

Abstract
Macrophage intracellular pathogen killing is defective in cystic fibrosis (CF), despite abundant production of reactive oxygen species (ROS) in lung tissue. Burkholderia species can cause serious infection in CF and themselves affect key oxidase components in murine non-CF cells. However, it is unknown whether human CF macrophages have an independent defect in the oxidative burst and whether Burkholderia contributes to this defect in terms of assembly of the NADPH oxidase complex and subsequent ROS production. Here we analyze CF and non-CF human monocyte-derived macrophages (MDMs) for ROS production, NADPH assembly capacity, protein kinase C (PKC) expression, and calcium release in response to PMA and CF pathogens. CF MDMs demonstrate a nearly 60% reduction in superoxide production following PMA stimulation compared to non-CF MDMs. Although CF MDMs generally have increased total NADPH component protein expression, they demonstrate decreased expression of the calcium-dependent PKC conventional subclass α/β leading to reduced phosphorylation of NADPH oxidase components p47phox & p40phox in comparison to non-CF MDMs. Ingestion of B. cenocepacia independently contributes to and worsens the overall oxidative burst deficits in CF MDMs compared to non-CF MDMs. Together, these results provide evidence for inherent deficits in the CF macrophage oxidative burst due to decreased phosphorylation of NADPH oxidase cytosolic components that are augmented by Burkholderia. These findings implicate a critical role for defective macrophage oxidative responses in persistent bacterial infections in CF and create new opportunities for boosting the macrophage immune response in order to limit infection.
Keywords: ROS, phagocyte, bacteria, calcium
INTRODUCTION
Cystic fibrosis (CF) is a multi-organ disease caused by defects in chloride transport due to defective cystic fibrosis transmembrane regulator (CFTR) conductance (1). Clinically, patients characteristically suffer from recurrent polymicrobial lung infections with eventual respiratory demise (2). Members of the Burkholderia cepacia complex cause fatal septicemia and rapid outbreaks in patients with CF (3–5), and their multi-drug resistant phenotype has rendered many antibiotic regimens ineffective. B. cenocepacia is thought to be the most virulent species of the complex, and is associated with poor post-transplant survival in chronically infected patients leading to exclusion from life-saving lung transplant eligibility (6–10). In addition to playing an important role in CF, Burkholderia species (including B. cenocepacia and B. multivorans) are increasingly recognized as the causative agents of serious hospital acquired infections in non-CF patients (11–13). The inability of patients with and without CF to clear Burkholderia is a major factor in the disease course and is predicated on the inability of macrophages to kill ingested B. cenocepacia or B. multivorans (14). The vital role of macrophages in CF host-pathogen interactions has been highlighted recently by several groups (14–19). Although macrophage-mediated clearance of B. cenocepacia is defective in CF (20, 21), little is known regarding differences when compared to clearance of the less clinically virulent B. multivorans (8, 22, 23).
Intracellular killing of other pathogens besides Burkholderia is also defective in CF, despite abundant neutrophil-mediated production of reactive oxygen species (ROS) in lung tissue (24, 25). CF airways are characterized by constitutive ROS production that is dependent on both failed bacterial clearance and defective CFTR, and can be alleviated with autophagy stimulation (26). However, it remains unclear why intracellular pathogen killing in CF macrophages is defective, despite the abundant production of ROS in lung tissue (24, 25). Macrophages are more resistant to damage from ROS-induced oxidative stress compared to neutrophils and monocytes (27), therefore intracellular organisms may persist within macrophages even in environments of continuous host inflammatory production such as the CF lung, providing a potential replicative niche for Burkholderia.
In addition to CF macrophage deficits in host immune responses, Burkholderia can scavenge ROS (28) and affect key oxidase components (29, 30) in non-CF cells. Generation of ROS by assembly of the NADPH in macrophages in response to pathogens is a fundamental host defense strategy. Despite this, little is known about the impact of CFTR on NADPH assembly and activation in human macrophages and no studies have demonstrated an inherent defect in ROS production in human CF macrophages. In addition, how generation of an oxidative burst occurs in CF macrophages after initial contact with Burkholderia is not clear, including assembly of the NADPH oxidase complex and subsequent production and function of ROS, despite the fact that these macrophages do indeed ingest and harbor bacteria. In murine macrophages, B. cenocepacia delays association of the NADPH oxidase complex with macrophage vacuoles and can disrupt NADPH oxidase assembly, but these events have not been studied in human macrophages. Assembly of the NADPH oxidase requires translocation of phosphorylated p47phox, p40phox, p67phox, and Rac from the cytoplasm to the phagosomal membrane (31). B. cenocepacia specifically affects the activation of Rac in murine macrophages (30).
We undertook this study to more clearly characterize ROS production and assembly of the NADPH oxidase in human CF macrophages in response to an ROS agonist, Burkholderia and other stimuli, with hopes of identifying new targets for drug development against Burkholderia and other chronic infections in CF. We demonstrate for the first time that human CF macrophages inherently have a reduced oxidative burst in response to PMA. Ingestion of Burkholderia cenocepacia independently contributes to and worsens this defect in CF macrophages compared to non CF macrophages. Finally, CF macrophages demonstrate decreased expression of the protein kinase C (PKC) conventional subclass leading to decreased activation of NADPH oxidase components p47phox and p40phox in comparison to non-CF macrophages. Together, these results provide evidence for deficits in the CF macrophage oxidative burst due to decreased phosphorylation of NADPH oxidase cytosolic components and a subsequent reduction in NADPH oxidase activation in CF. These findings implicate a critical role for defective macrophage oxidative responses in persistent bacterial infections in patients with CF.
MATERIALS AND METHODS
Bacterial Strains
Macrophages were infected with RFP-expressing B. cenocepacia strain k56-2, B. multivorans strain FC-445, or Pseudomonas aeruginosa PA01 for 1–2 h prior to treatments based on known intracellular uptake time (14, 21, 32). The B. cenocepacia strain is representative of an epidemic clinical strain from the ET12 lineage (33). Bacteria were reproducibly grown in Luria-Bertani media over 24 h. For paraformaldehyde (PFA) experiments, pelleted bacteria were re-suspended in 4% PFA for 30 min and washed 5 times with DPBS to remove PFA.
Ethics statement
All human subjects were recruited as approved by the Institutional Review Board of Nationwide Children’s Hospital. All subjects underwent written consent for the procedures including all adult subjects provided informed consent, and a parent or guardian of any child participant provided informed consent on their behalf along with written assent from children.
Human macrophage isolation and bacterial infection
Heparinized blood was obtained from 35 CF and 35 non-CF healthy controls. Subjects were excluded with a history of chronic immunosuppression including chronic steroid use, CFTR modulator use, or history of transplantation. Chronic azithromycin use was allowed. Peripheral monocytes were separated from whole blood using Lymphocyte Separation Medium (Corning, 25-072-CV). Isolated monocytes were re-suspended in RPMI (Gibco, 22400-089) plus 10% human AB serum (Lonza, 14-490E) and differentiated for 5 days at 37°C into monocyte-derived macrophages (MDMs) (21, 34). MDMs were isolated, placed in monolayer culture, and infected at bacterial multiplicity of infection (MOI) ranging from 2–10 based on prior studies on the impact of bacterial infection on ROS production (14, 21, 32). The THP-1 monocytic line was used in preliminary experiments to optimize conditions prior to studies with human MDMs. THP-1 cells were grown in 10% fetal bovine serum (Thermo scientific) in RPMI. THP-1 cells were treated with 200nM PMA (Calbiochem) and 30ng/mL GM-CSF (R&D Systems, 415-ML-050) to differentiate cells into macrophages. Media was replenished with 30ng/mL GM-CSF the next day and the THP-1 derived macrophages were allowed to mature 5 days before experimentation. THP-1 cells were then treated with the CFTR inhibitor Inh-172 for 24 h prior to experimentation.
Immunoblotting
MDMs were infected at a MOI of 50 for 30 min for studies of ROS production. Supernatants were removed post treatment and the cells were washed twice with PBS (Fisher). The cells were lysed in lysis buffer (HEPES, MgCl, EGTA, KCL, NP-40) with protease inhibitor (Roche Applied Science, 10-519-978-001). Then, 30 ug of protein was separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Membranes were immunoblotted for calreticulin (Enzo Life Sciences, ADI-SPA-600), phospo-p40phox (Cell Signaling, 4311), total p40phox (Abcam, ab137691), phospho-p47phox (donated by Jamel El-Benna), total p47phox (Life Technologies, A16636), p67phox (Santa Cruz, SC-15342), Rac2 (Abcam, ab2244), gp91phox (Santa Cruz, SC-5827), p22phox (Santa Cruz, SC-20751),) p-PKC α/β II (Cell Signaling, 9375P), p-PKC δ (Cell Signaling, 9374P), and p-PKC ζ/λ (Cell Signaling, 9378P). Protein bands were detected with HRP-conjugated secondary antibodies and visualized using ECL reagents (Life Sciences, RPN2106). Membrane and cytosolic fractionations were prepared via manufacturer kit instructions (Thermo Fisher, #78840).
