

Abstract
Acetaminophen (APAP) is a widely used analgesic drug that is frequently co‐administered with caffeine (CAF) in the treatment of pain. It is well known that APAP may cause severe liver injury after an acute overdose. However, the understanding of whether and to what extent CAF inhibits or stimulates APAP‐induced hepatotoxicity in humans is still lacking. Here, a multiscale analysis is presented that quantitatively models the pharmacodynamic (PD) response of APAP during co‐medication with CAF. Therefore, drug‐drug interaction (DDI) processes were integrated into physiologically based pharmacokinetic (PBPK) models at the organism level, whereas drug‐specific PD response data were contextualized at the cellular level. The results provide new insights into the inhibitory and stimulatory effects of CAF on APAP‐induced hepatotoxicity for crucially affected key cellular processes and individual genes at the patient level. This study might facilitate the risk assessment of drug combination therapies in humans and thus may improve patient safety in clinical practice.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ The co‐administration of APAP with CAF may potentiate and diminish APAP‐induced toxicity in mice and rats, respectively. However, the understanding of the effect of CAF on APAP in humans is still not well understood.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Here, we present a model‐based investigation of the impact of CAF on APAP‐induced toxicity during comedication of both drugs in humans.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ The study provides new insights into the inhibitory and stimulatory effects of CAF on APAP‐induced toxicity in humans by considering PK and PD interactions between CAF and APAP at the organism and the cellular level, respectively. Thereby, relative PD effects of CAF on PD responses of APAP were quantitatively described for significantly affected key cellular processes and individual genes.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ The concept presented in this study might facilitate the understanding of PK and PD interactions caused by drug combination therapies at patient level and thus may improve patient safety in clinical practice.
Acetaminophen (APAP)1 is a widely used over‐the‐counter drug with analgesic and antipyretic activities. In therapeutic applications, APAP is an effective and safe drug mostly used in the treatment of pain. However, in humans, acute overdosing of APAP increases the risk of hepatotoxic events leading to severe liver damage or even to death.1 The specific molecular mechanisms underlying APAP‐induced hepatotoxicity are still not well understood. However, it was suggested that an accumulation of N‐acetyl‐p‐benzoquinone imine (NAPQI), which is supposed to be the reactive intermediate of APAP,2 causes the toxic reactions.1, 3 NAPQI is a phase I metabolite of APAP that is mostly formed by cytochrome P450 (CYP) enzymes, in particular CYP1A2, CYP2E1, and CYP3A4.2 When APAP is administered at toxic doses, the conjugation of NAPQI with glutathione and the subsequent conversion to APAP cysteine (APAPC) is decreased, which leaves NAPQI as potential binding partner for proteins within the cell.4 Furthermore, APAP and its metabolites are involved in active drug transport processes across extracellular and intracellular membranes mediated by the adenosine triphosphate‐binding cassette (ABC) transporters, in particular ABCB1 and ABCG2.5, 6
Caffeine (CAF) is a stimulant of the central nervous system and is daily consumed in hot or cold beverages. CYP enzymes, particularly CYP1A2 and CYP2E1, are predominantly involved in the metabolism of CAF.7 Moreover, CAF showed inhibitory effects on active drug transport mediated by ABCB1.5 CAF is often administered as combination therapy in the treatment of pain because CAF is supposed to enhance the analgesic effects evoked by APAP or other analgesic agents.8, 9, 10 In this regard, CAF may alter APAP pharmacokinetic (PK) processes at the organism level8, 11 and may influence APAP‐induced pharmacodynamic (PD) responses at the cellular scale.10 In this context, CAF and APAP may thus be considered as perpetrator and victim drug, respectively.12 Notably, the unintentional co‐administration of CAF together with other drugs is mostly unavoidable, because coffee is one of the most popular drinks in the world.
In clinical practice, simultaneous administration of multiple drugs is often a standard treatment. In such combination therapies, drug‐drug interactions (DDIs) may inevitably occur and may potentially have a substantial impact on the PK behavior and the resulting PD effect of the administered drugs eventually leading to additive, synergistic, or antagonistic drug effects.3, 8, 10, 11, 13
In vitro drug response data measured at toxic concentrations may help to investigate the cellular effects induced by different drugs in cellular assays. However, a major challenge of such in vitro experiments is the translatability to patients. Recently, we have developed an integrative multiscale approach called PBPK‐based in vivo contextualization of in vitro toxicity data (PICD)14 that allows the translation of such in vitro findings to an in vivo context by coupling in vitro toxicity data with whole‐body physiologically based pharmacokinetic (PBPK) models (Figure 1).
Figure 1.
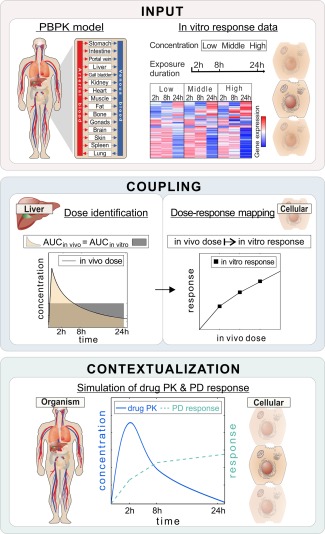
Basic concept of PBPK‐based in vivo contextualization of in vitro toxicity data (PICD). INPUT: Drug‐specific physiologically based pharmacokinetic (PBPK) models are developed at the organism level, whereas in vitro response data of compound‐treated primary hepatocytes are analyzed at the cellular level.28 COUPLING: In vivo doses are identified that are directly related to in vitro drug exposure (area under the curve (AUC)in vivo = AUCin vitro). Time‐dependent dose‐response curves are generated by mapping in vivo doses to in vitro response data. CONTEXTUALIZATION: pharmacodynamic (PD) responses over time are predicted for simulated pharmacokinetic (PK) profiles following drug administration of specific dose levels by use of time‐dependent dose‐response curves.
PBPK modeling allows a mechanistic description of absorption, distribution, metabolism, and elimination (ADME) processes governing the fate of a drug within the body (Supplementary Figure S1). PBPK models are particularly well suited for extrapolation to different dosage regimens and, moreover, to consider DDIs of co‐administered drugs influencing their ADME processes and hence altering their concentration‐time courses within the blood or the organs.15
In recent studies, the concomitant administration of APAP and CAF in rats and mice resulted in either a potentiation or a reduction of APAP‐induced hepatotoxicity, respectively.3, 4, 16, 17, 18 A possible explanation for these observations is the impact of CAF on the formation of NAPQI either due to inhibitory or stimulatory effects. Results obtained in rat and mice liver microsomes suggested an involvement of CAF on APAP metabolism mediated by CYP enzymes.4, 16, 19 In rat liver microsomes, for instance, co‐administration of CAF led to a reduced or an accelerated NAPQI formation dependent on the applied concentrations by affecting CYP enzymes, such as CYP1A1, CYP3A2, or CYP2E1.16 However, the interaction of CAF with APAP in humans, particularly at toxic dose levels, is still not well understood.
