

Abstract
It is widely believed that targeting the tumour-initiating cancer stem cell (CSC) component of malignancy has great therapeutic potential, particularly in therapy-resistant disease. However, despite concerted efforts, CSC-targeting strategies have not been efficiently translated to the clinic. This is partly due to our incomplete understanding of the mechanisms underlying CSC therapy-resistance. In particular, the relationship between therapy-resistance and the organisation of CSCs as Stem-Progenitor-Differentiated cell hierarchies has not been widely studied. In this review we argue that modern clinical strategies should appreciate that the CSC hierarchy is a dynamic target that contains sensitive and resistant components and expresses a collection of therapy-resisting mechanisms. We propose that the CSC hierarchy at primary presentation changes in response to clinical intervention, resulting in a recurrent malignancy that should be targeted differently. As such, addressing the hierarchical organisation of CSCs into our bench-side theory should expedite translation of CSC-targeting to bed-side practice. In conclusion, we discuss strategies through which we can catch these moving clinical targets to specifically compromise therapy-resistant disease.
Background
Tumours are heterogeneous collections of cells, only some of which are capable of initiating tumourigenesis. In many different types of malignancy, these ‘tumour-initiating’ cells have been shown to display the stem cell-like properties of self-renewal, differentiation and the development of (malignant) tissues. This has led to tumour-initiating cells being collectively referred to as ‘Cancer Stem Cells’ (CSCs), and interest in targeting cancer stemness as a clinical strategy. CSCs have been shown to be highly-resistant to conventional cancer therapies such as chemotherapy and radiotherapy. While the targeting of CSC mechanisms has been shown to reduce therapy-resistance in many cell culture models, this has not been successfully translated to the clinic. In this review we will discuss successes and limitations in targeting CSC therapy-resistance mechanisms. We will argue that clinical-failure in this area may be partly due to a poor understanding of the plastic nature of the complex hierarchies into which CSCs are organised in vivo. Finally, we will conclude by arguing that clinical translation will be hastened by an appreciation of therapy-resistant CSC populations as moving, rather than fixed clinical targets.
Stem cells, hierarchies, development, growth and repair
Stem cells (SCs) are defined as cells that can self-renew, produce different cell types during a cell division process known as ‘differentiation’, and re-generate the tissues from which they were generated [Reviewed in 1]. These properties are not shared by non-SCs [2]. SCs have the capacity for long-term proliferation in the undifferentiated state to perpetuate the SC pool throughout life (self-renewal). Depending on the body’s requirements, SCs can produce two undifferentiated cells through symmetrical self-renewal or two differentiated cells through symmetrical differentiation. Additionally, SCs often produce one undifferentiated cell and one differentiated cell simultaneously, in a process referred to as ‘asymmetric division’. The function of asymmetric division is to retain the pool of self-renewing cells while producing differentiating cells [3–5]. SCs use extensive rounds of self-renewal and differentiation to produce de novo tissues in the embryo and for growth and repair of tissues post-embryonically.
SCs are primarily characterised by their potency, a term used to refer to the number of cell and tissue types they can produce through differentiation. SCs are broadly categorised as Embryonic SCs (ESCs) and adult SCs. ESCs are found in the inner cell mass of the developing blastocyst and their primary function is to produce the tissues that compromise the body [6–8]. This property is referred to as pluripotency, which is defined as the ability to produce cells representative of all three germ layers (endoderm, mesoderm and ectoderm [9]). In contrast, adult SCs are located within specific niches in each adult tissue and function to produce new cells for growth and repair. Adult SCs are generally multipotent, which refers to their ability to generate several related cell types of relevance to their location. The best studied examples of the adult SC are the bone marrow SCs (BMSCs) of which there are two types: haematopoietic SCs, which produce the different types of blood cell, and mesenchymal stem/stromal cells (MSCs), which produce bone-related structural cells such as adipocytes, chondrocytes and osteoblasts [10].
In recent years it has become clear that SCs produce their differentiated progeny through one or more intermediaries known as (‘committed’) ‘Progenitors’. Progenitors are themselves SCs (can self-renew and differentiate), and are the work horses of tissuegenesis. However, progenitors are less potent than the parent SC that produces them, and in healthy tissues have a more limited proliferation potential [Reviewed in 1]. The concept of hierarchical arrangement of SCs was first described in bone marrow research. It is now known that HSCs and MSCs generate their repertoire of cell types through a number of intermediaries [11–14]. For example HSCs produce lymphoid and myeloid progenitors, which respectively develop to produce lymphocytes and myeloid cells such as red blood cells, neutrophils and macrophages. The Stem-Progenitor-Differentiated cell model has complicated SC analysis and, in particular, the identification and isolation of novel SCs. This is because it is now understood that most tissues contain multiple different SC types acting independently and inter-dependently. Unfortunately for CSC research, tumour tissue is similarly complicated, which has hindered clinical translation of CSC-targeting.
Cancer stem cells, hierarchies and tumourigenesis
The concept of the CSC dates back to the study of the gonadal tumour ‘Embryonal Carcinoma’ (EC) by pathologists in the late 1890s [15, 16]. In describing the tissues within ECs as a disorganised caricature of the embryo, it was proposed that these tumours developed from ESC-like pluripotent cells. With the development of immune-compromised animal models in the 1960s, Kleinsmith and Pierce [9] showed that a single EC (stem) cell, the malignant counterpart of an ESC [15, 16], was sufficient for tumourigenesis. Although the term CSC was not used at the time, this was the first experimental proof of a CSC, and was pivotal to the Leukaemia SC (LSC) work that led to the declaration of the CSC theory and the current intense interest in CSCs. EC research was complimented by extensive LSC research, where the term CSC was first described. Following identification of LSCs by Bonnet and Dick [17] as the tumour-initiating cells of acute myeloid leukaemia, CSCs were later isolated from solid tumours, such as breast cancer and brain tumours [18, 19]. Since these seminal publications, CSCs have been isolated from many solid cancers, including lung, colon, prostate, ovarian cancer and melanoma, among others [20–24]. It is now well-established that tumour-initiating cells from many, if not all, malignancies share some of the properties of SCs [1, 25, 26]. Today, CSCs are defined by the SC properties of self-renewal, differentiation and the ability to efficiently (from low cell numbers) re-generate their original malignancy in vivo. Additionally, CSCs are known to resist standard interventions such as chemotherapy and radiotherapy [Reviewed in 27].