DCF assay
The oxidative burst was measured by a 2′,7′-dichlorofluorescein (DCF) assay, (Life Technologies, D399) using relative fluorescent units (RFUs). MDMs were adhered to 96 well plates at 4E6 cells/well in duplicate for 2 h, and repleted in Dulbecco’s PBS + 10 mM HEPES + 1 mg/ml human serum albumin + 0.1% glucose (DPBS-HHG). After 30 min incubation at 37°C, 10%DCF was added to the wells for 30 min at 37°C. A stimulus such as PMA was then added to macrophages and fluorescence was measured at a 485 Excitation wavelength and a 515 Emission wavelength every 2 min for 2 h. In preliminary experiments, PMA was used at varying concentrations, with 200µM determined to be the optimal concentration for DCF experiments. 1-Oleoyl-2-acetyl-sn-glycerol (OAG, Sigma) was used at a concentration of 20µM for PKC induction.
Cytochrome C assay
Superoxide production was measured by a cytochrome C (CytC, Sigma) assay using relative absorbance (35). MDMs were adhered to 24 well plates at 4E6 PBMC/ml, and repleted in DPBS-HHG. Cells were stimulated with PMA, bacteria (MOI 10), and/or medium-only control in duplicate wells in the presence of 80 µM CytC and 500 U/ml catalase (Sigma) at 37°C for 2 h. CytC reduction indicative of superoxide production was measured by subtracting the absorbance at 550 nm of control wells treated with 300 U/ml superoxide dismutase (SOD, Sigma, S5395) from test well values. Un-stimulated cell background values were subtracted from treated wells.
Confocal Microscopy
Two million MDMs were cultured on 12 mm glass cover slips in 24-well tissue culture plates and infected synchronously with bacteria at an MOI of 2–10. Nuclei were stained with the DAPI blue for imaging. MDMs were fluorescently labeled with either Phospho-p40phox (Cell Signaling, 4311) or phospho-p47phox (Assay Biotech, A1171). Confocal microscopy was performed using an Axiovert 200M inverted epifluorescence microscope equipped with the Apotome attachment for improved fluorescence resolution and an Axiocam MRM CCD camera (Carl Zeiss Inc., Thornwood, NY). At least 100 MDMs were scored for each condition. All experiments were performed in triplicate or quadruplicate.
Calcium flux
MDMs were re-suspended at 1E7 cells per mL in Hanks Buffered Salt Solution with Ca2+ and MgCl2 (HBSS, HyClone, SH 30268.02), 1% FBS (HyClone, SH30071.03, Thermo Scientific), and 4mM Probenecid (Invitrogen, P36400). MDMs were treated with 1:100 anti-human CD-16 Brilliant Violet 605TM (BioLegend, 302040) and 1:100 anti-human CD-14-APC-Cy7 (BioLegend, 325620) to identify macrophage populations. The MDMs were incubated at 37°C for 30 min with 4µg/mL fluorescent dye Fluo-3 AM (Life Technologies, F1242), washed twice and re-suspended in cell loading HBSS medium at 1E7 cells/mL. MDMs were stimulated with 1µg/mL Ionomycin (Sigma-Aldrich, 124222), 1µM Platelet Activating Factor (PAF, Sigma-Aldrich, P4904), bacteria (MOI 10) or PMA (Calbiochem, 524400). The intensity of intracellular Ca2+ in individual cells was assessed by flow cytometer (LSR II, BD) measuring the fluorescence emission of Fluo-3 in the FL-1 channel over 200s. Data were analyzed using FlowJo software (Tree Star. San Carlos, California).
Statistical Analysis
All statistical analyses were performed using GraphPad Prism software (version 6.0). Two sample t-tests or Mann-Whitney U tests were used for comparisons of independent samples. Paired t-tests or Wilcoxon signed-rank tests were used for within patient comparisons. Statistical significance was defined as P value <0.05. Age and gender matched healthy controls were used in the analysis.
RESULTS
Subject characteristics
Human donor characteristics are summarized in Table 1. There were more CF males (60.0%) compared to controls (34.3%) available for sampling, and both populations were entirely Caucasian. On average, CF patients had moderate lung disease with a mean FEV1 % predicted of 65.8 ± 24.7%. Most CF patients were pancreatic insufficient (83.3%) and had at least one copy of the F508del mutation (87.6%). Pseudomonas aeruginosa was the predominant respiratory pathogen isolated on the most recent respiratory cultures for the CF patients.
Table I.
Patient Demographics
| Non-CF (n=35) | CF (n=35) | |
|---|---|---|
| Mean age (years ± st. dev) | 34.7 ± 10.8 | 29.1 ± 10.1 |
| Males | 34.3% | 60.0% |
| Caucasian | 100.0% | 100.0% |
| Genotype | ||
| F508del homozygous | N/A | 54.3% |
| F508del heterozygous | N/A | 34.3% |
| Other | N/A | 11.4% |
| Pancreatic insufficiency | N/A | 83.3% |
| Mean FEV1 (% predicted)1 | N/A | 65.8 ± 24.7 |
| P. aeurginosa colonization | N/A | 80% |
| MRSA colonization1 | N/A | 37.1% |
Footnote:
. Abbreviations used: FEV1, Forced expiratory volume in 1 second; MRSA, methicillin-resistant Staphylococcus aureus
CF macrophages have an inherent deficit in NADPH oxidase activity
It is unclear if CF macrophages have basal deficits in oxidative killing since some studies have suggested functional ROS responses to pathogens such as P. aeruginosa (36), while others have demonstrated deficient ROS production (37). Importantly, a comprehensive assessment of human CF macrophage oxidase assembly has not been performed. Therefore, we isolated human MDMs from stable CF and non-CF donors and analyzed them for their basal capacity to produce an oxidative burst in response to PMA, an analog of diacylglycerol (DAG) and a soluble, intracellular activator of the NADPH oxidase via PKC. The oxidative burst was measured by a DCF assay using RFUs of cellular oxidation through hydrogen peroxide-mediated pathways. CF MDMs demonstrated a significant decrease in the oxidative burst in response to PMA compared to non-CF MDMs (p value = 0.002, Figures 1A, B). This finding was then confirmed with a second specific assay of superoxide production (cytochrome C assay). Superoxide production was measured in response to mock or PMA after 60 min stimulation by measuring the SOD inhibitable reduction of exogenous cytochrome C. CF MDMs demonstrated a nearly 60% reduction in superoxide production in response to PMA compared to non-CF MDMs (Figure 1C, 1D).
Figure 1. CF MDMs have reduced ROS production in response to PMA.
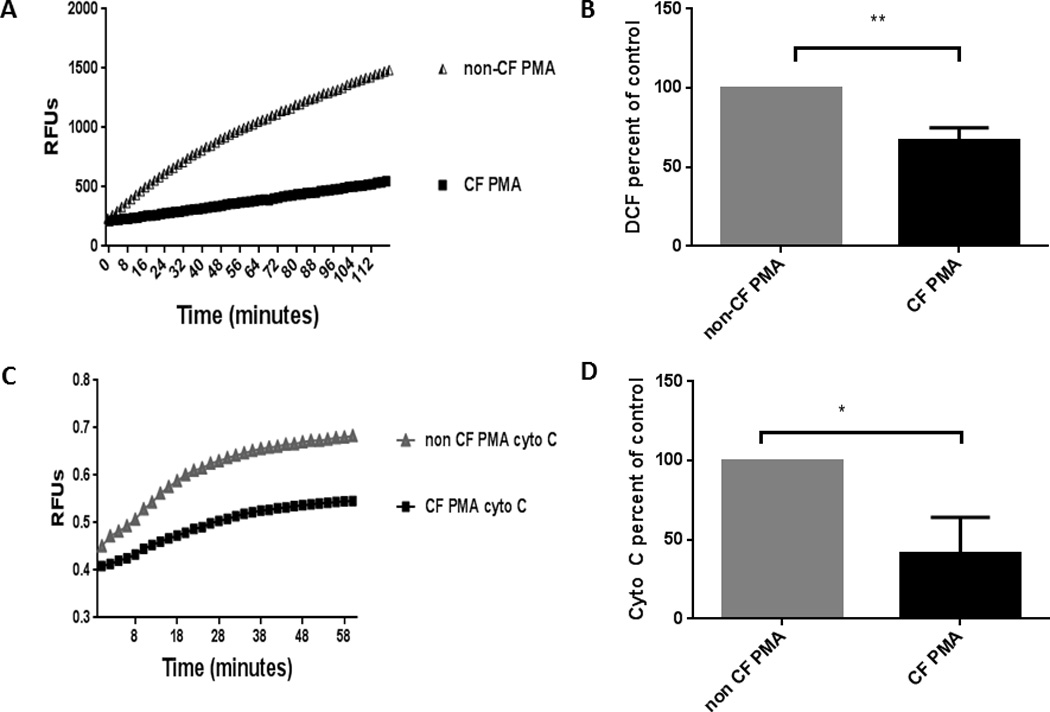
A) CF and non-CF MDMs were treated with PMA for 30 min and assessed for ROS production using RFUs via a DCF assay. Representative assay of n=6. B) Summed end-point analysis of 1A experiments expressed as %ROS production at 2 h for CF MDMs relative to control non-CF MDMs in response to PMA, P value = 0.002. C) Cells were treated with PMA and/or medium only control in triplicate wells in the presence of 500 U/ml catalase and 80 µM CytC. CytC reduction, indicative of superoxide production, was measured after 60 min of stimulation by subtracting the absorbance at 550 nm of control wells treated with 300 U/ml SOD from test well values. Unstimulated cell background values were subtracted from test conditions, and all values were set relative to the positive control. Representative experiment of n=3. D) Summed end-point analysis of 1C experiments, results are expressed as percentage of non-CF PMA-stimulated cells for three independent experiments, P value = 0.01.