The aim of this study was a model‐based investigation of the PK and PD interactions of CAF on APAP‐induced toxicity during comedication in humans, through the consideration of drug interactions at the organism level, and the contextualization of drug‐specific PD response data at the cellular level, respectively. PD responses of CAF and APAP were therefore predicted for an in vivo situation by the application of PICD14 thereby coupling in vitro toxicity data with drug‐specific PBPK models. To validate the PBPK models, simulated drug concentrations of APAP, CAF, and their main metabolites (APAPC, acetaminophen cysteine; APAPG, acetaminophen glucuronide; APAPS, acetaminophen sulfate; PX, paraxanthine; TP, theophylline; and TB, theobromine) were first assessed with clinical PK profiles from several studies obtained for different dosage regimens.8, 11, 20, 21, 22, 23, 24, 25, 26, 27 Using an additive PD response model, the influence of CAF on APAP‐induced hepatotoxicity was analyzed for key cellular processes and individual genes. Dose escalation studies were finally performed to evaluate the transition from desired therapeutic effects to undesired toxic events, thereby quantitatively describing clinically relevant situations.
METHODS
Predicting the individual pharmacodynamic responses of acetaminophen and caffeine
PICD allows a quantitative description of drug responses at the patient level by integrating in vitro toxicity data from Open TG‐GATEs28 into whole‐body PBPK models14 (Supplementary Methods). In short, the basic concept of PICD is the identification of in vivo doses such that the simulated drug exposure in the interstitial space of the liver is equal to the in vitro drug exposure of the in vitro assay (Figure 1). The identified in vivo doses were mapped to the in vitro drug response data to quantitatively describe PD responses for different dose levels applied in vivo (Figure 1).14 Here, PICD was applied separately on single doses of APAP and CAF to predict PD responses for genes (defined as log2 fold change) and key cellular processes (defined as mean absolute log2 fold change of all involved genes) at 8 hours and 24 hours. Note that in TG‐GATEs, in vitro response data of APAP and CAF was measured at these timepoints.28 Bioavailability values calculated from the developed PBPK models were used (APAP = 92%; CAF = 100%) to consider oral administration.
Modeling the pharmacodynamic response of acetaminophen during co‐administration with caffeine
PICD was applied for a co‐administration of APAP and CAF with a relative dose ratio of 1000:130, according to therapeutic indications.8, 9 When both drugs were given concomitantly, the PICD‐based PD response of APAP ( ) were adjusted according to its changed concentration‐time profile caused by the competitive inhibition of CAF on ABCB1‐mediated and CYP2E1‐mediated transport and metabolization of APAP, respectively.5, 7, 16, 19 Furthermore, the predicted PD response of CAF ( ) was considered separately. The total PD response of APAP during co‐administration with CAF was thus calculated as follows:
| (1) |
where represents a gene or a key cellular process, represents the timepoint, and represents the oral dose level.
An additive PD response model was used here to calculate the PD response of APAP for a co‐administration with CAF, because the in vitro data were only available for single drug administration, such that potential synergistic or antagonistic effects induced by comedication of both drugs beyond pure additional effects could not be described. The relative PD effect of CAF co‐administered with APAP compared to the PD response predicted for single administration of APAP alone ( ) was computed as follows:
| (2) |
Note that a positive or negative relative PD effect value means that CAF increases and decreases the PD response of APAP, respectively, whereas a value of zero indicates no effect of CAF.
RESULTS
Whole‐body physiologically based pharmacokinetic models of acetaminophen and caffeine
At first, reference PBPK models for APAP and CAF were established by using clinical PK data (Supplementary Figure S2, Supplementary Table S1‐S5).8, 20, 21, 25 Twenty‐one biochemical processes were implemented in the PBPK models of CAF and APAP (Figure 2) to represent key metabolic reactions, active drug transport (Supplementary Table S2), as well as elimination processes (Supplementary Table S4).29, 30
Figure 2.
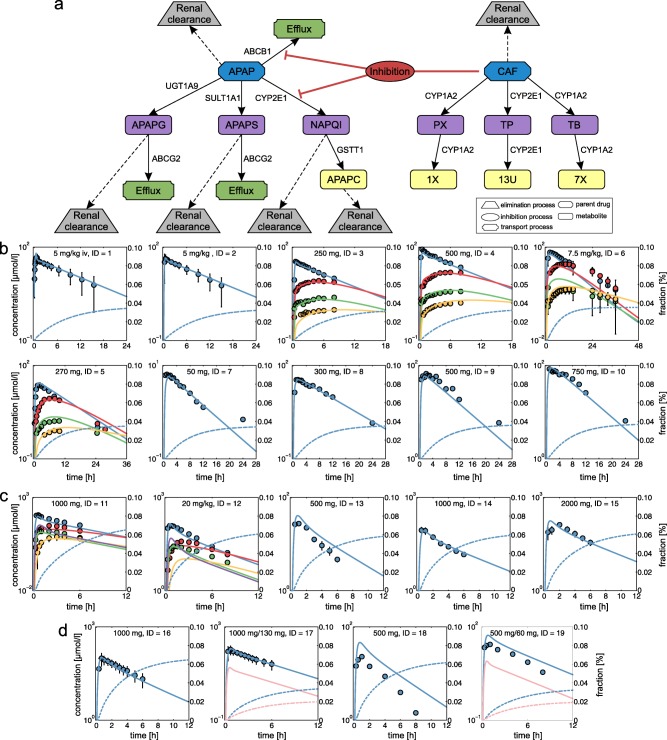
Physiologically based pharmacokinetic (PBPK) model development and validation. (a) Reaction diagram of twenty‐one biochemical processes implemented in the PBPK models of acetaminophen (APAP) and caffeine (CAF) illustrating active drug transport (green), metabolizing reactions for phase I (purple) and phase II (yellow) metabolites, kidney plasma clearance (gray), and inhibition processes (red). Metabolic enzymes and transporters are shown next to the respective reaction. (b) PBPK model of CAF (CAF = blue, PX = red, TB = green, TP = yellow). (c) PBPK model of APAP (APAP = blue, APAPG = red, APAPC = green, APAPS = yellow, NAPQI = purple). (d) PBPK model for single administration of APAP and for co‐administration of APAP and CAF (APAP = blue, CAF = pink). Simulated drug concentration‐time curves (lines) were assessed with experimental PK profiles (circles). Renal excretion rates were additionally simulated for APAP and CAF (dashed lines). Study IDs and dose levels of the experimental data are shown within each plot (Supplementary Table S5). APAPC, acetaminophen cysteine; APAPG, acetaminophen glucuronide; APAPS, acetaminophen sulfate; NAPQI, N‐Acetyl‐p‐benzoquinone imine; PX, paraxanthine; TB, theobromine; TP, theophylline; 13U, 1,3,dimethyluric acid; 7X, 7‐methylxanthine; 1X, 1‐methylxanthine.