In 2001, a seminal article by the Weissman group highlighted the need to develop CSC-targeting strategies to complement existing anti-cancer treatments [25]. It was proposed that the persistence of CSCs post-intervention was a likely explanation for recurrence. In the early days of CSC Theory, tumours were modelled as broadly consisting of a small population of tumour-initiating CSCs, surrounded by their differentiation progeny, which formed the bulk of the tumour. However, more recently this has been complicated by an appreciation that CSCs, like SCs, are organised hierarchically [Reviewed in 26]. The organisation of CSCs as Stem-Progenitor-Differentiated cell hierarchies was first described in LSCs [17]. However, in the 20 years since, relatively few hierarchies were characterised in specific malignancies. This was primarily due to the difficulty associated with identification and hierarchy-elucidation of specific CSCs from the heterogeneous population of CSCs found in tumours. Thankfully, rapid advances in technologies such as flow cytometry in recent years have permitted elucidation of CSCs in cancers such as skin, glioblastoma and liver [28–31]. Current models indicate that CSCs are organised as tree-like hierarchies (Fig. 1). At the top of the CSC hierarchy, the ‘Apex CSC’ is believed to spend very little time in an active state and, instead, primarily resides in a quiescent G0 state outside the cell cycle. In contrast, quiescence does not appear to be a property of progenitors CSCs. Mechanistically, it is believed that tumour-initiation involves three different stages (Fig. 1). First, apex CSCs enter a highly proliferative state, which results in the production of a population of lower potency Progenitor CSCs that is sufficient to continue tumour development. Secondly, apex CSCs enter quiescence while CSC progenitors undergo extensive asymmetric division to produce the mature, specialised cells that constitute the bulk of the tumour. Finally, CSC progenitor function slows while the tumour enters a process of maturation, where differentiated cell structures such as vasculature are completed. This may indicate a link between CSC activity and tumour grade: tumours that have not entered this maturation stage present as higher grade malignancies and are associated with worse prognosis due to their more proliferative phenotype.
Fig. 1.
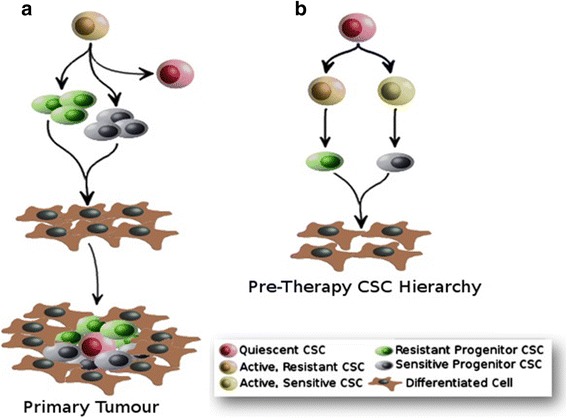
The Role of the CSC Hierarchy in Tumourigenesis. a Cancer Stem Cell (CSC) Theory indicates that tumourigenesis begins with rapid proliferation of stem cell-like tumour-initiating cells known as CSCs (Orange). Once a pool of CSCs has been established, less-potent ‘Progenitor CSCs’ are produced via differentiation, which our data indicate can be sensitive (white) or (green) resistant to conventional cancer therapies. These Progenitor CSCs differentiate to produce the mature cells (Brown) that constitute the bulk of the primary tumour. As the tumour becomes established, active CSCs (Orange) can enter a dormant state known as Quiescence (Red). b We propose an alternative model for hierarchical CSC structure where both CSCs and Progenitors can be sensitive or resistant to standard cancer therapies. Clinical-targeting of all CSC and Progenitor types is likely to compromise tumourigesis, which is an attractive clinical strategy. However, to efficiently treat refractory disease, this model suggests that it may be important to identify, model and target the specific therapy-resistant component(s) of the CSC hierarchy
Ovarian cancer hierarchy
An interesting and illustrative example of the complex organisation of CSCs as hierarchies is the elucidation of these aspects of ovarian cancer in recent years. To date, several groups have described ovarian CSCs (ovCSCs [32–36]). However, many of these ovCSCs express different CSC markers, which reflect their identification in different models of a malignancy that is now considered to be a collection of diseases. For example, Chen et al. [35] and Silva et al. [33] have described CD44+/CD117+ and ALDH+/CD133+ ovCSCs, respectively (CD: ‘Cluster of Differentiation’; ALDH: Aldehyde Dehydrogenase). Subsequently, several studies have described ovCSCs based on comparison of cells expressing specific markers [32–36]. Despite the diverse nature of the markers, many ovCSC studies were combined in a consensus model for ovarian cancer proposed by Burgos-Ojeda et al. [37]. In recent years, the first ovCSC hierarchy has been described [33, 38]. In this work, single cell asymmetric division (SCAD) assays [Reviewed in 1] were used to describe a four-population hierarchy based on the expression of the stem cell markers ALDH and CD133. In this Stem-Progenitor-Differentiated cell model, highly-tumourigenic ALDH+/CD133+ CSCs can produce less-tumourigenic ALDH+/CD133− Progenitor CSCs, which can in turn produce non-tumourigenic ALDH−/CD133− Differentiated CSCs.
Cancer stem cells and therapy-resistance
In early CSC Theory there was already an appreciation that CSCs were therapy-resistant [25]. The clear implication from this was that therapy-resistant CSCs could survive clinical intervention to regenerate recurrent disease through their tumour-initiation properties. It was proposed that therapy-resistant CSCs should be targeted in combination with conventional interventions as part of an overall anti-cancer strategy [25]. In subsequent years, this was largely interpreted as an indication that all CSCs were broadly resistant to all therapies. In the absence of models of CSC hierarchies, this was a reasonable view. This model was furthered by considerable evidence across many malignancies that indicated that whenever a CSC was identified, it displayed therapy-resistant properties. For example, it was found that cells positive for the CSC marker ALDH isolated from lung cancer cells lines demonstrated a high resistance to multiple chemotherapeutic agents (Cisplatin, Gemcitabine, Vinorelbine, Docetaxel, Doxorubicin and Daunorubicin) when compared to ALDH− cells [39]. These chemoresistant CSCs can then re-establish the tumour following chemotherapy. However, it is now understood that tumours are composed of one or more CSC hierarchies, which are composed of multiple CSC-types [40, 41]. With the development of improved analysis tools, new questions can be asked. For example, it is important for clinical intervention that the relative therapy-resistance properties of different types of CSC within the tumour are assessed. An improved understanding of the relationship between CSC hierarchies and therapy-resistance can only aid the development of improved technologies. For example, it is established that SCs and CSCs from the same tissue type share many of the same self-renewal and differentiation regulatory mechanisms, which makes it difficult to target CSCs without damaging the non-malignant SC pool [42]. In a striking example of this, inhibition of Wnt Signalling as an anti-cancer treatment had devastating effects on Wnt-regulated normal development in pre-clinical studies [41]. A better understanding of the relationship between CSCs and therapy-resistance is needed to target CSCs specifically.
