

Abstract
8-Oxoguanine (8-oxoG), a mutagenic DNA lesion generated under oxidative stress, differs from its precursor guanine by only two substitutions (O8 andH7). Human 8-oxoguanine glycosylase 1 (OGG1) can locate and remove 8-oxoG through extrusion and excision. To date, it remains unclear how OGG1 efficiently distinguishes 8-oxoG from a large excess of undamaged DNA bases. We recently showed that formamidopyrimidine–DNA glycosylase (Fpg), a bacterial functional analog of OGG1, can selectively facilitate eversion of oxoG by stabilizing several intermediate states, and it is intriguing whether OGG1 also employs a similar mechanism in lesion recognition. Here, we use molecular dynamics simulations to explore the mechanism by which OGG1 discriminates between 8-oxoG and guanine along the base eversion pathway. The MD results suggest an important role for kinking of the DNA by the glycosylase, which positions DNA phosphates in a way that assists lesion recognition during base eversion. The computational predictions were validated through experimental enzyme assays on phosphorothioate substrate analogs. Our simulations suggest that OGG1 distinguishes between 8-oxoG and G using their chemical dissimilarities not only at the active site, but also at earlier stages during base eversion, and this mechanism is at least partially conserved in Fpg despite lack of structural homology. The similarity also suggests that lesion recognition through multiple gating steps may be a common theme in DNA repair. Our results provide new insight into how enzymes can exploit kinetics and DNA conformational changes to probe the chemical modifications present in DNA lesions.
Keywords: 8-Oxoguanine (8-oxoG), base excision repair (BER), DNA repair, DNA damage recognition, enzyme kinetics, molecular dynamics
Graphical abstract
1. Introduction
The DNA integrity of living organisms is constantly under attack from reactive oxygen species (ROS). One major product of ROS is 8-oxoguanine (8-oxoG), which is generated by oxidation of guanine. 8-oxoG differs from guanine at only two positions: 8-oxoG has an O8 atom at C8 and anH7 atom at N7, whereas guanine has an H8 at C8 and a lone pair of electrons at N7 (Figure 1A). Like guanine, 8-oxoG can pair with cytosine in the Watson-Crick mode; however, 8-oxoG in the syn conformation can also form a stable Hoogsteen base pair with adenine (Figure 1B). Thus, during replication unrepaired 8-oxoG may lead to a G-C to T-A transversion mutation.1 Increase in the frequency of 8-oxoG lesions has been linked to cancer and aging.2–3
Figure 1.

A, the chemical structure of guanine and 8-oxoG. The structural difference between the two bases is highlighted in red. B, Watson-Crick pairing of 8-oxoG:C and Hoogsteen pairing of 8-oxoG:A.
8-oxoG is very stable when paired with cytosine in duplex DNA, and the 8-oxoG-containing DNA is almost indistinguishable from the undamaged duplex.4 Nevertheless, 8-oxoG can be detected and repaired via the base excision repair (BER) pathway which is initiated by DNA N-glycosylases that can remove their cognate bases and create apurinic/apyrimidinic (AP) sites. The human 8-oxoguanine-DNA glycosylase (OGG1) is one of the major glycosylases involved in the human BER pathway, and is responsible for removal of 8-oxoG from 8-oxoG:C base pairs in duplex DNA. Formamidopyrimidine-DNA glycosylase (Fpg, also known as MutM) is a functional analog of OGG1 in prokaryotes, but these two enzymes have no sequence or structural homology. Both OGG1 and Fpg search for 8-oxoG by 1D diffusion (sliding) along DNA in a Brownian manner, and the DNA is kinked at the interrogated site.5–7 When an 8-oxoG:C base pair is located, the lesion is everted from the DNA helix and inserted deeply into the catalytic pocket, followed by hydrolysis of the N-glycosidic bond. Such base eversion (also known as base flipping) is a common feature of diverse glycosylase–DNA complexes. The target nucleotide must rotate out of the double helix so that the glycosylase can accommodate the damaged base at the extrahelical catalytic pocket and hydrolyze the glycosidic bond. This process involves a series of conformational changes described as “pinch–push–plug–pull”8. In the case of the OGG1-8-oxoG system, three pre-excision steps have been suggested using stopped-flow kinetics: (i) initial encounter and extrusion of 8-oxoG (“pinch-push”), (ii) insertion of certain OGG1 residues into DNA (“plug”), and (iii) insertion of 8-oxoG into the active site of OGG1 which isomerizes for catalysis (“pull”).9
To date, it remains unclear how OGG1 so specifically and rapidly distinguishes the relatively rare 8-oxoG among a tremendous excess of undamaged G. It has been proposed that the Lys249+/Cys253− dipole helps to recognize the N7/O8 dipole of 8-oxoG10, but this dipole-dipole interaction is not essential since the double mutant Lys249Cys/Cys253Lys retains the ability to excise 8-oxoG.11 Crystallographic studies have suggested that OGG1 can distinguish 8-oxoG from G at the active site, by forming a discriminatory hydrogen bond between the carbonyl of Gly42 and the pyrrole N7 atom of 8-oxoG.10, 12 On the other hand, there is no evidence that direct contact between OGG1 and the O8 of 8-oxoG plays a role in damage recognition, and the Gly42/H7 hydrogen bond is the only direct interaction observed to distinguish 8-oxoG from G. However, biochemical studies have revealed several OGG1 substrates besides 8-oxoG that possess O8 but notH7, such as 2,6-diamino-4-oxo-5N-methylformamidopyrimidine (Me-FaPyG).13–14 These findings suggest that bothH7 and O8 may be exploited for lesion recognition, and thus there are probably some unidentified interactions between OGG1 and theH7/O8 atoms other than the single G42/H7 hydrogen bond that forms in the active site. In addition, the extremely fast diffusion rate of OGG1 sliding along DNA almost certainly excludes the possibility that OGG1 fully extrudes each interrogated DNA base into its active site.5 Our studies have suggested that Fpg can recognize 8-oxoG through multiple gating intermediates during eversion. Therefore, we hypothesize that the substrate recognition by OGG1 may occur not only at the active site, but also at one or more gating intermediates in the base eversion pathway. Unfortunately, early intermediates for the wild type system are likely to be infrequently populated and thus inaccessible to direct experimental characterization, hindering our understanding of the damage recognition mechanism by OGG1.