To consider the influence of CAF on APAP pharmacokinetics,8, 11 an inhibitory effect of CAF on CYP2E1‐mediated NAPQI formation and on ABCB1‐mediated active transport of APAP was mechanistically represented by incorporating competitive inhibition processes in the developed PBPK models.5, 7, 16, 19 Notably, PK simulations following co‐administration of APAP and CAF at toxic dose levels resulted in a decrease of NAPQI concentration in plasma (Figure 3), which is in accordance with experimental observations obtained in rat liver microsomes.16
Figure 3.
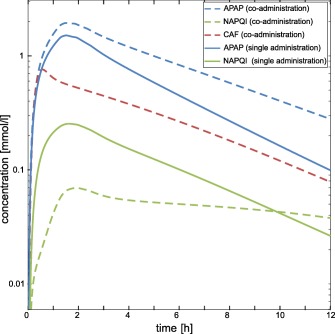
Pharmacokinetic (PK) simulations for single administration of acetaminophen (APAP) and co‐administration of caffeine (CAF). Plasma concentrations were simulated for APAP (blue) and NAPQI (green) following a single toxic dose of APAP (solid lines) and a co‐administered dose (dashed lines) of APAP and CAF (red).
After model establishment, the simulated drug concentrations in plasma showed an excellent agreement with in vivo PK data for both single doses of CAF and APAP alone as well as for concomitant administration of both drugs (Figure 2 , Supplementary Figure S3, Supplementary Table S7). The relative contribution of phase I CYP isoforms vs. phase II enzymes in the PBPK models of APAP and CAF was 70:30, and 100:0, respectively (Figure 2). The established reference PBPK models were validated for additional doses and individual subgroups by using clinical PK data from different studies not used for developing the reference PBPK models.11, 22, 23, 24, 26, 27 Note that all model parameters were left unchanged in this validation step except study parameters specifying the design of the clinical trials. Importantly, the validated PBPK models allow accurate predictions for different doses, because potential nonlinearities in ADME processes21, 31 are implicitly considered through the underlying model structure.
The validated PBPK models were next used in the application of PICD to predict PD responses induced by single administration of APAP alone as well as by co‐administration of APAP and CAF. Note that the PD response is based on transcriptome data from Open TG‐GATEs.28
Analyzing pharmacodynamic responses induced by single administration of acetaminophen and by co‐administration of caffeine
Drug‐specific PD responses following oral administration of APAP and CAF were predicted at the cellular level to investigate acute hepatotoxicity induced by a single toxic dose of APAP and by a co‐administered dose with CAF.
Relative PD effect values of CAF on APAP were therefore computed, which notably reflect both (i) the influence of CAF on the concentration‐time course of APAP at the organism level represented by competitive inhibition processes of CAF on ABCB1‐mediated and CYP2E1‐mediated transport and metabolization of APAP, respectively (PK interaction); (ii) the changed PD response of APAP at the cellular level implemented by additively contributing the PD response predicted for CAF (PD interaction).
A mean toxic dose of APAP (34 g) was identified in literature at a sublethal level,32, 33 whereas the dose level of CAF (4.4 g) was derived from a relative dose ratio of 1000:130, according to therapeutic indications used in drug combination therapy.8, 9, 10 Note that APAP and CAF are frequently co‐administered because CAF is supposed to enhance the analgesic effect of APAP.8, 9, 10
In the following, PD responses and relative PD effects induced by a single toxic dose of APAP and by a co‐administered dose of CAF were analyzed for genes expressed differentially (absolute fold change>1.5, Benjamini–Hochberg corrected P < 0.01) at 8 hours and 24 hours, and for key cellular processes significantly overrepresented (Benjamini–Hochberg corrected P < 0.01) at any timepoint. Note that these subsets were identified for both APAP and CAF.
Analysis of key cellular processes
Overall, the co‐administration of CAF led to a statistically significant perturbation (P < 0.01, two‐sample t‐test) of all considered key cellular processes (Figure 4). Analyzing PD responses at 8 hours following single administration of APAP revealed a substantial impact on cell cycle G1/S and G2/M checkpoint regulation as well as on liver necrosis, whereas a co‐administration of CAF resulted in a significantly increased PD response of these cellular processes by about 22%, 16%, and 43%, respectively (Figure 4). Note that an increased PD response of a key cellular process may result from both inhibition and activation of genes involved, because absolute log2 fold changes were considered for calculation purposes.
Figure 4.
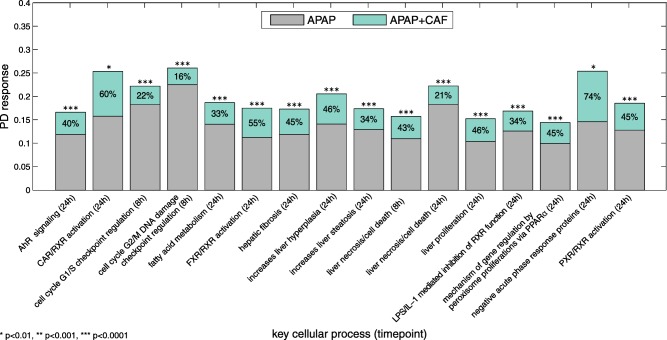
Pharmacodynamic (PD) response of key cellular processes. PD responses for significantly perturbed key cellular processes following drug administration of acetaminophen (APAP) as single toxic dose (gray) or co‐administered with caffeine (CAF; mint green). Percentages indicate relative PD effects of CAF.