Cancer stem cell hierarchies pose additional considerations for therapy-resistance
Modelling hierarchical CSC organisation is challenging. As a result, the relationship between CSC hierarchies and chemoresistance has, arguably, not been studied, which leaves several key questions open [1]. Specifically, these key questions are based upon our hypothesis that specific properties such as chemoresistance may be due to one or more specific members of a CSC hierarchy (Fig. 2). For example, which cell do we need to target: therapy-resistance might be a property of the apex CSC or the progenitor cell, or both? Are CSCs inherently resistant or do they adapt to therapy over time? How can we identify and specifically target therapy-resistant CSCs?
Fig. 2.
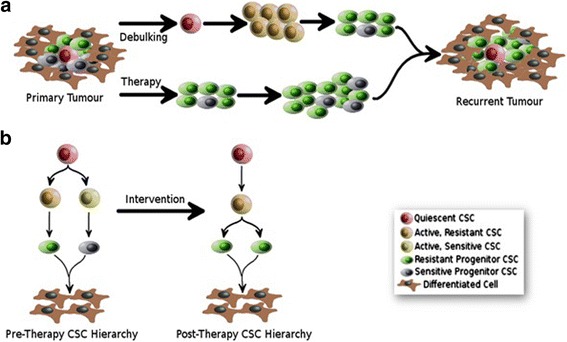
Clinical Implications of a Dynamic CSC Hierarchy Model. The model shown proposes that CSC hierarchies are altered by and adapt to clinical intervention, which poses additional consideration for clinical targeting of CSCs. a Many malignancies are treated by surgical removal of the tumour mass (debulking) and/or therapy (chemotherapy, radiation-therapy etc.). Contemporary CSC Theory suggests that debulking may cause an awakening of quiescent CSCs (Red). In parallel, anti-cancer therapies are likely to kill off therapy-sensitive CSCs (Yellow, White). This model proposes that this is likely to result in the formation of a recurrent tumour that is dominated by therapy-resistant CSCs (Orange) and Progenitors (Green). b This model proposes that CSC hierarchies are dynamic, particularly when challenged with therapeutic interventions. As a result, identification and targeting of specific therapy-resistant components within the malignancy may improve the treatment of recurrent disease
When considered in the context of the hierarchical organization of CSCs, the CSC(s) responsible for therapy-resistance must be characterized as either apex CSCs or progenitor CSCs, as this will determine the logical targeting strategy. Where these are apex CSCs, a direct target strategy is appropriate, as this should compromise the entire hierarchy. However, where these are found to be progenitor CSCs, the apex CSC that produces ‘therapy-resistant CSCs’ must also be identified, studied and targeted. By targeting both cell types simultaneously, treatments would ensure the removal of the CSC/progenitor responsible for therapy-resistant disease and the apex CSC responsible for their replacement. It is also important to note that the removal of active CSCs via chemotherapy stimulates quiescent CSCs into activity to drive further tumourigenesis [44, 45], Fig. 2]. To address this challenge, ideally both the active and quiescent CSC populations should be targeted. However, quiescent cells are by definition difficult to identify, isolate and study. An alternative for future treatments is a ‘Proliferate to Kill’ strategy, which aims to stimulate the quiescent CSC population, forcing them to re-enter the cell cycle, thus exposing them to standard therapeutic intervention [1].
CSCs employ a collection of therapy-resistance mechanisms
The description of CSC therapy-resistance mechanisms in different malignancies in recent years [46] suggest a generalised vision of CSC therapy-resistance composed of five components: A) Quiescence; B) Detoxification/Multi-Drug Resistance (MDR); C) Repair of damaged DNA; D) Survival; E) Adaptation. As it emerges that these traits can be shared by CSCs from different malignancies, it is important to consider the nature of the underlying biology driving these mechanisms. One prospect is regulation by stemness signalling pathways such as Wnt and Notch, which are known to regulate several key mechanisms in many different types of SC and CSC. We propose that it would be prudent to consider each generalised set of mechanisms, which are now discussed in detail, when generating CSC models and designing clinical targeting strategies.
Quiescent CSCs are insusceptible to conventional therapies
Contemporary SC Theory indicates that SCs are organised as hierarchies and that the apex SC at the top of the hierarchy can become quiescent [44, 47]. As quiescent cells reside outside the cell cycle they are broadly insusceptible to conventional therapies, which target the mitotic mechanisms of rapidly-dividing cells [48]. As such, ‘Quiescence’ is the first CSC therapy-resistance property. In more recent years, the description of two further mechanisms, DNA-monitoring and DNA repair during quiescence, offers further possibilities for the complexity of CSC therapy-resistance. In the first set of studies it has been shown that, rather than simply being inactive, quiescent SCs actively monitor DNA damage [49]. The strongest evidence for this mechanism is the demonstration that loss of key cell cycle regulators such as p53, p21, p57 or the Retinoblastoma protein impairs quiescence [50–53]. In addition, two related but separate studies have indicated that quiescent SCs are primed for re-entry in to the cycle in a process that involves DNA repair. Specifically, SCs have been shown to transition from G0 to G1 of the cell cycle via a ‘GALERT’ state that is primed for rapid cell cycle re-entry in response to stress [54]. Complementing this study, it has recently been shown that quiescent HSCs can accumulate DNA damage and that many of these damaged HSCs are repaired during cell cycle re-entry rather than being signalled to undergo apoptosis [55]. These studies suggest that the quiescent CSC may possess a collection of DNA repair mechanisms that offer protection against therapeutic intervention. Despite the difficulty associated with modelling non-dividing cells, there are indications that quiescent stem-like populations contribute to at least some tumours. For example, in primary ovarian tumours it was demonstrated that CD24+ CSCs were slower proliferating but more tumourigenic than bulk cells, suggesting a quiescent, tumourigenic CSC population [56]. In addition, quiescent CSCs that survive chemotherapy can re-enter the cell cycle and re-establish the tumour [44, 57, 58].