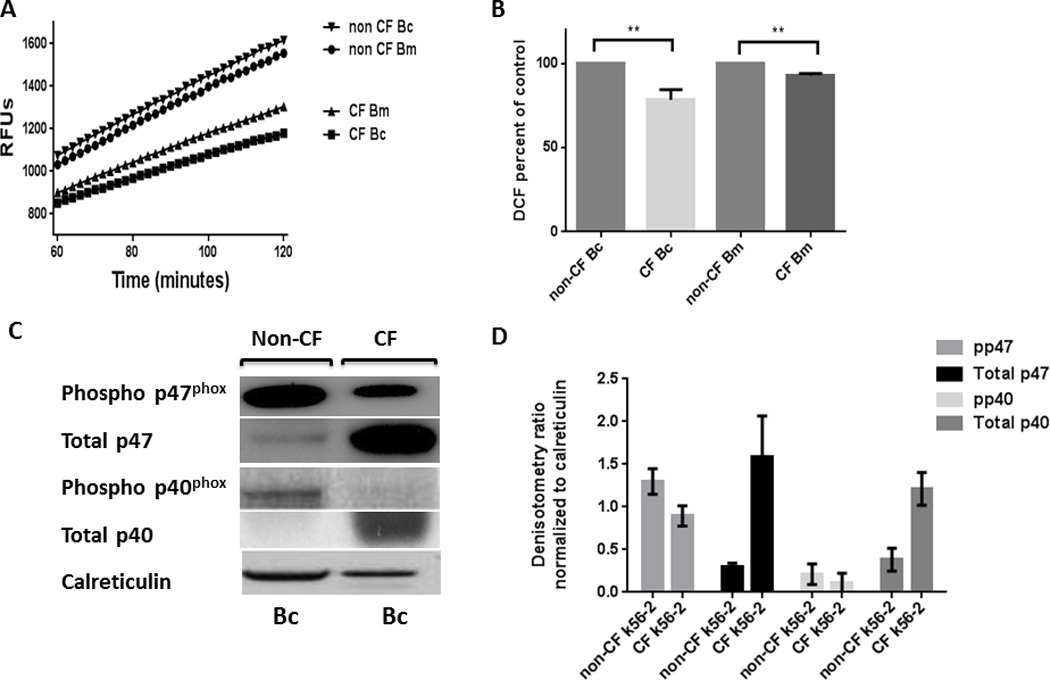
As a result of this observed difference in ROS generation, we next examined the amounts of essential NADPH oxidase proteins in cell lysates after 30 min exposure to PMA. Membrane component p22phox along with cytosolic components Rac2, total p40phox, and total p47phox were all increased in CF in response to PMA compared to non-CF MDMs (Figures 2A, B, C, D). Additionally all components except total p67phox and gp91phox were increased in untreated CF MDMs compared to non-CF (Figures 2A, B, C, D). We therefore next measured the expression of phosphorylated p40phox and p47phox, which are required to form an activated NADPH complex. In contrast to the total protein expression, there was a marked decrease in expression of phosphorylated p47phox and p40phox in CF in response to PMA compared to non-CF MDMs (Figures 2C, D). This finding was confirmed when examining the translocation of p47phox from the cytosolic to membrane-bound fraction. Expression of membrane-bound p47phox was decreased in CF compared to non-CF (Figure 3A). Additionally, phosphorylated p47phox expression was restored to non-CF levels in a subject receiving treatment with the CFTR modulator combination ivacaftor/lumacaftor (Figure 3B). Together, these results provide evidence for a deficit in the CF macrophage oxidative burst characterized by accumulation of total NADPH complex proteins, but decreased phosphorylation of key cytosolic components required for NADPH oxidase assembly.
Figure 2. Abnormal NADPH expression in CF.
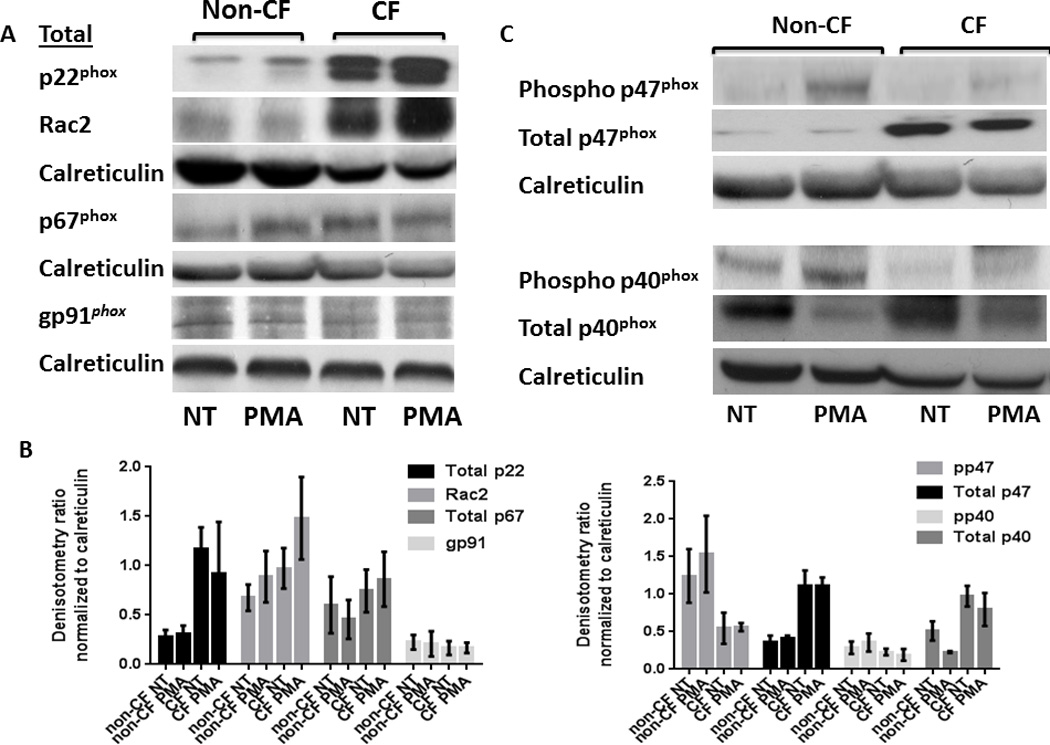
CF MDMs have increased total NADPH component proteins. A) Western blots of CF and non-CF MDM cell lysates for total amounts of NADPH components with/without 30 min treatment with PMA. Representative image of > 5 experiments. B) Densitometric analysis of ≥ 3 Western blots per condition in 2A, normalized to the loading control calreticulin. C) CF MDMs have decreased phosphorylation of cytosolic NADPH components. Western blots of CF and non-CF cell lysates for phosphorylated NADPH components after 30 min treatment with/without PMA. Representative image of > 5 experiments. D) Densitometric analysis of ≥ 3 Western blots per condition in 2C, normalized to the loading control calreticulin.
Figure 3. p47phox fails to translocate to plasma membranes in CF.
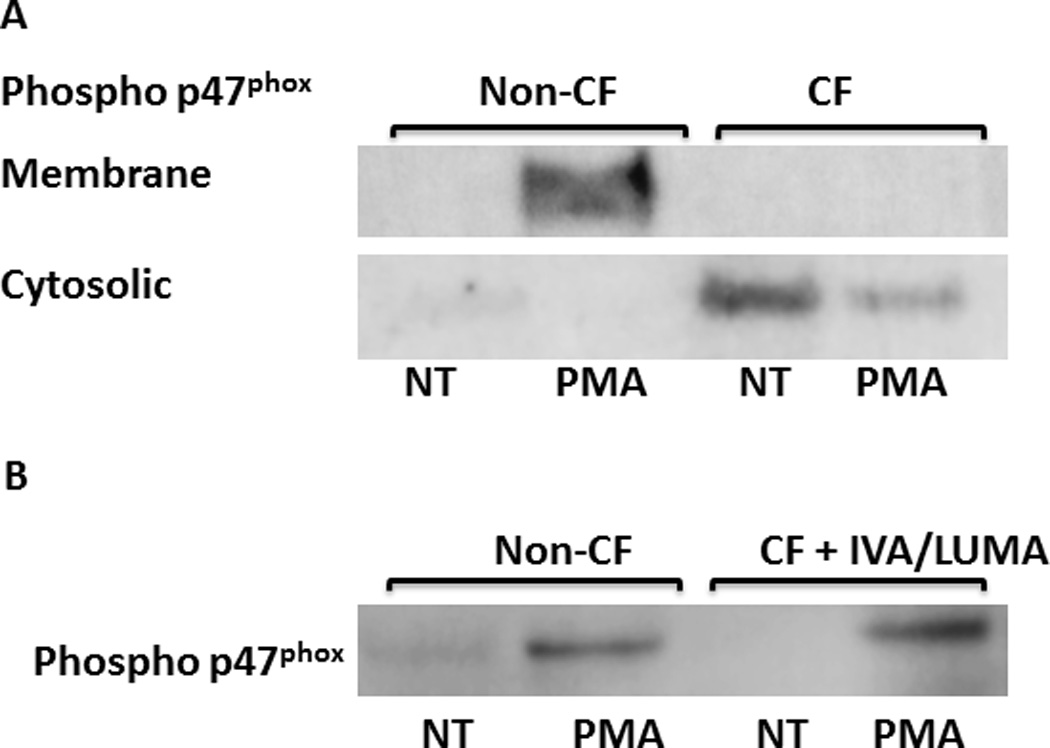
A) Western blots of CF and non-CF MDM cell lysates for phosphorylated p47phox in cytosolic and plasma membrane fractions, with/without 30 min treatment with PMA. Representative image of 4 experiments. B) Western blot of CF and non-CF cell lysates for phosphorylated p47phox after 30 min treatment with/without PMA. CF subject was receiving treatment with the CFTR modulator ivacaftor/lumacaftor, n=1.
