond the GI tract. Further work to determine the mechanistic underpinnings of these defects, and potential therapeutic targets, is warranted.
Microbiome
Over the last decade, many lines of investigation have revealed the influence of the microbiome on human health and disease[52]. These studies have demonstrated both spatial and temporal variability of the microbiome within and amongst individuals, along with demonstrating dysbioses that may contribute directly to disease pathogenesis. Historically, investigations into an infectious cause for HAEC have failed to identify a single, responsible organism, although viral and bacterial etiologies have been suggested. However, recent studies in mouse models of HSCR/HAEC have identified broader changes in the intestinal microbiome that may contribute to HAEC. These advances have been made possible by next-generation sequencing technologies, which are culture-independent.
Two studies have used mouse models to examine the role of the microbiome in HAEC. Using the EdnrB−/− model, Ward, etl.al., demonstrated increasing diversity of the microbiome over time (from post-natal day 7–24), with a greater increase in the EdnrB−/− versus control animals[31]. Using principal coordinate analyses, which allow visualization of how the microbiome “clusters”, the HSCR animals clearly demonstrated distinct microbiota from the controls at all ages. When specific bacterial species were examined, the HSCR animals had higher levels of Bacteroidetes and Firmicutes than the control animals. They also noted decreased Lactobacillus species over time in both HSCR and control animals. In another study, using the EdnrB−/− model, survival could be extended to 36 days by changing to a liquid diet and the addition of oral antibiotics[28], further supporting a role for the microbiome in the development of HAEC. Another group has employed the EdnrBNCC−/− model in a similar fashion[35]. They noted similar compositions of the microbiota between HSCR animals and controls at an early time point, with a shift towards “dysbiosis” occurring prior to the onset of HAEC. Additionally, unlike the findings in the EdnrB−/− model, they noted a decrease in Lactobacillus in HSCR animals preceding HAEC. They also observed increases in Bacteroidetes and Clostridium species, and noted that only HSCR animals demonstrated Escherichia coli in their microbiome samples. Finally, unlike models of inflammatory bowel disease, they noted increased species diversity at the time of enterocolitis, suggesting distinct mechanisms of intestinal inflammation between HAEC and IBD.
Culture-independent techniques to study the microbiome have recently been applied to human HSCR/HAEC patients as well. DeFilippo, etl.al., followed a single child with HSCR over a five month time period, obtaining 15 fecal samples[53]. While the majority of clustering they observed was explained by the passage of time, they did note progression of microbiota changes from “pre-enterocolitis”, through acute HAEC, and on to remission phases. Yan, etl.al., applied a similar approach to four patients with HSCR, two with HAEC and two without[54]. They found that the patients with HAEC clustered separately from those without HAEC. Additionally, similar to the EdnrBNCC−/− finding[35], they noted increased species diversity in the HAEC patients. Finally, another group has examined both the bacterial and fungal microbiome in a group of 19 HSCR patients, half with a history of HAEC[55]. They noted HAEC patients had increased fungal diversity, with increased Candida species and reduced Malassezia and Saccharomyces species, suggesting the microbiota changes contributing to HAEC may extend beyond bacteria.
Genetics
From the accumulating basic science evidence, it is tempting to postulate that a specific genotypic mutation confers a higher risk for development of HAEC.
Nucleotide oligomerisation domain 2 (NOD2), which is a pattern recognition receptor, has been associated with Crohn’s disease and graft versus host disease[56]. In the intestine, NOD2 is involved in post-natal PP development and its deficiency results in overgrowth of lymphoid tissues in the gut[57]. However, a study of human patients with HSCR showed that while 19.2% of patients were carriers of NOD2 mutations, none of the patients that developed HAEC carried NOD2 variants[58].
Integrin beta-2 (ITGB2) is involved in cell-surface mediated signaling and is required for leukocyte extravasation from the blood into tissues. ITGB2 has been associated with chronic colitis conditions, and participates in T-cell development. In a study of blood and colonic tissue samples from 33 patients with HSCR, 66% of patients demonstrated ITGB2 variants[59]. Of these, 13 patients had experience episodes of HAEC, suggesting a molecular target for further investigation.