At 24 hours, high PD responses of APAP administered alone were found in particular for fatty acid metabolism, liver necrosis, as well as for the promotion of hepatic steatosis, and for negative acute phase response proteins (Figure 4). These key cellular processes were additionally affected by 33%, 21%, 34%, and 74%, respectively, due to the co‐administration of CAF (Figure 4). Furthermore, the activation of CAR/RXR, FXR/RXR, and PXR/RXR heterodimers as well as the inhibition of the RXR function mediated by LPS/IL‐1 were strongly perturbed by APAP and were further significantly induced by 60%, 55%, 45%, and 34%, respectively, when CAF was given concomitantly (Figure 4). Moreover, a relative PD effect of about 45% was observed for CAF on PD responses of APAP inducing hepatic fibrosis, liver proliferation, and liver hyperplasia at 24 hours (Figure 4).
Analysis of individual genes
When analyzing the impact of single administration of APAP and co‐administration together with CAF on individual genes, the analyzed genes were additionally subdivided into their corresponding functional classes to allow a functional interpretation. In this context, a positive and negative PD effect value means that CAF increases or reduces the PD responses of APAP at the cellular level.
Comparing the PD responses of APAP at both timepoints following single administration revealed both an increased inhibition and activation of individual genes after 8 hours independently from the considered functional classes (Figure 5, Supplementary Figure S4). Likewise, calculated PD effects of CAF showed only minor changes on significantly perturbed genes at 24 hours in contrast to observations at 8 hours (Figure 5, Supplementary Figure S4).
Figure 5.
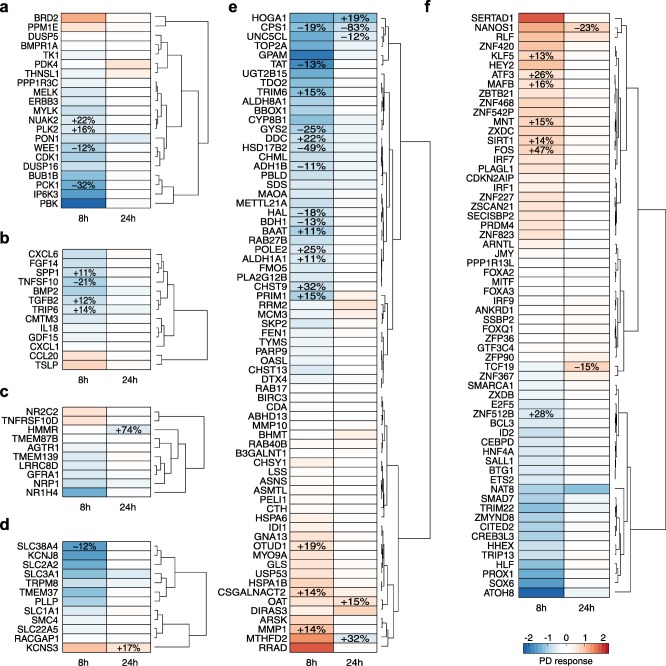
Pharmacodynamic (PD) response of individual genes. PD responses of significantly perturbed genes following drug administration of acetaminophen (APAP) as single toxic dose and correspondent PD effects induced by co‐administration of caffeine (CAF). Genes were classified according to their functional classes. Relative PD effect values indicated by percentages were only shown for highly regulated genes (absolute fold change>1.5 and absolute relative PD effect>10%). (a) Kinase/Phosphatase. (b) Cytokine/Growth factor. (c) Receptor. (d) Ion channel/Transporter. (e) Metabolic enzyme. (f) Transcription/Translation regulator.
At 8 hours, APAP induced the inhibition of several genes belonging to different functional classes, among which the following were found to be noteworthy due to a substantial impact of one or both drugs: the kinases PBK, PCK1, and IP6K3 (Figure 5 a); the cytokines TNFSF10 and CXCL6 (Figure 5 b); the ligand‐dependent nuclear receptor NR1H4 (Figure 5 c); the ion channel KCNJ8 and the transporter SLC38A4 (Figure 5 d); the metabolic enzymes GPAM and TAT (Figure 5 e); the transcription factor ATOH8 (Figure 5 f); and CDC20 and RTP3 (Supplementary Figure S4). On the other hand, a few genes were highly activated, for instance, the kinase BRD2 (Figure 5 a), the metabolic enzymes MMP1, MTHFD2, and RRAD (Figure 5 e), the transcription regulator FOS, and SERTAD1 (Figure 5 f), as well as the histone cluster HIST2H2BE and HIST1H2BD (Supplementary Figure S4). At 24 hours, only a few genes were substantially activated or inhibited by APAP, such as the kinase PDK4 (Figure 5 a), the receptor HMMR (Figure 5 c), the ion channel KCNS3 (Figure 5 d), the metabolic enzymes HOGA1, CPS1, and MTHFD2 (Figure 5 e), the transcription regulators TCF19 (Figure 5 f), as well as cyclin E2 (Supplementary Figure S4).
Analyzing PD effects of CAF on APAP elucidated both a reduced inhibitory effect (21–83%) of APAP on specific genes, such as TNFSF10, PCK1, CPS1, GYS2, HSD17B2, FST, and CLRN3, as well as an enhanced inhibitory effect (19–74%) on other genes, such as NUKA2, HMMR, HOGA1, DDC, CHST9, ZNF512B, RTP3, and CDC20 (Figure 5, and Supplementary Figure S4). The activation of APAP on individual genes was mostly potentiated by CAF, particularly on FOS and ATF3 by 47% and 26%, respectively (Figure 5 f).
In this gene‐level analysis, PD responses of significantly perturbed genes induced by a single toxic dose of APAP and the corresponding PD effects provoked by a co‐administration of CAF were analyzed. Besides the identification of genes crucially affected by APAP, inhibitory and stimulatory effects of CAF on APAP were thereby investigated.
Dose escalation study: Transition from therapeutic to toxic conditions
In the dose escalation study, an exemplary set of genes (at 8 hours: ATF3, PCK1, TNFSF10, SLC38A4, HSD17B2, FOS, and ZNF512B; at 24 hours: HMMR, KCNS3, CPS1, CCNE2, and CDC20; at both timepoints: DTL) and key cellular processes (at 8 hours: regulation of cell cycle G1/S and G2/M DNA damage checkpoint; at 24 hours: activation of CAR/RXR heterodimer, and liver hyperplasia) were next analyzed, which were substantially affected by a single toxic dose of APAP and by a co‐administered dose of CAF (Figure 5 , Supplementary Figure S4). The dose escalation study was performed on these genes and key cellular processes to quantitatively explore the transition from desired therapeutic effects to undesired toxic events (Figure 6). In this regard, the initial therapeutic dose was stepwise increased by 1,000 mg until the considered toxic dose level was reached, thereby simultaneously monitoring PD responses following single administration of APAP and its co‐administration with CAF.
