

Abstract
The goal of this study was to explore the relationships between tenofovir (TFV) and emtricitabine (FTC) disposition and markers of biologic aging, such as the frailty phenotype and p16INK4a gene expression. Chronologic age is often explored in population pharmacokinetic (PK) analyses, and can be uninformative in capturing the impact of aging on physiology, particularly in human immunodeficiency virus (HIV)‐infected patients. Ninety‐one HIV‐infected participants provided samples to quantify plasma concentrations of TFV/FTC, as well as peripheral blood mononuclear cell (PBMC) samples for intracellular metabolite concentrations; 12 participants provided 11 samples, and 79 participants provided 4 samples, over a dosing interval. Nonlinear mixed effects modeling of TFV/FTC and their metabolites suggests a relationship between TFV/FTC metabolite clearance (CL) from PBMCs and the expression of p16INK4a, a marker of cellular senescence. This novel approach to quantifying the influence of aging on PKs provides rationale for further work investigating the relationships between senescence and nucleoside phosphorylation and transport.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Although profound biologic changes have been described with human aging, it is currently unknown if aging in HIV‐infected subjects affects antiretroviral drug disposition, particularly agents metabolized in immune cells, such as the nucleos(t)ide reverse transcriptase inhibitors TFV and FTC.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study aimed to explore both chronologic age and aging biomarkers' effects on TFV and FTC PK, including that of their intracellular metabolites.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE?
☑ The expression of a tumor suppressor gene, p16INK4a, explains observed variability of intracellular metabolite clearance of TFV and FTC, using nonlinear mixed effects modeling in infected participants of various chronologic ages.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This work provides the impetus for quantifying aging biomarkers when determining if the aging process affects PK and pharmacodynamics to reflect biology, rather than only assessing chronologic age.
The majority of human immunodeficiency virus (HIV)‐infected people in the United States is now 50 years old or older, the age typically considered “old” in this population for a variety of immunologic and functional reasons. Whether HIV infection accelerates the aging process is a subject of intense scientific debate; what is clear, however, is that older patients with HIV are at increased risk for morbidity and mortality compared with noninfected age‐matched controls.1, 2 Current US guidelines recommend starting antiretroviral therapy in all HIV‐infected patients regardless of CD4 T‐cell count, and particularly in older patients.3 The same regimens used in younger patients are currently recommended for older patients, while being mindful of drug‐drug interactions and comorbid conditions, although aging may affect physiology that affects drug disposition. Tenofovir (TFV), administered as tenofovir disoproxil fumarate (TDF), and emtricitabine (FTC), are the recommended nucleos(t)ide reverse transcriptase inhibitor backbones for most first‐line antiretroviral regimens.3 Both drugs undergo renal elimination, with dosing recommendations in place for patients with decreased creatinine clearance (CL).4 Both are metabolized inside target cells to the active phosphorylated metabolites, tenofovir diphosphate (TFV‐dp) and emtricitabine triphosphate (FTC‐tp). The catabolic processes are well described, whereas the anabolic processes for these metabolites are largely unknown.5 It is also unknown what effects aging on the cellular level, such as senescence, may have on the formation and elimination of these metabolites.
In most patients, renal function declines predictably with age, with a reduction in glomerular filtration rate of about 1% per year.6 Therefore, a decrease in CL may occur with renally eliminated drugs, such as TFV and FTC. Although severe toxicity is rare, subclinical decline in proximal tubule function may be present in up to 20% of patients receiving TFV.7 Older age is a known risk factor for increased TFV renal toxicity, and electrolyte changes due to renal damage may play a role in bone toxicity as well.8 Patients >45 years of age receiving TFV experience significantly greater declines in renal function than younger patients, a decrease 15 mL/min per year vs. 11 mL/min in patients <30 years old.9 Modest evidence suggests that cellular activation may increase intracellular phosphorylase activity in the elderly,10 potentially resulting in increased toxicity.11 The active intracellular phosphorylated forms of TFV and FTC have not been studied in older patients.12, 13, 14
Human aging is a complex process with multiple stages and methods of quantifying disability and dysfunction. Frailty is a commonly used marker of aging, and has been described using multiple physical assessments, including development of a phenotype.15 Although not fully validated in HIV‐infected patients, links to development of the phenotype and increased morbidity and mortality in HIV‐infected patients have been shown.16, 17 In older literature, frailty has primarily been cited as the cause of age‐related altered drug CL not due to well‐established changes in kidney function that decrease CL of renally eliminated drugs.18, 19, 20, 21, 22 On a cellular level, the tumor suppressor gene p16INK4a is a marker of senescence and its expression in peripheral blood T‐cells has been shown to be a biomarker of aging.23 As chronologic age alone does not capture either of these phenomena, the goal of this investigation was to quantify markers of biologic aging in HIV‐infected participants along the age spectrum receiving TDF/FTC, and use nonlinear mixed effects modeling to test the hypothesis that frailty and cellular senescence decrease the CL of TFV/FTC and their metabolites.
METHODS
Clinical study conduct
HIV‐infected adults (≥18 years old) receiving TFV/FTC 300/200 mg with either efavirenz (EFV) 600 mg or atazanavir/ritonavir (ATV/r) 300/100 mg once daily for ≥2 weeks were recruited from the UNC HealthCare Infectious Diseases Clinic (Chapel Hill, NC) and the Cone Health Regional Center for Infectious Diseases (Greensboro, NC). The study protocol was approved by the UNC Biomedical Institutional Review Board, as well as the Moses Cone Hospital Institutional Review Board (Clinicaltrials.gov NCT01180075).