In an effort to further our understanding of how genotype is linked to phenotype, the Hirschsprung Disease Research Collaborative has gathered geneticists, pediatric surgeons, pediatricians, pathologists, and gastroenterologists together. This group is enrolling children and families worldwide in order to perform a global genetic analysis with deeply phenotyped clinical information. The goal of this research is to provide improved genetic counseling for patients and families and to discover possibilities for improved and individualized treatment.
Treatment
Based on the findings in basic science and pilot human studies, a few recent efforts have been made to advance our approach to HAEC treatment. El-Sawaf, etl.al., randomized children undergoing surgery for HSCR to receive either probiotic or placebo postoperatively[60]. While they did not observe a difference in the incidence of HAEC (28.3% overall) or recurrent HAEC (19–21%), this may be a limitation of the choice of probiotic formulation, rather than the overall treatment strategy. In contrast, Wang, etl.al., similarly randomized patients to receive probiotic for a duration of 4 weeks[61]. Over the subsequent three months, the probiotic group demonstrated decreased incidence and severity of HAEC, along with decreased circulating pro-inflammatory cytokines. These studies suggest that strategies aimed at altering susceptibility factors for HAEC may hold therapeutic potential.
Conclusions
Since its original description by Harald Hirschsprung 130 years ago, HAEC remains a common complication of HSCR, and carries significant morbidity and potential mortality. Recent advances in our mechanistic understanding of HAEC have focused on mucosal immunity, the role of the intestinal microbiome, and the role of genetics in determining disease phenotype (Figure 2). Initial efforts have been made to translate these discoveries into therapeutic measures, with mixed success. Moving forward, further basic science efforts combined with collaborative translational efforts will be required to advance our understanding and treatment of HAEC.
Figure 2.
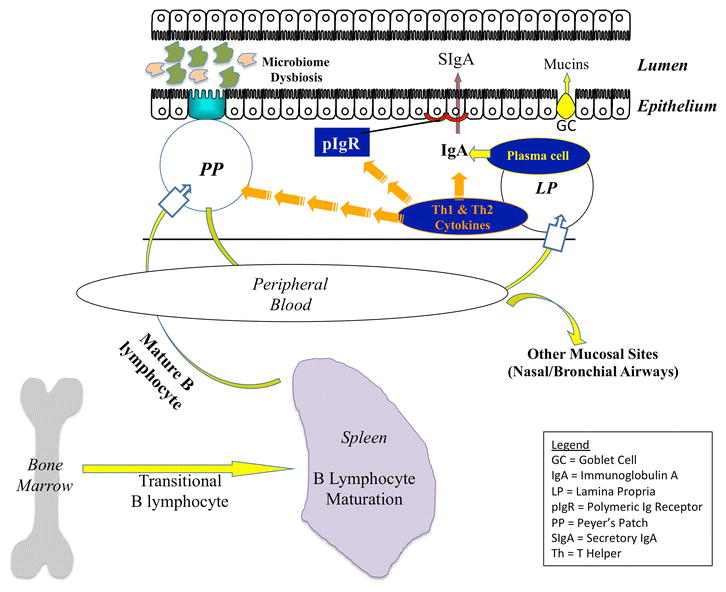
Emerging Concepts in HAEC Pathogenesis. Recent studies in the EdnrB−/− and EdnrBNCC−/− mouse, along with human studies, have implicated facets of mucosal immunity (encompassing innate and adaptive immunity, as well as epithelial barrier defense mechanisms) and microbiome dysbiosis in the development of HAEC.
Acknowledgments
This work was supported by the National Institutes of Health (K08DK098271) and the American College of Surgeons (George H.A. Clowes Award) to AG.
