

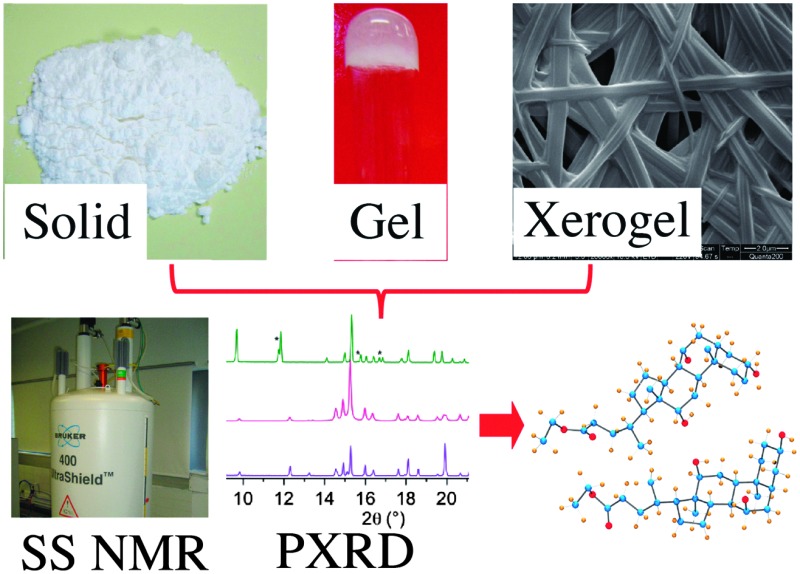
 The emergence of NMR crystallography provides a unique opportunity to study solids, gels and xerogels, thereby providing ample information to elucidate molecular packing in the native gel. This review details the importance as well as the application of solid state NMR spectroscopy combined with other analytical tools to study gels derived from low molecular mass organo- and hydrogelators.
The emergence of NMR crystallography provides a unique opportunity to study solids, gels and xerogels, thereby providing ample information to elucidate molecular packing in the native gel. This review details the importance as well as the application of solid state NMR spectroscopy combined with other analytical tools to study gels derived from low molecular mass organo- and hydrogelators.
Abstract
Since its invention more than six decades ago, nuclear magnetic resonance (NMR) spectroscopy has evolved as an inevitable part of chemical as well as structural analysis of small molecules, polymers, biomaterials and hybrid materials. In the solution state, due to the increased viscosity of complex viscoelastic fluids such as gels, liquid crystals and other soft materials, the rate of molecular tumbling is reduced, which in turn affects the chemical shift anisotropy, dipolar and quadrupolar interactions. As a consequence the solution state NMR spectra show broad lines, and therefore, extracting detailed structural information is a challenging task. In this context, solid state (SS) NMR has the ability to distinguish between a minute amount of polymorphic forms, conformational changes, and the number of non-equivalent molecules in an asymmetric unit of a crystal lattice, and to provide both qualitative as well as quantitative analytical data with a short-range order. Therefore, SS NMR has continued to evolve as an indispensable tool for structural analysis and gave birth to a new field called NMR crystallography. Solid state cross polarization (CP) and high resolution (HR) magic angle spinning (MAS) NMR spectroscopy has been used to study weak interactions in polymer gels. However, the application of SS NMR spectroscopy to study gels derived from low molecular weight gelators has been limited until recently. In this review, we will focus on the importance of solid state NMR spectroscopy in understanding and elucidating the structure of supramolecular gels derived from low molecular weight gelators with selected examples.
Introduction
Weak non-covalent interactions are ubiquitous in nature and are present in a wide variety of systems ranging from small organic and inorganic molecules, organometallics, polymers, proteins, virus particles and nucleic acids.1–3 Even though an isolated non-covalent interaction is relatively weak compared to that of a covalent bond, the cumulative energies of multiple interactions can be huge thereby controlling numerous structural and functional properties of molecules in their solid as well as solution states.4,5 For example, the unusual properties of water, the double helical structure of DNA and protein folding are primarily driven by non-covalent interactions.6,7 In addition to the traditional and long studied hydrogen bonding, van der Waals interactions, London dispersion forces, charge transfer complexation, and electrostatic interactions, several other interactions such as halogen bonding have recently been investigated.8 The ability of these weak interactions to generate well-ordered superstructures at multiple length scales has been instrumental in molecular recognition, protein–ligand interaction, host–guest chemistry, catalysis and energy storage materials.9,10 They have also proved to be an integral part in designing self-assembled soft materials.11 Among soft materials, self-assembled supramolecular organo- and hydrogels derived from low molecular weight organic compounds have been growing rapidly over the past 25 years, due to their unique ability to encapsulate and immobilize solvents at extremely low concentrations. The hydrogels as well as organogels have shown potential applications in tissue engineering, biomedicine, regenerative medicine, optoelectronics, sensors, catalysis and material science.12–19 Studies towards the rational design of low molecular weight gelators as well as their structural understanding are growing continuously.20 It has been hypothesized and experimentally demonstrated that gel formation is due to self-assembled fibrillar networks (SAFINs) of gelators and the hypothesis is accepted by the scientific community in general.15 However, there remained some ambiguities, which are either partially answered or remained challenging to address. Evidence for the presence of fibrillar network structures has been provided using cryo-transmission electron microscopy (TEM), environmental scanning electron microscopy (ESEM) and atomic force microscopy (AFM) studies.13,15,16 At the same time, direct information about packing in the native gel and the molecular interactions involved in gelation remained a challenge.
A systematic analysis of the existence and evaluation of the nature of molecular and supramolecular interactions in the solid state is often approached using single crystal and powder X-ray powder diffraction, small-angle X-ray scattering (SAXS) or small angle neutron scattering (SANS) analysis.13 However, in a number of cases the compounds under investigation fail to form suitable crystals for X-ray single crystal diffraction or the environment under study is unsuitable for X-ray diffraction. Moreover, the above requirements also restrict the studies of amorphous materials, powder samples, polymers and soft materials. Nuclear magnetic resonance (NMR) spectroscopy, on the other hand, has played a significant role in the analysis and structural characterization of small organic and inorganic molecules, hybrids, macrostructures, biomolecules and single cells.21 Notably, the initial attempts have been to develop and apply the methods in solution state experiments.22 However, the possibility of using nuclear absorption for solid crystals has been realized soon after its invention.23 Since then NMR spectroscopy has continued to evolve as an inevitable part of chemical analysis, structural chemistry and molecular dynamics studies of small organic molecules, polymers, and biomolecules as well as inorganic materials. Coupled with rapid progress in the field of NMR spectroscopy, solid state (SS) cross polarization (CP) and high resolution (HR) magic angle spinning (MAS) NMR spectroscopy is becoming an integral part of structural chemistry, material science and biology.24–26 Solid state NMR spectroscopy provides a unique opportunity to study non-crystalline materials, amorphous powders, liquid crystals, gels and even living cells.27 Owing to its ability to distinguish the number of different polymorphs, conformational isomers and non-equivalent molecules present in an asymmetric unit of a crystal lattice, this technique has emerged as a complementary tool to X-ray diffraction.28 Furthermore, solid state NMR is a non-destructive and non-invasive method, and as a result the samples can be recovered completely and used for further analysis.
Historically, in 1948 Pake in his classic experiment observed a fine structure in the nuclear magnetic resonance absorption for protons, while studying crystalline gypsum (CaSO4·2H2O).29 This experiment proved that NMR spectra for solids can provide useful structural information and is a potential complementary tool to X-ray diffraction. In another experiment, Andrew et al. solved the ambiguity regarding the position of hydrogens in urea using solid state NMR.30 Since then a number of inorganic salts and organic crystals have been attempted. The historical development of SS NMR is beyond the scope of this review and has been reviewed elsewhere.23–25
Over the past six decades solid state NMR has progressed continuously and is becoming a routine tool in structural chemistry and biology.26 This is attributed to the seminal paper by Schaefer and Stejskal in 1976, which combined magic angle (54.74°) spinning (MAS), cross polarization (CP) from proton-to-carbon and high power proton decoupling commonly known as CP MAS NMR.31
The above developments have led to a new field in solid state science, known as NMR crystallography. Though there was some hesitation to accept the term “NMR crystallography” by the crystallographic community, it was for the first time clarified by Elena et al. in their paper,32 and a reference footnote stating, “Interestingly, Crystallography is often assimilated today to X-ray studies on single crystals, due to the phenomenal success of this method. Crystallography is obviously a much wider discipline, defined (according to the Encyclopedia Britannica) as “the branch of science that deals with discerning the arrangement and bonding of atoms in crystalline solids and with the geometric structure of crystal lattices.” Since the powders we study here are microcrystalline, the term NMR Crystallography appears natural.”
Since 2000, there has been rapid progress within the field of NMR crystallography (Fig. 1).33,34 In this review, we briefly mention representative examples from the literature, especially how SS NMR coupled with other techniques has been used to study the self-assembled low molecular weight gels and gelators. Furthermore, we discuss how to derive the packing patterns of low molecular weight gelators in their solid and native gel states.
Fig. 1. The number of publications vs. year of publications using keywords “NMR crystallography” and “supramolecular gels” as entered in SciFinder as of 21.04.2016. Note: keywords such as molecular gels, organogels and hydrogels lead to 115, 1705, 71952 hits respectively. * The numbers for 2016 are incomplete.