Calcium-dependent PKC α/β expression is decreased in CF
PKC activation is a key upstream signaling event in activation of the NADPH oxidase. In human non-CF monocytes the PKC isoform delta (PKCδ) is required for p47phox phosphorylation and translocation to the cell membrane to enable NADPH activation (38); however PKC isoform activation has not been examined in CF, or specifically in human CF macrophages. There are three major classes of PKC isoforms (39). The conventional sub-class is made up of PKC-α (PRKCA), PKC-β1 (PRKCB), PKC-β2 (PRKCB), and PKC-γ (PRKCG). The novel sub-class consists of PKC-δ (PRKCD), PKC-δ1 (PRKD1), PKC-δ2 (PRKD2), PKC-δ3 (PRKD3), PKC-ε (PRKCE), PKC-η (PRKCH) and PKC-θ (PRKCQ). Finally, the atypical sub-class consists of PKC-ι (PRKCI), PKC-ζ (PRKCZ), PK-N1 (PKN1), PK-N2 (PKN2), PK-N3 (PKN3). Therefore we examined representative isoforms from each class in lysates from CF and non-CF MDMs at baseline and during PMA stimulation. CF MDMs specifically demonstrated decreased expression of phospho-PKC α/β compared to non-CF (Figures 4A, B), representing decreased expression of the conventional PKC sub-class. There was no difference in atypical or novel isoforms (Figures 4A, B). In order to determine the importance of the reduction in the PKC conventional isoform sub-class on subsequent ROS production, we incubated CF MDMs with the PKC conventional sub-class inducer OAG and measured the oxidative burst in response to PMA. CF MDMs demonstrated a significant increase in ROS production when pre-incubated with OAG in comparison to untreated CF MDMs (fold change 1.58 ± 0.35, p = 0.047, Figures 4C, D). Addition of OAG did not increase ROS production in non-CF macrophages compared to PMA alone (not shown).
Figure 4. PKC conventional subclass expression is decreased in CF MDMs.
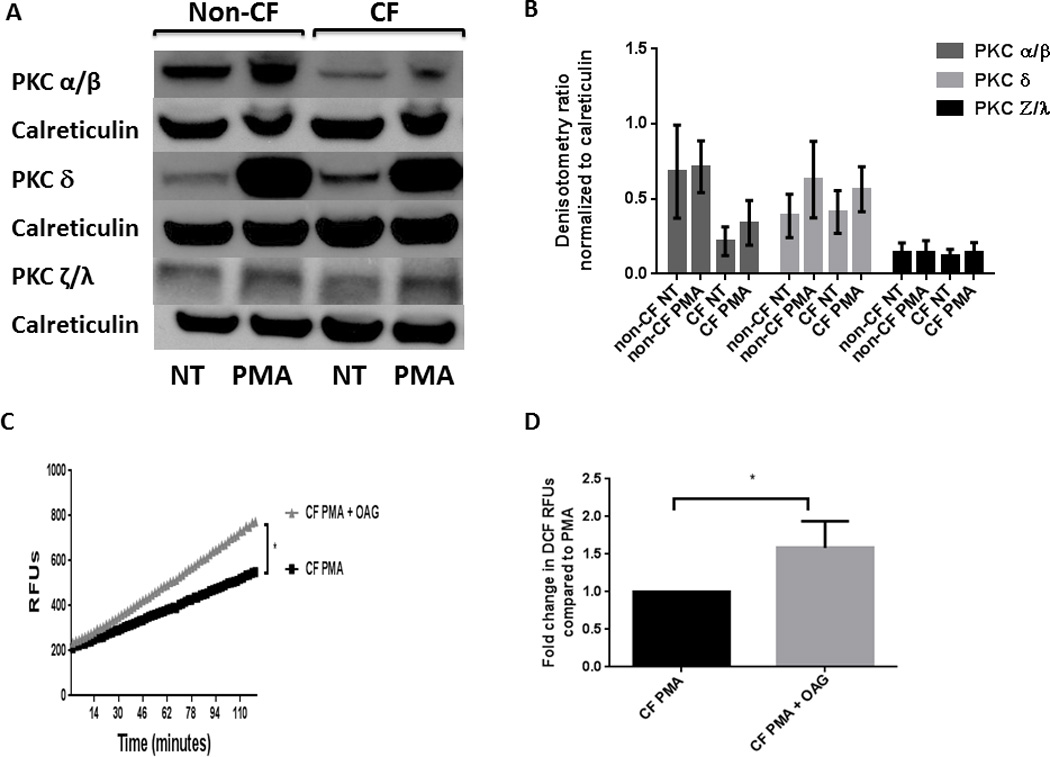
A) Expression of phosphorylated representatives from the three PKC subclasses (conventional: α/β, novel:δ, and atypical:ζ/λ) was determined by Western blotting of cell lysates from CF and non-CF MDMs after 30 min exposure to PMA. Representative image of 3 experiments. B) Densitometric analysis of 3 Western blots per condition in 3A, normalized to the loading control calreticulin. C) PKC conventional agonist OAG increases ROS production in CF. Representative image of DCF assay in CF MDMs stimulated with PMA alone, or PMA pre-incubated with OAG for 1 h. D) Summed end-point analysis at 2 h of the experiments in 3B; results are expressed as a percentage of CF PMA-stimulated cells for four independent experiments, P= 0.047.
Due to the observed differences in PKC isoform expression, we next examined for differences in PKC isoform activators. The conventional class requires DAG, calcium and phospholipid for activation, in contrast to the novel class which requires DAG but not calcium, and the atypical class which requires neither DAG nor calcium (39). Therefore, we measured calcium influx in MDMs during stimulation with known calcium activators (Ionomycin, PAF) as controls compared to the addition of PMA. There was no difference in calcium production in response to Ionomycin or PAF between CF and non-CF macrophages (Figure 5). However, there was significantly less calcium production in CF MDMs in response to PMA (p value = 0.03, Figures 5A, B). Taken together, these results indicate that CF macrophages have decreased activation of the oxidative burst due to decreased calcium-dependent PKC α/β activation.
Figure 5. Calcium influx is decreased in CF macrophages during PMA exposure.
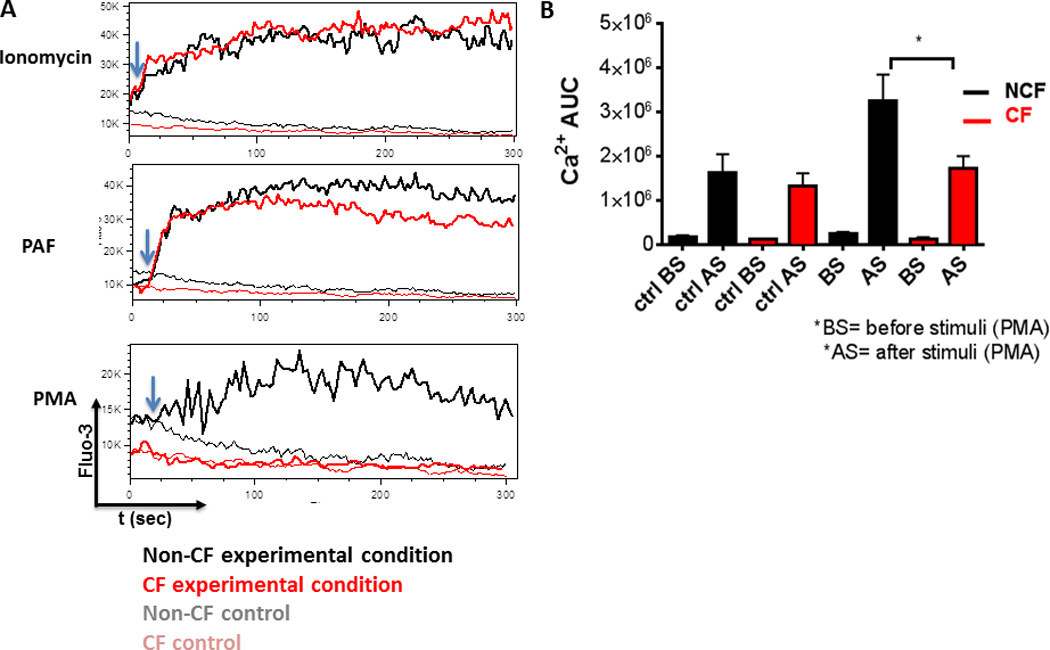
Macrophages were stimulated with either Ionomycin, PAF or PMA, and the increase in cytosolic Ca2+ in individual macrophages was assessed by flow cytometry measuring the fluorescence emission of Fluo-3 over 200 s. A) Representative images of responses to experimental agents and no treatment (control) in CF and non-CF MDMs. B) Summed analysis of calcium influx for PMA experiments in 4A. Influx calculated as the area under curve before and after PMA stimulus, n=3, P value = 0.030. Not shown are Ionomycin P value = 0.59 and PAF P value =0.35.
