

Abstract
Morphine shows large interindividual variability in its pharmacokinetics; however, the cause of this has not been fully addressed. The variability in morphine disposition is considered to be due to a combination of pharmacogenetic and physiological determinants related to morphine disposition. We previously reported the effect of organic cation transporter (OCT1) genotype on morphine disposition in pediatric patients. To further explore the underlying mechanisms for variability arising from relevant determinants, including OCT1, a physiologically based pharmacokinetic (PBPK) model of morphine was developed. The PBPK model predicted morphine concentration‐time profiles well, in both adults and children. Almost all of the observed morphine clearances in pediatric patients fell within a twofold range of median predicted values for each OCT1 genotype in each age group. This PBPK modeling approach quantitatively demonstrates that OCT1 genotype, age‐related growth, and changes in blood flow as important contributors to morphine pharmacokinetic (PK) variability.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ There is a lack of mechanistic understanding of the underlying factors causing the large variability in morphine PKs.
WHAT QUESTION DOES THIS STUDY ADDRESS?
☑ This is the first study using PBPK modeling to demonstrate the impact of OCT1 genotype and liver blood flow on morphine CL after intravenous administration.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ The pediatric morphine PBPK model showed OCT1 genotype‐dependent and age‐dependent morphine disposition. In addition, sensitivity analysis showed that morphine CL is influenced by changes in cardiac output, which links to hepatic blood flow.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This study provides an example of the utility of PBPK modeling to quantitatively assess the multiple factors contributing to variability, including transporter‐related PG effects, in drug disposition.
Morphine is widely used for pain management in pediatric and adult patients. However, clinicians continue to struggle with finding the right dose due to our incomplete understanding of the factors predicting the large variability in morphine pharmacokinetics (PKs) among patients. The variability in morphine disposition is considered to result from a combination of pharmacogenetic and physiological determinants, whereas in pediatric patients the age‐dependent developmental changes also need to be considered.1, 2 Application of physiologically‐based pharmacokinetic (PBPK) modeling is recognized as an informative approach to explore the combined effects of the system parameter3 (e.g., physiology and genetics) and drug parameters (e.g., physicochemical and in vitro PK properties). Therefore, the aim of this study was to develop a PBPK model of morphine to assess the multiple determinants contributing to variability in morphine disposition.
After intravenous administration, morphine is mainly eliminated through the liver with ∼10% of drug excreted unchanged in the urine.4, 5 Morphine enters hepatocytes via passive diffusion and transporter‐mediated pathways. Morphine is a known substrate of the organic cation transporter 1 (OCT1, gene name SLC22A1), which is expressed in the sinusoidal membrane of human hepatocytes.6 Previously, we showed the impact of OCT1 genotype on morphine clearance (CL) in pediatric patients.7 After the uptake of morphine into hepatocytes, morphine is metabolized by uridine 5′‐diphosphate glucuronosyltransferase (UGT)2B7.8, 9, 10 Because UGT2B7 is also expressed in the kidneys,11 the kidneys could also contribute to morphine metabolism. Based on these findings, OCT1 transporter activity was considered one of the important factors in explaining variability in morphine disposition among patients, in addition to UGT2B7 metabolism and renal excretion.
In this study, a PBPK model of morphine was developed to assess the impact of multiple contributing factors to variability in morphine disposition by implementing OCT1‐mediated hepatic uptake transport as well as UGT2B7 metabolism and renal excretion. The predictive performance of the PBPK model was tested for OCT1 contribution based on OCT1‐genotype‐dependent morphine PKs by comparing predicted with observed clinical data. In addition, the contribution of other potential determinants, such as hepatic blood flow level and UGT2B7 activity, on morphine CL was accessed through the sensitivity analysis based on PBPK modeling and simulations.
MATERIALS AND METHODS
PBPK model development in adults
Simcyp simulator software version 14.1 (Simcyp, Sheffield, UK) was used to develop the morphine PBPK model. This simulator enables the transcription of physicochemical parameters (e.g., molecular weight, lipophilicity) and in vitro data (e.g., kinetic parameters of enzyme and transporter functions) into in vivo data, based on in vitro‐in vivo extrapolation methods. In turn, these transcribed parameters are incorporated with anatomic and physiological data, in order to simulate concentration‐time profiles of the drug based on the trial design, as described in the reports by Jamei et al.12, 13 In this study, the relevant physicochemical parameters, in vitro, and in vivo data of morphine were collected from the literature. The morphine‐specific data collected are summarized in Table 1.4, 5, 6, 14, 15, 16, 17, 18 The morphine compound file was developed and validated in the adult Simcyp Simulator model (consisting of adult systems parameters) before being used in the Pediatric Simcyp Simulator model, which considers age‐dependent anatomic and physiological changes. The extrapolation processes for morphine transport and UGT2B7‐mediated metabolism are described in detail in the Supplementary Methods. Regarding the prediction of morphine distribution, a full PBPK model based on the method by Rodgers et al.19 and Rodgers and Rowland20 was used in the simulations. The permeability‐limited liver model was used to describe the liver distribution process.21, 22
Table 1.