NMR crystallography of gels or gelators utilizes a combination of techniques to gain complete structural details. One or more approaches have been followed and can be summarized as shown in Fig. 2. It is important to note that “NMR crystallography” based on various solid state NMR techniques combined with XRD is aimed for the study of crystalline structures. However, SS NMR is also useful for the characterization of non-crystalline amorphous materials including liquid crystals, supramolecular gels and other soft materials where XRD has limited resolution. In the case of supramolecular gels SS NMR combined with various microscopic (SEM, TEM, AFM, optical) techniques can provide a useful correlation between gels, xerogels and precipitated solid materials. Importantly, GIPAW calculation methods useful for crystalline structures are not suitable in the case of gels as they lack periodicity in their structures. However, combined solid-state NMR with first principles calculations has shown that due to high sensitivity, SS NMR provides certain advantages to study systems that are disordered or lack periodicity as well as materials where some dynamics are present.33 More importantly, it has been shown that the presence of monomorphic and polymorphic gel networks can be detected in a hydrogel matrix.34
Fig. 2. (A) The gelation mechanism of low molecular weight gelators. Reproduced with permission from ref. 43 copyright © 2010 Royal Society of Chemistry; (B) various steps involved in NMR crystallography of gels and gelators.

Solid state NMR of gels
Gels (polymeric and low molecular mass) are complex viscoelastic fluids either physically or chemically cross-linked with highly entangled networks. The increased viscosity of the medium restricts tumbling thereby reducing the molecular motion. This also affects chemical shift anisotropy, and dipolar and quadrupolar interactions. As a result, the solution state NMR spectra of gels/viscous liquids display broad signals. Therefore, it is a challenge to extract the information related to chemical and structural details. However, in 1985 Ford et al. in their experiment used polystyrene swollen in CDCl3 and demonstrated that solid state MAS NMR spectra can be obtained at 4 kHz spinning frequency.35 Ginter et al. reported the possibility of using MAS NMR for polyethylene oxide (PEO, MW 3800) hydrogel (Fig. 3).36
Fig. 3. 13C MAS solid state NMR spectra of polyethylene oxide (PEO, 1). (a) 13C MAS NMR spectrum of gel containing 70% PEO and 30% of water at 4 kHz; (b) 13C MAS NMR spectrum of gel containing 70% PEO and 30% of water in a static rotor and (c) 13C NMR spectrum of 5% PEO solution in the liquid state probe. Reproduced with permission from ref. 36 Copyright © 1989 Elsevier Inc.