CSCs can detoxify in response to chemotherapy via multi-drug resistance efflux pumps
It is now established that many CSCs possess MDR mechanisms that function to detoxify the cell in response to chemotherapy. Multi-Drug Resistance is a collective term for efflux mechanisms that allow chemotherapy drugs to be pumped out of the cell before DNA damage can occur [59]. The best characterised MDR proteins are the members of the ‘ABC (ATP-Binding Cassette) transporter’ family, which were originally described in bacterial anti-biotic resistance [60]. It was subsequently found that many cell types possess ABC transporters. ABC transporters have been described in many types of cancer cells, where they act to efflux a wide array of chemotherapeutic drugs [61–63]. Furthermore, ABC transporters have been shown to be associated with chemoresistance of CSCs in several malignancies, including ovarian, breast, colon, and non-small cell lung cancers [64–67]. For example, the ABC transporters ABCB1 and ABCG2 have been associated with drug resistance in ovCSCs [64, 68]. As such, MDR is the second CSC therapy-resistance mechanism. Unfortunately, attempts to develop clinical-targeting of MDR mechanisms have not been successful to date [69, 70]. It appears that targeting specific ABC transporters leads to activation of redundant ABC transports to continue the MDR mechanism, which has hindered their exploitation as clinical targets [71].
CSCs repair therapy-induced DNA damage via an enhanced DNA damage response
The third broad category of CSC therapy-resistance mechanism is the repair of DNA damage induced by chemotherapy and/or radiotherapy. In addition to those DNA repair mechanisms expressed by quiescent CSCs (described above), active cells can elicit a ‘DNA Damage Response’ (DDR [Reviewed in 72, 73]). There is now a broad appreciation that many therapy-resistant CSCs display elevated capacity for detection and repair of DNA damage, which allows them to survive via resistance to DNA damage induced by cancer therapies. Platinum-based chemotherapy is based upon forcing the formation of intra- and inter-strand DNA crosslinks (ICLs) upon the rapidly-diving cancer cell. ICL-formation disrupts chromatin structure, resulting in stalling of the replication fork and activation of several DDR pathways. In sensitive cells, the formation of multiple ICLs is beyond the cells DDR capacity. As a result, unrepaired DNA lesions cause cell-cycle arrest via apoptosis either directly or following DNA replication during the S phase of the cell cycle [74]. Pathways that have been shown to be involved in platinum-induced DNA damage include the Nuclear Excision Repair (NER) and the FA/BRCA pathway, each of which are now described in detail below. Similar DDR mechanisms are responsible for resistance to DNA-damage induced by radiotherapy. In contrast, taxane-based chemotherapies target microtubule dynamics during mitosis. Targeted microtubules cannot be repaired by the cell, which results in apoptosis via the G2/M checkpoint. Taxane-resistance is due to mutations in the tubulin sub-units that comprise microtubules [71], rather than therapy-resistance mechanisms. As such, taxane-resistance is not discussed in this review.
Nucleotide excision repair
Intra-strand crosslinks, the most abundant lesion, are repaired via NER [75], a DDR mechanism that is often overexpressed in therapy-resistance disease. Mechanistically, NER involves recognition and excision of single-strand DNA damage. The remaining undamaged single-strand DNA is used as a template for DNA synthesis, which is followed by ligation to complete the DDR process. Excision is facilitated by the ERCC1 (excision repair cross – complementation group 1) protein. Overexpression of ERCC1 contributes to platinum-resistance in many cancers, such as ovarian, non-small cell lung and testicular cancers [76–78]. Recent studies indicate a link between overexpression of NER proteins and to CSC chemoresistance [79]. However, this has not definitively been demonstrated. Overexpression of ERRC1 may be a mechanism of CSC therapy-resistance, as elevated mRNA and protein levels of ERRC1 have been shown in platinum-resistant oral CSCs [80]. However, Wang et al. [81] have demonstrated that NER protein levels are unrelated to platinum-sensitivity in ovarian cancer. Furthermore, knockdown of NER factors compromised NER efficiency, but caused only a minimal effect on platinum-sensitivity [81].
The fanconi anemia/BRCA DNA damage repair pathway
The detection and repair of ICLs also involves the cooperation of the Fanconia Anemia (FA) and Breast Cancer 2 (BRCA) pathways, which are now often referred to as the FA/BRCA pathway. In this mechanism, DNA damage is detected and removed by the FA pathway, and DNA synthesis subsequently completed by the BRCA-regulated homologous recombination (HR) pathway [72, 73]. FA is an uncommon disorder characterised by congenital abnormalities, progressive bone marrow failure, and cancer susceptibility [82, 83]. FA was found to be due to abnormalities in a novel DDR mechanism that became known as the FA pathway. Subsequently, the FA pathway was found to be associated with the development of cancers in FA patients [84] and therapy-resistance in cancer generally [85, 86]. The FA/BRCA pathway begins with recognition of ICLs by the FA complementation group complex (FANC), which is composed of multiple FA proteins (A, B, C, D1, D2, E, F, G, I, J, L, M and N). The connection between the FA pathways was originally highlighted by studies showing that FANCD1 and BRCA2, and FANCS and BRCA1 are the same genes. Upon ICL detection, the FA pathway is activated by the monoubiquitination of FANCD2 and FANCI. FANCD2 is subsequently targeted to the damaged chromatin site where it interacts with the BRCA2 protein, a key regulator of HR [Reviewed in 72, 73]. This interaction appears to be required for normal HR and ICL repair. The molecular relationship between monoubiquitinated FANCD2 and BRCA2 in the HR component of DDR is not known: it is speculated that the proteins may cooperate in the timed release of RAD51 at sites of DNA repair [87, 88].