Subjects were recruited for two separate sampling schemes, sparse and intensive. Both underwent a screening visit to assess their medication adherence and medical history prior to providing blood samples at four time points (predose, and then 2 hours, 4–6 hours, and 10–14 hours postdose) or 11 time points. Sparse sampling times were optimized based on the intensive pharmacokinetic (PK) study; detailed descriptions of methods and results for this group have been published.24 Briefly, to optimize collection times, a two‐compartment model with first‐order absorption and linear CL was fit to each drug using S‐ADAPT with the S‐ADAPT TRAN pre‐ and postprocessing package.25 The mean parameter estimates and their variability were used in designing the sampling scheme. D‐optimal sampling design (ADAPT5; Biomedical Simulations Resource, University of California at Los Angeles, Los Angeles, CA) was used to select windows of sampling times practical and feasible for study conduct, optimizing the CL for each drug. For each drug, four optimal times were selected, and then times were aligned as closely as possible for each drug in the regimen (TFV, FTC, and EFV; or TFV, FTC, and ATV/r) in order to capture informative regions of the PK profiles. The optimal times were aligned so one scheme could be used regardless of regimen. Power calculations, using a two‐sided test with a significance level of 0.05, for a regression analysis of age effects on drug CL, a sample size of 36 provides ability to detect a correlation of 0.55 (30% variation) with 90% power.
At each sampling time, subjects provided 18 mL of whole blood for extracellular and intracellular drug concentrations; at one visit, subjects underwent frailty phenotyping and provided additional whole blood for pharmacogenomics, cytokine concentrations, and p16INK4a expression analysis. For intensive subjects, frailty phenotyping was performed at screening, and cytokine and p16INK4a samples were not collected. Subjects completed the protocol in one to three study visits, completing an adverse event questionnaire at each visit; all intensive subjects completed all in a single visit. Sparse‐sampling subjects with evening doses temporarily switched to morning to accommodate study visits; the switch was undertaken gradually over several days, and subjects dosed at the new time consistently for at least 5 days prior to providing samples. Study staff provided subjects with detailed dosing calendars and consulted with the subject's clinic provider prior to initiating the change. If subjects desired a return to their previous dosing time, a calendar was also provided to gradually switch back. Intensive subjects did not switch dosing times for their overnight visits.
Subjects were included in the study if they: demonstrated adherence, defined as: <3 doses missed in the last 30 days, with no missed doses in the 3 days immediately prior to sampling, by participant self‐report on provided dosing calendars. Subjects were asked to record the times of medication dosing for 7 days prior to the first visit, and for 3 days prior to subsequent study visits; actual dosing times were used in PK analysis. Included subjects were not anemic by Division of Acquired Immunodeficiency Syndrome (National Institute of Allergy and Infectious Diseases, National Institutes of Health) grade 1 criterion (hemoglobin <10 mg/dL), and had no unstable medical conditions or Division of Acquired Immunodeficiency Syndrome grade 2 or higher laboratory abnormalities, with the exception of total bilirubin concentrations for subjects receiving ATV/r. Subjects were included if their creatinine CL, as calculated by the Cockroft‐Gault formula using total body weight,26 was >30 mL/min. Women of childbearing potential underwent a urine pregnancy testing prior to providing samples at each visit, as pregnancy was exclusionary. If a subject did not have an available CD4+ count and HIV viral load available in their clinic record within 90 days of enrollment, these laboratory results were obtained upon study initiation.
Frailty phenotyping was conducted per Fried et al.,15 and consisted of questions regarding unintentional weight loss, perceived effort of daily living, grip strength, walk time for 15 feet, and the Minnesota Leisure Activity Questionnaire. Each component has defined cutoff values, and if a subject's result for a component falls into the range defined as frail, this is a positive component. For example, if a subject's walk time exceeds the cutoff time based on height, this would be considered a positive component; if the time were less than the cutoff time, then it would be considered a negative component. Components are not weighted; the final result of this phenotyping is the sum of the positive components, and can range from 0–5. Frailty was defined as having three or more positive components of the phenotype; prefrail was defined as having one to two positive components; nonfrail was defined as having no positive components. Testing was conducted by staff of the NC TraCS Institute Bionutrition Core for consistency. Frailty (3 or more positive components) and age <55 years were exclusionary for the intensive‐sampling group (i.e., only older, nonfrail subjects were included in this group).
Analytical methods
Analytical methods have been previously described.27 All drug concentrations were determined in the UNC Center for AIDS Research Clinical Pharmacology and Analytical Chemistry Laboratory. The laboratory participates in national and international proficiency testing of its methods, and consistently achieves >95% accuracy in this testing. For the TFV and FTC assay in plasma, the lower limit of quantification of the assay is 10 ng/mL, and within‐ and between‐day precision ranged from 1.7–3.7% and 3.7–5.2%, for FTC and TFV, respectively. For the TFV‐dp and FTC‐tp assay in peripheral blood mononuclear cells (PBMCs), the lower limit of quantification is 0.2 ng/mL for TFV‐dp and 2 ng/mL for FTC‐tp. For each PBMC sample, a sample‐specific lower limit of quantification is determined based on the cell count of the sample, and reported as femtomoles/million cells. Inter‐run calibration standards and quality controls were within 15% acceptance criteria for precision and accuracy for all analytes.