Figure 6.
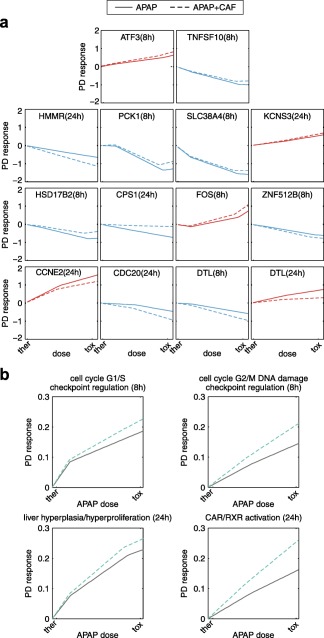
Dose escalation study. Pharmacodynamic (PD) responses were predicted following single administration of acetaminophen (APAP; solid lines) and co‐administration of APAP and caffeine (CAF; dashed lines). The doses were stepwise increased from therapeutic (ther) to toxic (tox) dose levels. (a) Individual genes. (b) Key cellular processes.
Analyzing dose‐response curves for single genes revealed that the co‐administration of CAF at high doses near the toxic range resulted in the strongest impact on PD responses of APAP, as expected, in comparison to doses around the therapeutic range (Figure 6 a). However, opposing PD effects of CAF were observed with regard to diminishing or enhancing the regulatory effects of APAP. The up‐regulation or down‐regulation of the kinase PCK1, the dehydrogenase HSD17B2, the synthase CPS1, or cyclin E2, which were strongly induced by high doses of APAP, were attenuated by co‐administration of CAF. In contrast, perturbations of APAP on the transcription regulators FOS, ATF3, and ZNF512B, the transmembrane receptor HMMR or CDC20 were obviously increased by CAF (Figure 6 a). Interestingly, studying PD effects of CAF on DTL, which is involved in the detection of DNA damage and repair mechanisms, elucidated an enhanced inhibitory effect of APAP at 8 hours, whereas the upregulation at 24 hours was rigorously reduced (Figure 6 a).
Next, dose‐response curves for an exemplary set of four key cellular processes were analyzed (Figure 6 b). During the escalation from therapeutic to toxic doses, a high perturbation of APAP on the cell cycle checkpoints G1/S and G2/M was observed at 8 hours, which was additionally increased due to a co‐administration of CAF (Figure 6 b). At 24 hours, gradually increasing the therapeutic to the toxic dose led to a significant perturbation of APAP on genes associated with an increased hyperplasia of the liver and with the activation of the CAR/RXR heterodimer that transcriptionally activates the promoters of CYP2B and CYP3A gene expression (Figure 6 b).34 Moreover, these key cellular processes were additionally affected when both drugs were administered concomitantly (Figure 6 b).
This dose escalation study allows the simultaneous investigation of cellular perturbations induced by single administration of APAP or co‐administration with CAF to quantitatively describe drug‐induced changes in clinically relevant situations, which hence may have important implications for dose decisions in the future.
Investigating the effect of caffeine on the analgesic action of acetaminophen under therapeutic conditions
From a therapeutic perspective, CAF is expected to increase the analgesic effect of APAP in humans.8, 9, 10 To explore this effect in our model, PD responses on pain‐related genes35 were predicted for single‐administration and co‐administration of APAP and CAF, respectively, by applying dose levels up to a maximum daily dose (APAP: 4,000 mg, Drugs.com; APAP/CAF: 4,000 mg/520 mg)8 (Figure 7). Here, CAF showed a slight but significant effect on the PD response of APAP on pain‐related genes, particularly at 8 hours (Figure 7 a). Note that the therapeutic PD response was here analogously calculated as before for the key cellular processes (mean absolute log2 fold change of all involved genes). PD responses were further investigated for an exemplary set of six genes (LIF, CCL2, IL18, NR1I2, SCN9A, and CACNA2D2), which are involved in pain modulation as well as in pain conduction and synaptic transmission. It was found that CAF slightly enhances the inhibitory effect of APAP on these pain‐related genes (Figure 7 b). Interestingly, the effect of CAF at 24 hours was most prominent on the chemokine CCL2 that is supposed to mediate the activation of pain pathways.35
Figure 7.
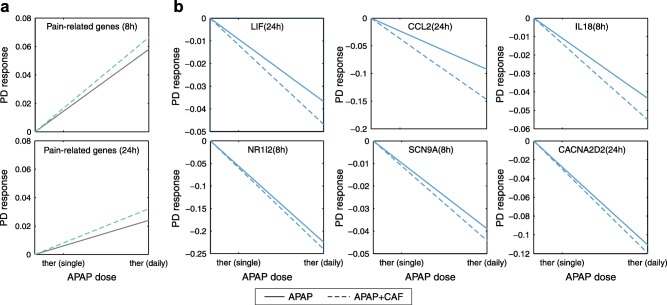
Pharmacodynamic (PD) response of genes associated to pain. The PD response of genes associated with pain were predicted following single administration of acetaminophen (APAP; solid lines) and co‐administration of APAP and caffeine (CAF; dashed lines). The doses were stepwise increased from a therapeutic single dose to the maximum daily dose. (a) PD response of a set of pain‐related genes. (b) PD response of individual genes involved in pain modulation as well as in pain conduction and synaptic transmission.
DISCUSSION
In this article, the impact of CAF on APAP‐induced hepatotoxicity was investigated at patient level by evaluating the effects of CAF on the PK and PD behavior of APAP induced by comedication of both drugs.
A mean toxic dose level of APAP was identified by collecting nonfatal toxic doses from public databases.32, 33 The co‐administered dose of CAF was selected according to relative dose ratios applied in combination therapy,8, 9 and was found to be in the lower range of clinical observations about sublethal acute poisoning32 indicating a low toxic potential caused by the single dose of CAF. Hence, the in vivo doses used here reflect clinically relevant situations for which toxic events were observed.
Drug interaction processes between CAF and APAP were considered in the PBPK models by incorporating competitive inhibition in ADME‐related processes36 to reflect the inhibitory effect of CAF on ABCB1‐mediated active transport of APAP, and on CYP2E1‐mediated metabolization of APAP to NAPQI, respectively (Figure 2 a).5 APAP‐induced hepatotoxicity may occur due to an accumulation of NAPQI.4 Here, simulations of NAPQI concentrations were significantly decreased when both drugs were administered concomitantly (Figure 3), which is in accordance with experimental observations from rat liver microsomes.16 This might indicate a favorable effect of the co‐administration of CAF for the reduction of acute liver failure induced by extensive exposure of APAP.