Crystallography studies have provided important structural clues related to base eversion. The end of the eversion pathway is represented by a crystal structure of fully extrahelical 8-oxoG occupying the active site pocket of an OGG1 sequence mutated to prevent substrate turnover (PDB ID: 1EBM12). A putative early intermediate (PDB ID: 2I5W15) along the base eversion pathway showed an opened G:C pair of which the G is slightly flipped-out towards the major groove, stabilized by a hydrogen bond to an 8-oxoG adjacent to the interrogated base. In a putative later intermediate (PDB ID: 1YQK10), the target G is extrahelical but rejected from the active site, instead residing at an adjacent “exo-site”. 8-oxoG has also been trapped in an exo-site when the active site is sterically obstructed by a Gln319Phe mutation (PDB ID: 2NOF16).
It should be noted that these intermediate structures employed an engineered crosslink (between the cytosine of the interrogated pair and Cys149, mutated from WT Asn149) to force OGG1 to interrogate a non-cognate G:C pair. Cys149 invades the DNA helix from the minor groove and forms a disulfide bond with the cytosine, displacing the target base from its position in the duplex. Furthermore, the details of the crosslink influences the extent of resulting eversion; the Cys149 crosslink shifted the target base to the exo-site, while a distal crosslink at position 292 (PDB ID: 3IH717) resulted in complete eversion of undamaged G into the active site. Thus the crystal structures provide valuable insight, but lack information on the energetics of eversion in the biological system.
The major groove eversion mechanism implied by some of these crystal structures is consistent with reports for other glycosylases. For example, the crystal structure of uracil DNA glycosylase (UDG) suggested a major groove eversion mechanism, as UDG, like OGG1, binds DNA from the minor groove and the minor groove path is sterically hindered18. Also, computational studies on the Fpg-DNA complex strongly indicate that the eversion of 8-oxoG through the major groove is energetically more favorable than through the minor groove19. Thus, it is plausible that OGG1 also everts 8-oxoG through a major groove pathway.
To understand how OGG1 recognizes 8-oxoG against G, and evaluate potential similarities to Fpg, we energetically and structurally characterized the base eversion pathway in OGG1 through the use of molecular dynamics (MD) and enzyme kinetics with modified substrates that cannot form the apparently important transient interactions detected by MD. This combined approach has a unique advantage of connecting energy, structure and dynamics with high spatial (i.e. atomic) and temporal resolution. Following the protocol of our previous study on the Fpg system19, here we compare the free energy and structure of 8-oxoG and G eversion to understand the damage recognition mechanism of OGG1. We found that in addition to the active site discrimination against 8-oxoG and G, the system also provides several earlier stages of potential damage recognition, and the mechanism is similar to that observed for Fpg including a mechanistic role for the crystallographically observed bending of the interrogated DNA.
2. Methods
2.1 Software, force fields and parameters
The SANDER module in the Amber 10 and Amber 11 suites of programs20–21 was used for molecular dynamics (MD) simulations. For all systems the ff99SB force field22 was used with the parmbsc023 adjustment for DNA α,γ backbone parameters. The parameters for 8-oxoG were obtained from Miller et al.24 The TIP3P25 explicit solvent water model was used to solvate the protein-DNA complexes.
2.2 Calculations of the base eversion paths using PNEB
As with our previous work for Fpg19, we used the PNEB variant26 of the nudged elastic band method27. This approach finds a minimum energy pathway between two given states (endpoints). In this work, one endpoint is defined as OGG1 interrogating an intrahelical, unopened 8-oxoG:C or G:C base (the intrahelical state), whereas the other endpoint is that with the target base inserted into the active site pocket of OGG1 (the in-pocket state). In order to direct the sampling of the base eversion path along the major groove, additional intermediate structures were used to seed the initial path, similar to what we did for Fpg19. Because using one seeding intermediate failed to generate a major groove path in OGG1, here we used two seeding intermediates (see additional details in Supporting Information). For both the 8-oxoG-complex and the G-complex, the initial base eversion path was generated by linking 32 replicas: 10 replicas of the equilibrated intrahelical structure model, 10 replicas of the equilibrated in-pocket structure, and 6 replicas of each of the two intermediate guiding seeds. Parameters for running PNEB were same as used in our previous study19, except as noted in SI.
The 1EBM12, 2I5W15 and 1YQK10 crystal structures were considered reasonable candidates for input structures. They represent the everted end state and two putative intermediate states of base eversion, respectively: in the 1EBM structure, the 8-oxoG is everted into the active site of OGG1 (in-pocket state); in the 2I5W structure, the target G is slightly everted towards the major groove, stabilized by a hydrogen bond with the neighboring 8-oxoG; 1YQK is a putative later intermediate in which the target G samples an extrahelical exo-site along the major groove path.
For intermediates to seed the major groove pathway for 8-oxoG, we used 2I5W and 1YQK. For G, we elected to use only 2I5W and not 1YQK to seed the path. Crystallographic studies have suggested that 1YQK is a relatively stable intermediate state for an extrahelical G, thus we used 1YQK as independent validation that the simulated G eversion sampled a pathway consistent with experimental data. A second guiding seed for G was generated by performing 50ps MD simulation on the equilibrated 2I5W based intermediate, forcing the target G to further evert to 9.5 Å of the eversion reaction coordinate (see below) using 10 kcal·mol−1·Å−2 restraint.
Currently, there is no available crystallographic structure representing a fully intrahelical endpoint due to the use of crosslinks to create artificially specific interrogation structures. Therefore, a model was generated by computationally modifying an existing OGG1 structure. The 2I5W structure was considered the best candidate since the target base is only slightly everted, thus we expected that it would be relatively straightforward to force the base to be fully intrahelical. The modification was performed after initial equilibration of the 8-oxoG-complex model of 2I5W. Detailed procedures of generating the input structures for NEB can be found in SI.