Summary of physicochemical parameters, in vitro and in vivo data of morphine from the literature
| Parameter | Value |
|---|---|
| Molecular weight (g/mol) | 285.3414 |
| Log P | 0.7714 |
| pKa (proton on N) | 7.9315 |
| pKa (phenolic H) | 9.6315 |
| Fraction unbound in plasma | 0.6214 |
| Blood‐to‐plasma ratio | 1.0816 |
| Plasma binding protein | Human serum albumin (assumed) |
| Full PBPK model | |
| Vss (Rodgers and Rowland,19 L/kg) | 3.6 |
| Apparent Vd after i.v. administration (L/kg) | 4.017 |
| Elimination | |
| Enzyme kinetics (HLM) a | |
| UGT2B7/3MG | |
| Km (µM) | 115.814 |
| Vmax (pmol/min/mg microsomal protein) | 9,25014 |
| UGT2B7/6MG | |
| Km (µM) | 115.814 |
| Vmax (pmol/min/mg microsomal protein) | 1,91714 |
| Total CL (L/hr) | 84 b |
| Urine excretion ratio (%) | 10 c |
| Renal CL estimate (L/hr) | 8 |
| Permeability limited liver model (PerL) | |
| Transporter kinetics (HEK293 transfected cells) | |
| OCT1 Km (µM) | 3.4 ± 0.3 (mean ± SEM)6 |
| OCT1*1 (Wild) Jmax (pmol/min/mg lysate protein) | 29.0 ± 2.76 |
| OCT1*2 (420del) Jmax (pmol/min/mg lysate protein) | 7.21 ± 0.76 |
| OCT1*3 (61Cys) Jmax (pmol/min/mg lysate protein) | 6.25 ± 0.96 |
CL, clearance; HEK293, human embryonic kidney 293; HLM, human liver microsome; Km, kinetic metabolite; OCT1, organic cation transporter‐1; PBPK, physiologically based pharmacokinetic; UGT, uridine 5′‐diphosphate glucuronosyltransferase; Vmax, maximum velocity; Vss, volume of distribution at steady state.
aAssumed fu,mic= 1. bTotal CL was recalculated using AUC (ng/mL*hr) after i.v. administration and morphine free base dose (mg).18 cUrinary excreted ratio of unchanged morphine was assumed to be 10% from the data of package insert (http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202515s000lbl.pdf) and the report by Osborne et al.4 and Hasselstrom and Sawa.5
PBPK model validation using clinical data in adults
The predictive performance of the developed adult PBPK model was tested using reported PK data in adult healthy volunteers and patients with cancer.17, 18, 23, 24, 25, 26 All simulations were conducted based on the trial design used in the respective clinical studies with specific information entered on dose, administration route, subject demographics, age range, proportion of women, and trial size. As for the simulation trial size, a total of 400 virtual individuals were simulated to reasonably capture the expected variability within the population.27 These parameter settings in the virtual clinical trial simulations were defined as close as possible to those in the original clinical studies. Details on the parameters used for each simulation in this study are summarized in Supplementary Table S1.
Clinically observed PK data were used to evaluate the predictive performance of the developed PBPK model. When numeric data were not reported, they were extracted from graphs in the original publications with GetData Graph Digitizer version 2.26 (getdata‐graph‐digitizer.com). The evaluation of the developed PBPK models was performed by visual predictive check and by comparison of the observed and predicted values of the various PK parameters. In the visual predictive check, the observed systemic drug concentration‐time profiles were overlaid with the simulated profiles.
PBPK modeling in children, including OCT1 transporter genetic contribution
The PK simulations of morphine in children were conducted using the Simcyp Pediatric platform version 14 that takes age‐dependent anatomic and physiological changes into account (i.e., age‐dependent organ size, cardiac output, microsomal protein concentration per organ, plasma protein concentration, hematocrit level, UGT2B7 protein expression level, and glomerular filtration rate).27, 28 The Simcyp Pediatric simulator generates age‐dependent anatomic and physiological parameters as system parameters for virtual pediatric subjects based on the equation describing the relationship between subject's age and each parameter reported by Johnson et al.,28 except the age‐dependent OCT1 protein expression. Regarding age‐dependent OCT1 protein expression, Prasad et al.29 reported that protein expression of OCT1 was not significantly different between children and older age groups, such as adolescents and adults; but there was a difference in neonates and infants compared with the older age groups.29 It was also reported that the mRNA expression level in the group of 7 years and older was similar to the adult level in human liver.30 In predictive performance testing using our previously reported clinical PK data obtained from pediatric patients older than 6 years of age,7 it was assumed that the OCT1 protein expression level was not different between children and adults. The age‐dependent UGT2B7 expression in liver microsomal protein was described with the following equation up to 20 years of age in the Simcyp simulator31: fraction of adults = 0.0453 × age (years) + 0.089. Last, the age‐dependent UGT2B7 expression in kidney microsomal protein was assumed the same as in the liver.
Previously published pharmacokinetic‐pharmacogenetic (PK‐PG) data7 were used to evaluate the predictive performance of the developed pediatric PBPK model and the PG contribution of OCT1 to morphine disposition, as the OCT1 transporter genotype was available for each pediatric patient. The maximum uptake rate for morphine transport in hepatocytes (Jmax, pmol/min/million hepatocytes) estimates, for each OCT1 genotype, was defined as the mean of Jmax' (pmol/min/mg lysate protein) of the OCT1 variants in the OCT1‐overexpression cell system, as shown in Table 1.4, 5, 6, 14, 15, 16, 17, 18 For example, the Jmax' estimate for OCT1*1/*2 was calculated as the mean of Jmax' of *1 variant (29 pmol/min/mg lysate protein) and Jmax *2 variant (7.21 pmol/min/mg lysate protein); (29.0 + 7.21)/2 = 18.1 pmol/min/mg lysate protein, as summarized in Table 2.6 The Michaelis‐Menten constant (Km) value of 3.4 µM for the OCT1*1 genotype was taken from the publication of Tzvetkov et al.6 In this study, the same Km value of 3.4 µM was used across three OCT1 genotypes to avoid overparameterization because expressed variant proteins did not show a significant difference in the Km (affinity) of morphine uptake.6 Using these kinetic parameters for OCT1‐mediated morphine transport, the PK profiles of morphine were simulated for each OCT1 genotype in virtual pediatric subjects using yearly increments from age 6–16 years. The proportion of women in the virtual pediatric population was set at 50%, and the size of the trial simulation was 400 subjects, with 20 trials of 20 subjects performed for each age group. In addition, dose was set at 0.15 mg morphine base/kg administered by bolus injection. Area under the curve to infinity (AUC∞) was calculated using the linear trapezoidal method. The CL estimates from the simulated PK profiles (calculated as dose of morphine divided by AUC∞) were compared with observed CL data from the clinical PK‐PG study.7
Table 2.