Several ADME processes were further included in the PBPK models of APAP and CAF to describe the processes governing a drug PK with a high level of detail (Figure 2). The PBPK models were carefully validated and showed excellent agreement with experimental data from literature obtained for different dosage regimens in human clinical studies8, 11, 20, 21, 22, 23, 24, 25, 26, 27 (Figure 2 , Supplementary Figure S3, Supplementary Table S7). This validation step ensures reliable predictions of PK profiles following drug administration of doses ranging from therapeutic to toxic levels.
The previously established multiscale approach PICD allows a quantitative description of drug‐induced toxicity at patient level14 (Figure 1) by coupling whole‐body PBPK models (Figure 2) with in vitro toxicity data from Open TG‐GATEs,28 an exceptional large‐scale toxicogenomics database. In this study, PD responses were analyzed at 8 hours and 24 hours because in vitro response data for CAF had only been measured at these timepoints.28 Although estimating PD responses induced by multiple dosing would also be very interesting, only single drug administration was considered here because no adequate in vitro response data for repeated dosing was available.28 Instead of using time‐resolved gene expression profiles28 to represent PD responses at the cellular level, PICD also allows contextualizing in vitro drug response data obtained at different omics levels, like proteomics or metabolomics, as well as incorporating other functional or clinical endpoints.
Here, PICD was applied for single administration of APAP and CAF, respectively, as well as for co‐administration of both drugs. To predict PD responses of APAP in combination therapy with CAF, both the PK and PD interaction of CAF were considered: (i) the implemented competitive inhibition processes of CAF led to an altered PK profile of APAP and consequently to a changed PD response when applying PICD (PK interaction); (ii) the predicted PD response of CAF was added separately to the PD responses of APAP (PD interaction). Although the co‐administration of different drugs may lead not only to additive drug effects, but also to synergistic or antagonistic effects, an additive PD response model was used because no adequate in vitro data were available to identify or differentiate potential synergism or antagonism.
PD responses of APAP and correspondent PD effects of CAF were evaluated for single genes and key cellular processes, which were significantly affected by both drugs. Among other things, it was found that a single toxic dose of APAP highly affected the G1/S and G2/M DNA damage checkpoint of the cell cycle at 8 hours, which was further increased by the co‐administration of CAF (Figure 4). At 24 hours, CAF strongly enhanced the effect of APAP on heterodimerization of the receptors CAR and RXR, which transcriptionally induced the expression of P450 enzymes, as well as bilirubin and thyroid hormone metabolism (Figure 4). The transcription regulator FOS was substantially upregulated at 8 hours by a single‐dose administration of APAP (Figure 5). This observation is in agreement with earlier experimental results in which FOS expression was induced by APAP in MCF‐7 breast cancer cells.36 Moreover, the PD effect of CAF revealed an enhancement on the activation of FOS, which may potentiate the APAP‐induced hepatotoxicity, because FOS seems to favor the development of toxic events.37, 38
The validated PBPK models and the generic application of PICD here allowed considering drug interactions between APAP and CAF and monitoring PD responses induced by single or co‐administration of several doses. In the dose escalation study, the therapeutic dose was stepwise increased until the considered toxic dose was reached, thereby investigating the transition from desired drug effects to adverse events. The predicted dose‐response curves provided insights into the inhibitory or stimulatory effects of APAP and enabled to check whether these regulatory effects were enhanced or diminished by co‐administration of CAF. For DTL, which supports the detection of DNA damage and repair, an increased and decreased impact of CAF on the inhibitory and stimulatory effect of APAP was found at 8 hours and 24 hours, respectively (Figure 6 a). This might indicate a potentiation of the APAP‐induced hepatotoxicity, because DTL plays an essential role in the detection of DNA damage. However, a potential reduction of the toxicity caused by APAP would also be possible, because a decreased expression of DTL might also be related to a reduced DNA damage after drug exposure within the cell.
In a further dose escalation study, the effect of CAF on the analgesic action on APAP was investigated under therapeutic conditions. It was found that CAF slightly enhances the inhibitory effects of APAP on genes involved in pain perception and modulation (Figure 7). These results may explain the observed increase in the clinical efficacy of APAP at the cellular scale induced by co‐administration of CAF.8, 9, 10
In conclusion, the impact of CAF on APAP‐induced hepatotoxicity was here investigated in humans by simultaneously considering drug effects of CAF on APAP at both the PK and the PD level. It was shown that CAF has a significant effect on APAP‐induced hepatotoxicity due to a co‐administration of both drugs. Key results demonstrate, on the one hand, that CAF might favor a reduction of APAP‐induced hepatotoxicity in humans at the PK and the PD levels by reducing the concentrations of NAPQI, which is supposed to be the reactive metabolite of APAP,2 as well as by positively regulating genes playing an essential role in the development of toxicity, respectively. On the other hand, CAF might also potentiate APAP‐induced toxicity by affecting crucial genes, such as FOS, that may support the activation of cell death pathways. Although key outcomes of the study demonstrated inhibitory and stimulatory effects of CAF on APAP, the question if CAF potentiates or diminishes the hepatotoxicity caused by extensive exposure of APAP partly remains open. To adequately address this question, more in vitro data would be required, such as measurements of other omics levels, like proteomics or metabolomics, as well as in vitro response data obtained in an appropriate cell system after simultaneous exposure to multiple drugs. This would obviously help to improve the understanding of the molecular mechanisms following co‐administration of APAP and CAF, and would clearly facilitate to discover potential synergistic or antagonistic drug effects. As presented here, dose escalation studies might further enhance the development of safe and efficient dosage regimens in drug combination therapy. Moreover, the concept used to consider DDIs at the PK and PD level is generically applicable for different drug combinations in clinically relevant situations. Hence, this might help to explore the PK and PD interactions caused by drug combination therapies at the patient level and thus may improve patient safety in clinical practice.
Supporting information
Supplementary information accompanies this paper on the CPT: Pharmacometrics & Systems Pharmacology website (http://psp-journal.com)
Supplementary Model Files
Supplementary Methods
Supplementary Figure S1 Schematic representation of a multiscale whole‐body physiologically based pharmacokinetic model. Schematic representation of a multiscale whole‐body PBPK model including 15 different tissues and organs that are connected by blood flow. Subcompartmentalization into blood cells, blood plasma, interstitial, and intracellular space is exemplarily presented for a default compartment.
Supplementary Figure S2 Workflow of physiologically based pharmacokinetic (PBPK) model development and validation. Workflow of PBPK model development and validation, including experimental data and modeling steps for (a) single administration of acetaminophen (APAP), (b) single administration of caffeine (CAF), and (c) co‐administration of APAP and CAF.