They compared the 13C NMR spectra of 5 wt% PEO solution in water in its solution state with that of 70 wt% PEO gel in water by placing the gel in a 7 mm (od) zirconia rotor. The 13C NMR spectra in the solid state under static conditions and magic angle spinning at 4 kHz and the resonance patterns of methylene carbon at 69 ppm were compared. The solid state 13C MAS NMR spectra displayed a feature similar to the solution NMR spectra but with broad and overlapping signals, suggesting that there are more than one co-existing phases in the gel state. It was attributed to the presence of two different phases viz. mobile and immobile components present in the gel.
Similarly, Kobayashi et al. reported an extended study on polyvinyl alcohol (PVA, 2) gel and extracted the structural details of mobile and immobile components present in the gel (Fig. 4).37 The chemical shift values and splitting patterns of 13C signals arising from –CH2 and –CH in the solution state, gel state and solid state were compared. The solution of PVA in D2O showed the splitting of –CH and –CH2 into multiplets, which is attributed to their stereochemical configuration. The –CH peaks which are split into triads are assigned as mm, mr and rr (m = meso, r = racemic). The –CH2 signal splitting arises from the tetrad configuration. Interestingly, in the 13C NMR spectrum of gel, –CH shows a triplet and –CH2 showed a broad peak (Fig. 4B(b)). The 13C pulse-saturation transfer (PST) MAS NMR of PVA gels showed similar features as that of the solution state NMR of PVA gel signals (Fig. 4B(c)). It was suggested that the above observation was due to the fact that only mobile components are visible in the gels under PST MAS conditions. The solid state 13C CP MAS NMR spectrum of solid PVA, displayed –CH carbon with three splitting peaks. Notably, the chemical shift difference between these peaks was considerably larger than the splitting due to stereochemical configuration. This is due to splitting by the number of intramolecular hydrogen bonds between the neighbouring hydroxyl groups (Fig. 2B and 4A). The peaks show the chemical shifts depending on the number of hydrogen bonds involved. Important observations were made by recording the 13C CP MAS NMR of PVA gel, wherein it was found that there are splitting patterns, which resemble the 13C NMR of PVA gel as well as that of the 13C CP MAS NMR of the solid PVA. Therefore, 13C CP MAS has the ability to detect both mobile and immobile components. It was further shown that upon increasing the concentration of PVA the 13C CP MAS NMR tends to behave more like that of a solid as the stereochemical configurations are decreased. PVA gel with a concentration of 35%, behaved similar to solid in its spectral features. It is important to note that 13C CP MAS NMR relies on the principle of magnetization transfer from protons to carbons via a dipolar coupling mechanism. This process is highly effective in immobile components such as solids but the efficiency is greatly reduced for mobile components. In such situations a combination of techniques which do not rely on magnetization transfer instead utilizes direct single pulse 13C MAS NMR is useful as they provide information related to mobile components.
Fig. 4. Chemical structure of (A) polyvinyl alcohol (PVA, 2a) and various hydrogen bonding interactions (2b); (B) (a) solution state 13C NMR spectrum of PVA/D2O solution; (b) solution state 13C NMR spectrum in the gel state; (c) solid state 13C PST MAS NMR spectrum of the gel; (d) solid state 13C CP MAS NMR spectrum of the gel (e) 13C CP MAS NMR spectrum of the solid; (C) 67.8 MHz 13C CP MAS NMR spectrum of PVA in the gel state: (a) 9.1% PVA gel prepared by freeze–thaw cycles; (b–d) 11.8, 13.8 and 35.0% PVA gels prepared by vaporizing water from the gel and (D) 67.8 MHz 13C CP MAS NMR spectrum of PVA gel at –50 °C. Reproduced with permission from ref. 37 Copyright © 1995 American Chemical Society.

An extensive review on solid state NMR of polymer gels is beyond the scope of this review. However, the above studies form the basis for the investigation of many supramolecular gels. In particular, the solution state NMR spectroscopy of low molecular weight gels has been discussed based on the concept of mobile and immobile components as well as the components which are bound to the gel network and free molecules.38 Recently, DOSY NMR experiments and HRMAS studies have been shown to support this hypothesis.39 The details of such studies have been reviewed elsewhere.40 Therefore, the following part of the review we will focus towards the solid state NMR of low molecular weight gelators with a special emphasis on CP MAS NMR techniques.
Unlike polymeric gels, the low molecular weight gels are composed of supramolecularly self-assembled fibrillar networks of small molecules (M w < 3 kDa, Fig. 2A). Importantly, a number of gels are studied based on their xerogels or aerogels, and packing patterns are deciphered based on single crystal structures.40 However, most of the gelators do not undergo crystallization. Therefore, the packing pattern leading to the process of gelation is still poorly understood and there is a debate on the presence of 3D networks in the native gel and its similarity/identity to those observed in the xerogels. This question arises because upon sample preparation, during evaporation of the solvent or under experimental conditions (high vacuum in SEM, TEM), there may be a change in the structure and as a result the morphology in the gel state may differ significantly from that of the xerogel. There exist a few reports in the literature, in which suggestions have been made regarding the mode of the packing pattern and a correlation between the bulk solid and xerogels mostly evidenced by powder X-ray diffraction (PXRD), small angle neutron scattering (SANS), and small angle X-ray scattering (SAXS) techniques.40 Most of the gelators do not undergo crystallization and therefore, a complete understanding and clear evidence of the gelation process remained undisclosed. However, a few reports which describe a correlation between the single crystal and the interactions which lead to gelation have been reported in the literature. More importantly, gelation conditions (concentration, temperature and solvent) are significantly different from the crystallization conditions. Therefore, packing patterns in the gel or xerogel may differ drastically from the crystal structure. One way to overcome this ambiguity is by combining more than one analytical techniques, which are complementary to each other. In this context, solid state NMR spectroscopy provides useful information when combined with X-ray powder diffraction and other complementary tools. Taira et al. reported the gelation of alkylpyrdinium derivatives (Fig. 3a–c and 5) in the presence of α-cyclodextrin (α-CD).41 By combining the MALDI-TOF results with 13C CP MAS of NMR spectroscopy, the authors concluded that the gelation is due to pseudorotaxane formation.
Fig. 5. Pseudorotaxane based gelators: (A) Chemical structures of alkylpyridinium halides and α-cyclodextrin, (B) pseudorotaxane formation leading to gelation: (C) 13C CP MAS NMR spectra of (a) α-CD; (b) xerogel of 3b and α-CD, and (c) 3b. Peaks with an asterisk are assigned to C-1 and C-4 with a conformationally strained glycoside linkage. Reproduced with permission from ref. 41 Copyright © 2009 Royal Society of Chemistry.