To employ these crystal structures in our eversion model, we reverted all OGG1 sequences back to wild type (active enzyme, with no crosslinks) and also made the DNA sequence consistent among all structures, adopted from 1EBM (5′-GGGGTAGACCTGGAC-3′, 5′-GTCCAXGTCTACCCC-3′; “X” is the interrogation position, using 8-oxoG in the 8-oxoG-complex or G in the G-complex). Note that while the 2I5W crystal structure had an 8-oxoG proximal to the interrogation site, all simulations described here with 8-oxo-G have it located only in the interrogation site.
2.3 Calculation of the potentials of mean force (PMFs) for base eversion
Umbrella sampling (US) was used to obtain the PMF as a function of the eversion distance (defined in Figure 2, with further details provided in SI). In order to further enhance conformational sampling, Hamiltonian replica exchange molecular dynamics28 was employed. The US protocol was otherwise similar to those in our previous studies using conventional umbrella sampling29–30. For both the 8-oxoG-complex and the G-complex, two independent sets of initial structures were taken from the PNEB production trajectories and the US was performed for 2.5ns per window in an NVT ensemble, repeated for each set of initial structures. US windows were evenly spaced along the eversion distance with 0.3 Å intervals (Figure S1), restrained by 10 kcal·mol−1·Å −2 umbrella potential. The time step was 2 fs and exchanges between neighboring windows were attempted every 10 ps. The temperature was maintained at 310K using a Langevin thermostat31 with a 75.0 ps−1 collision frequency. The eversion distance values were recorded every time step and then analyzed using the weighted histogram analysis method (WHAM).32 The difference between the profiles obtained from two independent US runs were used to estimate the lower bound of uncertainty (precision) in the data. The zero of energy is arbitrary in molecular mechanics, and only relative energies on PMF curves are meaningful. Therefore, absolute energies on the PMFs for different systems cannot be directly compared. Following our previous work on Fpg, the intrahelical endpoint of each PMF was set to 0 kcal/mol to allow the PMFs to highlight differences in the energetic profiles of 8-oxo-G and G as eversion proceeds.19
Figure 2.

Eversion distance is defined as the distance between the center of mass of the heavy atoms of the base pairs flanking the target base pair (colored in red) and the center of mass of the heavy atoms of the target base (colored in blue). This distance was used as the reaction coordinate for base eversion.
2.4 Enzyme activity assay
Oligonucleotides of the sequence 5′-CTCTCCCTpsTpsCXpsCpsTCCTTTCCTCT-3′ containing 8-oxoG (X) and a phosphorothioate linkage at one of “ps” positions (or all regular linkages), as well as the complementary strand 5′-AGAGGAAAGGAGCGAAGGGAGAG-3′, were synthesized from commercially available monomers (Glen Research, Sterling, VA) using an ASM-700 synthesizer (Biosset, Novosibirsk, Russia). The oxoG-containing strand was 32P-labeled using T4 polynucleotide kinase (Biosan, Novosibirsk, Russia) and annealed to a 1.5-fold molar excess of the complementary strand. E. coli Fpg and human OGG1 were purified from the respective overproducing E. coli strains as described.33–34 The reaction mixture (20 μl) included 50 mM Tris–HCl (pH 7.5), 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 100 nM 8-oxoG-containing duplex, and 10 nM OGG1 or Fpg. The reaction was allowed to proceed for 20 min (OGG1) or 10 min (Fpg) at 37°C. For OGG1, the reaction was terminated by adding NaOH to 50 mM and heating for 1 min at 95°C, immediately neutralized with HCl, mixed with 0.5 volumes of the loading dye containing 80% formamide, and heated again for 1 min. For Fpg, the reaction was terminated by adding an equal volume of the loading dye and heating for 1 min at 95°C. The products were separated by electrophoresis in 20% polyacrylamide gel containing 8 M urea and quantified using Molecular Imager FX (Bio-Rad, Hercules, CA).
3. Results and discussions
3.1 Simulation strategy
We hypothesize that 8-oxoG recognition by OGG1 occurs in a series of steps, including those prior to complete insertion of target base into the active site. To test this, we structurally and energetically compared the eversion of 8-oxoG and G in OGG1, which are the processes of base eversion from inside DNA (intrahelical state) to the active site of OGG1 (in-pocket state). The base eversion process in OGG1 has been found to occur on the millisecond timescale9 which is unlikely to be adequately sampled by currently affordable conventional MD simulations. We used pathway mapping and umbrella sampling simulations to investigate the free energy profile for 8-oxoG and G eversion in Fpg; the results indicated that 8-oxoG is stabilized by Fpg at several intermediate stages of eversion, whereas the G eversion is energetically unfavored.19, 35 Here, we used the same methods to characterize the eversion paths for OGG1, permitting comparison of the recognition mechanisms of these 2 proteins that carry out comparable functions despite lack of structural similarity.
3.2 Free energy profiles of eversion of 8-oxoG and G in OGG1
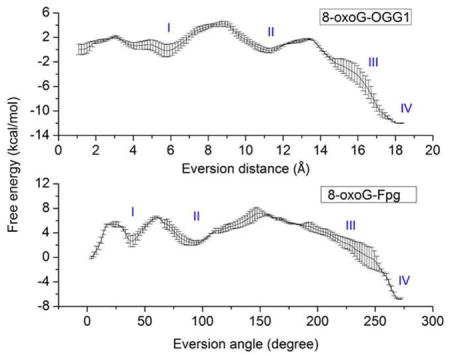
Using the protocols described in Methods, we generated and compared the free energy profiles along the eversion pathways of 8-oxoG and G (Figure 3). Crystallographic studies have proposed a major groove path for OGG110, 15–16, 36, and OGG1’s functional analog Fpg also prefers base eversion through the major groove19. Thus, in this work we focus only on comparing eversion and recognition through the major groove. The intra-helical position is represented by the local minimum near 1 Å of eversion distance, while complete eversion into the OGG1 active site corresponds to the minimum near 18 Å. The 8-oxoG-complex favors the in-pocket everted state by about 12 kcal/mol with respect to the intrahelical state, while for the G-complex, the in-pocket state is energetically similar to the intra-helical state. In addition, the energetic barrier to eversion of 8-oxoG is only ~4 kcal/mol, while G encounters a substantially higher energetic barrier (~7–8 kcal/mol). A higher kinetic barrier to extrusion of G vs. 8-oxoG was also observed in Fpg35, consistent with the fact that 8-oxoG is the cognate substrate for OGG1 and Fpg whereas G is not.