Whole blood (7 mL), collected in K2EDTA tubes, was used to isolate CD3+ T‐cells and measure p16INK4a expression by Taqman quantitative reverse transcriptase‐polymerase chain reaction using previously published methods in HIV‐infected subjects.23
Pharmacokinetic analysis
Dataset preparation, exploration, and visualization were performed using R‐software version 3.1.1 (www.r-project.org). FTC, FTC‐tp, TFV, and TFV‐dp concentration‐time data in plasma and PBMCs were analyzed using nonlinear mixed effect modeling in NONMEM 7.3 (ICON Development Solutions, Ellicott City, MD). A stochastic approximation expectation maximization with simulated annealing was used with ADVAN5 (FTC/FTC‐tp) or ADVAN6 (TFV/TFV‐dp) to model linear PKs. Data were analyzed as micromoles/L. The steady‐state feature in NONMEM was used, and participants’ self‐reported dose times were input for seven doses prior to their first visit and three prior doses for subsequent visits. The PKs of FTC and TFV in plasma, and FTC‐tp and TFV‐dp intracellularly were studied sequentially, in a stepwise manner, with models involving FTC and TFV optimized first, and then combined with their respective phosphorylated metabolites. All parameters were simultaneously estimated for the final model.
Several structural models were evaluated to characterize plasma concentrations. One, two, and three‐compartment models with linear CL and first‐order absorption were tested. For each of these models, proportional, additive, and proportional/additive combination error models were evaluated to describe the residual variability. Additionally, the correlation between the errors for the two analytes measured at each time point in each model was investigated by using the L2 data item. An exponential error model was used to describe interindividual variability (IIV). Selection of the structural PK model, IIV, and residual error model was driven by the data, biological plausibility, and minimization of the objective function, successful convergence, goodness‐of‐fit plots, Eigenvalue ratio of the covariance step, and precision of the parameter estimates. When comparing nested models, a decrease in objective function was considered significant (P < 0.01; df = 1). For non‐nested model comparisons, the Akaike Information Criteria was used for model selection, as calculated in the following formula:
To incorporate the intracellular triphosphate concentrations into the model, both linear and nonlinear functions were tested. Based on the anabolism of these species, the models assume that the triphosphates are fully contained within the cellular compartment until it is cleared by either cell death and subsequent nucleotide recycling or an unknown catabolic process. Although some single‐dose data suggest recycling of these metabolites,28 our data did not lend itself to a mathematical description of these phenomenon. Linear kinetic transitions were initially modeled by using a fractional amount of clearance (FRAC) parameter to estimate the fractional component of the total plasma CL into PBMCs, and CL out of the central compartment. Other linear models were investigated, such as estimating the parent drug CL into PBMC compartment driven by central plasma concentration. Nonlinear functions were also investigated, but due to sparsity of the data and steady‐state sampling of one dose, multiple parameter estimates were highly correlated.
Once the base model was fully developed and stable, the influence of subject covariates on PK parameter estimates was evaluated. Potential covariates were first inspected for potential relationships to variance components by graphical exploration of individual empirical Bayesians estimates and potential covariates. Of the potential covariates that showed a relationship to empirical Bayesians estimates, only covariates that made physiologic sense progressed to a formal forward addition/backward elimination stepwise covariate search. Covariates were tested using either an exponential or power model (Eqs. (1) and (2)) for continuous covariates based on visual inspection of relationship and biological plausibility.
| (1) |
| (2) |
Where TVPi is the typical value of a parameter (P) for the ith individual, COVi is the value of the covariate in the same individual, θ1 is the typical value for an individual with median covariate value, and θ2 is estimated effect of the covariate on P. All continuous covariates were centered at the median value for the dataset. Categorical covariates were modeled with a linear relationship (Eq. (3)):
| (3) |
Where TVPi is the typical value of a parameter (P) for the ith individual, INDi is an indicator variable equal to 0 or 1, and θ1 is the typical value for an individual without the covariate (INDi = 0), and θ2 is fractional change in typical value if covariate is present (INDi = 1).
A covariate was retained if the effect was biologically plausible, led to a reduction in the IIV, and resulted in a significant reduction in the objection function value. During “forward addition” of each covariate into the base model, a decrease in the objective function value of ≥3.84 points (P = 0.05, degrees of freedom = 1) was considered significant and the covariate was included in the model. For “backward elimination” a worsening (increase) of the objective function value by ≥6.64 points (P = 0.01; df = 1) when single covariates were removed from the model led to retention of the covariate in the model. Missing covariates for individuals were imputed as having the median value of that covariate in population for continuous covariates and were imputed as the standard population for categorical covariates.
Model evaluation and validation
The final combined model was validated using visual predictive check and bootstrap methods. To generate visual predictive check plots, 1,000 datasets were simulated using the parameter estimates from the final model. From the simulated plasma and intracellular concentrations, the 5th, 50th and 95th percentile (90% prediction interval) along with associated 95% confidence interval around those percentiles were constructed at each time point and overlaid with the observed concentrations to construct visual predictive check plots for plasma and PBMCs. If 90% of the observed concentrations fell within the predictive interval, this demonstrated the adequacy of the model. A nonparametric bootstrap method with repeated random sampling with replacement from the original dataset was carried out 500 times. Model stability was assessed by a convergence rate of bootstrap datasets of ≥90%. The parameter estimates generated from the bootstrap analysis along with their associated 95% confidence intervals were then compared with the model estimated PK parameters.