In more detail, the 13C CP MAS NMR spectrum of xerogel derived from a mixture of 3b : α-CD (1 : 2) hydrogel (Fig. 5C) did not show 13C NMR signals at 81 and 98 ppm, which are unique to C-1 and C-4 carbons of free α-CD. This suggests that xerogel is composed of pseudorotaxane. Therefore, the original hydrogel is composed of pseudorotaxane and not free CD (Fig. 5B). However, there was no evidence based on solid state NMR of the native gel.
In this direction Schoonbeek et al. studied the properties of 1,2-bis-urea benzene derivatives (4a–g, Fig. 6) and compared the solution NMR, solid state 13C CP MAS NMR of the solid gelator and CP MAS NMR of toluene-d 8 gel (Fig. 6B).42 There were no significant changes in the position of 13C NMR signals between solution at various concentrations, solid and gel states. It was attributed to the fact that there exists an intramolecular H-bonding within the gelator molecules in their solution state and also strong solvent–solute interactions, which essentially have the same effect as strong inter- and intramolecular interactions in the solid and gel state.
Fig. 6. (A) Chemical structures of bis-urea benzene derivatives; (B) comparison of solution, gel and solid state 13C NMR spectra of 4a: (a) 61.1 mM in DMSO-d 6; (b) 5.4 mM in CDCl3; (c) 47.7 mM in CDCl3; (d) 13C CP MAS NMR of the solid and (e) toluene-d 8 gel of 4a. Reproduced with permission from ref. 42 © Wiley-VCH.

More importantly, both the carbonyls were found to be equivalent in solution, gel and solid states. Furthermore, the phenyl carbons in the solid state showed no splitting; however, the butyl carbons did show splitting in their solid state NMR spectra. Though there is no clear explanation for this observation, this might be a result of a severe disorder in the flexible butyl chains. Even in this work no conclusive evidence was obtained from the gel state due to poor spectral resolution.
In 2010, Nonappa et al. have shown that by combining solid state 13C CP MAS NMR, X-ray powder diffraction and thermoanalytical techniques, the packing patterns in the gel and xerogel states can be elucidated.43 This study represents one of the first examples of a gel studied in its native gel state (using benzene, previously deuterated solvents were used). This forms the basis for the discussion in the following part of the manuscript. Using simple esters of cholic acid, a number of gelators were prepared (Fig. 7).44 The gel formation was observed only for ethyl 5b, propyl 5c, allyl 5d and propargyl 5e esters of cholic acid. The methyl derivative 5a readily formed crystals, whereas the butyl derivative 5f remained in the solution in the tested solvents. Based on SEM, AFM and polarizing optical microscopy images of the xerogles it was found that the gelators undergo self-assembly into 300 nm (diameter) fibers with indefinite length. Furthermore, the direct evolution of fibers was supported using optical microscopy imaging.43 In order to gain the interactions involved in gel formation and packing patterns the solution state NMR of the gel, and the solid state 13C CP MAS NMR of the xerogels and native gels were combined with X-ray powder diffraction. The 13C CP MAS NMR of the synthetic solid and the xerogel displayed a doublet resonance pattern (Fig. 7B), which was a consequence of two non-equivalent molecules in the asymmetric unit. Furthermore, using very careful sample preparation the solid state 13C CP MAS NMR spectra of gels were compared. Interestingly, the gels when spun at 4, 5 and 8 kHz showed a similar resonance pattern as that of the xerogel (Fig. 8A).
Fig. 7. Cholic acid ester based gelators: (A) chemical structures of gelators and non-gelators; (B) solid state13C CP MAS NMR of xerogel and recrystallized samples of ethyl cholate 5d from different solvents. (C) 13C CP MAS NMR spectra of 5a–5f, indicating that all the gelators show a doublet resonance pattern, whereas 5a forms a solvate and 5f is relatively less crystalline. (Note: asterisk indicates the signals arising from solvent molecules). Reproduced with permission from ref. 43 Copyright © 2010 Royal Society of Chemistry.

Fig. 8. (A) Schematic representation showing the location and preparation of a gel sample for the CP MAS experiment and the 13C CP MAS NMR spectra of benzene-d 6 gel of 5b at (a) 5 kHz and (b) 4 kHz; and (c) benzene gel of 5b; (B) experimental PXRD patterns of 5a–f. The similarity of the patterns of 5b and 5c indicates isostructural features on their packing modes. A similar trend can be observed between the patterns of 5d and 5e; (C) X-ray structure of 5b showing two molecules in an asymmetric unit. Carbon (blue), oxygen (red) and hydrogen (gold). Reproduced with permission from ref. 43 © Royal Society of Chemistry.

The above observation suggested that the packing pattern in the gel state and the xerogel is similar. Using X-ray powder diffraction indexing and Rietveld refinement, the solid state structure of the gelator was solved (Fig. 8B and C). This represents the first example of a gelator crystal structure solved by combining the solid state NMR and X-ray powder diffraction analysis. As revealed by 13C CP MAS studies, the crystal structure displayed two non-equivalent molecules in an asymmetric unit of a crystal lattice. More importantly, the non gelators either formed solvates or amorphous solids. Interestingly, all the gelators followed similar packing patterns, whereas the non-gelators showed a singlet resonance pattern. This suggests that the packing patterns are important in determining whether a given molecule undergoes crystallization into single crystals, solvates, amorphous solids or gels.
Noponen et al. reported the organogelation of bile acid-l-methionine methyl ester conjugates and compared the solid state single crystal X-ray structure, 13C CP MAS NMR and X-ray powder diffraction patterns (Fig. 9).45 Compounds 6a–c underwent crystallization leading to quality single crystals suitable for structural determination. However, only 6b and 6c showed the ability to form gels in aromatic solvents.
Fig. 9. (A) Chemical structures of bile acid-l-methionine methyl ester conjugates and ORTEP10 plots of asymmetric units of compounds 6a–c; (B) temperature-dependent diffraction patterns of xerogel of compound 6a together with a simulated pattern of the low-temperature single crystal structure 6a; (C) overlays of (a) molecular structures and (b) packing modes of single crystal X-ray structure (dark structure) and structure solved using XRPD (light structure) presented along the b-axis of the room-temperature structure 6a. For clarity, the sulfur atom (s1) of room temperature structure is presented in a ball-style and hydrogen atoms are omitted. Reproduced with permission from ref. 45 © Royal Society of Chemistry.