Activation of the FA/BRCA pathway culminates in repair of ICLs, UV-induced dimers, and double-strand breaks [89, 90], which plays an important role in the acquisition of drug resistance in many cancers, such as multiple myeloma, glioma, cervical and ovarian cancer [91–94]. Recent studies indicate that overexpression of FA/BRCA pathway proteins is also linked to CSC chemoresistance. For example, Meng et al. [34] demonstrated that platinum resistant ALDH+ ovarian cancer cells express elevated levels of FA/BRCA DNA repair proteins. It is well-established that loss of function BRCA mutations leads to increased susceptibility to the development of cancer, particularly in hereditary disease. In these cases, cancer cell therapy-resistance becomes dependent upon related, functional DDR mechanisms, which can be targeted to improve clinical outcomes [Reviewed in 95]. For example, inhibition of DDR components known as ‘PARPs’ (poly ADP ribose polymerases) can enhance therapy-sensitivity in some patients [Reviewed in 95]. More recently it has been shown that functional over-expression of BRCA is associated with therapy-resistance in non-inherited disease [96–99]. Some evidence is emerging that FA/BRCA inhibitors such as the FDA approved Bortezomib can improve chemoresponse in otherwise refractory disease [100, 101].
CSCs employ anti-apoptotic mechanisms to promote survival in response to clinical intervention
The accumulation of substantial DNA damage results in the activation of apoptotic mechanisms, which must be inhibited in therapy-resistant cells. For example, it is now well-established that evasion of apoptosis is one of the major mechanisms of ovarian cancer associated platinum-resistance [102, 103]. Anti-apoptosis mechanisms contribute to therapy-resistance directly by blocking cell death and indirectly by providing time for enhanced DDR mechanisms to repair therapy-induced DNA damage. These anti-apoptotic mechanisms include the extrinsic and intrinsic pathways, and the tumour suppressor protein p53, each of which will now be discussed. Collectively, these represent a fourth, pro-survival mechanism for CSC therapy-resistance.
The extrinsic apoptotic pathway
The extrinsic apoptotic pathway is characterised by detection of death signals by ‘Death Receptors’. Death receptors are cell-surface expressed, and belong to the superfamily of tumour necrosis factor receptors (TNF-R) that are activated by TNF family ligands. The best-characterized death receptors include FAS (APO-1/CD95), TNF receptor 1 (TNFRI), TNF-related apoptosis-inducing ligand-receptor 1 (TRAIL-R1) and TRAIL-R2. Upon stimulation by the FAS ligand, FAS trimerises, which allows intracellular binding of the adaptor-protein FADD to the receptor. FADD in turn serves to recruit pro-caspases-8 and −10 into the complex, which ultimately activates downstream caspases, leading to apoptosis [104, 105]. Cancer cells have evolved numerous strategies to resist drug-induced cell death via the extrinsic pathway. For example, surface expression of death receptors have been shown to be downregulated or absent in drug resistant tumours, including platinum-resistant ovarian cancer [106]. Upregulation of FAS has been shown to reverse platinum-resistance in ovarian cancer [107]. While FAS plays a pro-apoptotic role in SCs, it appears to promote survival in CSCs [108]. Furthermore, upregulation of the adaptor-protein FADD has been shown to sensitise ovarian cancer cells to platinum treatment [109]. Although little is known yet about evasion of apoptosis as a possible CSC chemoresistance mechanism, data indicates that CSCs resist cell death via the extrinsic pathway. For example, an upregulation of TRAIL-R1 leads to chemoresistance in colon CSCs [110]. Several mechanisms have been identified in mammalian cells for the induction of apoptosis. These mechanisms include factors that lead to perturbation of the mitochondria leading to leakage of cytochrome C or factors that directly activate members of the death receptor family. Epothilones are a new group of compounds with action mechanisms similar to taxanes. In an ovarian cancer model Epothilone A and B have been shown to induce cell death via a decrease in mitochondrial membrane potential [111]. This may represent a future clinical strategy.
The intrinsic apoptotic pathway
The intrinsic apoptotic pathway induces cell death by affecting mitochondrial permeability, which leads to the release of cytochrome C to activate caspases. Among its many regulators, the pathway is primarily regulated by members of the BCL-2 family [112, 113]. The protein BCL-2, the founding member of the BCL-2 family, primarily mediates its pro-survival effects by binding to the pro-apoptotic partners BCL2-associated-X-protein (BAX) and BCL-2 homologous antagonist killer (BAK), which decreases release of cytochrome C from the mitochondria. BCL-2 family members are overexpressed in many solid tumours and have been linked to tumourigenesis, cancer cell survival and chemoresistance [109, 114, 115]. Recent studies show that BCL-2 overexpression is also linked to CSC chemoresistance. For example Madjd et al. [116] showed that the protein BCL-2 is highly expressed in CD44+/CD24-/low breast CSCs. To date, little is known about the mechanism, but CSC research suggests that these proteins can affect chemoresistance through induction by other signalling pathways required for CSC survival. For example, Ma et al. [117] demonstrated that BCL-2 induction by AKT1 may be a mechanism by which CSCs can mediate chemoresistance.
The role of p53 in apoptosis
The tumour suppressor p53 acts as the overall regulator of the activation of apoptotic processes in response to the detection of unrepairable DNA damage [118, 119]. Like BRCA, ‘loss of function’ p53 mutations are associated with the development of various malignancies, while its over-expression in cancers with functional DDR mechanisms is associated with therapy-resistance. In addition, numerous chemotherapy drugs exert their function through targeting p53-related signalling pathways. Therapy-induced DNA damage leads to activation of p53, which binds to the regulatory sequences of a number of target genes to initiate a program of cell cycle arrest and DDR. If the drug-induced damage cannot be repaired completely, over-activation of p53 leads to tumour growth stagnation or even apoptosis via the induction of the intrinsic and extrinsic apoptosis pathways [120–122]. Loss of p53 function occurs during the development of most, but not all, tumour types. This results in tumour cells being able to escape drug-induced apoptosis [122]. A downregulation of p53 has been shown in platinum-resistant ovarian cancer [109]. However, recent studies show an alternative mechanism where p53 overexpression is linked to CSC chemoresistance. For example, it was shown that overexpression of p53 sensitises glioblastoma CSCs to drug treatment trough enhanced apoptosis [123].