RESULTS
Participant disposition and demographics
Ninety‐one participants contributed data to the analysis; 60 received TDF/FTC/EFV, and 31 received TDF/FTC/ATV/r. Six participants on each regimen completed intensive sampling. Participant demographics are presented in Table 1; the median age was 49 years, 3 participants demonstrated frailty, and 19 were prefrail. The demographics of the intensively sampled participants have been previously published.24
Table 1.
Participant demographics, all regimens and by antiretroviral background regimen
| Characteristics | Total (n = 91) | EFV arm (n = 60) | ATV/r arm (n = 31) |
|---|---|---|---|
| Age, years | 49 (22–73) | 48 (22–73) | 49 (24–61) |
| HIV duration, years | 10 (1–31) | 10.5 (1–31) | 10 (1–24) |
| BMI, kg/m2 | 28.3 (17.3–44.3) | 27.2 (17.3–44.3) | 30.3 (20.2–40.4) |
| CrCL, mL/min | 107 (43–227) | 108 (43–200) | 100 (67–227) |
| Log2 (p16INK4a) | 2.0 (0.16–3.9) | 2.0 (0.20–2.8) | 2.2 (0.16–3.9) |
| CD4 count, cells/mm3) | 671 (10–1,724) | 662 (10–1,724) | 692 (375–1,501) |
| Female | 30 (33) | 18 (30) | 12 (39) |
| African American | 53 (58) | 34 (57) | 19 (61) |
| White | 32 (35) | 22 (37) | 10 (32) |
| Other race | 6 (7) | 4 (7) | 2 (6) |
| Total frailty markers | |||
| 0 | 65 (72) | 44 (73) | 21 (68) |
| 1 | 14 (15) | 7 (12) | 7 (23) |
| 2 or more | 8 (9) | 5 (8) | 3 (10) |
All participants received tenofovir disoproxil fumarate 300 mg and emtricitabine 200 mg by mouth once daily. Data are presented as median (range) or number (percentage).
ATV/r, atazanavir/ritonavir; BMI, body mass index; CrCL, creatinine clearance, as calculated by the Cockroft‐Gault equation; EFV, efavirenz; HIV, human immunodeficiency virus.
Pharmacokinetic modeling
Figure 1 shows the model schematics of the TFV/TFV‐dp model and the FTC/FTC‐tp model. The absorption of both FTC and TFV from the gastrointestinal tract was modeled as first‐order input to a central compartment. The link model was described by a unidirectional flow from plasma to the PBMC compartment. The initial approach to linking plasma to PBMC was to estimate the fraction of total drug CL into the PBMC compartment, and the fraction cleared out of the central compartment. This required estimating plasma CL and FRAC term, which represented the fraction of CL to each compartment. This methodology worked well for the FTC/FTC‐tp model, but for TFV/TFV‐dp model, the FRAC estimate was not stable and highly sensitive to starting estimates, and highly correlated to CL out of the PBMC. An alternate approach used the plasma TFV concentrations to drive the TFV‐dp concentrations in PBMC. As volume of the PBMC compartment is unidentifiable for both models, this parameter was fixed to a value of one for both models, such that the CL out of the PBMC was modeled as CLpbmc* concentration of FTC‐tp/TFV‐dp. For both drugs, an exponential error model and a proportional error model were used to describe the IIV and residual variability, respectively. An attempt was made to estimate correlation between the random effects associated with the IIV and the correlation between residual error associated with different analytes within an individual, but did not improve the objective function, and the estimated correlation was <20%, so this was removed from the model. For TFV/TFV‐dp, four samples yielded concentrations below the limit of quantification, and two samples for FTC/FTC‐tp yielded below the limit of quantification concentrations (<1% of total observations). One additional sample did not have sufficient volume to be analyzed. These samples were treated as missing in the dataset.
Figure 1.
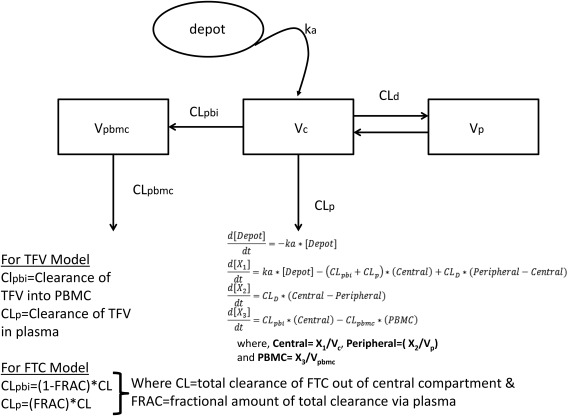
Model schematics. The same basic structural model was used for both drugs: first‐order oral absorption with plasma concentrations described by a linear two‐compartment model with clearance from the central compartment, with the central compartment linked to the peripheral blood mononuclear cell (PBMC) metabolite compartment by a linear process. Differences between tenofovir (TFV) and emtricitabine (FTC) are noted, and differential equations are provided. ka, absorption rate constant; Vc, volume of the central plasma compartment; CLp, clearance from the central compartment; CLd, intercompartmental clearance; Vp, volume of the peripheral plasma compartment; CLpbi, clearance from plasma to the PBMC; Vpbmc, volume of the PBMC compartment, fixed to 1 for both models; CLpbmc, clearance out of the PBMC compartment; X1, mass of drug in the central compartment; X2, mass of drug in the peripheral compartment; X3, mass of drug in the PBMC compartment.