Figure 3.

Comparison of computed free energy profiles for base eversion in the 8-oxoG complex (black) and the G complex (red). Four potential 8-oxoG-recognizing steps are labeled as I, II, III, and IV. Error bars reflect differences between two independent runs.
The free energy profiles show significant differences between 8-oxoG and G at four regions along the base eversion paths: 3–6 Å, 9–12 Å, 14–17 Å, and 17–18 Å of eversion distance (labeled in Figure 3). These locations suggest multiple intermediate states of energetic and, presumably, structural discrimination between 8-oxoG and G, through which we may gain insight into how damage recognition occurs. The details of these four states are discussed below.
Initial opening of the 8-oxoG:C base pair occurs at an eversion distance of 3 Å (where 8-oxoG loses all hydrogen bonds with the opposite C), requiring only 1–2 kcal/mol (Figure 3). These barriers are significantly lower than those we calculated for 8-oxo-G:C and G: C pairs in the context of duplex DNA37, probably due to the destabilizing effects of the Y203 wedge as observed in our intrahelical model (see SI for details). This is also consistent with our previous work showing that buckling induced by the wedge intercalation in Fpg destabilizes the intrahelical state.38 The energetic cost of breaking the G:C or 8-oxoG:C pairs are comparable, indicating no preferential opening of damaged DNA. Notably, the energy cost for the 8-oxoG:C opening in OGG1 is comparable to that in Fpg,19 and also comparable to the energy barriers of OGG1/Fpg sliding along DNA (0.5 to 2 kcal/mol),5 but is significantly lower than G opening in the context of B-DNA, which has a ~10 kcal/mol barrier.39 Thus, OGG1 and Fpg probably promote base eversion to faciliate base readout during fast sliding.
The free energy landscapes of the two systems start to diverge shortly after the base pair has been opened, indicating an opportunity for damage recognition early in eversion (Figure 3, step I). When 8-oxoG disengages from the paired cytosine and is slightly everted (3~6 Å), it is stabilized by a hydrogen bond between theH7 and the second 5′ phosphate (the phosphate of the 5′ nucleotide, hereafter referred to as p1) (Figure 4). This damage-specific interaction corresponds to the free energy minimum at 6 Å eversion; whereas for the G-complex, the same phosphate exerts electrostatic repulsion with the lone pair of electrons on N7, contributing to the energy rise at 4~6 Å eversion (Figure 3). The data suggest that the discriminatory interaction with 8-oxoG and G provided by p1 constructs an early checkpoint for 8-oxoG damage which could be more efficient than selection only in the active site. To pass through this unfavorable state, G has to overcome a ~6 kcal/mol energetic barrier (Figure 3), a significant impediment during the fast lesion search by OGG1, and thus G probably rapidly returns to the duplex after minimal eversion followed by continued sliding of OGG1.
Figure 4.

A, the DNA structure in eversion step I of Figure 3, with an eversion distance of 6.1 Å. The black dots indicate the hydrogen bond between the N7 of 8-oxoG and the OP2 of p1. Hydrogen atoms are present but most are not shown for clarity. B, comparison of change during eversion of average distance between the OP2 of p1 and the N7 of 8-oxoG (black) and G (red). The error bars reflect the difference between two independent runs.
It should be noted that the base-p1 interactions here require a bent DNA; in a standard, unbent B-DNA, such interactions are unlikely due to the larger distance between the base and p1 as compared to that in OGG1 (Figure 5). This suggests that DNA bending may have a functional role by recruiting the phosphodiester backbone to read the status of N7. This hypothesis is supported by recent studies indicating that a mismatch at the 5′-neighboring position of 8-oxoG strongly decreased the rate of 8-oxoG removal,40–41 and here we suggest that those mismatches may interrupt the favorable 8-oxoG/p1 interaction by repositioning the p1 phosphate and thus hinder 8-oxoG eversion.
Figure 5.

Superimposition of the bent DNA (purple) from Step I and standard B-DNA duplex (yellow, generated by 3DNA42). Hydrogen atoms are present but most are not shown for clarity. In the magnified region, the B-DNA is in yellow with 8-oxoG and p1 in orange; the DNA from step I is purple with 8-oxoG and p1 colored by atom. The difference in geometries is consistent with the distance data shown in Figure 1B.
When 8-oxoG is everted to a midway point (11 Å eversion, Step II), it forms an additional hydrogen bond to p1 (Figure 6A). G can also form the same hydrogen bonds to p1, but the hydrogen-bonding distances between 8-oxoG and p1 are considerably shorter than those between G and p1 (Figure 6B), suggesting that the p1 may have stronger interactions with 8-oxoG than with G. This is probably because 8-oxoG at the midway point is also stabilized by the hydrogen bond between O8 of 8-oxoG and the backbone amide linking His270 and Val269 (Figure 6A, C). On the other hand, His270 does not stabilize G in this state, since G lacks O8. This hampers ideal positioning of G for hydrogen bonding, and the resulting energy minimum is significantly shallower than that of 8-oxoG complex. This suggest an early potential checkpoint for 8-oxoG, again facilitated through recruitment of the phosphodiester backbone via DNA bending. At the energy barrier between steps I and II (near 9 Å eversion in Figure 3), the structures for 8-oxoG and G are similar, with both adopting a repulsive interaction between p1 and O6 (Figure S2).
Figure 6.

A, Structure of the 8-oxoG-complex in step II, with an eversion distance of 11.6 Å. The hydrogen bonds stabilizing 8-oxoG are depicted as dotted lines. B, comparison of distances between p1 and the nitrogen atoms at the Watson-Crick face of the 8-oxoG (black/green) and G (orange/blue) as a function of eversion. C, comparison of distances between the backbone nitrogen of H270 and the O8 of 8-oxoG (black) or the H8 of G (red). The error bars reflect differences between two independent US runs.