Supplementary Figure S3 Physiologically based pharmacokinetic (PBPK) model assessment. Observed vs. predicted plots, RMSD value and the coefficient of determination (R2) determined by comparing experimental pharmacokinetic (PK) data with simulated drug concentration‐time profiles. Study identifications (IDs) and dose levels of the experimental data are shown within each plot (Supplementary Table S5). (a) PBPK model of caffeine (CAF). (b) PBPK model of acetaminophen (APAP). (c) PBPK model for single administration of APAP and for co‐administration of APAP with CAF.
Supplementary Figure S4 Pharmacodynamic (PD) response for individual genes. PD responses of significantly perturbed genes following drug administration of acetaminophen (APAP) as single toxic dose and correspondent PD effects induced by co‐administration of caffeine (CAF). Genes were classified into the functional class “other.” Relative PD effect values indicated by percentages were only shown for highly regulated genes (absolute fold change >1.5 and absolute relative PD effect >10%).
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Model Files S1 Caffeine
Supplementary Model Files S2 Acetaminophen
Acknowledgments
The authors acknowledge financial support by the European Union Seventh Framework Programme HeCaToS (FP7/2007‐2013) under the grant agreement number 602156.
Conflict of Interest
L.K. is an employee of Bayer Technology Services GmbH, the company developing the PBPK modeling tools PK‐Sim and MoBi.
Author Contributions
C.T., H.C., V.B., L.M.B., and L.K. wrote the manuscript. C.T. and L.K. designed the research. C.T. performed the research. C.T. analyzed the data. C.T. and H.C. developed the PBPK models.
References
- 1. Nelson, S.D. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin. Liver Dis. 10, 267–278 (1990). [DOI] [PubMed] [Google Scholar]
- 2. Walubo, A. , Barr, S. , Abraham, A.M. & Coetsee, C. The role of cytochrome‐P450 inhibitors in the prevention of hepatotoxicity after paracetamol overdose in rats. Hum. Exp. Toxicol. 23, 49–54 (2004). [DOI] [PubMed] [Google Scholar]
- 3. Sato, C. & Izumi, N. Mechanism of increased hepatotoxicity of acetaminophen by the simultaneous administration of caffeine in the rat. J. Pharmacol. Exp. Ther. 248, 1243–1247 (1989). [PubMed] [Google Scholar]
- 4. Jaw, S. & Jeffery, E.H. Interaction of caffeine with acetaminophen. 1. Correlation of the effect of caffeine on acetaminophen hepatotoxicity and acetaminophen bioactivation following treatment of mice with various cytochrome P450 inducing agents. Biochem. Pharmacol. 46, 493–501 (1993). [DOI] [PubMed] [Google Scholar]
- 5. Wishart, D.S. et al DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 34(Database issue), D668–D672 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mazaleuskaya, L.L. , Sangkuhl, K. , Thorn, C.F. , FitzGerald, G.A. , Altman, R.B. & Klein, T.E. PharmGKB summary: pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet. Genomics 25, 416–426 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gu, L. , Gonzalez, F.J. , Kalow, W. & Tang, B.K. Biotransformation of caffeine, paraxanthine, theobromine and theophylline by cDNA‐expressed human CYP1A2 and CYP2E1. Pharmacogenetics 2, 73–77 (1992). [DOI] [PubMed] [Google Scholar]
- 8. Renner, B. et al Caffeine accelerates absorption and enhances the analgesic effect of acetaminophen. J. Clin. Pharmacol. 47, 715–726 (2007). [DOI] [PubMed] [Google Scholar]
- 9. Palmer, H. , Graham, G. , Williams, K. & Day, R. A risk‐benefit assessment of paracetamol (acetaminophen) combined with caffeine. Pain Med. 11, 951–965 (2010). [DOI] [PubMed] [Google Scholar]
- 10. Sawynok, J. & Yaksh, T.L. Caffeine as an analgesic adjuvant: a review of pharmacology and mechanisms of action. Pharmacol. Rev. 45, 43–85 (1993). [PubMed] [Google Scholar]
- 11. Iqbal, N. , Ahmad, B. , Janbaz, K.H. , Gilani, A.U. & Niazi, S.K. The effect of caffeine on the pharmacokinetics of acetaminophen in man. Biopharm. Drug Dispos. 16, 481–487 (1995). [DOI] [PubMed] [Google Scholar]
- 12. Prueksaritanont, T. et al Drug‐drug interaction studies: regulatory guidance and an industry perspective. AAPS J. 15, 629–645 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coors, A. & De Meester, L. Synergistic, antagonistic and additive effects of multiple stressors: predation threat, parasitism and pesticide exposure in Daphnia magna. J. Appl. Ecol. 45, 1820–1828 (2008). [Google Scholar]
- 14. Thiel, C. , Cordes, H. , Conde, I. , Castell, J.V. , Blank, L.M. & Kuepfer, L. Model‐based contextualization of in vitro toxicity data quantitatively predicts in vivo drug response in patients. Arch. Toxicol.; e‐pub ahead of print 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou, D. , Bui, K. , Sostek, M. & Al‐Huniti, N. Simulation and prediction of the drug‐drug interaction potential of naloxegol by physiologically based pharmacokinetic modeling. CPT Pharmacometrics Syst. Pharmacol. 5, 250–257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee, C.A. , Thummel, K.E. , Kalhorn, T.F. , Nelson, S.D. & Slattery, J.T. Inhibition and activation of acetaminophen reactive metabolite formation by caffeine. Roles of cytochromes P‐450IA1 and IIIA2. Drug Metab. Dispos. 19, 348–353 (1991). [PubMed] [Google Scholar]
- 17. Raińska, T. et al Caffeine reduces the hepatotoxicity of paracetamol in mice. J. Int. Med. Res. 20, 331–342 (1992). [DOI] [PubMed] [Google Scholar]
- 18. Sato, C. , Izumi, N. , Nouchi, T. , Hasumura, Y. & Takeuchi, J. Increased hepatotoxicity of acetaminophen by concomitant administration of caffeine in the rat. Toxicology 34, 95–101 (1985). [DOI] [PubMed] [Google Scholar]
- 19. Nouchi, T. , Lasker, J.M. & Lieber, C.S. Activation of acetaminophen oxidation in rat liver microsomes by caffeine. Toxicol. Lett. 32, 1–8 (1986). [DOI] [PubMed] [Google Scholar]
- 20. Blanchard, J. & Sawers, S.J. Comparative pharmacokinetics of caffeine in young and elderly men. J. Pharmacokinet. Biopharm. 11, 109–126 (1983). [DOI] [PubMed] [Google Scholar]