The crystallization studies were carried out in acetonitrile and the solid state 13C CP MAS NMR spectra of recrystallized samples were compared with the benzene-d 6 and toluene-d 8 gels. It was found that compound 6a shows a similar spectral pattern for recrystallized sample in acetonitrile as well as native gel, suggesting that the gelator molecules have a similar packing pattern in their gel state and in single crystals. This was further supported using the X-ray powder diffraction patterns of xerogels. The experimental diffraction patterns of xerogels were compared with that of the simulated powder diffraction patterns from the single crystal X-ray structure. On the other hand, compound 6b showed a different spectral pattern for the single crystals and native gel (Fig. 10). This suggests that the packing mode in the gel state significantly differed from that of the single crystal structure for 6b.
Fig. 10. (A) 13C NMR spectra of compound 6b; (a) solution state; (b) 13CP MAS NMR spectrum of the synthesis product recrystallized from acetonitrile; (c) 13C CP MAS NMR spectrum of 2% (w/v) benzene gel. (B) 13C NMR spectra of compound 6c; (a) solution state; (b) 13C CP MAS NMR spectrum of the synthesis product recrystallized from acetonitrile and (c) 13C CP MAS NMR spectrum of 4% (w/v) toluene-d 8 gel. Reproduced with permission from ref. 45 Copyright © 2010 Royal Society of Chemistry.

In another study, Ikonen et al. reported the mono- and diketal derivatives of bile acids using pentaerythritol, catechol and 2,3-dihydroxy naphthalene.46 The monoketal derivatives of pentaerythritol displayed gelation ability in organic solvents (Fig. 11A), while the diketals from pentaerythritol as well as the ketals from catechol and 2,3-dihydroxy naphthalene formed single crystals. Gelator 7a was recrystallized from various solvents (benzene, toluene, p-xylene, chlorobenzene, acetonitrile and acetone) and studied using 13C CP MAS NMR spectroscopy. The solid state 13C CP MAS NMR spectra were compared with respective solution state 13C NMR spectral data. A systematic analysis of 13C CP MAS NMR spectra of compound 7a recrystallized from toluene, p-xylene, and chlorobenzene revealed a doublet resonance pattern indicating the presence of two crystallographically independent molecules per asymmetric unit (Fig. 11B(c)). Interestingly, the sample crystallized from benzene, was found to contain two different polymorphic forms. The minor polymorphic form was found to display similar spectral patterns as the ones crystallized from the other aromatic solvents (Fig. 11B(d)), whereas the major form was a benzene solvate. Furthermore, recrystallization of 7a from non-gelling solvents such as acetonitrile or acetone displayed a doublet resonance pattern. However, the doublet resonance patterns were not observed for all the carbons and the signals from pentaerythritol carbons were not well resolved, presumably due to a severe disorder in that part of the gelator molecule. More importantly, recrystallization of gelator 7a from gel-forming solvents displayed well resolved signals in their 13C CP MAS NMR spectra suggesting a highly ordered packing system in the gel state.
Fig. 11. (A) Chemical structures of mono- and diketal derivatives of bile acids. (B) 13C NMR spectrum of 7a in CDCl3 (a) and 13C CP MAS NMR spectra of 7a crystallized from acetonitrile (b), toluene (c), and benzene (d). Reproduced with permission from ref. 46 Copyright © 2010 Royal Society of Chemistry.

Svobodová et al. reported the stigmasterol-aminoacid (glycine, l-leucine and l-phenyl alanine) conjugates as stimuli responsive gelators (Fig. 12).47 The hydrochloride salts of the conjugates underwent self-assembly leading to organogelation upon dissolving in a number of alcoholic and aromatic solvents. The 13C CP MAS NMR spectra were recorded for the synthetic solids and the spectral patterns were compared with that of the gels as well xerogels. The structural studies were performed for 8a and 8b. The 13C CP MAS NMR of the synthetic solid showed to be crystalline in nature but the signals were relatively broad. However, a careful analysis of the13C CP MAS NMR spectral data of 8a revealed three signals for carbonyl carbon at 169.88, 169.34 and 168.76 ppm suggesting the presence of more than one polymorphic forms (Fig. 12B). This was further confirmed by performing the 15N CP MAS NMR experiment, which indicated the presence of more than one 15N signals. Interestingly, the 13C CP MAS NMR of the xerogel prepared from the benzene gel of 8a showed better crystallinity compared to that of the synthetic solid (Fig. 12B(e)). More importantly a doublet resonance pattern with carbonyl signals at 169.85 and 168.92 ppm was observed. This suggests that there are two crystallographically independent molecules in an asymmetric unit.
Fig. 12. (A) Chemical structures of stigmasterol–amino acid conjugates and protonation and deprotonation of stigmasteryl phenylalaninate in tetrachloromethane; (B) 13C CP MAS NMR of (a) 8a solid; (b) 8b solid, and xerogels of 8b obtained from (c) benzene gel of 8a; (d) from CCl4 gel of 8a, and (e) CCl4 gel of 8b (4% w/v). (C) 13C solid state NMR of (a) 8b solid and (b) xerogel from 1-butanol gel of 8b. Reproduced with permission from ref. 47, Copyright © 2011 Elsevier Inc.