Jmax estimates for each OCT1 transporter genotype
| Group | Genotype | Jmax estimate (pmol/min/mg lysate protein) |
|---|---|---|
| Wild | *1 × *1 | 29.0 |
| Heterozygotes | *1 × *2 | 18.1 |
| *1 × *3 | 17.6 | |
| Homozygotes | *2 × *2 | 7.21 |
| *2 × *3 | 6.73 |
Jmax was estimated as described in the Method section, based on the reported OCT1 variant‐dependent kinetic parameters.6
Jmax, maximal flux value; OCT1, organic cation transporter‐1.
Sensitivity analysis of cardiac output and UGT2B7 activity on morphine CL in virtual pediatric and adult subjects
Sensitivity analyses focusing on cardiac output and UGT2B7 intrinsic activity were conducted to explore their potential impact on morphine CL. The cardiac output value, implemented in the Simcyp platform, was modified to assess its impact on morphine CL. Cardiac output values were changed from 100% to 25% of the default value (set at 100%) for healthy pediatric or adult populations in the Simcyp platform. The age‐dependent cardiac output was defined as a function of age and body surface area, as described by Johnson et al.28
The effect of UGT2B7 activity on morphine CL was also tested. Several variants in UGT2B7 have been identified as potentially contributing to the disposition of known substrates, such as morphine.32 However, to date, the effects of several reported UGT2B7 variants on morphine glucuronidation have not been fully addressed. Regarding the most common UGT2B7 nonsynonymous single nucleotide polymorphism (UGT2B7*2), it was reported that UGT2B7*2 was not associated with altered morphine 3 and 6‐glucuronidation in adult liver microsomes (n = 53).33 A comprehensive PG study found that diplotypes containing haplotype #4 in UGT2B7 resulted in a significant increase in the formation of morphine 3 and 6‐glucuronidation (45% and 56%, respectively) compared with diplotypes without the haplotype #4.34 Based on the observed activity changes as a result of UGT2B7 genetic variants, the maximum velocity of UGT2B7‐mediated morphine glucuronidation in human liver microsomes was scaled from 50–150% of the default value (set at 100%), as shown in Table 1, 4, 5, 6, 14, 15, 16, 17, 18 for the simulations in virtual pediatric and adult healthy subjects.
The PK profiles of morphine were simulated with modified cardiac output and UGT2B7 intrinsic activity in virtual pediatric and adult subjects. In the simulations, the OCT1 genotype was considered and OCT1*1/*1, *1/*2, and *2/*2 were used as the representation of wild type, heterozygotes, and homozygotes, respectively. In the trial design, age was set at 6 years old for pediatric subjects and 30–40 years for adult subjects. The proportion of women were set at 50%, and a total number of 400 simulations (n = 20 virtual subjects × n = 20 trials) was performed for both virtual pediatric and adult subjects. The CL of morphine was calculated from the simulation result, as described earlier.
RESULTS
Morphine PBPK model development and validation in adults
An initial PBPK model was developed using morphine‐specific physicochemical parameters and in vitro and in vivo data, as summarized in Table 1.4, 5, 6, 14, 15, 16, 17, 18 Morphine was assumed to be transported into hepatocytes via passive diffusion and OCT1 transporter‐mediated uptake. The parameters related to the hepatic transport of morphine are the passive diffusion CL and the scaling factor of OCT1 activity for in vitro‐in vivo extrapolation, as implemented in the morphine PBPK model. The impact of these two parameters on morphine concentration‐time profiles was evaluated by exploratory sensitive analysis using PBPK modeling (Supplementary Figure S1). The changes in both parameters showed a direct effect on the morphine PK simulation results. The estimation of these parameters was conducted via fitting as part of the development of the morphine PBPK model because the passive diffusion CL and the scaling factor of OCT1 activity were not available. The passive diffusion CL and the scaling factor of OCT1 activity were estimated to be 0.0034 mL/min/million hepatocytes and 5.1 (mg lysate protein/million cells), respectively, using data from six individual subjects. After these estimated parameters for the hepatic uptake transport component were implemented into the model, the PK simulation of morphine was conducted using the same trial design as used for the parameter estimation process (Supplementary Table S1). The simulation result of the morphine concentration‐time profiles are shown in Figure 1 a 17, 18, 23, 24, 25, 26 together with observed clinical data from six individual adult subjects. The predicted AUC∞ and CL estimates were 49.4 ± 10 ng/mL*hr and 17.7 ± 3.8 mL/min/kg, respectively (mean ± SD; Table 3, 18, 25). These values were close to the observed AUC∞ values of 45.9 ± 7.7 ng/mL*hr and CL of 17.9 ± 7.3 mL/min/kg, and the ratio of predicted to observed data was 1.1 and 0.99, respectively. Regarding the range of trial means, minimum to maximum was 41.9–64.6 ng/mL*hr for AUC∞ and 13.8–21.6 mL/min/kg for CL (virtual 67 trials with 6 subjects).
Figure 1.