Live B. cenocepacia further reduces the oxidative burst in CF
Members of the Burkholderia cepacia complex can affect NADPH oxidase assembly (30, 40) in murine models and scavenge ROS (29), and have been shown to cause increased virulence in CF through avoidance of macrophage killing (14, 21). However, it is unknown how these members affect human CF macrophage oxidative responses, or if there are differential responses between the most predominant strains affecting patients with CF: B. cenocepacia and B. multivorans. Therefore, we next asked if ROS responses are further suppressed in CF macrophages with the addition of Burkholderia species. Human MDMs were infected with B. cenocepacia clinical isolate strain k56-2 and B. multivorans clinical isolate strain FC-445 at MOIs between 2 and 50. CF MDMs demonstrated a significantly reduced oxidative burst in response to B. cenocepacia, and to a lesser extent B. multivorans, compared to non-CF MDMs (B. cenocepacia p value =0.005, B. multivorans p value =0.008, Figures 6A, 6B). Although Burkholderia can delay the onset of the oxidative burst in murine models (40), we observed a significant reduction in ROS production in MDMs at early time points and this persisted over a 6 hour infection (6 hour not shown).
Figure 6. Burkholderia species further reduce ROS production in CF MDMs.
A) CF MDMs have reduced ROS production as measured by the DCF assay in response to 30 min infection with Burkholderia cenocepacia (Bc) and Burkholderia multivorans (Bm) compared to non-CF, representative image of n=6. B) Summed end-point analysis of 5A experiments, results are expressed as a percentage of non-CF bacteria-stimulated MDMs for six independent experiments. Bc P value =0.005, Bm P value =0.008, n=6. C) CF MDMs have reduced phosphorylation of p40phox and p47phox during Bc infection. Western blots of cell lysates for phosphorylated and total NADPH components after 30 min infection with Bc. Representative image of > 3 experiments. D) Densitometric analysis of 3 Western blots per condition in 5C, normalized to the loading control calreticulin.
Due to the previously noted differences in p40phox and p47phox phosphorylation in CF macrophages in response to PMA, we examined phosphorylation during B. cenocepacia infection. MDMs were infected with B. cenocepacia for 30 min, cell lysates collected, and Western blotting performed for total and phosphorylated p40phox and p47phox. CF MDMs demonstrated decreased phosphorylation of p40phox and p47phox and increased total p40phox and p47phox during B. cenocepacia infection in comparison to non-CF MDMs (Figures 6C, D). These findings are consistent with untreated and PMA-stimulated MDMs findings as shown in Figure 2. This finding was further assessed using confocal microscopy. Decreased co-localization of phosphorylated p40phox and p47phox with B. cenocepacia phagosomes was observed in CF MDMs in comparison to non-CF (Figures 7A, B, C). Additionally, confocal microscopy demonstrated a 25.0% reduction in k56-2 co-localization with p47phox (p < 0.0001) and a 24.3% co-localization reduction of p40phox in CF MDMs compared to non-CF (p = 0.0001). Overall, we interpret the further decrease in the oxidative burst with Burkholderia in CF MDMs to indicate an additive effect with the inherent defect on ROS inhibition in these cells.
Figure 7. CF MDMs have decreased co-localization with phospho-p47phox.
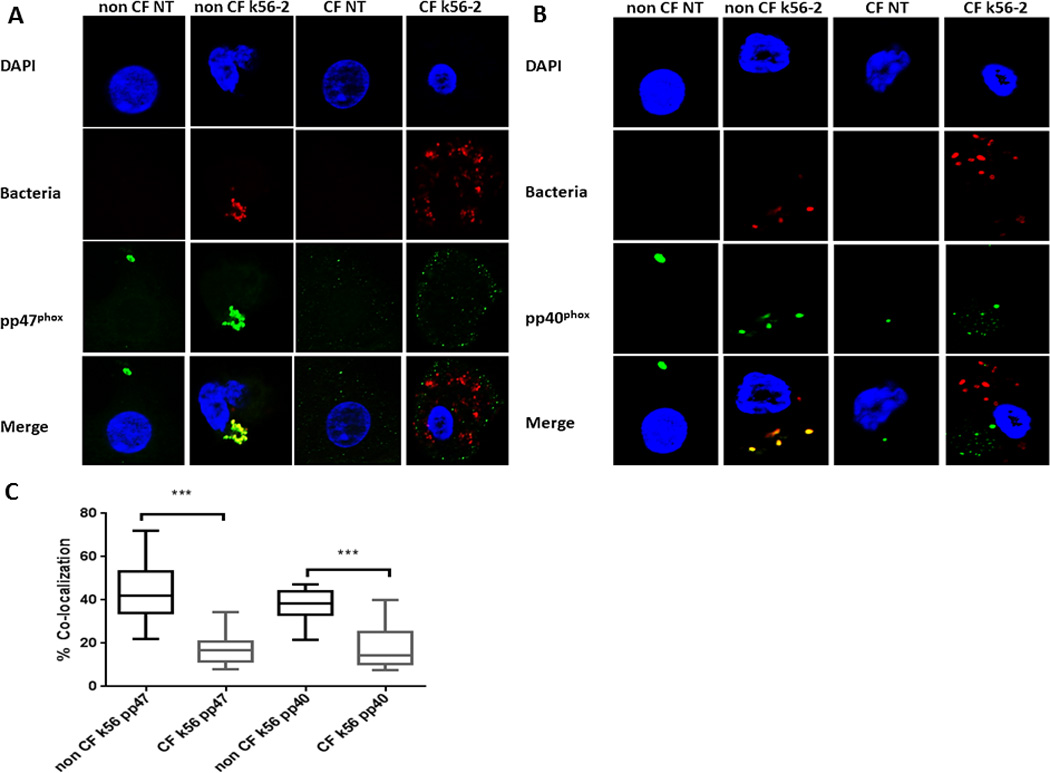
A) Confocal microscopy images of CF and non-CF MDMs with and without infection with B. cenocepacia for 30 min. The macrophage nucleus is stained blue with DAPI, B. cenocepacia (k56-2) is shown in red, phosphorylated p47phox is shown in green, and bacteria co-localized with p47phox are yellow in the merged image. N=3. B) CF MDMs have decreased co-localization with phospho-p40phox. Confocal microscopy images of CF and non-CF MDMs with and without infection with B. cenocepacia. The macrophage nucleus is stained blue with DAPI, B. cenocepacia (k56-2) is shown in red, phosphorylated p40phox is shown in green, and bacteria co-localized with p40phox are yellow in the merged image. N=3. C) Summed scoring of bacterial co-localization from 6A and 6B. 100 MDMs scored per condition. p47phox P value < 0.0001, p40phox P value = 0.001.
In order to determine if the reduced ROS production in response to B. cenocepacia was dependent on a live bacterial factor versus a viability independent factor, CF MDMs were incubated with PFA killed B. cenocepacia and production of ROS compared to that of live bacteria. There was an increased oxidative burst in response to PFA killed B. cenocepacia in both CF and non CF MDMs in comparison to live B. cenocepacia for both host cell types, however CF MDMs had a significantly lower response compared to non-CF MDMs (Figures 8A, B). Next, the ability of B. cenocepacia to suppress the oxidative burst in response to a secondary stimulus was tested, as might occur in a patient with CF facing multiple pathogens after infection with B. cenocepacia. Both CF and non-CF MDMs had a reduced response to PMA when infected with B. cenocepacia one h prior to PMA stimulus (Figures 8C, D). Taken together, these data suggest that a factor produced from live bacteria is at least partially responsible for B. cenocepacia’s suppression of ROS responses, which are additive in CF MDMs with basal deficits in ROS generation, but can also suppress secondary responses in non-CF MDMs.
Figure 8. Impact of bacterial viability, antecedent bacterial infection, and bacterial spp. on ROS production.