CSC hierarchies can adapt to survive clinical intervention
The principles governing the relationship between CSC hierarchies and therapy-resistance are poorly understood. Our most recent work has highlighted that therapy-resistance is not the property of all CSCs within a hierarchy: therapy-resistance can be CSC- and drug-type specific (Ffrench et al. Unpublished). Furthermore, we have found that progenitor CSCs can adapt to platinum-treatment by altering their potency (producing a different cell type via differentiation). This data indicates that, in at least some cases, CSC models contain more than one type of CSC, some of which are therapy-sensitive and some therapy-resistant. This data suggests a model (Fig. 2) where the treatment-naïve CSC hierarchy can contain therapy-sensitive and therapy-resistant CSCs. Post-intervention, sensitive CSCs are killed off, which results in a recurrent tumour and CSC hierarchy with increased numbers of therapy-resistant CSCs. In addition, removal of the tumour by surgical de-bulking may awaken quiescent CSCs to synergistically enhance the production of the therapy-resistant recurrent malignancy. This may explain why initial pre-clinical success has not broadly translated to the clinic (Fig. 2). As such, it is important to identify, study and target the specific CSCs responsible for specific types of therapy-resistance within specific malignancies.
Altered CSC-related signalling pathways in therapy-resistance
SC function is regulated by a number of signalling pathways such as Wnt and Notch [1], which are commonly dysregulated in therapy-resistant CSCs [124]. However, targeting of these pathways can have adverse effects on similarly-regulated non-malignant SC pools. The involvement of stemness signalling pathways such as Wnt and Notch in CSCs from multiple different types of malignancy is now described and suggests an important role in CSC therapy-resistance. It is tempting to speculate that these stemness signalling pathways may facilitate coordinated CSC therapy-resistance mechanisms such as quiescence, detoxification (MDR), repair (DDR), survival (anti-apoptosis) and adaptation.
The Wnt signalling pathway has been shown to play an important role in the maintenance of SCs and lineage differentiation in a wide array of tissues and organs [125]. Altered expression of this pathway has been shown in many cancers, such as ovarian, breast and colon [126–128]. While the precise mechanism has not been fully elucidated, elevated levels of Wnt Signalling modulator β-catenin have been shown to actively contribute to platinum-resistance in ovarian cancer cells [129]. Hepatic CSCs have been shown to exhibit enhanced platinum-resistance that could be reversed by lentiviral microRNA knockdown of β-catenin [130]. It is not clear how Wnt signalling mediates CSC chemoresistance but upregulation of ABC transporters presents one potential mechanism: ABCG2 resistance to both platinum and taxanes could be reversed by β-catenin siRNA knockdown in c-kit+ ovCSCs [131]. Furthermore, it was shown that inhibition of the Wnt signalling pathway, in combination with platinum, induced cytotoxicity and cell cycle arrest in a higher percentage of primary ovarian samples than with single drug treatment [126]. Because of the molecular similarity between SCs and CSCs from the same region of the body it is very difficult to target CSCs without damaging the non-malignant SC pool, a side-effect that would have devastating growth and repair consequences for the patient. In a stark example of this, Wnt inhibition as an anti-cancer treatment has been shown to have devastating effects on Wnt-regulated normal intestine development in pre-clinical studies [43].
The Notch signalling pathway plays a role in both CSC maintenance and drug resistance in many malignancies [Reviewed in 1]. For example, Notch proteins have been identified to be upregulated in ovCSCs [132]. Furthermore, in the same study a γ-secretase inhibitor (GSI), which inhibits Notch signalling, was shown to erase CSCs and increase tumour sensitivity to platinum-agents. Most importantly, it was found that platinum/GSI combination compared to single treatment was more effective to eliminate both CSCs and the bulk of tumour cells, indicating that a dual combination targeting both populations is needed for tumour eradication. In addition, the group found that the Cisplatin/GSI combination therapy has a synergistic cytotoxic effect in Notch-dependent tumour cells by enhancing the DNA-damage response, G2/M cell-cycle arrest, and apoptosis [132]. The superior effectiveness of a combination of chemotherapy drug and Notch-targeting CSC-inhibitor (GSI) compared to monotherapy in an otherwise refractory disease, has also been reported in pancreatic metastasis [133]. As such, it is emerging as an early paradigm for how CSC-targeting should be conducted as part of an overall treatment [1].
In the absence of other evidence, stemness signalling pathways appear to be likely candidates for a role in coordinating the multiple responses of therapy-resistant CSCs to clinical intervention. We propose that all of the mechanisms described in this section should be considered during analysis of CSC models. An improved understanding of the mechanisms of CSC therapy-resistant may facilitate development of novel treatment strategies. This is discussed in detail later. However, before they can be analysed, CSC models must be built.
CSC discovery: identifying the therapy-resistant components of CSC hierarchies
While it is well-accepted that CSC-targeting must be assessed as a novel therapeutic avenue, little progress has been made in improving survival rates in the clinic. We believe that this is partially due to our failure to identify specific therapy-resistant component in the context of CSC hierarchies. We hypothesise that it cannot simply be assumed that each component of a CSC hierarchy contributes to therapy-resistance equally. Addressing this, we have recently built a novel, four-component CSC hierarchy and assessed platinum- and taxane-resistance in each, individual CSC type. Our data indicate that platinum-resistance is the property of only one CSC-type within that hierarchy and that all four components are taxane-sensitive. Additionally, we found that repeated exposure to platinum results in an altered hierarchy with altered plasticity, with a different CSC type taking the ‘Apex’ position at the top of the hierarchy (Ffrench et al. Unpublished). These findings impose new considerations upon CSC research, which are now discussed in the context of our perspective on CSC Discovery and clinical targeting.
CSC discovery
We propose that therapy-resistant CSC Discovery should be conducted in paired therapy-sensitive and therapy-resistant models. In our opinion, this is one of most over-looked factors in CSC Discovery and is a large determinant of success [1]. For a detailed description of our approach to CSC Discovery the reader is referred to [1]. Briefly, CSC Discovery should begin with a model that addresses a clinical problem. For example, if a particular malignancy is characterised by high levels of mortality due to platinum-resistance, CSC Discovery is best undertaken in paired cell models of platinum-sensitivity and platinum-resistance in that malignancy. Many available cell line models are therapy-naïve and can be easily rendered therapy-resistant by exposure to increasing doses of the therapy over time. While these cell lines are not a perfect facsimile of in vivo disease, many studies have demonstrated that they are readily available sources for CSC Discovery. Broadly, CSCs have been successfully identified through screening for SC markers such as ALDH and ‘Cluster of Differentiation’ (CD44, CD133 etc.), and SC properties such as efflux (Hoechst dye efflux ‘side population’ assay) and non-adherent growth (‘spheroid formation’) assays. Once identified and validated, these CSCs are available for in vivo validation in clinical samples. Subsequently, CSC hierarchy relationships can be elucidated as now described.