References
- 1.Hirschspring H. Jahrbuch Kinderheikunde. 1888. Constipation in newborns due to dilation and hypertrophy of the colon. [Google Scholar]
- 2.Raffensperger JG. Hirschsprung’s Disease: A Historical Review. Bull Soc Sci Med Grand Duche Luxemb. 1987;124:31–36. [PubMed] [Google Scholar]
- 3.Engum SA, Grosfeld JL. Long-term results of treatment of Hirschsprung’s disease. Semin Pediatr Surg. 2004;13:273–285. doi: 10.1053/j.sempedsurg.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 4.Kenny SE, Tam PKH, Garcia-Barcelo M. Hirschsprung’s disease. Semin Pediatr Surg. 2010;19:194–200. doi: 10.1053/j.sempedsurg.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Baxter KJ, Bhatia AM. Hirschsprung’s disease in the preterm infant: implications for diagnosis and outcome. Am Surg. 2013;79:734–738. [PubMed] [Google Scholar]
- 6.Duess JW, Hofmann AD, Puri P. Prevalence of Hirschsprung’s disease in premature infants: a systematic review. Pediatr Surg Int. 2014;30:791–795. doi: 10.1007/s00383-014-3540-8. [DOI] [PubMed] [Google Scholar]
- 7.Downey EC, Hughes E, Putnam AR, et al. Hirschsprung disease in the premature newborn: a population based study and 40-year single center experience. J Pediatr Surg. 2015;50:123–125. doi: 10.1016/j.jpedsurg.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 8.Austin KM. The pathogenesis of Hirschsprung’s disease-associated enterocolitis. YSPSU. 2012;21:319–327. doi: 10.1053/j.sempedsurg.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Frykman PK, Short SS. Hirschsprung-associated enterocolitis: prevention and therapy. YSPSU. 2012;21:328–335. doi: 10.1053/j.sempedsurg.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foster P, Cowan G, Wrenn EL. Twenty-five years“ experience with Hirschsprung”s disease. J Pediatr Surg. 1990;25:531–534. doi: 10.1016/0022-3468(90)90566-r. [DOI] [PubMed] [Google Scholar]
- 11.Rescorla FJ, Morrison AM, Engles D, et al. Hirschsprung’s disease. Evaluation of mortality and long-term function in 260 cases. Arch Surg. 1992;127:934–41. doi: 10.1001/archsurg.1992.01420080068011. discussion 941–2. [DOI] [PubMed] [Google Scholar]
- 12.Harrison MW, Deitz DM, Campbell JR, Campbell TJ. Diagnosis and management of Hirschsprung’s disease. A 25 year perspective. AJS. 1986;152:49–56. doi: 10.1016/0002-9610(86)90138-8. [DOI] [PubMed] [Google Scholar]
- 13.Pini-Prato A, Rossi V, Avanzini S, et al. Hirschsprung’s disease: what about mortality? Pediatr Surg Int. 2011;27:473–478. doi: 10.1007/s00383-010-2848-2. [DOI] [PubMed] [Google Scholar]
- 14.Dalkey N, Helmer O. An experimental application of the Delphi method to the use of experts. Management science 1963 [Google Scholar]
- 15.Pastor AC, Osman F, Teitelbaum DH, et al. Development of a standardized definition forHirschsprung’s-associated enterocolitis:a Delphi analysis. J Pediatr Surg. 2009;44:251–256. doi: 10.1016/j.jpedsurg.2008.10.052. [DOI] [PubMed] [Google Scholar]
- 16.Brearly S, Armstrong GR, Nairn R, et al. Pseudomembranous colitis: a lethal complication of Hirschsprung’s disease unrelated to antibiotic usage. J Pediatr Surg. 1987;22:257–259. doi: 10.1016/s0022-3468(87)80341-x. [DOI] [PubMed] [Google Scholar]
- 17.Marty TL, Seo T, Matlak ME, et al. Gastrointestinal function after surgical correction of Hirschsprung’s disease: long-term follow-up in 135 patients. J Pediatr Surg. 1995;30:655–658. doi: 10.1016/0022-3468(95)90682-7. [DOI] [PubMed] [Google Scholar]