3.3 Phosphate p1 is critical for the activity of OGG1
Since our simulations predicted important transient interactions of the p1 phosphate with 8-oxoG during the eversion, we analyzed the effect of the replacement of this phosphate with a phosphorothioate linkage. Phosphorothioates keep the negative charge of phosphates but have a bulkier, more hydrophobic, easier to polarize, and less electronegative sulfur atom replacing one of the non-bridging oxygens; as a result, charge is mostly concentrated on the remaining oxygen while the sulfur is nearly neutral. All these differences are expected to alter the interactions of the internucleotide linkage with its surroundings, allowing us to probe experimentally the important interactions suggested by the computational results on the biologically relevant DNA systems. Analogous simulations of phosphorothioates would involve significant effort in the development of a modified DNA backbone force field, and was therefore considered to be beyond the scope of this work.
We substituted p2, p1, p−1, or p−2 phosphates with phosphorothioates (no p0 substitution was made since 8-oxoG phosphorothioate is presently not available synthetically) and investigated the activity of OGG1 on these substrates. The products were treated with hot alkali to digest all abasic sites and thus follow the base excision reaction only rather than the combined base excision/β-elimination. A substitution at p1 significantly reduced the activity of OGG1 both in the multiple-turnover (Figure 7) and single-turnover mode (Figure S3). Notably, in the structure of OGG1 bound to 8-oxoG-containing DNA,12 p1 forms no direct contact with the protein, suggesting that this effect is not due to destabilization of the Michaelis complex. The other three substitutions decreased the activity about twofold (Figure 7), likely because we used racemic mixtures of R- and S-phosphorothioates, only one of which conserved the bonds with the protein. Moreover, Fpg tested on this set of substrates showed the same preferential inhibition by the p1-to-prosphorothioate substitution (Figure S3); in this case, too, p1 does not form bonds with the protein when the lesion is fully everted.33 Unlike in the well-known blockage of nuclease hydrolysis by phosphorothioates, p1 does not participate in the reaction chemistry of glycosylases, so the inhibition of OGG1 and Fpg is most likely of conformational nature as suggested by the computational model.
Figure 7.

Activity of OGG1 on phosphorothioate-substituted 8-oxoG-containing substrates. See Methods for details.
3.4 Role of the exo-site
The 1YQK10 crystal structure represents a putative intermediate with G occupying an exo-site, but the biological relevance of this intermediate is unclear due to the artificial nature of the crosslink. As discussed in Methods, the exo-site structure was not used as input in the PNEB calculation for the G eversion pathway. Nevertheless, we observed that multiple independent G complex PNEB path simulations sampled this intermediate at an eversion distance of ~14.5–16.5 Å (Figure 8), supporting the relevance of the exo site notwithstanding its observation in a crosslinked structure, and also suggesting that the modeled pathway is reasonable, since it sampled an experimental intermediate structure that was not used as input for the calculations. The simulation data also help rationalize the sensitivity of the conformation observed in the crystal structures to the position of the crosslink, since the exo-site and in-pocket states are seen to be nearly degenerate in free energy.
Figure 8.

Superposition of the crystal structure 1YQK (DNA in yellow and protein in pink) and the structure sampled during the simulated base eversion (the DNA colored by atom and protein in green). Both structures were superimposed using protein heavy atoms. Hydrogen atoms are present but most are not shown for clarity.
In our simulations, the backbone amide of Ile152 and the N7 atom of G form a weak hydrogen bond in the exo-site (~3.7 Å average distance, Figure 9D), very similar to the distance in the 1YQK crystal structure (~4.2 Å). Additional stabilization in the exo-site comes from stacking of the π-face of G with the His270 aromatic ring as seen in simulations (Figure 9C), although the high B-factors10 for the His270 side chain in 1YQK suggest this may also be a weak interaction. Nevertheless, when G loses these weak interactions and enters the active site, it has to overcome a modest energetic barrier. 8-oxoG also samples the exo-site, wherein O8 forms a hydrogen bond to the ε-amino group of Lys249 (Figure 9A,B); the resulting O8-Lys249 interaction thus stabilizes 8-oxoG in the exo-site. Due to lack of O8 on G, the favorable O8-Lys249 interaction is not observed in the G complex (Figure 9B) and thus this interaction helps to further distinguish 8-oxoG and G. Notably, Lys249Gln and Lys249Cys/Cys253Lys mutants retain the ability of extruding 8-oxoG to the active site11–12, suggesting that a lysine in position 249 may not be imperative for 8-oxoG eversion. However, the long, flexible and polar side chains of Gln249 or Lys253 may retain the ability to hydrogen bond to the O8 of 8-oxoG, facilitating the eversion of 8-oxoG but presumably to a lesser extent due to reduced charge on the protein side chain. The hypothesis is supported by pre-steady-state kinetic experiments, which indicated that the Lys249Gln mutant of OGG1 can evert 8-oxoG but does so more slowly than the wild-type protein.43
Figure 9.

A, Structure indicating the DNA interaction with Lys249 in step III, with an eversion distance of 14.8 Å. The 8-oxoG-stabilizing hydrogen bond is depicted as a dotted line. B, comparison of distances between the Lys249 ε-amino and the O8 of 8-oxoG (black) or the H8 of G (red). C, structure of the G complex in step III, with an eversion distance of 13.9 Å. G is stabilized by the stacking with His270 and a hydrogen bond with Ile152. D, comparison of distances between the backbone nitrogen of Ile152 and the N7 of 8-oxoG (black) or G (red). Error bars reflect the differences between two independent runs.