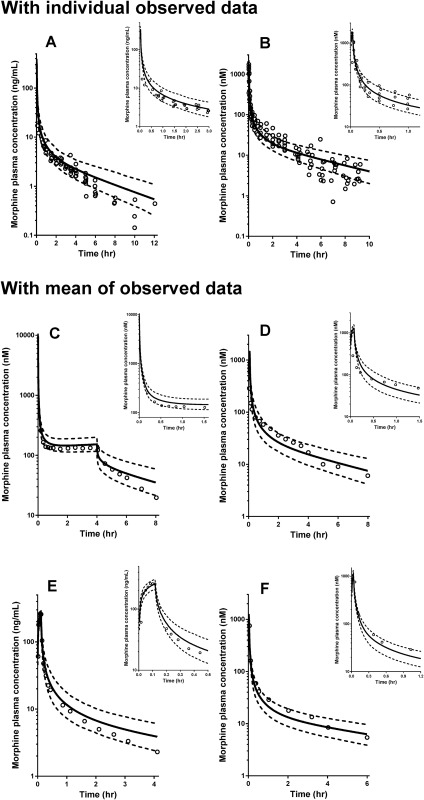
Observed and simulated concentration‐time profiles of morphine with the adult physiologically based pharmacokinetic (PBPK) model. Open circles represent the observed data from reported clinical studies: Hoskin et al.18; Lötsch et al.23; Lötsch et al.24; Stuart‐Harris et al.25; Dershwitz et al.26; Säwe et al.17 Details on parameter settings used for each simulation in this study are summarized in Supplementary Table S1.
Table 3.
Comparison of predicted and observed morphine pharmacokinetics parameters in healthy adults
| PK parameters (mean ± SD) a | ||
|---|---|---|
| Model development | AUC∞ (ng/mL*hr) | CL (mL/min/kg) |
| Prediction | 49.4 ± 10 | 17.7 ± 3.8 |
| Observed (n = 6)18 | 45.9 ± 7.7 b | 17.9 ± 7.3 c |
| Ratio d | 1.1 | 0.99 |
| PK parameters (mean ± SD) a | ||
| Model verification | AUC∞ (nM*hr/70 kg) e | CL (mL/min/70kg)e |
| Prediction | 352 ± 133 | 1,232 ± 260 |
| Observed (n = 6)25 | 290 ± 67 | 1,584 ± 408 |
| Ratio | 1.2 | 0.78 |
AUC∞, area under the curve to infinity; CL, clearance; PK, pharmacokinetic.
aMean ± SD of total of 402 simulations (67 virtual trials with 6 subjects). bAUC was corrected for the actual free‐based dose. cCL estimate based on dose as morphine base. dRatio of predicted to observed data. ePredicted PK parameters were normalized to 70 kg because reported PK parameters were standardized to a weight of 70 kg.
This PBPK model was defined as the base model of morphine for adults. After that, the evaluation of the developed base model was conducted using data reported in five different clinical studies in adults using the same trial design in the simulations as used in the original clinical studies (Supplementary Table S1). The simulation results of the morphine concentration‐time profiles are shown in Figure 1 b–f together with the actual observed data (individual observations in Figure 1 b; mean observational data in Figure 1 c–f). Almost all observed data fell within the 5th to 95th percentile range of the simulations. Among the five clinical trial datasets used for the model validation, morphine PK parameter estimates for AUC∞ and CL were available from the study by Stuart‐Harris et al.25 (Figure 1 d). The predicted AUC∞ and CL estimates were 352 ± 133 nM*hr/70 kg and 1,232 ± 260 mL/min/70 kg, respectively (mean ± SD; Table 3, 18, 25). These predicted data were within a twofold window of the reported mean AUC∞ values of 290 ± 67 nM*hr/70 kg and CL of 1,584 ± 408 mL/min/70 kg, and the ratio of predicted to observed data was 1.2 and 0.78, respectively. Regarding the range of trial means, minimum to maximum was 233–549 nM*hr/70 kg for AUC∞ and 993–1,558 mL/min/70 kg for CL (virtual 67 trials with 6 subjects).
The effect of OCT1 genotype on morphine CL in pediatric patients
The OCT1‐mediated morphine transport activity was implemented into the PBPK model to describe the morphine uptake in hepatocytes. As part of the model validation process focusing on the adequacy of OCT1 activity for hepatic transport of morphine, morphine CL for each OCT1 genotype was simulated with the Simcyp Pediatric platform. In vitro OCT1 transporter activity (i.e., Jmax) was defined as the average value corresponding to each OCT1 variant in the in vitro expression cell system used in this study. The simulated CL values are shown in Figure 2, 7 together with observed data in individual pediatric patients. The observed CL values were within a twofold range of the median predictions in wild‐type subjects (*1/*1), in heterozygotes (at least one *1 allele), and in homozygotes. There was one 7‐year‐old patient who was heterozygous (*1/*2) and who showed a 2.1‐fold higher CL compared to the median of the simulated CL data in the 7‐year age group with the same *1/*2 genotype. The simulated concentration‐time profiles in 6‐year‐old virtual subjects with each representative OCT1 genotype are shown in Figure 3, 7 overlaid with observed data from individual pediatric patients with the same age/genotype. The PBPK model simulations demonstrated the impact of OCT1 genotype on the morphine concentration‐time profiles (Figure 3 e).
Figure 2.
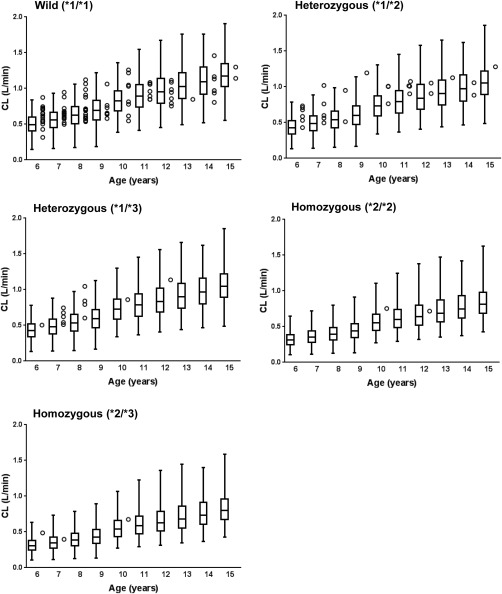
Comparison between predicted and observed morphine clearance (CL) for each organic cation transporter (OCT1) transporter genotype in pediatric subjects aged 6–15 years old. The predicted CL values are represented by box‐and‐whisker plots. The 25th percentile, median, and 75th percentile are represented by the bottom, middle, and top of the boxes, respectively. The minimum and maximum values are represented by the vertical lines arising from each box. Each closed circle represents observed individual data.7
Figure 3.