In addition to the above studies, the authors studied the xerogel obtained from the CCl4-gel, which displayed a singlet resonance pattern. This observation revealed that the synthetic solid was a mixture of polymorphs and can be separated by changing the solvents. Furthermore, the comparison of 13C CP MAS NMR of CCl4 native gel with that of the xerogel confirmed that both the gel and xerogels have similar packing patterns. The above observation was further supported using the morphological features of xerogels obtained using SEM, which suggest that they are highly solvent dependent. On the other hand, the 13C CP MAS NMR spectrum of 8b displayed a doublet resonance pattern. The comparison of 13C CP MAS NMR and 15N CP MAS NMR of the 1-butanol gel of 8b with that of xerogel confirmed the similarity between synthetic solid, xerogel and the gel packing patterns.
In another study, Svobodová et al. reported the gelation properties of aromatic-linker-steroid A(LS)2 type gelators containing pyridine-2,6-dicarboxylic acid as a linker and cholersterol glycinate as the steroidal moiety (Fig. 13).48–50 The molecules also underwent metal complexation induced gelation, leading to metallosupramolecular gels. The molecular packing of gelator 9a and its metal complexes was performed using combined 13C CP MAS NMR and X-ray diffraction experiments. In this study 13C CP MAS NMR spectral patterns of compound 9a crystallized from chloroform and the xerogels from pentan-1-ol gel were compared (Fig. 13). 13C CP MAS NMR spectra of the synthetic solid 9a (Fig. 13C(a)) and its xerogel from pentan-1-ol (Fig. 13C(b)) gel displayed similarities in their resonance pattern suggesting similar molecular packing. The broad signals in the spectrum of the xerogel of 9a + Ag(i) from pentan-1-ol (Fig. 13C(c)) due to its amorphous nature and was supported by X-ray powder diffraction analysis. The 13C CP MAS NMR spectrum of the xerogel of 9a + Zn(ii) complex from pentan-1-ol gel (Fig. 13C(d)) displayed crystalline nature. However, the spectral patterns deviated from the other xerogels indicating a different packing mode.
Fig. 13. (A) Chemical structures of metallosupramolecular gelators. (B) Structure of compound 9a, a gelator of the A(LS)2 type, with marked non-covalent interaction sites. (C) 13C CP MAS NMR spectra of (a) solid of 9a from CHCl3; (b) xerogel of 9a from pentan-1-ol; (c) xerogel of 9a + Ag(i) from pentan-1-ol and (d) xerogel of 9a + Zn(ii) from pentan-1-ol. Reproduced with permission from ref. 48, Copyright © 2012 Royal Society of Chemistry.

Alanne et al. reported bisphosphonate hydrogelators 10a–10d (Fig. 14) and used solid state 13C CP MAS and 31P MAS NMR spectroscopy to compare the solid state structural properties and packing patterns in the gel, xerogel and synthetic solids (Fig. 14A–D).51 The authors concluded that the 13C CP MAS NMR of the synthetic solids, and the gels resembled each other but differed from that of the xerogels. However, a close examination of the 13C CP MAS NMR gives an interesting overview which was not taken into consideration in this work. Careful analysis of the 13C NMR spectra of the synthetic product, xerogel and gels shows that they all differ in their spectral pattern. Comparing the solid state NMR spectra of synthetic solid and xerogels suggests that there exists more than one form. Interestingly, the two 13C signals appearing in the gel state resemble that of the peaks at around 25 ppm signals (Fig. 14B–D) in their solid state. This suggests that there exists more than one phase in the gel such as mobile and immobile components. Interestingly, the third signal disappeared in the gel state. On other hand, the intensities of 13C signals at around 25 ppm are reversed in the gel state compared to that of the xerogel. This suggests that all the three states have different packing patterns. However, the X-ray powder patterns were not supportive for this argument, and it is not a surprise since several examples are known in the literature where powder X-ray diffraction failed to detect a minor polymorphic form in the solid state compared to solid state NMR. In particular, CP MAS NMR is very sensitive in identifying a minute amount of polymorphic forms. On the other hand, similar spectral patterns in 31P CP MAS NMR were also shown. For example, for gelator molecule 10a, there is a significant difference between the synthetic solid and the xerogels. This is in correlation with that of the 13C CP MAS NMR studies.
Fig. 14. Bisphosphonate hydrogelators: (A) chemical structures of hydrogelators; (B–E) 13C CP MAS NMR spectra of (a) synthetic solid; (b) xerogel and (c) gel (4 or 5 w/v%) of 10a–10d; (F) 31P CP MAS NMR spectra of (a) synthetic solid; (b) xerogel and (c) the hydrogel of 10a–10d. Reproduced with permission from ref. 51, Copyright © 2013 Royal Society of Chemistry.

This suggests that a simple correlation between the synthetic solid, xerogel and gels is not always straightforward. In such cases more extended studies such as 2D solid state NMR spectroscopy will be useful for correlation spectroscopy. Therefore, the study of gels and gelators using solid state NMR is still developing and there is more scope to apply this technique and develop into more complex systems.
Polymorphism is an important property of organic compounds and can significantly alter the physical and chemical properties.
Solid state NMR has been extensively utilized to identify and characterize polymorphs.28 A given gelator molecule may contain various polymorphs or solvates, which can affect the gelation properties. It has been shown that a recrystallized form of commercial caffeine has the ability to undergo gelation in alcoholic as well as aromatic organic solvents (Fig. 15).
Fig. 15. Caffeine as a gelator. (a) Chemical structure of caffeine, theophylline and theobromine; (b) photograph of gels derived from caffeine; (c) optical microscopy image of xerogel; (d) microcrystalline network formation and (e) 13C CP MAS NMR of commercial caffeine, recrystallized form and xerogels. Reproduced with permission from ref. 52, Copyright © 2016 by the authors.

Using 13C CP MAS solid state NMR, it was demonstrated that the commercial caffeine contains more than one form. When recrystallized from ethanol, it results in an anhydrous form, which undergoes self-assembly into microcrystalline networks and immobilizes the solvents.52
Conclusions
In conclusion both solid state NMR spectroscopy and supramolecular gels are continuously evolving areas of research with new applications. Solid state NMR coupled with other complementary techniques such as X-ray diffraction, SAXS, SANS, FT-IR, Raman spectroscopy and electron microscopy known as NMR crystallography is becoming an important tool to study molecules, materials and nanostructures in a wide variety of forms. While the pharmaceutical industries concentrate on polymorphs, solvates and salts, the utilization of SS NMR in materials science is growing rapidly. Solid state NMR of gels is not straightforward as there are several disadvantages, especially, due to high spinning speed. This might result in the disintegration of gels due to the centrifugal force generated upon spinning. Therefore, low spinning speeds are generally recommended, which might lead to poor resolution and overlapping spinning side bands. On the other hand, there is a growing interest in developing gels based on new supramolecular interactions such as subcomponent self-assembly and halogen bonding.8,50 Solid state NMR is extremely useful for studying such new interactions and multicomponent assemblies.
Acknowledgments
We acknowledge Academy of Finland's Centre of Excellence for Molecular Engineering of Biosynthetic Hybrid Materials Research (HYBER).
Biographies