3.5 Recognition in the active site
At an eversion distance near 17~18 Å (Step IV), the system forms the Gly42/N7 hydrogen bond that is the hallmark of the in-pocket state (Figure 10A, B). For 8-oxoG, the free energy of the system drops dramatically from step III in exo-site to step IV in active site (Figure 3), suggesting that 8-oxoG is better stabilized in the active site than in the intermediate exo-site. A rationale for at least part of this energetic difference is suggested by the 8-oxoG complex crystal structure 1EBM, which reveals a discriminatory hydrogen bond between the backbone carbonyl of Gly42 and the protonated N7 of 8-oxoG.12 For the G complex, the in-pocket state was also captured experimentally (PDB 3IH7) by using a distal artificial crosslink (Ser292Cys) to force OGG1 to interrogate a G:C.17 In that crystal structure, G occupies almost the same position as does the in-pocket 8-oxoG seen in 1EBM, despite the unfavorable interaction between the backbone carbonyl of Gly42 and the lone pair of electrons on N7 of G.17 This is probably an artificial effect induced by the distal crosslink, as suggested by previous uncrosslinked MD simulations indicating that G was unstable in the 3IH7 crystallographic in-pocket position.17 In our simulations, the in-pocket endpoint for G was not generated from 3IH7; instead, it was generated from 1EBM with the in-pocket 8-oxoG changed to G. Nevertheless, we also observed that G becomes retracted from the active site even when placed there, and the backbone of Gly42 rotates to avoid the lone pair electrons on N7 of G (Figure 10C). Similar conformational changes for G were also observed in quantum mechanics/molecular mechanics (QM/MM) simulations previously performed to investigate the active site preference for 8-oxoG or G.10 These observations are reinforced by failure of OGG1 to catalyze base excision of G even when it is forcibly presented in the active site by crosslinking; this catalytic checkpoint mechanism is still unclear17. Our data suggest that although Gly42 provides the sole interaction that structurally discriminates 8-oxoG and G in the active site, it may be strong enough to keep G from entering the active site as deeply as does 8-oxoG (Figure 10C). Experimental studies have shown that the Gln315Phe, Gln315Trp, Cys253Ile, and Cys253Leu mutations, which perturb the active site disposition of 8-oxoG but not expel it altogether, can severely reduce the catalytic activity of OGG1,16, 44 thus suggesting that the catalysis of base excision by OGG1 requires very precise positioning of the reacting moieties. Therefore, the unfavorable interaction of Gly42 may prevent G from achieving the optimal position for catalysis, and thus the active site geometry constitutes the final damage checkpoint of OGG1.
Figure 10.

A. Structure of the 8-oxoG-complex in step IV from Figure 3, with an eversion distance of 16.3 Å. B. Distance between the carbonyl of G42 and the N7 of either G (red) or 8-oxoG (black). C. Position of 8-oxoG (green) and G (blue) in the active site, the rest of DNA is shown in grey. The two structures are superimposed on the protein backbone (yellow). The Gly42:8-oxoG h-bond is depicted with a black dotted line. The eversion distances of the 8-oxoG and the G structures are 16.3 Å and 15.8 Å, respectively.
Discussion
Previous studies have found considerable similarities in the damage recognition mechanisms of OGG1 and Fpg: both kink the DNA and insert a bulky wedge to disrupt stacking of the target base pair, both extrude 8-oxoG into the active site where the N7/H7 of 8-oxoG is recognized by the enzyme, and both insert several plug residues into the DNA to stabilize the extrahelical state, including an arginine residue that recognizes the Watson-Crick interface of the orphaned cytosine.12, 45
By comparing the data from this work and our previous work,35 we find additional important evidence for a shared damage recognition mechanism, including the nature of several early checkpoints prior to full eversion. The free energy profiles for 8-oxoG eversion in OGG1 and Fpg show a similar pattern (Figure 11): they have two minima in the first half of eversion (Step I and II) and a global minimum at the extrahelical endpoint (step IV); between steps II and IV is an area with significant energy drop (step III). For both enzymes, these four steps correspond to four potential checkpoints for 8-oxoG, because in each step specific interactions are made to the sites modified in the lesion (N7/H7 or the O8). Notably, such specific interactions are similar in pattern between OGG1 and Fpg (Figure 12): in step I, the slightly everted 8-oxoG is stabilized by the N7/p1 hydrogen bond; in step II, the Watson-Crick edge of 8-oxoG hydrogen-bonds to p1 while the O8 hydrogen-bonds to a polar side chain (Asn173 in Fpg and H270 in OGG1); in step III, a catalytic residue (Pro1 in Fpg and Lys249 in OGG1) contacts the O8 of 8-oxoG; in step IV, 8-oxoG is specifically recognized by a hydrogen bond made to the protonated N7.
Figure 11.

Free energy profiles for 8-oxoG eversion in OGG1 and Fpg. Four distinct steps of eversion are labeled as I, II, III and IV. Note that the progress variables on the X axes are not the same in the two studies, thus alignment of minima between systems is arbitrary. The error bars reflect differences between two independent runs.
Figure 12.

Simulation structures at Steps I, II, III, IV along the 8-oxoG eversion pathway in Fpg and OGG1. H-bonds that contact the O8 or the protonated N7 atom of 8-oxoG are indicated by green dotted lines.
Thus, although OGG1 and Fpg are different in overall structure, they employ similar early gates for structural and energetic discrimination that likely facilitate rapid damage recognition on the fast timescale needed for genome-level scanning of DNA. When a glycosylase samples a base pair, it enters a kinetic competition between the sampling path with the wedging and plugging residues fully inserted and a 1D diffusion path that requires their withdrawal from the base stack. The energy difference of ~5.5 kcal/mol at the earliest eversion intermediate (step I) corresponds to ~104-fold G vs 8-oxoG discrimination, effectively allowing the enzyme to quickly reject the non-damaged base and resume scanning with minimal time wasted for unproductive encounters. Interestingly, in Fpg, step I contributes little to the selectivity, but step II (~10.8 kcal/mol difference) provides ~5×107-fold discrimination against G.