Building a therapy-resistant hierarchy
The rapid advances in flow cytometry-based technology in recent years mean that building CSC hierarchies is now much more achievable than it has been in the past. In this regard, we have found the SCAD assay particularly useful [1]. In this assay, single cells positive and negative for identified SC markers are allowed to form colonies, which are then tested for the presence of the marker: where both positive and negative cells are found in the colonies, this is suggestive of SC properties. Importantly, SCAD assays can easily be scaled to include two or more markers. For example, where two SC markers (A and B) are identified in screens, a four component hierarchy (A+/B+, A−/B−, A+/B−, A−/B+) can easily be built. Characterising of the colonies formed allows the relationship between each CSC type to be established, and an accurate model of the CSC hierarchy to be identified, which may include the apex CSCs, and inter-related and independent progenitors [1]. For example, and SCAD approach has recently been used to describe an ovCSC hierarchy based on the expression of ALDH and CD133 [38]. In this model, highly tumourigenic ALDH+CD133+ CSCs give rise to somewhat less tumourigenic ALDH+CD133− CSC/progenitors, which in turn give rise to non-tumourigenic ALDH−CD133− cells. Once validated in standard xenograft-formation assays, the platinum-resistant component of this hierarchy could be identified, and the mechanism identified as now described.
Identification of the therapy-resistant component of a CSC hierarchy, and its mechanism
The standard approach to the identification of therapy-resistant cells is to test populations for survival following treatment with chemotherapy or radiotherapy. As such, measuring the relative contribution to resistance of each cell type in a CSC hierarchy appears to be a daunting task. However, flow cytometry’s ability to accurately measure the relative proportion of sub-populations within a CSC hierarchy provides an elegant solution. This approach can be adapted for CSC hierarchy research purposes by simply assaying the post-treatment CSC hierarchy for the presence of its original SC markers via flow cytometry. This approach easily facilitates quantification of the relative therapy-resistance of each individual CSC type within the hierarchy, which is a strong indication of their proportional contributions to therapy-resistance. A therapy-sensitive model can be assessed pre- and post- acute treatment, and the resistance of specific CSC populations highlighted by an increase in their respective proportions. Furthermore, chronic treatment of the sensitive model over time can be used to generate a paired resistant model, which can be reanalysed before and after acute treatment. Such a matrix of comparisons will point towards the identity of the intrinsically and adaptively resistance cell populations within the models being investigated.
This analysis can be carried out on both the entire hierarchy or individual CSC types present in sufficient numbers to allow purification for assays. As a caveat, we have noted that the quantitative tolerance of resistant CSC types can be higher when assayed in isolation compare to within the hierarchy as a whole. This is likely due to the absence of pro-apoptotic signalling from therapy-sensitive CSCs that are absent when a therapy-resistant CSC is assayed in isolation. Once identified, the specific mechanisms expressed by individual therapy-resistant CSC types can be identified using standard gene expression assay or RNAseq approaches. For example, our most recent work has highlighted elevated expression of MDR, DDR and anti-apoptotic mechanisms in platinum-resistant CSCs compared to platinum-sensitive CSCs from the same hierarchy (Ffrench et al. Unpublished). As a final consideration, we have found that specific MDR, DDR and anti-apoptotic mechanisms are expressed in treatment-naïve and platinum-adapted models, as discussed above. It is clear that this observation would likely have been missed had our work been undertaken in a single model. This is strong evidence for the undertaking of CSC Discovery in paired therapy-sensitive and therapy-resistant models.
Clinical perspective: towards targeting of therapy-resistant CSCs
Early CSC Theory highlighted several principles on which clinical targeting of CSCs should be based. At the time, there was great hope that CSC targeting in the clinic could dramatically improve the prognosis of refractory disease. In spite of this, the translation of CSC-targeting to the clinic has not been as efficient as had been hoped. In this section we will describe some of the clinical implications of our contemporary understanding of CSC Theory. In particular, we will discuss how our clinical-targeting strategies must allow for the model of CSC hierarchies as moving, rather than static, targets.
Clinical considerations from early CSC theory
CSC theory as presented by Reya et al. [25] highlighted several clinical implications of relevance to our understanding of how CSCs influence therapy-resistance. Firstly, CSCs were identified as clear clinical targets due to their role in primary, metastatic and recurrent disease. Secondly, low numbers of CSCs persisting post-intervention were sufficient to drive recurrent disease. As the CSCs responsible for this repopulation were by definition therapy-resistant, it was suggested that this property would be passed on to the entire progeny of the tumour reinitiating CSCs. This theory, therefore, could account for the higher levels of therapy-resistance found in recurrent disease compared to primary tumours. Thirdly, CSCs were originally thought to be rapidly-dividing and thus particularly susceptible to standard interventions, which are designed to target rapidly dividing (cancer) cells. The fact that this was not the case in the clinic provided evidence that CSCs possessed strong therapy-resistance properties, which were postulated as mechanisms such as enhanced MDR and DDR. Finally, one of the most contentious suggestions at this point was that CSCs were not simply one of many different types of tumour-initiating cell but were the only known tumour-initiating cell. Indeed, to this day an efficient, tumour-initiating cell de-void of stem cell properties has not been reported to our knowledge. This implication presents CSCs as the primary culprit for circulating tumour cells that facilitate metastasis. As such, targeting CSCs should produce the additional benefit of reducing metastasis. These concepts were widely accepted by the cancer research community, which held out great hope that CSC targeting could be developed as part of a combined cancer treatment strategy, which would be particularly suited to refractory disease. Strategically, it was believed that CSCs should be targeted during primary treatment to prevent metastasis and recurrence and the development of therapy-resistance.
This model suggested a strategy where cancer treatment could be improved by removing stemness properties from CSCs globally. This so called ‘Forced Differentiation’ approach has been best examined using differentiation morphogen all-trans retinoic acid (ATRA), which has being explored in a variety of haematological malignancies, including myelodysplastic syndromes, multiple myeloma and chronic myelogenous leukemia [134, 135]. Alone or in combination with other therapies, ATRA is also being investigated in a variety of solid tumours including prostate cancer, breast cancer and glioma [136–138]. Although pharmacological doses of retinoids have proven effective in the treatment of haematological malignancies [139], clinical trials in the prevention and treatment setting in a number of solid tumours have failed to show significant benefit to date [68, 140]. For example, clinical trials in breast cancer have shown moderate potential, but consistently low response rates of below 30% [141, 142]. This is one example that illustrates how inefficient CSC-targeting approaches have been to date. This suggests that targeting CSCs globally may not be an efficient strategy in all but a few, specific malignancies. In contrast, our contemporary understanding of CSCs suggest that specific targeting strategies may be more successful, which is now discussed in detail.