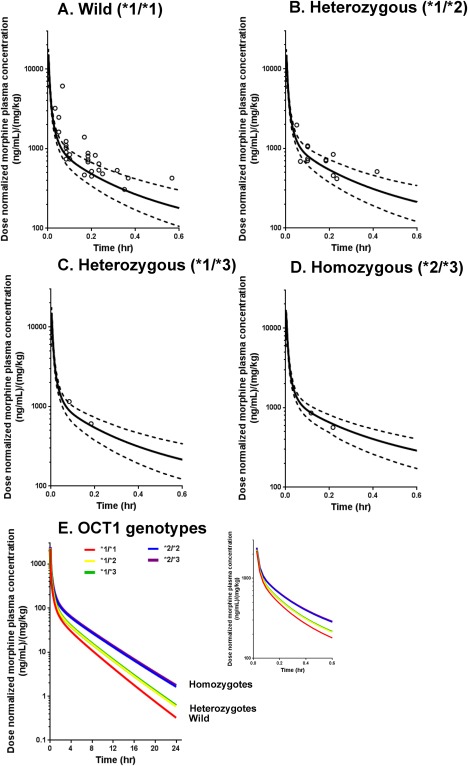
Pediatric physiologically based pharmacokinetic (PBPK) model predicted vs. observed concentration‐time profiles of morphine in children. The simulation of morphine concentration‐time profile was conducted for each organic cation transporter (OCT1) genotype (a) for wild *1/*1 type, (b) for *1/*2 heterozygous, (c) for *1/*3 heterozygous, (d) for *2/*3 homozygous, and (e) for all OCT1 genotype tested in this study with time range up to 24 hours, according to the Method section. The simulation results shown in a to d were overlaid with observed data (open circles).7 Solid and dashed lines represent the mean and 5th/95th percentiles of simulation results, respectively.
The impact of cardiac output and UGT enzyme activity on morphine CL in virtual pediatric and adult subjects
To assess the impact of changes in cardiac output and UGT2B7 enzyme activity on morphine CL, values were changed from 25% to 100% for cardiac output and from 50% to 150% for UGT enzyme activity, in which 100% represents a normal value in a subject (Figure 4). Morphine CL was sensitive to changes in cardiac output in both adult and pediatric subjects. In adults, reducing cardiac output to 50% of normal resulted in a decrease in morphine CL of 33% for OCT1 wild type, 30% for OCT1 *2 heterozygotes, and 24% for OCT1 *2 homozygotes, respectively. In children, the results were a decrease in morphine CL of 29% for OCT1 wild type, 26% for *2 heterozygotes, and 19% for *2 homozygotes, respectively.
Figure 4.
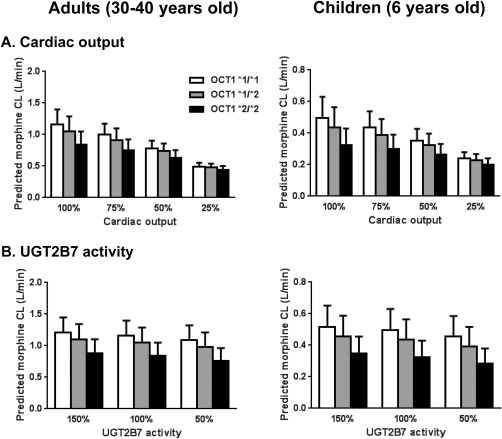
Impact of cardiac output (a) and uridine 5′‐diphosphate glucuronosyltransferase (UGT)2B7 activity (b) on predicted morphine clearance (CL) in virtual children and adults. Data are presented as mean ± SD of the simulation results. OCT1, organic cation transporter.
Changes in UGT2B7 enzyme activity resulted in a smaller impact on morphine CL compared to those for cardiac output changes in both adult and pediatric subjects. In adult subjects, a reduction in UGT2B7 enzyme activity of 50% resulted in a decrease in morphine CL of 6% for OCT1 wild type, 7% for OCT1 *2 heterozygotes, and 9% for *2 homozygotes, respectively. In pediatric subjects, the results were a decrease in morphine CL of 9% for OCT1 wild type, 10% for OCT1*2 heterozygotes, and 13% for OCT1*2 homozygotes, respectively.
DISCUSSION
There is a lack of mechanistic understanding of the important factors contributing to the large variability in morphine PKs observed in adult and pediatric patients. In this study, variability in morphine disposition was postulated to be due to multiple contributing factors, such as PG and physiological determinants, as well as age‐dependent developmental changes in pediatric patients. In order to assess the impact of these determinants, a whole body PBPK model of morphine was developed with morphine specific in vitro kinetic parameters for the hepatic uptake transporter OCT1, UGT2B7 enzyme activity, and renal excretion. Because a PBPK model includes system parameters, the model also allows us to assess the effects of hepatic blood flow changes on morphine CL.
Morphine is known to be water‐soluble with low lipophilicity (log P = 0.77; Table 1, 4, 5, 6, 14, 15, 16, 17, 18) and this will make it more difficult for the drug to pass through the membrane phospholipid bilayer; in agreement with this morphine has shown poor passive membrane permeability in HEK293 cells.6 We previously identified the PG contribution of OCT1, an uptake transporter at the sinusoidal membrane of human hepatocytes, to morphine CL in pediatric patients after intravenous administration. These findings indicate that the hepatic transport of morphine is mediated by OCT1 in addition to passive diffusion. As another clinical observation related to morphine CL, it was reported that the morphine CL after noncardiac surgery was higher than that after cardiac surgery in neonates and infants.35 These two clinical observations made us hypothesize that OCT1‐genotype and hepatic blood flow could be key determinants of variability in morphine disposition.