- 18.Fujimoto T. Natural history and pathophysiology of enterocolitis in the piebald lethal mouse model of Hirschsprung’s disease. J Pediatr Surg. 1988;23:237–242. doi: 10.1016/s0022-3468(88)80730-9. [DOI] [PubMed] [Google Scholar]
- 19.Demehri FR, Halaweish IF, Coran AG, Teitelbaum DH. Hirschsprung-associated enterocolitis: pathogenesis, treatment and prevention. Pediatr Surg Int. 2013;29:873–881. doi: 10.1007/s00383-013-3353-1. [DOI] [PubMed] [Google Scholar]
- 20.Lyonnet S, Bolino A, Pelet A, et al. A gene for Hirschsprung disease maps to the proximal long arm of chromosome 10. Nat Genet. 1993;4:346–350. doi: 10.1038/ng0893-346. [DOI] [PubMed] [Google Scholar]
- 21.Romeo G, Ronchetto P, Luo Y, et al. Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung’s disease. Nature. 1994;367:377–378. doi: 10.1038/367377a0. [DOI] [PubMed] [Google Scholar]
- 22.Amiel J, Sproat-Emison E, Garcia-Barcelo M, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45:1–14. doi: 10.1136/jmg.2007.053959. [DOI] [PubMed] [Google Scholar]
- 23.Veiga-Fernandes H, Coles MC, Foster KE, et al. Tyrosine kinase receptor RET is a key regulator of Peyer’s Patch organogenesis. Nature. 2007;446:547–551. doi: 10.1038/nature05597. [DOI] [PubMed] [Google Scholar]
- 24.Schuchardt A, D’Agati V, Larsson-Blomberg L, et al. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367:380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- 25.Nagy N, Goldstein AM. Endothelin-3 regulates neural crest cell proliferation and differentiation in the hindgut enteric nervous system. Developmental Biology. 2006;293:203–217. doi: 10.1016/j.ydbio.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 26.Gariepy CE, Cass DT, Yanagisawa M. Null mutation of endothelin receptor type B gene in spotting lethal rats causes aganglionic megacolon and white coat color. Proc Natl Acad Sci USA. 1996;93:867–872. doi: 10.1073/pnas.93.2.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Druckenbrod NR, Powers PA, Bartley CR, et al. Targeting of endothelin receptor-B to the neural crest. genesis. 2008;46:396–400. doi: 10.1002/dvg.20415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng Z, Dhall D, Zhao L, et al. Murine model of Hirschsprung-associated enterocolitis. I: Phenotypic characterization with development of ahistopathologic grading system. J Pediatr Surg. 2010;45:475–482. doi: 10.1016/j.jpedsurg.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao L, Dhall D, Cheng Z, et al. Murine model of Hirschsprung-associated enterocolitis II:Surgical correction of aganglionosis does noteliminate enterocolitis. J Pediatr Surg. 2010;45:206–212. doi: 10.1016/j.jpedsurg.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng Z, Wang X, Dhall D, et al. Splenic lymphopenia in the endothelin receptor B-null mouse: implications for Hirschsprung associated enterocolitis. Pediatr Surg Int. 2010;27:145–150. doi: 10.1007/s00383-010-2787-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward NL, Pieretti A, Dowd SE, et al. Intestinal aganglionosis is associated with early and sustained disruption of the colonic microbiome. Neurogastroenterology & Motility. 2012;24:874–e400. doi: 10.1111/j.1365-2982.2012.01937.x. [DOI] [PubMed] [Google Scholar]