In summary, we have investigated the damage recognition mechanism of OGG1 by energetically and structurally comparing the base eversion of 8-oxoG and G. Based on the free energy profiles as well as the structural analysis that is linked to the energetic differences, we found that OGG1 facilitates 8-oxoG eversion while also hindering G eversion at multiple steps during base eversion, including stages earlier than those suggested by crystal structures of the final everted state. Thus, during rapid scan of DNA damage, OGG1 could potentially discriminate 8-oxoG from G even when the base is merely slightly everted. Interestingly, in comparable simulations for the Fpg system35, the potential checkpoints for 8-oxoG exhibit similar geometries compared to OGG1, suggesting that the damage recognition mechanism may be partly conserved between these two functional analogs, even though they have no sequence or structural similarities.
Supplementary Material
Acknowledgments
The authors thank the Laufer Center for Physical and Quantitative Biology at Stony Brook University for access to computational resources and support, and an NSF Petascale Computational Resource (PRAC) Award from the NSF (OCI-1036208) to CS.
Funding sources. This work was supported by the National Institutes of Health grant R01CA017395. Biochemical experiments were supported by the Russian Science Foundation grant 14-24-00093 to D.O.Z. CS gratefully acknowledges support from Henry and Marsha Laufer.
Footnotes
Supporting information includes additional computational and experimental methods details, supporting figures, and a table of DNA properties. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Grollman AP, Moriya M. Mutagenesis by 8-Oxoguanine - an Enemy Within. Trends in Genetics. 1993;9(7):246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286(5446):1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T. Instability and Decay of the Primary Structure of DNA. Nature. 1993;362(6422):709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 4.Crenshaw CM, Wade JE, Arthanari H, Frueh D, Lane BF, Nunez ME. Hidden in Plain Sight: Subtle Effects of the 8-Oxoguanine Lesion on the Structure, Dynamics, and Thermodynamics of a 15-Base Pair Oligodeoxynucleotide Duplex. Biochemistry. 2011;50(39):8463–8477. doi: 10.1021/bi201007t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc Natl Acad Sci U S A. 2006;103(15):5752–7. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banerjee A, Santos WL, Verdine GL. Structure of a DNA glycosylase searching for lesions. Science. 2006;311(5764):1153–7. doi: 10.1126/science.1120288. [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Haushalter KA, Lieber CM, Verdine GL. Direct visualization of a DNA glycosylase searching for damage. Chem Biol. 2002;9(3):345–50. doi: 10.1016/s1074-5521(02)00120-5. [DOI] [PubMed] [Google Scholar]
- 8.Stivers JT. Site-specific DNA damage recognition by enzyme-induced base flipping. Prog Nucleic Acid Res Mol Biol. 2004;77:37–65. doi: 10.1016/S0079-6603(04)77002-6. [DOI] [PubMed] [Google Scholar]
- 9.Kuznetsov NA, Koval VV, Nevinsky GA, Douglas KT, Zharkov DO, Fedorova OS. Kinetic conformational analysis of human 8-oxoguanine-DNA glycosylase. J Biol Chem. 2007;282(2):1029–38. doi: 10.1074/jbc.M605788200. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee A, Yang W, Karplus M, Verdine GL. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature. 2005;434(7033):612–8. doi: 10.1038/nature03458. [DOI] [PubMed] [Google Scholar]
- 11.Dalhus B, Forsbring M, Helle IH, Vik ES, Forstrom RJ, Backe PH, Alseth I, Bjoras M. Separation-of-function mutants unravel the dual-reaction mode of human 8-oxoguanine DNA glycosylase. Structure. 2011;19(1):117–27. doi: 10.1016/j.str.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 12.Bruner SD, Norman DP, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403(6772):859–66. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 13.Hamm ML, Gill TJ, Nicolson SC, Summers MR. Substrate specificity of Fpg (MutM) and hOGG1, two repair glycosylases. J Am Chem Soc. 2007;129(25):7724–5. doi: 10.1021/ja0716453. [DOI] [PubMed] [Google Scholar]
- 14.Asagoshi K, Yamada T, Terato H, Ohyama Y, Monden Y, Arai T, Nishimura S, Aburatani H, Lindahl T, Ide H. Distinct repair activities of human 7,8-dihydro-8 oxoguanine DNA glycosylase and formamidopyrimidine DNA glycosylase for formamidopyrimidine and 7,8-dihydro-8-oxoguanine. Journal of Biological Chemistry. 2000;275(7):4956–4964. doi: 10.1074/jbc.275.7.4956. [DOI] [PubMed] [Google Scholar]
- 15.Banerjee A, Verdine GL. A nucleobase lesion remodels the interaction of its normal neighbor in a DNA glycosylase complex. Proc Natl Acad Sci U S A. 2006;103(41):15020–5. doi: 10.1073/pnas.0603644103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radom CT, Banerjee A, Verdine GL. Structural characterization of human 8-oxoguanine DNA glycosylase variants bearing active site mutations. J Biol Chem. 2007;282(12):9182–94. doi: 10.1074/jbc.M608989200. [DOI] [PubMed] [Google Scholar]
- 17.Crenshaw CM, Nam K, Oo K, Kutchukian PS, Bowman BR, Karplus M, Verdine GL. Enforced presentation of an extrahelical guanine to the lesion-recognition pocket of the human 8-oxoguanine DNA glycosylase, hOGG1. J Biol Chem. 2012;287(30):26916–24928. doi: 10.1074/jbc.M111.316497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384(6604):87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- 19.Bergonzo C, Campbell AJ, de Los Santos C, Grollman AP, Simmerling C. Energetic Preference of 8-oxoG Eversion Pathways in a DNA Glycosylase. J Am Chem Soc. 2011;133(37):14504–6. doi: 10.1021/ja205142d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Case DA, TAD, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 11. University of California; San Francisco: 2011. [Google Scholar]
- 21.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, Merz KM, Wang B, Hayik S, Roitberg A, Seabra G, Kolossvary I, Wong KF, Paesani F, Vanicek J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Mathews DH, Seetin MG, Sagui C, Babin V, Kollman PA. AMBER 10. University of California; San Francisco: 2008. [Google Scholar]