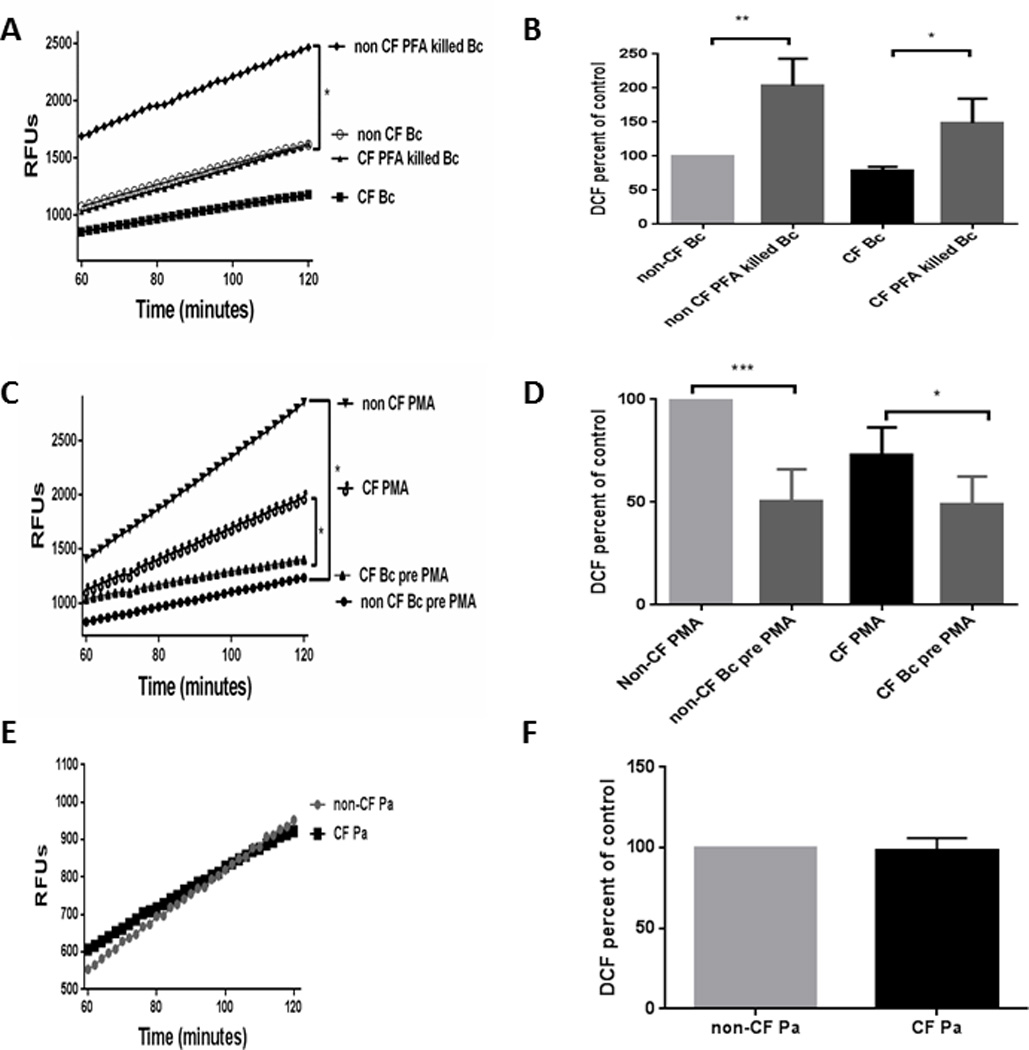
A) Increased ROS production as measured by the DCF assay is observed in response to paraformaldehyde killed Bc in comparison to live Bc in CF (P value = 0.028) and non-CF MDM s (P value = 0.006) n=3. B) Summed end-point analysis of 8A experiments, results are expressed as % ROS production at 2h of CF MDMs relative to non-CF MDMs with B. cenocepacia. C) Burkholderia species decrease ROS production as measured by the DCF assay in response to PMA in both CF (P value = 0.05) and non-CF MDMs (P value = 0.0003) when infected for 1 h prior to PMA exposure. n=4. D) Summed end-point analysis of 8C experiments, results are expressed as % ROS production at 2h of CF MDMs relative to non-CF MDMs with PMA. E) CF and non CF MDMs have equal ROS production in response to 30 min infection with Pseudomonas aeruginosa (Pa), as measured by the DCF assay. P value = 0.70, n=3. F) Summed end-point analysis of 8E experiments, results are expressed as % ROS production at 2h of CF MDMs relative to non-CF MDMs with P. aeruginosa.
Finally, we examined the oxidative burst in response to another common CF pathogen, P. aeruginosa, which has been previously shown to not affect the respiratory burst in CF macrophages (36). There was no difference in ROS production in response to P. aeruginosa between CF and non-CF MDMs (Figures 8E, F), confirming previous findings. When combined with the B. cenocepacia and PMA data, this would suggest that CF MDMs have deficits in intracellular PKC-mediated activation of the oxidative burst in response to certain pathogenic stimuli, which may explain differential handling of pathogens in CF.
Discussion
In recent years we have gained improved understanding of underlying deficits in the host immune responses of patients with CF beyond a previous emphasis on continued neutrophil overproduction in the lung (14, 41–45). Despite this increased knowledge and recent advances in CF care including CFTR modulators (46, 47), patients with CF remain burdened by chronic, multi-drug resistant bacterial infections. With a continued dearth in the development of novel antimicrobials (48) combined with increasing antibiotic resistance worldwide (49, 50), it remains more critical than ever to determine how bacteria avoid host immune defenses in CF in order to generate new approaches to therapy. To this end, we have discovered a novel defective pathway in CF macrophages independent of pathogens involving calcium-dependent PKC activation of the oxidative burst. This deficit in macrophage oxidative killing is further exaggerated by specific bacteria such as B. cenocepacia, which may in part explain the increased prevalence of Burkholderia infections in CF.
The generation of ROS in CF has been well studied in CF neutrophils, but remains less characterized in macrophages, where intracellular bacteria may reside and avoid host defenses. While CF neutrophils have been shown to have adequate ROS production (51), this has not been fully explored in the setting of chronic airway infections where non-CF biofilm-entrapped neutrophils demonstrate an ineffective oxidative burst (52), and CF sputum neutrophils demonstrate reduced ROS capacity compared to blood neutrophils, highlighting potential differences in activated versus basal states and differences in tissue compartments (53). Human CF MDMs have a normal ROS response to P. aeruginosa (18) which we have confirmed, but characterization of human macrophage NADPH assembly has not been performed in CF. Previously murine CF alveolar macrophages were shown to sequester gp91phox in ceramide-containing platforms, preventing the release of ROS (37). We found that accumulation of multiple NADPH components occurred in CF macrophages, but not gp91phox. This finding implicates the involvement of multiple NADPH components in human CF macrophage dysfunction, and not just specifically gp91phox, further highlighting differences from murine studies.
Importantly, despite the increased presence of NADPH components, there was decreased phosphorylation of p40phox and p47phox, resulting in defective assembly of an activated NADPH complex. While p40phox phosphorylation is not required for oxidase activation by PMA in neutrophils from patients with chronic granulomatous disease where known abnormalities in NADPH assembly occur (54, 55), assembly defects in CF macrophages involving this component may be different. The phosphorylation deficit was predicated upon a decrease in calcium-dependent PKC α/β expression. This result is in contrast to non-CF monocytes where PKCδ is required for p47phox phosphorylation (38), but correlates with a recent study showing restoration of CF macrophage microbicidal functions with OAG-mediated calcium signaling (56). Additionally, conventional PKC expression is implicated in macrophage control of intracellular pathogens such as Mycobacterium tuberculosis (57) and we now demonstrate PKC to be deficient in the handling of B. cenocepacia infection in CF MDMs, lending support to the notion of dysregulated PKC control of intracellular pathogens in CF. Our findings are consistent with an early study that showed chemiluminescent defects in PKC-mediated actions in CF neutrophils (58). p47phox contains 8 phosphorylation sites and determining which of these may be involved will be the subject of further research. Importantly, deficits in p47phox phosphorylation were reversed in a patient with CF receiving treatment with the CFTR modulator combination Ivacaftor/Lumacaftor, demonstrating the importance of functional CFTR in NADPH assembly.
B. cenocepacia further exaggerated the reduction in ROS production seen in CF macrophages. B. cenocepacia can down-regulate ROS production when grown in biofilm culture only (59), and delay association of the NADPH oxidase complex within murine macrophage vacuoles (40), but B. cenocepacia-induced ROS production in human CF macrophages has not been studied previously. Additionally, we observed an early and persistent decrease in ROS production in CF macrophages rather than a time-delayed increased in ROS in macrophages as was shown in one murine study. Furthermore, we found that Rac2 protein expression is elevated in CF macrophages independent of Burkholderia infection, emphasizing differences compared to a murine study involving Rac1 (30). However, there are multiple signaling pathways for Rac activation which will require further examination in the context of B. cenocepacia infection in humans with CF.
There have been no macrophage studies (CF or otherwise) regarding B. multivorans and the oxidative burst. B. multivorans demonstrated a decreased impact on ROS generation in CF macrophages compared to B. cenocepacia, which may explain the differences in virulence observed clinically between the two species. B. cenocepacia was able to decrease subsequent ROS production to a secondary stimulus in both CF and non-CF macrophages, highlighting a potential connection between CF and chronic granulomatous disease (60), both with defective ROS and an unusual tendency for B. cenocepacia infection compared to immunocompetent hosts.
Both CF and non-CF macrophages demonstrated increased ROS to PFA-killed B. cenocepacia, suggesting that exposed cell wall components of B. cenocepacia are at most only partly responsible for the active subversion of macrophage ROS responses. The exact identity of a viability-associated factor in B. cenocepacia that is responsible for decreased ROS production is not known at this time, but will be an area of continued research. Exopolysaccharides from B. cenocepacia have been shown to scavenge neutrophil ROS (29), as well as the ability of B. cenocepacia to down-regulate the tricarboxylic acid cycle when grown in a biofilm (59), both of which may provide future areas to target.
In summary, human CF macrophages have an inherent reduction in ROS production that is worsened by B. cenocepacia. This deficit is caused by a calcium-dependent decrease in expression of the PKC conventional subclass α/β leading to decreased activation of the NADPH oxidase in comparison to non-CF macrophages. These findings implicate a critical role for defective macrophage oxidative responses in persistent bacterial infections in patients with CF.
Acknowledgments
The authors kindly thank Dr. Jamel El-Benna for providing the anti-phospho-p47 antibody. The authors thank Dr. Jane Burns at the CF Isolate Core at Seattle Children’s Research Institute for provision of the B. multivorans strain (NIH P30 DK089507) and Dr. Miguel Valvano for B. cenocepacia strains.
The study was supported by CTSA grant UL1TR001070, NIH 1K08AI108792-01A1, and Cystic Fibrosis Foundation grant KOPP14I0.