Final fitted model parameters, along with relative SEs and associated bootstrap 95% confidence intervals, are provided in Table 2. Although other investigations have suggested that TFV‐dp disposition is affected by protease inhibitors,29 this model did not identify background regimen as a significant covariate, although it was tested. Therefore, data from participants on EFV and ATV/r were comodeled.
Table 2.
Model output for both the TFV/TFV‐dp and FTC‐tp models
| TFV/TFV‐dp model | FTC/FTC‐tp model | ||||
|---|---|---|---|---|---|
| Parameter (units) | Final NONMEM estimate (RSE%) | Bootstrap 95% CI | Parameter (units) | Final NONMEM estimate (RSE%) | Bootstrap 95% CI |
| Fixed effects | |||||
| ka (1/hr) | 0.623 (40) | 0.100–1.50 | ka (1/hr) | 0.298 (9) | 0.217–0.413 |
| Vc (L) | 186 (59) | 10.0–462 | Vc (L) | 19.7 (11) | 6.35–31.6 |
| CLp (L/hr) | 43.2 (4) | 37.1–47.1 | CL (L/hr) | 15.6 (2) | 15.0–17.3 |
| CLd (L/hr) | 176 (20) | 0.09–274 | CLd (L/hr) | 7.77 (13) | 3.82–11.8 |
| Vp (L) | 800 (17) | 567–1000 | Vp (L) | 98.5 (5) | 83.6–398 |
| CLpbmc (L/hr) | 2.20*10−3 (116) | 1.80*10−3, 5.30*10−3 | CLpbmc (L/hr) | 2.70*10−2 (2) | 1.50*10−2, 4.30*10−2 |
| CLpbi (L/hr) | 2.27*10−3 (116) | 1.00*10−3, 5.40*10−3 | FRAC | 0.986 (0) | 0.977–0.992 |
| Lag time (hr) | 0.472 (2) | 0–0.968 | |||
| Covariate effects | |||||
| p16INK4a on CLpmbc a | 0.460 (23) | 0.278–0.665 | p16INK4a on CLpmbc a | 0.495 (21) | 0.276–0.754 |
| CrCL on CLp b | 0.698 (20) | 0.403–1.02 | CrCL on CLp b | 0.551 (22) | 0.279–0.811 |
| Age on Vc c | 2.47 (38) | 0.500–4.98 | |||
| IIV | |||||
| IIV CLp | 37.4 (8) [6%] | 30.0–43.6 | IIV CL | 34.6 (9) [8%] | 24.5–41.2 |
| IIV Vc | 136 (27) [31%] | 87.7–300 | IIV Vc | 67.8 (30) [29%] | 22.4–140 |
| IIV Vp | 34.6 (49) [65%] | 20.0–74.2 | IIV CLpbmc | 53.8 (8) [6%] | 41.2–60.8 |
| IIV CLpbmc | 33.2 (82) [46%] | 28.2–50.9 | Covariance Vc∼CL | −0.07 (63) | −0.20 to 0.16 |
| IIV CLpbi | 44.7 (41) [25%] | 26.5–50.0 | Covariance Vc∼CLpbmc | 0.13 (84) | −0.04 to 0.62 |
| Covariance CLp∼Vc | 0.22 (58) | −0.36 to 0.68 | Covariance CL∼CLpbmc | 0.05 (80) | 0.01–0.09 |
| Correlation CLp∼Vc | 0.434 | – | Correlation Vc∼CL | −0.397 | – |
| Correlation Vc∼CLpbmc | 0.411 | – | |||
| Correlation CL∼CLpbmc | 0.248 | – | |||
| Residual error | |||||
| Plasma (proportional) | 28.3 (10) | 24.5–33.2 | Plasma (proportional) | 38.7 (10) | 31.6–44.7 |
| PBMC (proportional) | 38.7 (8) | 36.1–42.4 | PBMC (proportional) | 42.4 (11) | 37.4–50.0 |
Data are presented as population means and bootstrapped 95% CIs; IIV parameters and proportional error parameters are reported as %CV. The relationship between model parameters fitted with covariance relationships are presented as covariances. Shrinkage of IIV is indicated in brackets.
CI, confidence interval; CrCL, creatitine clearance; FRAC, fractional amount of clearance; FTC/FTC‐tp, emtricitabine/emtricitabine triphosphate; IIV, interindividual variability; PBMC, peripheral blood mononuclear cell; RSE, relative standard error; TFV/TFV‐dp, tenofovir/tenofovir diphosphate.
aCLpbmc = CLpbmc,pop EXP(COEF*(log2p16INK4a−2.04)), where CLpbmc,pop represents the population estimate of PBMC clearance, COEF is the covariate coefficient, log2p16INK4a is the log‐base‐2 transformed expression level of the marker, and 2.04 is the median value of this marker. bCLp = CLp,pop * (CrCL/107.24)^COEF, where CLp,pop represents the population estimate of the central clearance, COEF is the power coefficient, CrCL is the subject's estimated creatinine clearance, and 2.04 is the population median estimated creatinine clearance. cVc = Vc,pop * (Age/49)^COEF, where Vc,pop represents the population estimate of the central volume of distribution, COEF is the power coefficient, age is the subject's age, and 49 is the population median age.