- 21. Kaplan, G.B. et al Dose‐dependent pharmacokinetics and psychomotor effects of caffeine in humans. J. Clin. Pharmacol. 37, 693–703 (1997). [DOI] [PubMed] [Google Scholar]
- 22. Lelo, A. , Birkett, D.J. , Robson, R.A. & Miners, J.O. Comparative pharmacokinetics of caffeine and its primary demethylated metabolites paraxanthine, theobromine and theophylline in man. Br . J. Clin. Pharmacol. 22, 177–182 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tang‐Liu, D.D. , Williams, R.L. & Riegelman, S. Disposition of caffeine and its metabolites in man. J. Pharmacol. Exp. Ther. 224, 180–185 (1983). [PubMed] [Google Scholar]
- 24. Newton, R. , Broughton, L.J. , Lind, M.J. , Morrison, P.J. , Rogers, H.J. & Bradbrook, I.D. Plasma and salivary pharmacokinetics of caffeine in man. Eur. J. Clin. Pharmacol. 21, 45–52 (1981). [DOI] [PubMed] [Google Scholar]
- 25. Shinoda, S. , Aoyama, T. , Aoyama, Y. , Tomioka, S. , Matsumoto, Y. & Ohe, Y. Pharmacokinetics/pharmacodynamics of acetaminophen analgesia in Japanese patients with chronic pain. Biol. Pharm. Bull. 30, 157–161 (2007). [DOI] [PubMed] [Google Scholar]
- 26. Prescott, L.F. Kinetics and metabolism of paracetamol and phenacetin. Br. J. Clin. Pharmacol. 10Suppl2, 291S–298S (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rawlins, M.D. , Henderson, D.B. & Hijab, A.R. Pharmacokinetics of paracetamol (acetaminophen) after intravenous and oral administration. Eur. J. Clin. Pharmacol. 11, 283–286 (1977). [DOI] [PubMed] [Google Scholar]
- 28. Igarashi, Y. et al Open TG‐GATEs: a large‐scale toxicogenomics database. Nucleic Acids Res. 43(Database issue), D921–D927 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Willmann, S. , Lippert J., Sevestre, M. , Solodenko, J. , Fois, F. , & Schmitt, W. PK‐Sim®: a physiologically based pharmacokinetic “whole‐body” model. Biosilico. 4, 121–124 (2003). [Google Scholar]
- 30. Kuepfer, L. , Niederalt, C. , Wendl, T. , Schlender, J. , Willmann, S. , Lippert, J. et al Applied Concepts in PBPK modeling: How to build a PBPK/PD model. CPT Pharmacometrics Syst. Pharmacol. 5, 516–531 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sahajwalla, C.G. & Ayres, J.W. Multiple‐dose acetaminophen pharmacokinetics. J. Pharm. Sci. 80, 855–860 (1991). [DOI] [PubMed] [Google Scholar]
- 32. Clemedson, C. , Kolman, A. & Forsby, A. The integrated acute systemic toxicity project (ACuteTox) for the optimisation and validation of alternative in vitro tests. Altern. Lab. Anim. 35, 33–38 (2007). [DOI] [PubMed] [Google Scholar]
- 33. Hoofnagle, J.H. , Serrano, J. , Knoben, J.E. & Navarro, V.J. LiverTox: a website on drug‐induced liver injury. Hepatology 57, 873–874 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen, S. , Wang, K. & Wan, Y.Y. Retinoids activate RXR/CAR‐mediated pathway and induce CYP3A. Biochem. Pharmacol. 79, 270–276 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Foulkes, T. & Wood, J.N. Pain Genes Fisher E.M.C. editor. PLoS Genet. 4, e1000086 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gadd, S.L. , Hobbs, G. & Miller, M.R. Acetaminophen‐induced proliferation of estrogen‐responsive breast cancer cells is associated with increases in c‐myc RNA expression and NF‐kappaB activity. Toxicol. Sci. 66, 233–243 (2002). [DOI] [PubMed] [Google Scholar]
- 37. Fernandez, M. , Pirondi, S. , Antonelli, T. , Ferraro, L. , Giardino, L. & Calzà, L. Role of c‐Fos protein on glutamate toxicity in primary neural hippocampal cells. J. Neurosci. Res. 82, 115–125 (2005). [DOI] [PubMed] [Google Scholar]
- 38. Gillardon, F. , Skutella, T. , Uhlmann, E. , Holsboer, F. , Zimmermann, M. & Behl, C. Activation of c‐Fos contributes to amyloid beta‐peptide‐induced neurotoxicity. Brain Res. 706, 169–172 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information accompanies this paper on the CPT: Pharmacometrics & Systems Pharmacology website (http://psp-journal.com)
Supplementary Model Files
Supplementary Methods
Supplementary Figure S1 Schematic representation of a multiscale whole‐body physiologically based pharmacokinetic model. Schematic representation of a multiscale whole‐body PBPK model including 15 different tissues and organs that are connected by blood flow. Subcompartmentalization into blood cells, blood plasma, interstitial, and intracellular space is exemplarily presented for a default compartment.
Supplementary Figure S2 Workflow of physiologically based pharmacokinetic (PBPK) model development and validation. Workflow of PBPK model development and validation, including experimental data and modeling steps for (a) single administration of acetaminophen (APAP), (b) single administration of caffeine (CAF), and (c) co‐administration of APAP and CAF.
Supplementary Figure S3 Physiologically based pharmacokinetic (PBPK) model assessment. Observed vs. predicted plots, RMSD value and the coefficient of determination (R2) determined by comparing experimental pharmacokinetic (PK) data with simulated drug concentration‐time profiles. Study identifications (IDs) and dose levels of the experimental data are shown within each plot (Supplementary Table S5). (a) PBPK model of caffeine (CAF). (b) PBPK model of acetaminophen (APAP). (c) PBPK model for single administration of APAP and for co‐administration of APAP with CAF.
Supplementary Figure S4 Pharmacodynamic (PD) response for individual genes. PD responses of significantly perturbed genes following drug administration of acetaminophen (APAP) as single toxic dose and correspondent PD effects induced by co‐administration of caffeine (CAF). Genes were classified into the functional class “other.” Relative PD effect values indicated by percentages were only shown for highly regulated genes (absolute fold change >1.5 and absolute relative PD effect >10%).
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Model Files S1 Caffeine
Supplementary Model Files S2 Acetaminophen