Abbreviations used
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane regulator
- DAG
diacylglycerol
- DCF
2′,7′-dichlorofluorescein
- MDM
monocyte-derived macrophages
- MOI
multiplicity of infection
- OAG
1-Oleoyl-2-acetyl-sn-glycerol
- PAF
Platelet Activating Factor
- PFA
paraformaldehyde
- PKC
protein kinase C
- RFU
relative fluorescent units
- ROS
reactive oxygen species
REFERENCES
- 1.Collawn JF, Matalon S. CFTR and lung homeostasis. Am J Physiol Lung Cell Mol Physiol. 2014;307:L917–L923. doi: 10.1152/ajplung.00326.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sherrard LJ, Tunney MM, Elborn JS. Antimicrobial resistance in the respiratory microbiota of people with cystic fibrosis. Lancet. 2014;384:703–713. doi: 10.1016/S0140-6736(14)61137-5. [DOI] [PubMed] [Google Scholar]
- 3.Govan JR, Brown PH, Maddison J, Doherty CJ, Nelson JW, Dodd M, Greening AP, Webb AK. Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis. Lancet. 1993;342:15–19. doi: 10.1016/0140-6736(93)91881-l. [DOI] [PubMed] [Google Scholar]
- 4.Sun L, Jiang RZ, Steinbach S, Holmes A, Campanelli C, Forstner J, Sajjan U, Tan Y, Riley M, Goldstein R. The emergence of a highly transmissible lineage of cbl+ Pseudomonas (Burkholderia) cepacia causing CF centre epidemics in North America and Britain. Nat Med. 1995;1:661–666. doi: 10.1038/nm0795-661. [DOI] [PubMed] [Google Scholar]
- 5.Walsh NM, Casano AA, Manangan LP, Sinkowitz-Cochran RL, Jarvis WR. Risk factors for Burkholderia cepacia complex colonization and infection among patients with cystic fibrosis. J Pediatr. 2002;141:512–517. doi: 10.1067/mpd.2002.127665. [DOI] [PubMed] [Google Scholar]
- 6.Alexander BD, Petzold EW, Reller LB, Palmer SM, Davis RD, Woods CW, Lipuma JJ. Survival after lung transplantation of cystic fibrosis patients infected with Burkholderia cepacia complex. Am J Transplant. 2008;8:1025–1030. doi: 10.1111/j.1600-6143.2008.02186.x. [DOI] [PubMed] [Google Scholar]
- 7.De Soyza A, Meachery G, Hester KL, Nicholson A, Parry G, Tocewicz K, Pillay T, Clark S, Lordan JL, Schueler S, Fisher AJ, Dark JH, Gould FK, Corris PA. Lung transplantation for patients with cystic fibrosis and Burkholderia cepacia complex infection: a single-center experience. J Heart Lung Transplant. 2010;29:1395–1404. doi: 10.1016/j.healun.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 8.Murray S, Charbeneau J, Marshall BC, LiPuma JJ. Impact of burkholderia infection on lung transplantation in cystic fibrosis. Am J Respir Crit Care Med. 2008;178:363–371. doi: 10.1164/rccm.200712-1834OC. [DOI] [PubMed] [Google Scholar]
- 9.Olland A, Falcoz PE, Kessler R, Massard G. Should cystic fibrosis patients infected with Burkholderia cepacia complex be listed for lung transplantation? Interact Cardiovasc Thorac Surg. 2011 doi: 10.1510/icvts.2011.271874. [DOI] [PubMed] [Google Scholar]
- 10.Egan TM, Detterbeck FC, Mill MR, Bleiweis MS, Aris R, Paradowski L, Retsch-Bogart G, Mueller BS. Long term results of lung transplantation for cystic fibrosis. Eur J Cardiothorac Surg. 2002;22:602–609. doi: 10.1016/s1010-7940(02)00376-7. [DOI] [PubMed] [Google Scholar]
- 11.Hanulik V, Webber MA, Chroma M, Uvizl R, Holy O, Whitehead RN, Baugh S, Matouskova I, Kolar M. An outbreak of Burkholderia multivorans beyond cystic fibrosis patients. J Hosp Infect. 2013;84:248–251. doi: 10.1016/j.jhin.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Satpute MG, Telang NV, Dhakephalkar PK, Niphadkar KB, Joshi SG. Isolation of Burkholderia cenocepacia J 2315 from non-cystic fibrosis pediatric patients in India. Am J Infect Control. 2011;39:e21–e23. doi: 10.1016/j.ajic.2010.10.034. [DOI] [PubMed] [Google Scholar]
- 13.Mann T, Ben-David D, Zlotkin A, Shachar D, Keller N, Toren A, Nagler A, Smollan G, Barzilai A, Rahav G. An outbreak of Burkholderia cenocepacia bacteremia in immunocompromised oncology patients. Infection. 2010;38:187–194. doi: 10.1007/s15010-010-0017-0. [DOI] [PubMed] [Google Scholar]
- 14.Assani K, Tazi MF, Amer AO, Kopp BT. IFN-gamma stimulates autophagy-mediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PLoS One. 2014;9:e96681. doi: 10.1371/journal.pone.0096681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brennan S, Sly PD, Gangell CL, Sturges N, Winfield K, Wikstrom M, Gard S, Upham JW. Alveolar macrophages and CC chemokines are increased in children with cystic fibrosis. Eur Respir J. 2009;34:655–661. doi: 10.1183/09031936.00178508. [DOI] [PubMed] [Google Scholar]
- 16.Bruscia EM, Zhang PX, Ferreira E, Caputo C, Emerson JW, Tuck D, Krause DS, Egan ME. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator−/− mice. Am J Respir Cell Mol Biol. 2009;40:295–304. doi: 10.1165/rcmb.2008-0170OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruscia EM, Zhang PX, Satoh A, Caputo C, Medzhitov R, Shenoy A, Egan ME, Krause DS. Abnormal trafficking and degradation of TLR4 underlie the elevated inflammatory response in cystic fibrosis. J Immunol. 2011;186:6990–6998. doi: 10.4049/jimmunol.1100396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Del Porto P, Cifani N, Guarnieri S, Di Domenico EG, Mariggio MA, Spadaro F, Guglietta S, Anile M, Venuta F, Quattrucci S, Ascenzioni F. Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS One. 2011;6:e19970. doi: 10.1371/journal.pone.0019970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartl D, Gaggar A, Bruscia E, Hector A, Marcos V, Jung A, Greene C, McElvaney G, Mall M, Doring G. Innate immunity in cystic fibrosis lung disease. J Cyst Fibros. 2012;11:363–382. doi: 10.1016/j.jcf.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 20.Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DH, Newland C, Rosales-Reyes R, Kopp B, McCoy K, Montione R, Schlesinger LS, Gavrilin MA, Wewers MD, Valvano MA, Amer AO. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy. 2011;7:1359–1370. doi: 10.4161/auto.7.11.17660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopp BT, Abdulrahman BA, Khweek AA, Kumar SB, Akhter A, Montione R, Tazi MF, Caution K, McCoy K, Amer AO. Exaggerated inflammatory responses mediated by Burkholderia cenocepacia in human macrophages derived from Cystic fibrosis patients. Biochem Biophys Res Commun. 2012;424:221–227. doi: 10.1016/j.bbrc.2012.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones AM, Dodd ME, Govan JR, Barcus V, Doherty CJ, Morris J, Webb AK. Burkholderia cenocepacia and Burkholderia multivorans: influence on survival in cystic fibrosis. Thorax. 2004;59:948–951. doi: 10.1136/thx.2003.017210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cystic Fibrosis Foundation Patient Registry. 2012 Annual Data Report to the Center Directors. Bethesda, Maryland: ©2013 Cystic Fibrosis Foundation; [Google Scholar]
- 24.Pongnimitprasert N, El-Benna J, Foglietti MJ, Gougerot-Pocidalo MA, Bernard M, Braut-Boucher F. Potential role of the "NADPH oxidases" (NOX/DUOX) family in cystic fibrosis. Annales de biologie clinique. 2008;66:621–629. doi: 10.1684/abc.2008.0285. [DOI] [PubMed] [Google Scholar]
- 25.Cohen TS, Prince A. Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat Med. 2012;18:509–519. doi: 10.1038/nm.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, Gavina M, Pulze L, Giardino I, Pettoello-Mantovani M, D'Apolito M, Guido S, Masliah E, Spencer B, Quaratino S, Raia V, Ballabio A, Maiuri L. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol. 2010;12:863–875. doi: 10.1038/ncb2090. [DOI] [PubMed] [Google Scholar]
- 27.Bauer M, Goldstein M, Christmann M, Becker H, Heylmann D, Kaina B. Human monocytes are severely impaired in base and DNA double-strand break repair that renders them vulnerable to oxidative stress. Proc Natl Acad Sci U S A. 2011;108:21105–21110. doi: 10.1073/pnas.1111919109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keith KE, Killip L, He P, Moran GR, Valvano MA. Burkholderia cenocepacia C5424 produces a pigment with antioxidant properties using a homogentisate intermediate. J Bacteriol. 2007;189:9057–9065. doi: 10.1128/JB.00436-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bylund J, Burgess LA, Cescutti P, Ernst RK, Speert DP. Exopolysaccharides from Burkholderia cenocepacia inhibit neutrophil chemotaxis and scavenge reactive oxygen species. J Biol Chem. 2006;281:2526–2532. doi: 10.1074/jbc.M510692200. [DOI] [PubMed] [Google Scholar]
- 30.Rosales-Reyes R, Skeldon AM, Aubert DF, Valvano MA. The Type VI secretion system of Burkholderia cenocepacia targets multiple Rho family GTPases disrupting the actin cytoskeleton and the assembly of NADPH oxidase complex in macrophages. Cell Microbiol. 2011 doi: 10.1111/j.1462-5822.2011.01716.x. [DOI] [PubMed] [Google Scholar]