In this study, the passive diffusion CL of morphine was estimated to be 0.0034 mL/min/million hepatocytes through the fitting approach using observed clinical data in healthy volunteers together with physicochemical and in vitro data. In a similar fashion, the scaling factor of OCT1 activity for the extrapolation of in vitro OCT1‐mediated intrinsic clearance of morphine from the in vitro cell system (HEK293) to hepatocyte was estimated to be 5.1 lysate protein/million hepatocytes. Based on these parameter estimates, morphine sinusoidal uptake transport activity (CL) by OCT1 was calculated to be more than 10 times higher than transport activity via passive diffusion. This observation indicated that the transport process of morphine into hepatocytes could be mainly driven by the OCT1 transporter. Although, empirically, the approach to model OCT1 by applying different Jmax values for each genotypes gives reasonable results, mechanistically, OCT1 genotype may not affect protein expression too much as this is a transporter process driven by an electrochemical gradient and by membrane potential, which we do not account for mechanistically in the current model.
In PBPK modeling, organ blood flow is a function of cardiac output. In the Simcyp Simulator platform, the total liver blood flow is calculated from the sum of hepatic arterial and hepatic portal vein blood flows, representing 6.5% and 19% of cardiac output, respectively.28 This study demonstrates that changes in cardiac output, which links to hepatic blood flow level, resulted in considerable alteration in morphine CL in both virtual pediatric and adult subjects. In our simulations, reducing cardiac output to 25% of normal resulted in a decrease in morphine CL of 48–58% in adults (depending on OCT1 genotype). Interestingly, the PBPK prediction of morphine CL well represents a reported clinical observation in adult patients with shock. The liver blood flow in these patients was reported to decrease to 30% compared with patients recovering from shock (287 mL/min vs. 870 ml/min).36 This reduction in blood flow resulted in a decrease in morphine CL to about 50% (137 min/min in patients with shock vs. 290 min/min in recovered patients).36 Thus, the developed PBPK model is able to reproduce morphine CL changes in relation to the changes in liver blood flow/cardiac output, as observed in critically ill patients. In this way, the advantage of PBPK modeling is to mechanistically explore likely underlying reasons for observed drug PK behavior in critically ill patients.
Morphine is reported to have a high extraction ratio of 0.7 after intravenous administration in healthy volunteers.37 For a drug with a high extraction ratio, in general, hepatic blood flow is the rate‐limiting factor for drug elimination through the liver, which we confirmed in this study. Furthermore, slight differences were observed in the influence of blood flow level and UGT2B7 activity on predicted morphine CL between pediatric subjects and adults. That is, the predicted morphine CL in adults was slightly more sensitive to changes in blood flow compared to that in pediatric subjects aged 6 years; however, the predicted morphine CL in pediatric subjects was slightly more sensitive to changes in UGT2B7 activity compared with that in adults. These differences might be due to a smaller hepatic extraction ratio in pediatric compared with adult subjects. Recently, it was reported that the hepatic extraction ratio of midazolam varies with age due to age‐dependent physiological changes.38
Regarding age‐dependent OCT1 protein expression and OCT1‐mediated intrinsic CL, Prasad et al.29 reported that protein expression of OCT1 was not significantly different between children and older age groups, such as adolescents and adults; but there was a difference in neonates and infants compared with the older age groups. In addition, there was no significant difference found in the mRNA expression level between children aged 7 and older and adults in human liver.29, 30 Accordingly, the developmental changes of the OCT1 transporter expression per milligram protein in the liver was not considered in this study because our previous clinical data used as a clinical reference included the age of pediatric patients ranged from 6–15 years.7 Recently, the Pediatric Transporter Working Group reported a lack of knowledge on ontogeny of drug transporters, including the OCT1 transporter.39 As a next step, the ontogeny profile of the OCT1 transporter is being investigated as part of morphine PK simulation studies in neonates and small infants.
One limitation of the current study is that we have not considered the role of transporters in morphine renal CL, although it is considered <10% of total elimination, there are circumstances in which this pathway could be of importance. The overall renal CL of morphine is around 8.4 L/h but the calculated fu*GFR will give a CL estimate due to filtration of ∼4.7 L/h, thus active tubular secretion of morphine must be occurring. As discussed earlier, morphine has a poor passive permeation across a lipid bilayer. However, the corresponding transporter at the apical side moving the compound from the renal cell toward the urine is currently not known, although there are several potential candidates. In fact, a series of morphine analogues have been investigated against organic cation and anion transporters in the rat.40 The data indicate that these morphine analogues are “polysubstrates” that interact with different renal organic cation and anion transporters. Further investigation on the contribution of renal transporters to morphine CL would improve the current PBPK model. On another note, the current morphine PBPK model can predict the morphine concentration‐time profile, but not the metabolite profiles for morphine‐3‐glucuronide (an inactive metabolite) and morphine‐6‐glucuronide (an active metabolite).41 In order to capture the exposure levels of morphine metabolites, we need to develop the combined PBPK models of morphine and its metabolites, which will be considered in a next study.
This study indicates that hepatic blood flow and OCT1‐dependent hepatic uptake are the rate‐determining processes for morphine entry into hepatocytes in vivo and explain the drug exposure in plasma after intravenous administration. Thus, OCT1 genotype has an influence on morphine CL. Similarly, changes in cardiac output, including age‐dependent changes in pediatric patients, could contribute to the variability in morphine disposition. This study illustrates the utility of the PBPK modeling approach to perform an assessment for alteration of drug CL due to multiple physiological parameters varying with age. Furthermore, the factors identified through this PBPK modeling would improve population PK models of morphine, which have already integrated body weight and/or age of patients, as additional physiologically informed covariates. This complementary usage of both PBPK modeling and population PK modeling will improve our mechanistic understanding of which factors are contributing to the observed variability in morphine PKs.