Nonappa
Nonappa received his MSc in Chemistry from the Mangalore University in 2003, Karnataka, India, and his PhD degree in 2008 under the guidance of Professor Uday Maitra at the Department of Organic Chemistry, Indian Institute of Science, Bangalore, India. He has worked on the synthesis of rare bile acids, and supramolecular gels. He is a recipient of Eli Lilly Asia outstanding thesis award. He pursued his postdoctoral research at the Department of Chemistry, University of Jyväskylä, Finland, on the solid state NMR of soft materials with Professor Erkki Kolehmainen. He is currently working at the Department of Applied Physics, Aalto University, Finland. His research interests are in vitro and in vivo colloidal self-assembly and electron tomography.

E. Kolehmainen
Erkki Kolehmainen was born in 1947 in Hankasalmi, Finland. He received his MSc degree in physical chemistry in 1973 from the University of Jyväskylä and his PhD degree in organic chemistry in 1990 from the University of Kuopio. At present Erkki Kolehmainen is working as a professor in organic chemistry, University of Jyväskylä. His research interests are focused on the structures and dynamics of organic, bio-organic, and metallo-organic molecules and soft materials studied by multinuclear magnetic resonance spectroscopy.
References
- Steiner T., Zhang Y., Yang Z., Yuan F., Gu H., Gao P., Xu B., Ikonen S., Nonappa, Kolehmainen E. Angew. Chem., Int. Ed. J. Am. Chem. Soc. CrystEngComm. 2002;2004;2010;4112612:48–76. 15028–15029, 4304–4311. [Google Scholar]
- Grantcharova V., Alm E. J., Baker D., Howrwich A. L. Curr. Opin. Struct. Biol. 2001;11:70–82. doi: 10.1016/s0959-440x(00)00176-7. [DOI] [PubMed] [Google Scholar]
- Neidle S., Oxford Handbook of Nucleic Acid Structure, Oxford University Press, Oxford, 1999. [Google Scholar]
- Meyers M. A. and Chen P.-Y., Biological Materials Science Biological Materials, Bioinspired Materials, and Biomaterials, Cambridge University Press, 2014. [Google Scholar]
- Whitesides G. M., Boncheva M. Proc. Natl. Acad. Sci. U. S. A. 2002;99:4769–4774. doi: 10.1073/pnas.082065899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehn J.-M. Angew. Chem., Int. Ed. 1988;27:89–121. [Google Scholar]
- Lehn J.-M., Mao C., Sun W., Seeman N. C. Angew. Chem., Int. Ed. Nature. 1990;1997;29386:1304–1319. 137–138. [Google Scholar]
- Metrangolo P., Neukirch H., Pilati T., Resnati G., Bertolani A., Pirrie L., Stefan L., Houbenov N., Haataja J. S., Catalano L., Terraneo G., Giancane G., Valli L., Milani R., Ikkala O., Resnati G., Metrangolo P. Acc. Chem. Res. Nat. Commun. 2005;2015;386:386–395. 5754. doi: 10.1021/ar0400995. [DOI] [PubMed] [Google Scholar]
- Applications of Supramolecular Chemistry, ed. H.-J. Schneider, CRC Press, Boca Raton, 2012. [Google Scholar]
- Ariga K. and Kunitake T., Supramolecular Chemistry – Fundamentals and Applications, Springer-Verlag, Heidelberg, 2006. [Google Scholar]
- Zhang S. Nat. Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- Hirst A. R., Escuder B., Miravet J. F., Smith D. K. Angew. Chem., Int. Ed. 2008;47:8002–8018. doi: 10.1002/anie.200800022. [DOI] [PubMed] [Google Scholar]
- Terech P., Smith W. G., Weiss R. G. J. Chem. Soc., Faraday Trans. 1996;92:3157–3162. [Google Scholar]
- Nonappa, Maitra U. Org. Biomol. Chem. 2008;6:657–669. doi: 10.1039/b714475j. [DOI] [PubMed] [Google Scholar]
- Zinic M., Vögtle M. F., Fages F. Top. Curr. Chem. 2005;256:39–76. doi: 10.1007/b107171. [DOI] [PubMed] [Google Scholar]
- Smith D. K., in Organic Nanostructures, ed. J. L. Atwood and J. W. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007, pp. 111–154. [Google Scholar]
- Weiss R. G. Langmuir. 2009;25:8369. doi: 10.1021/la901621x. [DOI] [PubMed] [Google Scholar]
- Estroff L. A., Hamilton A. D. Chem. Rev. 2004;104:1201–1218. doi: 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]
- Weiss R. G. J. Am. Chem. Soc. 2014;136:7519–7530. doi: 10.1021/ja503363v. [DOI] [PubMed] [Google Scholar]
- Lana Y., Corradinia M. G., Weiss R. G., Raghavan S. R., Rogers M. A. Chem. Soc. Rev. 2015;44:6035–6058. doi: 10.1039/c5cs00136f. [DOI] [PubMed] [Google Scholar]
- Larsen J.-H. A., Boebinger G. S., Comment A., Duckett S., Edison A. S., Engelke F., Griesinger C., Griffin R. G., Hilty C., Maeda H., Parigi G., Prisner T., Ravera E., Bentum J., Vega S., Webb A., Luchinat C., Schwalbe H., Frydman L., Freedberg D. I., Selenko P. Angew. Chem., Int. Ed. Annu. Rev. Biophys. 2015;2014;5443:9162–9185. 171–192. doi: 10.1002/anie.201410653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell E. M., Torrey H. C., Pound R. V., Bloch F., Hansen W. W., Packard M. E. Phys. Rev. Phys. Rev. 1946;1946;6969:37. 127. [Google Scholar]