For both drugs, the parent drug CL was well estimated, as seen by the narrow confidence intervals. The central volume estimates for both compounds had large confidence intervals, which were expected due to the sparsity of the data and the sampling scheme that did not target peak concentrations. For the TFV/TFV‐dp model, there was uncertainty in CL into and out of PBMCs. For both drugs, the mass transferred to PBMC is relatively small compared to CL from the central compartment; the FRAC to PBMC compartment estimate for FTC is 1.6%, and although not explicitly included in the final model for TFV, the fractional amount of drug transferred to PBMC compartment would be <1%. For both drugs, log2 p16INK4a expression in peripheral blood T cells was found to be a significant covariate for PBMC CL of the metabolites. As expected based on CL mechanisms, creatinine CL significantly influenced both TFV and FTC plasma CL. Additionally, in the FTC model, age was found to be a marginally significant covariate for central volume of distribution for the parent compound, but was not found to be a significant covariate on drug CL for either TFV or FTC. Frailty was also not found to be a significant covariate on CL or central volume; it was tested as both a binary outcome (zero positive components vs. 1+ positive components, lumping frail and prefrail) and a categorical variable (frail (3+ positive components), prefrail (1–2 positive components), or non‐frail (zero positive components)).
Routine diagnostic plots, including observed vs. individual predicted concentrations and population concentrations, and normalized prediction distribution error vs. observations and time are included in Supplementary Figures S1 and S2. Visual predictive checks for each analyte are provided in Figure 2 (TFV/TFV‐dp) and Figure 3 (FTC/FTC‐tp). These plots indicate both models adequately describe the data collected from the clinical study; some overlap in the predicted confidence intervals are seen near the end of the dosing interval, likely due to the scarcity of data in that region of the PK curve, as only the 12 intensively‐sampled participants provided data between 14 and 24 hours postdose.
Figure 2.
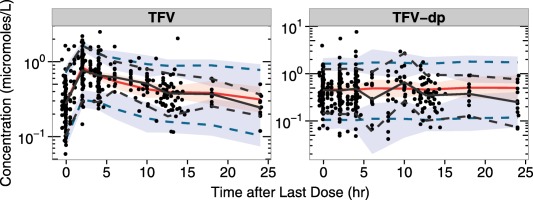
Tenofovir/tenofovir diphosphate (TFV/TFV‐dp) visual predictive checks. For each graph, time since the last dose in hours is on the x axis, with concentrations on the y axis. The black dots represent observed data. Percentiles of observations were presented in black, with solid lines for 50th percentiles and dotted lines for 5th and 95th percentiles. The 50th percentiles of predictions were presented in red solid lines, with 95% confidence intervals in red shaded areas. The 5th and 95th percentiles of predictions were presented in blue dotted lines, with 95% confidence intervals in blue shaded areas.
Figure 3.
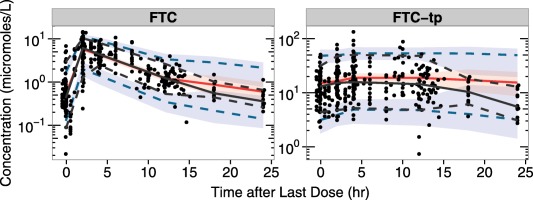
Emtricitabine/emtricitabine triphosphate (FTC/FTC‐tp) visual predictive checks. For each graph, time since the last dose in hours is on the x axis, with concentrations on the y axis. The black dots represent observed data. Percentiles of observations were presented in black, with solid lines for 50th percentiles and dotted lines for 5th and 95th percentiles. The 50th percentiles of predictions were presented in red solid lines, with 95% confidence intervals in red shaded areas. The 5th and 95th percentiles of predictions were presented in blue dotted lines, with 95% confidence intervals in blue shaded areas.
DISCUSSION
In this work, by quantifying aging‐related factors, such as p16INK4a and frailty, in a sample of HIV‐infected participants, new potential explanations for PK variability were uncovered. The covariate relationship between TFV‐dp and FTC‐tp CL and p16INK4a expression is novel, and is also associated with lower TFV‐dp and FTC‐tp exposures in these participants,27 as PBMC CL increases with higher expression of p16INK4a. This observation is consistent with our hypothesis that increased cellular activation, which may be associated with senescence, increases the kinase activity responsible for the metabolism of the intracellular species. The exact mechanism for this association remains to be elucidated, but could be related to effects of cellular senescence on transporter function in both uptake and efflux of the drugs and/or changes in the catabolic and anabolic processes for the metabolites inside the cell. Both TFV and FTC are transporter substrates, and recent work has demonstrated variability in transporter expression can affect intracellular PKs.30 As in previous models of TFV and FTC, creatinine CL is a significant covariate for TFV and FTC plasma CL.31, 32, 33, 34 Age and creatinine CL are correlated, and an independent effect of chronologic age above that of creatinine CL was not observed in either model.
Although earlier PK work demonstrated a link between frailty and drug clearance,18, 19, 20, 21, 22 our model did not indicate that such a relationship exists for these drugs or their metabolites. We did recruit low numbers of frail participants in this study, likely due to selection bias, rather than lack of frail patients in the clinics used for recruitment, as study participation required separate visits to the research center outside of clinical care. Given the cross‐sectional nature of this investigation, it is also possible that our markers of aging are confounded by other variables that we did not measure. Future work will aim to further refine the model by collecting samples from frail participants in the clinic, without additional visits. Given our results, and those of others, dosage adjustments beyond those recommended for renal dysfunction,4 based on age, frailty, or senescence, cannot be made at this time. The model results do provide useful insight into further mechanistic in vitro and in vivo research at the level of the aging and senescent cell, which can then be translated into the potential clinical effects on the whole organism.