- 32.Thiagarajah JR, Yildiz H, Carlson T, et al. Altered Goblet Cell Differentiation and Surface Mucus Properties in Hirschsprung Disease. PLoS ONE. 2014;9:e99944. doi: 10.1371/journal.pone.0099944.t001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frykman PK, Cheng Z, Wang X, Dhall D. Enterocolitis causes profound lymphoid depletion in endothelin receptor B- and endothelin 3-null mouse models of Hirschsprung-associated enterocolitis. Eur J Immunol. 2015;45:807–817. doi: 10.1002/eji.201444737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yildiz HM, Carlson TL, Goldstein AM, Carrier RL. Mucus Barriers to Microparticles and Microbes are Altered in Hirschsprung’s Disease. Macromol Biosci. 2015;15:712–718. doi: 10.1002/mabi.201400473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pierre JF, Barlow-Anacker AJ, Erickson CS, et al. Intestinal dysbiosis and bacterial enteroinvasion in a murine model of Hirschsprung’s disease. J Pediatr Surg. 2014;49:1242–1251. doi: 10.1016/j.jpedsurg.2014.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gosain A, Barlow-Anacker AJ, Erickson CS, et al. Impaired Cellular Immunity in the Murine Neural Crest Conditional Deletion of Endothelin Receptor-B Model of Hirschsprung’s Disease. PLoS ONE. 2015;10:e0128822. doi: 10.1371/journal.pone.0128822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brandtzaeg P, Kiyono H, Pabst R, Russell MW. Terminology: nomenclature of mucosa-associated lymphoid tissue. Mucosal Immunology. 2008;1:31–37. doi: 10.1038/mi.2007.9. [DOI] [PubMed] [Google Scholar]
- 38.Craig SW, Cebra JJ. Peyer’s patches: an enriched source of precursors for IgA-producing immunocytes in the rabbit. J Exp Med. 1971;134:188–200. doi: 10.1084/jem.134.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berlin C, Berg EL, Briskin MJ, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74:185–195. doi: 10.1016/0092-8674(93)90305-a. [DOI] [PubMed] [Google Scholar]
- 40.Berg EL, McEvoy LM, Berlin C, et al. L-selectin-mediated lymphocyte rolling on MAdCAM-1. Nature. 1993;366:695–698. doi: 10.1038/366695a0. [DOI] [PubMed] [Google Scholar]
- 41.Bowman EP, Kuklin NA, Youngman KR, et al. The intestinal chemokine thymus-expressed chemokine (CCL25) attracts IgA antibody-secreting cells. J Exp Med. 2002;195:269–275. doi: 10.1084/jem.20010670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uren TK, Johansen F-E, Wijburg OLC, et al. Role of the polymeric Ig receptor in mucosal B cell homeostasis. J Immunol. 2003;170:2531–2539. doi: 10.4049/jimmunol.170.5.2531. [DOI] [PubMed] [Google Scholar]
- 43.Imamura A, Puri P, O’Briain DS, Reen DJ. Mucosal immune defence mechanisms in enterocolitis complicating Hirschsprung’s disease. Gut. 1992;33:801–806. doi: 10.1136/gut.33.6.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gosain A, Barlow-Anacker AJ, Erickson CS, et al. Impaired Cellular Immunity in the Murine Neural Crest Conditional Deletion of Endothelin Receptor-B Model of Hirschsprung’s Disease. PLoS ONE. 2015;10:e0128822. doi: 10.1371/journal.pone.0128822.s001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosado MM, Aranburu A, Capolunghi F, et al. From the fetal liver to spleen and gut: the highway to natural antibody. 2009;2:351–361. doi: 10.1038/mi.2009.15. [DOI] [PubMed] [Google Scholar]
- 46.Zhao L, Cheng Z, Dhall D, et al. A novel corrective pullthrough surgery in a mouse model of Hirschsprung’s disease. J Pediatr Surg. 2009;44:759–766. doi: 10.1016/j.jpedsurg.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keita ÅV, Söderholm JD. The intestinal barrier and its regulation by neuroimmune factors. Neurogastroenterology & Motility. 2010;22:718–733. doi: 10.1111/j.1365-2982.2010.01498.x. [DOI] [PubMed] [Google Scholar]