- 31.Appelberg R. Neutrophils and intracellular pathogens: beyond phagocytosis and killing. Trends Microbiol. 2007;15:87–92. doi: 10.1016/j.tim.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 32.Kotrange S, Kopp B, Akhter A, Abdelaziz D, Abu Khweek A, Caution K, Abdulrahman B, Wewers MD, McCoy K, Marsh C, Loutet SA, Ortega X, Valvano MA, Amer AO. Burkholderia cenocepacia O polysaccharide chain contributes to caspase-1-dependent IL-1beta production in macrophages. J Leukoc Biol. 2011;89:481–488. doi: 10.1189/jlb.0910513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahenthiralingam E, Coenye T, Chung JW, Speert DP, Govan JR, Taylor P, Vandamme P. Diagnostically and experimentally useful panel of strains from the Burkholderia cepacia complex. J Clin Microbiol. 2000;38:910–913. doi: 10.1128/jcm.38.2.910-913.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schlesinger LS. Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J Immunol. 1993;150:2920–2930. [PubMed] [Google Scholar]
- 35.Crowther JE, Kutala VK, Kuppusamy P, Ferguson JS, Beharka AA, Zweier JL, McCormack FX, Schlesinger LS. Pulmonary surfactant protein a inhibits macrophage reactive oxygen intermediate production in response to stimuli by reducing NADPH oxidase activity. J Immunol. 2004;172:6866–6874. doi: 10.4049/jimmunol.172.11.6866. [DOI] [PubMed] [Google Scholar]
- 36.Cifani N, Pompili B, Anile M, Patella M, Diso D, Venuta F, Cimino G, Quattrucci S, Di Domenico EG, Ascenzioni F, Del Porto P. Reactive-oxygen-species-mediated P. aeruginosa killing is functional in human cystic fibrosis macrophages. PLoS One. 2013;8:e71717. doi: 10.1371/journal.pone.0071717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Li X, Grassme H, Doring G, Gulbins E. Alterations in ceramide concentration and pH determine the release of reactive oxygen species by Cftr-deficient macrophages on infection. J Immunol. 2010;184:5104–5111. doi: 10.4049/jimmunol.0902851. [DOI] [PubMed] [Google Scholar]
- 38.Bey EA, Xu B, Bhattacharjee A, Oldfield CM, Zhao X, Li Q, Subbulakshmi V, Feldman GM, Wientjes FB, Cathcart MK. Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J Immunol. 2004;173:5730–5738. doi: 10.4049/jimmunol.173.9.5730. [DOI] [PubMed] [Google Scholar]
- 39.Baier G, Wagner J. PKC inhibitors: potential in T cell-dependent immune diseases. Curr Opin Cell Biol. 2009;21:262–267. doi: 10.1016/j.ceb.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 40.Keith KE, Hynes DW, Sholdice JE, Valvano MA. Delayed association of the NADPH oxidase complex with macrophage vacuoles containing the opportunistic pathogen Burkholderia cenocepacia. Microbiology (Reading, England) 2009;155:1004–1015. doi: 10.1099/mic.0.026781-0. [DOI] [PubMed] [Google Scholar]
- 41.Yonker LM, Cigana C, Hurley BP, Bragonzi A. Host-pathogen interplay in the respiratory environment of cystic fibrosis. J Cyst Fibros. 2015;14:431–439. doi: 10.1016/j.jcf.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peters-Hall JR, Brown KJ, Pillai DK, Tomney A, Garvin LM, Wu X, Rose MC. Quantitative proteomics reveals an altered cystic fibrosis in vitro bronchial epithelial secretome. Am J Respir Cell Mol Biol. 2015;53:22–32. doi: 10.1165/rcmb.2014-0256RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang AC, Turvey SE, Alves MP, Regamey N, Tummler B, Hartl D. Current concepts: host-pathogen interactions in cystic fibrosis airways disease. European respiratory review : an official journal of the European Respiratory Society. 2014;23:320–332. doi: 10.1183/09059180.00006113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rieber N, Hector A, Carevic M, Hartl D. Current concepts of immune dysregulation in cystic fibrosis. The international journal of biochemistry & cell biology. 2014;52:108–112. doi: 10.1016/j.biocel.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 45.Keiser NW, Birket SE, Evans IA, Tyler SR, Crooke AK, Sun X, Zhou W, Nellis JR, Stroebele EK, Chu KK, Tearney GJ, Stevens MJ, Harris JK, Rowe SM, Engelhardt JF. Defective Innate Immunity and Hyper-Inflammation in Newborn CFTR-Knockout Ferret Lungs. Am J Respir Cell Mol Biol. 2014 doi: 10.1165/rcmb.2014-0250OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, Konstan MW, McColley SA, McCoy K, McKone EF, Munck A, Ratjen F, Rowe SM, Waltz D, Boyle MP. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. The New England journal of medicine. 2015;373:220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordonez C, Elborn JS. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. The New England journal of medicine. 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finch R. Regulatory opportunities to encourage technology solutions to antibacterial drug resistance. J Antimicrob Chemother. 2011;66:1945–1947. doi: 10.1093/jac/dkr259. [DOI] [PubMed] [Google Scholar]
- 49.Carlet J, Jarlier V, Harbarth S, Voss A, Goossens H, Pittet D. Ready for a world without antibiotics? The Pensieres Antibiotic Resistance Call to Action. Antimicrobial resistance and infection control. 2012;1:11. doi: 10.1186/2047-2994-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jarlier V, Carlet J, McGowan J, Goossens H, Voss A, Harbarth S, Pittet D. Priority actions to fight antibiotic resistance: results of an international meeting. Antimicrobial resistance and infection control. 2012;1:17. doi: 10.1186/2047-2994-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McKeon DJ, Cadwallader KA, Idris S, Cowburn AS, Pasteur MC, Barker H, Haworth CS, Bilton D, Chilvers ER, Condliffe AM. Cystic fibrosis neutrophils have normal intrinsic reactive oxygen species generation. Eur Respir J. 2010;35:1264–1272. doi: 10.1183/09031936.00089709. [DOI] [PubMed] [Google Scholar]
- 52.Jesaitis AJ, Franklin MJ, Berglund D, Sasaki M, Lord CI, Bleazard JB, Duffy JE, Beyenal H, Lewandowski Z. Compromised host defense on Pseudomonas aeruginosa biofilms: characterization of neutrophil and biofilm interactions. J Immunol. 2003;171:4329–4339. doi: 10.4049/jimmunol.171.8.4329. [DOI] [PubMed] [Google Scholar]
- 53.Houston N, Stewart N, Smith DS, Bell SC, Champion AC, Reid DW. Sputum neutrophils in cystic fibrosis patients display a reduced respiratory burst. J Cyst Fibros. 2013;12:352–362. doi: 10.1016/j.jcf.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 54.Dusi S, Nadalini KA, Donini M, Zentilin L, Wientjes FB, Roos D, Giacca M, Rossi F. Nicotinamide-adenine dinucleotide phosphate oxidase assembly and activation in EBV-transformed B lymphoblastoid cell lines of normal and chronic granulomatous disease patients. J Immunol. 1998;161:4968–4974. [PubMed] [Google Scholar]
- 55.Dusi S, Donini M, Rossi F. Mechanisms of NADPH oxidase activation: translocation of p40phox, Rac1 and Rac2 from the cytosol to the membranes in human neutrophils lacking p47phox or p67phox. Biochem J. 1996;314(Pt 2):409–412. doi: 10.1042/bj3140409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riazanski V, Gabdoulkhakova AG, Boynton LS, Eguchi RR, Deriy LV, Hogarth DK, Loaec N, Oumata N, Galons H, Brown ME, Shevchenko P, Gallan AJ, Yoo SG, Naren AP, Villereal ML, Beacham DW, Bindokas VP, Birnbaumer L, Meijer L, Nelson DJ. TRPC6 channel translocation into phagosomal membrane augments phagosomal function. Proc Natl Acad Sci U S A. 2015;112:E6486–E6495. doi: 10.1073/pnas.1518966112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Das S, Bhattacharjee O, Goswami A, Pal NK, Majumdar S. Arabinosylated lipoarabinomannan (Ara-LAM) mediated intracellular mechanisms against tuberculosis infection: involvement of protein kinase C (PKC) mediated signaling. Tuberculosis (Edinb) 2015;95:208–216. doi: 10.1016/j.tube.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 58.Graff I, Schram-Doumont A, Szpirer C. Defective protein kinase C-mediated actions in cystic fibrosis neutrophils. Cellular signalling. 1991;3:259–266. doi: 10.1016/0898-6568(91)90052-v. [DOI] [PubMed] [Google Scholar]
- 59.Van Acker H, Sass A, Bazzini S, De Roy K, Udine C, Messiaen T, Riccardi G, Boon N, Nelis HJ, Mahenthiralingam E, Coenye T. Biofilm-grown Burkholderia cepacia complex cells survive antibiotic treatment by avoiding production of reactive oxygen species. PLoS One. 2013;8:e58943. doi: 10.1371/journal.pone.0058943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, Osgood S, Yockey L, Darnell DN, Barnhart L, Daub J, Boris L, Rump AP, Anderson VL, Haney C, Kuhns DB, Rosenzweig SD, Kelly C, Zelazny A, Mason T, DeRavin SS, Kang E, Gallin JI, Malech HL, Olivier KN, Uzel G, Freeman AF, Heller T, Zerbe CS, Holland SM. Common severe infections in chronic granulomatous disease. Clin Infect Dis. 2015;60:1176–1183. doi: 10.1093/cid/ciu1154. [DOI] [PMC free article] [PubMed] [Google Scholar]