TFV disoproxil fumarate is likely to be largely replaced in the developed world, and possibly in the developing world, by TFV alafenamide, a novel formulation designed to increase intracellular TFV‐dp concentrations and minimize TFV plasma exposures35; understanding the ramifications of altered intracellular exposure in aging patients, who may have pre‐existing renal dysfunction and increased potential for adverse events, in addition to frailty and cellular senescence, will be critical to optimizing care of the older HIV‐infected patient receiving this formulation.
In summary, nonlinear mixed effects modeling demonstrates a significant covariate relationship between the CL of TFV‐dp and FTC‐tp from PBMCs and expression of the cellular senescence marker p16INK4a, although not with the Fried et al.15 frailty phenotype, in this sample of HIV‐infected subjects receiving TDF/FTC and assessed for presence of aging biomarkers. Measuring aging biomarkers should become standard when assessing the potential effects of age on PKs in the HIV‐infected population.
Supporting information
Supplementary information accompanies this paper on the CPT: Pharmacometrics & Systems Pharmacology website (http://psp-journal.com)
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
The authors wish to thank the IRB 09‐2120 study participants, as well as the staffs of the UNC Clinical and Translational Research Center (UL1RR02574), the UNC Health Care Infectious Diseases Clinic, and the Cone Health Regional Center for Infectious Diseases. We also thank Angela Kashuba for oversight of drug concentration analysis in the UNC Center for AIDS Research Clinical Pharmacology and Analytical Chemistry Laboratory, and Brian Maas for his assistance with graphical analysis. J.B.D. is supported by K23AI093156; funding for this work was provided in part by UL1RR02574 and the Society of Infectious Disease Pharmacists Young Investigator Research Award. J.W.C. is supported by the UNC Eshelman School of Pharmacy/GlaxoSmithKline PK/PD Fellowship. M.C., H.M.A.P., C.S., N.W., S.M., R.W., and M.G.H. are supported in part by the UNC Center for AIDS Research (P30AI050410). C.T. and N.E.S. are supported by R01‐AG024379‐10.
Conflict of Interest
Noncompartmental analysis of the intensive study has been previously published (ref. 15). A statistical analysis of noncompartmental analysis of the sparse data has been previously published (ref. 16). C.T. is now an employee of ViiV Healthcare and owns stock in GlaxoSmithKline. J.C. is now an employee of Parexel International.
Author Contributions
J.B.D. and J.W.C. wrote the manuscript. J.B.D., H.M.A.P., K.B.P., and A.F. designed the research. J.B.D., M.C., C.R.T., H.M.A.P., and K.B.P. performed the research. J.B.D., J.W.C., M.C., C.R.T., C.S., C.T., N.W., S.M., R.W., N.E.S., and A.F. analyzed the data. N.E.S. contributed new reagents/analytical tools.
This work was presented in part at the 16th International Workshop on Clinical Pharmacology of HIV & Hepatitis Therapy, May 27–29, 2015, Washington DC, Abstract P_43.
References
- 1. Hasse, B. et al Morbidity and aging in HIV‐infected persons: the Swiss HIV cohort study. Clin. Infect. Dis. 53, 1130–1139 (2011). [DOI] [PubMed] [Google Scholar]
- 2. Guaraldi, G. et al Premature age‐related comorbidities among HIV‐infected persons compared with the general population. Clin. Infect. Dis. 53, 1120–1126 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV‐1‐infected adults and adolescents. February 12, 2013, Department of Health and Human Services. <https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf>.
- 4.Gilead Sciences. Truvada (emtricitabne/tenofovir) Full U.S. prescribing information, Foster City, CA (2009). <http://www.gilead.com/medicines>.
- 5. Anderson, P.L. Emtricitabine‐tenofovir concentrations and pre‐exposure prophylaxis efficacy in men who have sex with men. Sci. Transl. Med. 4, 151ra125 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rowe, J.W. , Andres, R. , Tobin, J.D. , Norris, A.H. & Shock, N.W. The effect of age on creatinine clearance in men: a cross‐sectional and longitudinal study. J. Gerontol. 31, 155–163 (1976). [DOI] [PubMed] [Google Scholar]
- 7. Labarga, P. et al Kidney tubular abnormalities in the absence of impaired glomerular function in HIV patients treated with tenofovir. AIDS 23, 689–696 (2009). [DOI] [PubMed] [Google Scholar]
- 8. Woodward, C.L. et al Tenofovir‐associated renal and bone toxicity. HIV Med. 10, 482–487 (2009). [DOI] [PubMed] [Google Scholar]
- 9. Goeddel, L. , Crawford, K.W. , Moore, R. , Fine, D.M. & Flexner, C. Impact of age on renal function in patients receiving tenofovir. 11th International Workshop on Clinical Pharmacology and HIV Therapy; April 7–9, 2001; Sorrento, Italy, Abstract 38 (2001).