- Andrew E. R. Bull. Magn. Reson. 1995;17:16–20. [Google Scholar]
- Duer M. J., Stourton C. Magn. Reson. 1995;17:27–36. doi: 10.1006/jmre.1997.1243. [DOI] [PubMed] [Google Scholar]
- Andrew E. R., Szczesniak E. Prog. Nucl. Magn. Reson. Spectrosc. 1995;28:11–36. [Google Scholar]
- Su Y., Andreas L., Griffin R. G., Ladizhansky V. Annu. Rev. Biochem. Isr. J. Chem. 2015;2014;8454:465–497. 86–103. doi: 10.1146/annurev-biochem-060614-034206. [DOI] [PubMed] [Google Scholar]
- Jarvis M. C., Apperley D. C., Alia A., Ganapathy S., de Groot H. J. M., Warnet X. L., Arnold A. A., Marcotte I., Warschawski D. E. Plant Physiol. Photosynth. Res. Biophys. J. 1990;2009;2015;92102109:61–65. 415–425, 2461–2466. [Google Scholar]
- Vaughan R. W., Charpentier T., Sutrisno A., Huang Y., Harris R. K., Laws D. D., Bitter H.-M. L., Jerschow A., Dybowski C., Bai S., Nonappa, Lahtinen M., Ikonen S., Kolehmainen E., Kauppinen R. Annu. Rev. Phys. Chem. Solid State Nucl. Magn. Reson. Solid State Nucl. Magn. Reson. Analyst. Angew. Chem., Int. Ed. Anal. Chem. Cryst. Growth Des. 1978;2011;2013;2006;2002;2008;2009;29404913141809:397–419. 1–20, 1–11, 351–373, 3096–3129, 4295–4300, 4710–4719. [Google Scholar]
- Pake G. E. J. Chem. Phys. 1948;16:327–3367. [Google Scholar]
- Andrew E. R., Hyndman D. Faraday Sot. Discuss. 1955;19:195. [Google Scholar]
- Schaefer J., Stejskal E. O. J. Am. Chem. Soc. 1976;98:2013. [Google Scholar]
- Elena B., Pintacuda G., Mifsud N., Emsley L. J. Am. Chem. Soc. 2006;128:9555–9560. doi: 10.1021/ja062353p. [DOI] [PubMed] [Google Scholar]
- Harris R. K., Forbes J., Husted C., Oldfield E., Ashbrook S. E., McKay D. Solid State Sci. J. Am. Chem. Soc. Chem. Commun. 2004;1988;2016;611052:1025–1037. 1059–1065, 7186–7204. [Google Scholar]
- Ripmeester J. A., Wasylishena R. E., Macholl S., Tietze D., Buntkowsky G., Taulelle F., Bouchevreau B., Martineau C., Nagy-Smith K., Moore E., Schneider J., Tycko R. CrystEngComm. CrystEngComm. CrystEngComm. Proc. Natl. Acad. Sci. U. S. A. 2013;2013;2013;2015;151515112:8598. 8627–8638, 8613–8622, 9816–9821. [Google Scholar]
- Ford W. T., Mohganraj S., Hall H., O'Donnell D. J. J. Magn. Reson. 1985;65:156. [Google Scholar]
- Ginter D. M., Bell A. T., Radke C. J. J. Magn. Reson. 1989;81:217–219. [Google Scholar]
- Kobayashi M., Ando I., Ishiif T., Amiya S. Macromolecules. 1995;28:6677–6679. [Google Scholar]
- Escuder B., Llusar M., Miravet J. F. J. Org. Chem. 2006;71:7747–7752. doi: 10.1021/jo0612731. [DOI] [PubMed] [Google Scholar]
- Nonappa, Šaman D., Kolehmainen E. Magn. Reson. Chem. 2015;53:256–260. doi: 10.1002/mrc.4185. [DOI] [PubMed] [Google Scholar]
- Yu G., Yan X., Han C., Huang F. Chem. Soc. Rev. 2013;42:6697. doi: 10.1039/c3cs60080g. [DOI] [PubMed] [Google Scholar]
- Taira T., Suzaki Y., Osakada K. Chem. Commun. 2009:7027–7029. doi: 10.1039/b911667b. [DOI] [PubMed] [Google Scholar]
- Schoonbeek F. S., van Esch J. H., Hulst R., Kellogg R. M., Feringa B. L. Chem. – Eur. J. 2000;14:2633–2643. doi: 10.1002/1521-3765(20000717)6:14<2633::aid-chem2633>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Nonappa, Maitra U. Soft Matter. 2007;3:1428–1433. doi: 10.1039/b711010c. [DOI] [PubMed] [Google Scholar]
- Nonappa, Lahtinen M., Behera B., Kolehmainen E., Maitra U. Soft Matter. 2010;6:1748–1757. [Google Scholar]
- Noponen V., Nonappa, Lahtinen M., Valkonen A., Salo H., Kolehmainen E., Sievänen E. Soft Matter. 2010;6:3789–3796. [Google Scholar]
- Ikonen S., Nonappa, Valkonen A., Juvonen R., Salo H., Kolehmainen E. Org. Biomol. Chem. 2010;8:2784–2794. doi: 10.1039/c003228j. [DOI] [PubMed] [Google Scholar]
- Svobodová H., Nonappa, Wimmer Z., Kolehmainen E. J. Colloid Interface Sci. 2011;361:587–593. doi: 10.1016/j.jcis.2011.05.084. [DOI] [PubMed] [Google Scholar]
- Svobodová H., Nonappa, Lahtinen M., Wimmer Z., Kolehmainen E. Soft Matter. 2012;8:7840. [Google Scholar]
- Bunzen H., Kolehmainen E. Molecules. 2013;18:3745–3759. doi: 10.3390/molecules18043745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunzen H., Nonappa, Kalenius E., Hietala S., Kolehmainen E. Chem. – Eur. J. 2013;19:12978–12981. doi: 10.1002/chem.201302055. [DOI] [PubMed] [Google Scholar]
- Alanne A.-L., Lahtinen M., Löfman M., Turhanen P., Kolehmainen E., Vepsäläinen J., Sievänen E. J. Mater. Chem. B. 2013;1:6201. doi: 10.1039/c3tb20957a. [DOI] [PubMed] [Google Scholar]
- Nonappa, Kolehmainen E. Gels. 2016;2:9. doi: 10.3390/gels2010009. [DOI] [PMC free article] [PubMed] [Google Scholar]