Supporting information
Supplementary information accompanies this paper on the CPT: Pharmacometrics & Systems Pharmacology website (http://psp-journal.com)
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
This study was in part supported by Center for Clinical & Translational Science & Training (CCTST) T1 grant originated from the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1 TR001425.
Conflict of Interest
C.E. worked at Otsuka Pharmaceuticals in Japan until August 2014. T.N.J. and S.N. are employees of Simcyp Ltd (a Certara company). The other authors declared no conflict of interest.
Author Contributions
C.E., T.F., T.N.J., S.N., and A.A.V. wrote the manuscript. C.E., T.F., and A.A.V. designed the research. C.E., T.F., and S.S. performed the research. C.E., T.F., T.N.J., and S.N. analyzed the data. C.E. and T.F. contributed new reagents/analytical tools.
References
- 1. Anderson, B.J. & Holford, N.H. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab. Pharmacokinet. 24, 25–36 (2009). [DOI] [PubMed] [Google Scholar]
- 2. Vinks, A.A. , Emoto, C. & Fukuda, T. Modeling and simulation in pediatric drug therapy: application of pharmacometrics to define the right dose for children. Clin. Pharmacol. Ther. 98, 298–308 (2015). [DOI] [PubMed] [Google Scholar]
- 3. Maharaj, A.R. & Edginton, A.N. Physiologically based pharmacokinetic modeling and simulation in pediatric drug development. CPT Pharmacometrics Syst. Pharmacol. 3, e150 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Osborne, R. , Joel, S. , Trew, D. & Slevin, M. Morphine and metabolite behavior after different routes of morphine administration: demonstration of the importance of the active metabolite morphine‐6‐glucuronide. Clin. Pharmacol. Ther. 47, 12–19 (1990). [DOI] [PubMed] [Google Scholar]
- 5. Hasselström, J. & Säwe, J. Morphine pharmacokinetics and metabolism in humans. Enterohepatic cycling and relative contribution of metabolites to active opioid concentrations. Clin. Pharmacokinet. 24, 344–354 (1993). [DOI] [PubMed] [Google Scholar]
- 6. Tzvetkov, M.V. , dos Santos Pereira, J.N. , Meineke, I. , Saadatmand, A.R. , Stingl, J.C. & Brockmöller, J. Morphine is a substrate of the organic cation transporter OCT1 and polymorphisms in OCT1 gene affect morphine pharmacokinetics after codeine administration. Biochem. Pharmacol. 86, 666–678 (2013). [DOI] [PubMed] [Google Scholar]
- 7. Fukuda, T. et al OCT1 genetic variants influence the pharmacokinetics of morphine in children. Pharmacogenomics 14, 1141–1151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coffman, B.L. , Rios, G.R. , King, C.D. & Tephly, T.R. Human UGT2B7 catalyzes morphine glucuronidation. Drug Metab. Dispos. 25, 1–4 (1997). [PubMed] [Google Scholar]
- 9. Stone, A.N. , Mackenzie, P.I. , Galetin, A. , Houston, J.B. & Miners, J.O. Isoform selectivity and kinetics of morphine 3‐ and 6‐glucuronidation by human udp‐glucuronosyltransferases: evidence for atypical glucuronidation kinetics by UGT2B7. Drug Metab. Dispos. 31, 1086–1089 (2003). [DOI] [PubMed] [Google Scholar]
- 10. Achour, B. , Rostami‐Hodjegan, A. & Barber, J. Protein expression of various hepatic uridine 5'‐diphosphate glucuronosyltransferase (UGT) enzymes and their inter‐correlations: a meta‐analysis. Biopharm. Drug Dispos. 35, 353–361 (2014). [DOI] [PubMed] [Google Scholar]
- 11. Fisher, M.B. , Paine, M.F. , Strelevitz, T.J. & Wrighton, S.A. The role of hepatic and extrahepatic UDP‐glucuronosyltransferases in human drug metabolism. Drug Metab. Rev. 33, 273–297 (2001). [DOI] [PubMed] [Google Scholar]
- 12. Jamei, M. , Marciniak, S. , Feng, K. , Barnett, A. , Tucker, G. & Rostami‐Hodjegan, A. The Simcyp population‐based ADME simulator. Expert Opin. Drug Metab. Toxicol. 5, 211–223 (2009). [DOI] [PubMed] [Google Scholar]
- 13. Jamei, M. et al The simcyp population based simulator: architecture, implementation, and quality assurance. In Silico Pharmacol. 1, 9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krekels, E.H. et al From pediatric covariate model to semiphysiological function for maturation: part II‐sensitivity to physiological and physicochemical properties. CPT Pharmacometrics Syst. Pharmacol. 1, e10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaufman, J.J. , Semo, N.M. & Koski, W.S. Microelectrometric titration measurement of the pKa's and partition and drug distribution coefficients of narcotics and narcotic antagonists and their pH and temperature dependence. J. Med. Chem. 18, 647–655 (1975). [DOI] [PubMed] [Google Scholar]
- 16. Skopp, G. , Pötsch, L. , Ganssmann, B. , Aderjan, R. & Mattern, R. A preliminary study on the distribution of morphine and its glucuronides in the subcompartments of blood. J. Anal. Toxicol. 22, 261–264 (1998). [DOI] [PubMed] [Google Scholar]
- 17. Säwe, J. , Kager, L. , Svensson Eng, J.O. & Rane, A. Oral morphine in cancer patients: in vivo kinetics and in vitro hepatic glucuronidation. Br. J. Clin. Pharmacol. 19, 495–501 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoskin, P.J. , Hanks, G.W. , Aherne, G.W. , Chapman, D. , Littleton, P. & Filshie, J. The bioavailability and pharmacokinetics of morphine after intravenous, oral and buccal administration in healthy volunteers. Br. J. Clin. Pharmacol. 27, 499–505 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rodgers, T. , Leahy, D. & Rowland, M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate‐to‐strong bases. J. Pharm. Sci. 94, 1259–1276 (2005). [DOI] [PubMed] [Google Scholar]