- 10. Rea, I.M. , McNerlan, S.E. & Alexander, H.D. CD69, CD25, and HLA‐DR activation antigen expression on CD3+ lymphocytes and relationship to serum TNF‐alpha, IFN‐gamma, and sIL‐2R levels in aging. Exp. Gerontol. 34, 79–93 (1999). [DOI] [PubMed] [Google Scholar]
- 11. Anderson, P.L. , Kakuda, T.N. & Lichtenstein, K.A. The cellular pharmacology of nucleoside‐ and nucleotide‐analogue reverse‐transcriptase inhibitors and its relationship to clinical toxicities. Clin. Infect. Dis. 38, 743–753 (2004). [DOI] [PubMed] [Google Scholar]
- 12. Fletcher, C.V. et al Zidovudine triphosphate and lamivudine triphosphate concentration‐response relationships in HIV‐infected persons. AIDS 14, 2137–2144 (2000). [DOI] [PubMed] [Google Scholar]
- 13. Vourvahis, M. et al The pharmacokinetics and viral activity of tenofovir in the male genital tract. J. Acquir. Immune Defic. Syndr. 47, 329–333 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang, L.H. , Begley, J. , St. Claire, R.L. 3rd , Harris, J. , Wakeford, C. & Rousseau, F.S. Pharmacokinetic and pharmacodynamic characteristics of emtricitabine support its once daily dosing for the treatment of HIV infection. AIDS Res. Hum. Retroviruses 20, 1173–1182 (2004). [DOI] [PubMed] [Google Scholar]
- 15. Fried, L.P. et al Frailty in older adults: evidence for a phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 56, M146–M156 (2001). [DOI] [PubMed] [Google Scholar]
- 16. Desquilbet, L. et al HIV‐1 infection is associated with an earlier occurrence of a phenotype related to frailty. J. Gerontol. A Biol. Sci. Med. Sci. 62, 1279–1286 (2007). [DOI] [PubMed] [Google Scholar]
- 17. Desquilbet, L. et al Relationship between a frailty‐related phenotype and progressive deterioration of the immune system in HIV‐infected men. J. Acquir. Immune Defic. Syndr. 50, 299–306 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greenblatt, D.J. , Divoll, M.K. , Abernethy, D.R. , Ochs, H.R. , Harmatz, J.S. & Shader, R.I. Age and gender effects on chlordiazepoxide kinetics: relation to antipyrine disposition. Pharmacology 38, 327–334 (1989). [DOI] [PubMed] [Google Scholar]
- 19. Greenblatt, D.J. , Shader, R.I. & Harmatz, J.S. Implications of altered drug disposition in the elderly: studies of benzodiazepines. J. Clin. Pharmacol. 29, 866–872 (1989). [DOI] [PubMed] [Google Scholar]
- 20. Groen, K. et al The relationship between phenazone (antipyrine) metabolite formation and theophylline metabolism in healthy and frail elderly women. Clin. Pharmacokinet. 25, 136–144 (1993). [DOI] [PubMed] [Google Scholar]
- 21. Klotz, U. Pharmacokinetics and drug metabolism in the elderly. Drug Metab. Rev. 41, 67–76 (2009). [DOI] [PubMed] [Google Scholar]
- 22. Klotz, U. , Avant, G.R. , Hoyumpa, A. , Schenker, S. & Wilkinson, G.R. The effects of age and liver disease on the disposition and elimination of diazepam in adult man. J. Clin. Invest. 55, 347–359 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nelson, J.A. et al Expression of p16(INK4a) as a biomarker of T‐cell aging in HIV‐infected patients prior to and during antiretroviral therapy. Aging Cell. 11, 916–918 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dumond, J.B. et al Pharmacokinetics of two common antiretroviral regimens in older HIV‐infected patients: a pilot study. HIV Med. 14, 401–409 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bulitta, J.B. , Bingölbali, A. , Shin, B.S. & Landersdorfer, C.B. Development of a new pre‐ and post‐processing tool (SADAPT‐TRAN) for nonlinear mixed‐effects modeling in S‐ADAPT. AAPS J. 13, 201–211 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cockcroft, D.W. & Gault, M.H. Prediction of creatinine clearance from serum creatinine. Nephron 16, 31–41 (1976). [DOI] [PubMed] [Google Scholar]
- 27. Dumond, J.B. et al Tenofovir/emtricitabine metabolites and endogenous nucleotide exposures are associated with p16(INK4a) expression in subjects on combination therapy. Antivir. Ther. 21, 441–445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patterson, K.B. et al Penetration of tenofovir and emtricitabine in mucosal tissues: implications for prevention of HIV‐1 transmission. Sci. Transl. Med. 3, 112re4 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lahiri, C.D. et al Impact of protease inhibitors on intracellular concentration of tenofovir‐diphosphate among HIV‐1 infected patients. AIDS 29, 1113–1115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seifert, S.M. et al Transporter genetics and TFV‐DP/FTC‐TP cellular pharmacology in vivo. Conference on Retroviruses and Opportunistic Infections 2016. Poster 445. Boston, MA; February 2016.
- 31. Valade, E. et al Population pharmacokinetics of emtricitabine in HIV‐1‐infected adult patients. Antimicrob. Agents Chemother. 58, 2256–2261 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Valade, E. et al Modified renal function in pregnancy: impact on emtricitabine pharmacokinetics. Br. J. Clin. Pharmacol. 78, 1378–1386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baxi, S.M. et al Common clinical conditions – age, low BMI, ritonavir use, mild renal impairment – affect tenofovir pharmacokinetics in a large cohort of HIV‐infected women. AIDS 28, 59–66 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jullien, V. et al Population pharmacokinetics of tenofovir in human immunodeficiency virus‐infected patients taking highly active antiretroviral therapy. Antimicrob. Agents Chemother. 49, 3361–3366 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Antela, A. et al The role of tenofovir alafenamide in future HIV management. HIV Med. 17 Suppl 2, 4–16 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information accompanies this paper on the CPT: Pharmacometrics & Systems Pharmacology website (http://psp-journal.com)
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information