- 48.Teitelbaum DH, Caniano DA, Qualman SJ. The pathophysiology of Hirschsprung’s-associated enterocolitis: importance of histologic correlates. J Pediatr Surg. 1989;24:1271–1277. doi: 10.1016/s0022-3468(89)80566-4. [DOI] [PubMed] [Google Scholar]
- 49.Aslam A, Spicer RD, Corfield AP. Biochemical analysis of colonic mucin glycoproteins in children with Hirschsprung disease show disease specific alterations. Biochem Soc Trans. 1997;25:8S. doi: 10.1042/bst025008s. [DOI] [PubMed] [Google Scholar]
- 50.Mattar AF, Coran AG, Teitelbaum DH. MUC-2 mucin production in Hirschsprung’s disease: Possible association with enterocolitis development. J Pediatr Surg. 2003;38:417–421. doi: 10.1053/jpsu.2003.50071. [DOI] [PubMed] [Google Scholar]
- 51.Karlsson J, Pütsep K, Chu H, et al. Regional variations in Paneth cell antimicrobial peptide expression along the mouse intestinal tract. BMC Immunology. 2008;9:37. doi: 10.1186/1471-2172-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alverdy J, Gilbert J, DeFazio JR, et al. Proceedings of the 2013 A.S.P.E.N. Research workshop: the interface between nutrition and the gut microbiome: implications and applications for human health [corrected] JPEN J Parenter Enteral Nutr. 2014;38:167–178. doi: 10.1177/0148607113517904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Filippo C, Pini-Prato A, Mattioli G, et al. Genomics approach to the analysis of bacterial communities dynamics in Hirschsprung’s disease-associated enterocolitis: a pilot study. Pediatr Surg Int. 2010;26:465–471. doi: 10.1007/s00383-010-2586-5. [DOI] [PubMed] [Google Scholar]
- 54.Yan Z, Poroyko V, Gu S, et al. Biochemical and Biophysical Research Communications. Biochemical and Biophysical Research Communications. 2014;445:269–274. doi: 10.1016/j.bbrc.2014.01.104. [DOI] [PubMed] [Google Scholar]
- 55.Frykman PK, Nordenskjöld A, Kawaguchi A, et al. Characterization of Bacterial and Fungal Microbiome in Children with Hirschsprung Disease with and without a History of Enterocolitis: A Multicenter Study. PLoS ONE. 2015;10:e0124172. doi: 10.1371/journal.pone.0124172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 57.Barreau F, Meinzer U, Chareyre F, et al. CARD15/NOD2 Is Required for Peyer’s Patches Homeostasis in Mice. PLoS ONE. 2007;2:e523. doi: 10.1371/journal.pone.0000523.s004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lacher M, Fitze G, Helmbrecht J, et al. Hirschsprung-associated enterocolitis develops independently of NOD2 variants. J Pediatr Surg. 2010;45:1826–1831. doi: 10.1016/j.jpedsurg.2010.02.039. [DOI] [PubMed] [Google Scholar]
- 59.Moore SW, Sidler D, Zaahl MG. The ITGB2 immunomodulatory gene (CD18), enterocolitis, and Hirschsprung’s disease. J Pediatr Surg. 2008;43:1439–1444. doi: 10.1016/j.jpedsurg.2007.12.057. [DOI] [PubMed] [Google Scholar]
- 60.El-Sawaf M, Siddiqui S, Mahmoud M, et al. Probiotic prophylaxis after pullthrough for Hirschsprung disease to reduce incidence of enterocolitis: A prospective, randomized, double-blind, placebo-controlled, multicenter trial. J Pediatr Surg. 2013;48:111–117. doi: 10.1016/j.jpedsurg.2012.10.028. [DOI] [PubMed] [Google Scholar]
- 61.Wang X, Li Z, Xu Z, et al. Probiotics prevent Hirschsprung’s disease-associated enterocolitis: a prospective multicenter randomized controlled trial. Int J Colorectal Dis. 2015;30:105–110. doi: 10.1007/s00384-014-2054-0. [DOI] [PubMed] [Google Scholar]