- 20. Rodgers, T. & Rowland, M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 95, 1238–1257 (2006). [DOI] [PubMed] [Google Scholar]
- 21. Jamei, M. et al A mechanistic framework for in vitro‐in vivo extrapolation of liver membrane transporters: prediction of drug‐drug interaction between rosuvastatin and cyclosporine. Clin. Pharmacokinet. 53, 73–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neuhoff, S. , Yeo, K.R. , Barter, Z. , Jamei, M. , Turner, D.B. & Rostami‐Hodjegan, A. Application of permeability‐limited physiologically‐based pharmacokinetic models: part II – prediction of P‐glycoprotein mediated drug‐drug interactions with digoxin. J. Pharm. Sci. 102, 3161–3173 (2013). [DOI] [PubMed] [Google Scholar]
- 23. Lötsch, J. , Skarke, C. , Schmidt, H. , Liefhold, J. & Geisslinger, G. Pharmacokinetic modeling to predict morphine and morphine‐6‐glucuronide plasma concentrations in healthy young volunteers. Clin. Pharmacol. Ther. 72, 151–162 (2002). [DOI] [PubMed] [Google Scholar]
- 24. Lötsch, J. , Weiss, M. , Kobal, G. & Geisslinger, G. Pharmacokinetics of morphine‐6‐glucuronide and its formation from morphine after intravenous administration. Clin. Pharmacol. Ther. 63, 629–639 (1998). [DOI] [PubMed] [Google Scholar]
- 25. Stuart‐Harris, R. , Joel, S.P. , McDonald, P. , Currow, D. & Slevin, M.L. The pharmacokinetics of morphine and morphine glucuronide metabolites after subcutaneous bolus injection and subcutaneous infusion of morphine. Br. J. Clin. Pharmacol. 49, 207–214 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dershwitz, M. et al Pharmacokinetics and pharmacodynamics of inhaled versus intravenous morphine in healthy volunteers. Anesthesiology 93, 619–628 (2000). [DOI] [PubMed] [Google Scholar]
- 27. Johnson, T.N. & Rostami‐Hodjegan, A. Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Paediatr. Anaesth. 21, 291–301 (2011). [DOI] [PubMed] [Google Scholar]
- 28. Johnson, T.N. , Rostami‐Hodjegan, A. & Tucker, G.T. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin. Pharmacokinet. 45, 931–956 (2006). [DOI] [PubMed] [Google Scholar]
- 29. Prasad, B. et al Ontogeny of hepatic drug transporters as quantified by LC‐MS/MS proteomics. Clin. Pharmacol. Ther. 100, 362–370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klaassen, C.D. & Aleksunes, L.M. Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacol. Rev. 62, 1–96 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abduljalil, K. , Jamei, M. , Rostami‐Hodjegan, A. & Johnson, T.N. Changes in individual drug‐independent system parameters during virtual paediatric pharmacokinetic trials: introducing time‐varying physiology into a paediatric PBPK model. AAPS J. 16, 568–576 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.PharmGKB, The Pharmacogenomics Knowledgebase. What is the PharmGKB? <https://www.pharmgkb.org>.
- 33. Court, M.H. et al Evaluation of 3'‐azido‐3'‐deoxythymidine, morphine, and codeine as probe substrates for UDP‐glucuronosyltransferase 2B7 (UGT2B7) in human liver microsomes: specificity and influence of the UGT2B7*2 polymorphism. Drug Metab. Dispos. 31, 1125–1133 (2003). [DOI] [PubMed] [Google Scholar]
- 34. Innocenti, F. et al Single nucleotide polymorphism discovery and functional assessment of variation in the UDP‐glucuronosyltransferase 2B7 gene. Pharmacogenet. Genomics 18, 683–697 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lynn, A. , Nespeca, M.K. , Bratton, S.L. , Strauss, S.G. & Shen, D.D. Clearance of morphine in postoperative infants during intravenous infusion: the influence of age and surgery. Anesth. Analg. 86, 958–963 (1998). [DOI] [PubMed] [Google Scholar]
- 36. Macnab, M.S. , Macrae, D.J. , Guy, E. , Grant, I.S. & Feely, J. Profound reduction in morphine clearance and liver blood flow in shock. Intensive Care Med. 12, 366–369 (1986). [DOI] [PubMed] [Google Scholar]
- 37. Stanski, D.R. , Greenblatt, D.J. & Lowenstein, E. Kinetics of intravenous and intramuscular morphine. Clin. Pharmacol. Ther. 24, 52–59 (1978). [DOI] [PubMed] [Google Scholar]
- 38. Salem, F. , Abduljalil, K. , Kamiyama, Y. & Rostami‐Hodjegan, A. Considering age variation when coining drugs as high versus low hepatic extraction ratio. Drug Metab. Dispos. 44, 1099–1102 (2016). [DOI] [PubMed] [Google Scholar]
- 39. Brouwer, K.L. et al Human ontogeny of drug transporters: review and recommendations of the Pediatric Transporter Working Group. Clin. Pharmacol. Ther. 98, 266–287 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ullrich, K.J. & Rumrich, G. Morphine analogues: relationship between chemical structure and interaction with proximal tubular transporters – contraluminal organic cation and anion transporter, luminal H+/organic cation exchanger, and luminal choline transporter. Cell. Physiol. Biochem. 5, 290–298 (1995). [Google Scholar]
- 41. Penson, R.T. , Joel, S.P. , Bakhshi, K. , Clark, S.J. , Langford, R.M. & Slevin, M.L. Randomized placebo‐controlled trial of the activity of the morphine glucuronides. Clin. Pharmacol. Ther. 68, 667–676 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information accompanies this paper on the CPT: Pharmacometrics & Systems Pharmacology website (http://psp-journal.com)
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information