- 22.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins-Structure Function and Bioinformatics. 2006;65(3):712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, 3rd, Laughton CA, Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys J. 2007;92(11):3817–29. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller JH, Fan-Chiang CCP, Straatsma TP, Kennedy MA. 8-Oxoguanine enhances bending of DNA that favors binding to glycosylases. J Am Chem Soc. 2003;125(20):6331–6336. doi: 10.1021/ja029312n. [DOI] [PubMed] [Google Scholar]
- 25.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. Journal of Chemical Physics. 1983;79(2):926–935. [Google Scholar]
- 26.Bergonzo C, Campbell AJ, Walker RC, Simmerling C. A Partial Nudged Elastic Band Implementation for Use With Large or Explicitly Solvated Systems. International Journal of Quantum Chemistry. 2009;109(15):3781–3790. doi: 10.1002/qua.22405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jónsson H, Mills G, Jacobsen KW. Nudged elastic band method for finding minimum energy paths of transitions. In: Berne BJ, Ciccotti G, Coker DF, editors. Classical and Quantum Dynamics in Condensed Phase Simulations. World Scientific; 1998. p. 385. [Google Scholar]
- 28.Dashti DS, Roitberg AE. Optimization of Umbrella Sampling Replica Exchange Molecular Dynamics by Replica Positioning. Journal of Chemical Theory and Computation. 2013;9(11):4692–4699. doi: 10.1021/ct400366h. [DOI] [PubMed] [Google Scholar]
- 29.Song K, Campbell AJ, Bergonzo C, de los Santos C, Grollman AP, Simmerling C. An Improved Reaction Coordinate for Nucleic Acid Base Flipping Studies. Journal of Chemical Theory and Computation. 2009;5(11):3105–3113. doi: 10.1021/ct9001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacKerell AD, Banavali NK. Free energy and structural pathways of base flipping in a DNA GCGC containing sequence. Journal of Molecular Biology. 2002;319(1):141–160. doi: 10.1016/S0022-2836(02)00194-8. [DOI] [PubMed] [Google Scholar]
- 31.Loncharich RJ, Brooks BR, Pastor RW. Langevin Dynamics of Peptides - the Frictional Dependence of Isomerization Rates of N-Acetylalanyl-N′-Methylamide. Biopolymers. 1992;32(5):523–535. doi: 10.1002/bip.360320508. [DOI] [PubMed] [Google Scholar]
- 32.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules.1. The Method. Journal of Computational Chemistry. 1992;13(8):1011–1021. [Google Scholar]
- 33.Gilboa R, Zharkov DO, Golan G, Fernandes AS, Gerchman SE, Matz E, Kycia JH, Grollman AP, Shoham G. Structure of formamidopyrimidine-DNA glycosylase covalently complexed to DNA. Journal of Biological Chemistry. 2002;277(22):19811–19816. doi: 10.1074/jbc.M202058200. [DOI] [PubMed] [Google Scholar]
- 34.Kuznetsov NA, Koval VV, Zharkov DO, Nevinsky GA, Douglas KT, Fedorova OS. Kinetics of substrate recognition and cleavage by human 8-oxoguanine-DNA glycosylase. Nucleic Acids Res. 2005;33(12):3919–3931. doi: 10.1093/nar/gki694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Endutkin AV, Bergonzo C, Campbell AJ, de los Santos C, Grollman A, Zharkov DO, Simmerling C. A dynamic checkpoint in oxidative lesion discrimination by formamidopyrimidine–DNA glycosylase. Nucleic acids research. 2016;44(2):683–694. doi: 10.1093/nar/gkv1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S, Radom CT, Verdine GL. Trapping and structural elucidation of a very advanced intermediate in the lesion-extrusion pathway of hOGG1. Journal of the American Chemical Society. 2008;130(25):7784–7785. doi: 10.1021/ja800821t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng X, Kelso C, Hornak V, de los Santos C, Grollman AP, Simmerling C. Dynamic behavior of DNA base pairs containing 8-oxoguanine. J Am Chem Soc. 2005;127(40):13906–18. doi: 10.1021/ja052542s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuznetsov NA, Bergonzo C, Campbell AJ, Li H, Mechetin GV, de los Santos C, Grollman AP, Fedorova OS, Zharkov DO, Simmerling C. Active destabilization of base pairs by a DNA glycosylase wedge initiates damage recognition. Nucleic acids research. 2014;33(12):3919–3931. doi: 10.1093/nar/gku1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giudice E, Varnai P, Lavery R. Base pair opening within B-DNA: free energy pathways for GC and AT pairs from umbrella sampling simulations (vol 31, pg 1434; 2003) Nucleic Acids Research. 2003;31(10):2703–2703. doi: 10.1093/nar/gkg239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirpota OO, Endutkin AV, Ponomarenko MP, Ponomarenko PM, Zharkov DO, Nevinsky GA. Thermodynamic and kinetic basis for recognition and repair of 8-oxoguanine in DNA by human 8-oxoguanine-DNA glycosylase. Nucleic Acids Res. 2011;39(11):4836–4850. doi: 10.1093/nar/gkq1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sassa A, Beard WA, Prasad R, Wilson SH. DNA Sequence Context Effects on the Glycosylase Activity of Human 8-Oxoguanine DNA Glycosylase. J Biol Chem. 2012;287(44):36702–10. doi: 10.1074/jbc.M112.397786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Research. 2003;31(17):5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuznetsova AA, Kuznetsov NA, Ishchenko AA, Saparbaev MK, Fedorova OS. Step-by-step mechanism of DNA damage recognition by human 8-oxoguanine DNA glycosylase. Biochim Biophys Acta. 2014;1840(1):387–95. doi: 10.1016/j.bbagen.2013.09.035. [DOI] [PubMed] [Google Scholar]
- 44.Lukina MV, Popov AV, Koval VV, Vorobjev YN, Fedorova OS, Zharkov DO. DNA Damage Processing by Human 8-Oxoguanine-DNA Glycosylase Mutants with the Occluded Active Site. Journal of Biological Chemistry. 2013;288(40):28936–28947. doi: 10.1074/jbc.M113.487322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fromme JC, Verdine GL. DNA lesion recognition by the bacterial repair enzyme MutM. J Biol Chem. 2003;278(51):51543–8. doi: 10.1074/jbc.M307768200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.