Contemporary CSC theory suggests New considerations for clinical intervention
The important CSC-targeting principles highlighted by Reya et al. [25] provide the foundation upon which contemporary CSC Theory has been built. Early CSC theory suggested that tumour heterogeneity could be broadly divided in to two cell types, namely CSCs and differentiated cells. In the intervening years it has become clear that heterogeneity in at least some, and perhaps many or all, malignancies is due to the presence of multiple CSC types arranged as hierarchies [26]. There is clear evidence that the identification and targeting of a single CSC-type has potential as a clinical strategy. Perhaps the most striking example of this was described in ovarian cancer, where targeting of Notch signalling in ovCSCs resulted in platinum-based elimination of disease in an animal model of otherwise refractory disease [132]. However, a large collection of similar studies has not been produced in other malignancies. In addition, there have been few successes in the clinic. We propose that one of the key factors behind this failure is the complex hierarchical organisation of CSCs, which complicates clinical targeting. Rather than looking for a ‘needle in a haystack’, which was challenging enough, it appears that we must now locate and target a specific, clinically-relevant needle in a collection of similar needles within the haystack. Complicating this further, it appears that CSC hierarchies are altered by and adapt in response to our interventions, making them complex moving targets.
Clinical implications of CSC hierarchy dynamics
Contemporary CSC Theory proposes that tumour heterogeneity is due to the presence of multiple CSC Hierarchies within a single tumour [26]. In addition to the concepts described here, evidence is emerging for other forms of plasticity such as the conversion of non-CSCs to CSCs phenotypes in defined circumstances [143]. This more complex view of in vivo CSC organisation has important considerations for clinical targeting. The most obvious principle is that the apex CSC(s) should be targeted in order to compromise the entire hierarchy. As discussed above, apex CSCs might only be readily targetable when they are active during early tumourigenesis, as targeting the quiescent state is currently unachievable. This suggests a strategy where targeting of CSC progenitors should substantially compromise tumourigenicity. However, as quiescent (apex) CSCs are largely insusceptible to anti-mitotic cancer treatments, targeting CSC progenitors is likely to result in an awakening of the quiescent CSC pool and rapid regeneration of the tumour (Fig. 2). This may partially explain why successful primary intervention can be followed months later by substantial, aggressive recurrent disease.
This model of recurrent disease is based upon an assumption that awakened quiescent CSCs reconstitute the primary malignancy as recurrent disease. However, in our lab we have observed that therapeutic intervention can alter the CSC hierarchy, which has important considerations for clinical targeting. Our data indicate that only some components of a CSC hierarchy are resistant to platinum. As a result, after only a few days the hierarchy is altered: the sensitive CSC populations have been dramatically reduced in size, leaving a proportionally larger resistant CSC population (Fig. 2). As a result, after only a few days of platinum treatment the CSC hierarchy observed post-intervention is substantially different to the treatment-naïve model (Fig. 2). As such, we hypothesise that CSC hierarchies within the tumour are not ‘fixed targets’: in terms of CSC organisation, the primary tumour can be quite different to the recurrent tumour. This may partially explain why primary disease can be sensitive and recurrent disease resistant to the same therapy.
Conclusion
Catching moving targets: elucidation and targeting of dynamic CSC hierarchies
Considerable advances need to be made before efficient, clinically-relevant targeting of entire CSC hierarchies can be achieved. To date, very little research has been carried out to elucidate the relationship between CSC hierarchies and therapy-resistance. We have argued that therapy-resistance within CSC hierarchies can be highly specific: therapy-resistance can be the property of a specific type of CSC and not a global property of the hierarchy; CSCs that are resistant to one type of therapy (e.g. platinum) can be sensitive to another (e.g. taxanes); CSC hierarchies can be altered as sensitive populations are killed off by our interventions; this can result in substantial alterations to the CSC hierarchy, including the moving of a different CSC type to the apex position. While these observations must be tested and validated in other malignancies, this suggests that a broader, more specific view of CSC Discovery and targeting may be more successful that global targeting approaches. This type of specificity is in line with the personalised medicine approach, which has been highly successful in the clinic in malignancies such as breast cancer.
Dynamic CSC hierarchies are best modelled using paired-models of sensitive and resistant disease that are specific for different therapeutic-interventions. The obvious application for this is in the treatment of therapy-resistant breast cancer. In recent years, breast cancer treatment has been dramatically improved by the identification of sub-types within the disease that respond best to specific therapies. However, in contrast, the understanding of the specific CSC hierarchies associated with each sub-type of breast cancer is sparse. The identification and targeting of the specific CSC hierarchies responsible for therapy-resistance in different types of breast cancer may facilitate the development of improved treatment strategies. In addressing this, it is important to note that the CSC hierarchies in each sub-type may be quite different from one another and altered post-intervention. In addition, the success in breast cancer suggests that the specific targeting of sub-types within malignancies can enhance outcomes in other refractory malignancies. It seems clear that there is great potential for the exploitation of the specificities of CSC types within sub-types of malignancy as both bio-markers for early detection and targets for the treatment of refractory disease.
Acknowledgements
The authors work is supported by the following funding bodies, whom they would like to acknowledge and thank for their support: Science Foundation Ireland (JOL), the Health Research Board (HRB, Ireland. JOL), the Emer Casey Foundation (BF), Irish Cancer Society (JOL, MG).
Funding
Not applicable.
Availability of data and materials
Not applicable.
Authors’ contributions
CG, BF, JOL and MG all contributed to the writing of and concepts described in this review. All authors read and approved the final manuscript.
Author’s information
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors have consented to publication.
Ethics approval and consent to participate
Not applicable.
Contributor Information
Claudia Gasch, Email: claudia.gasch@web.de.
Brendan Ffrench, Email: brendan.ffrecn@gmail.com.
John J. O’Leary, Email: olearyjj@tcd.ie
Michael F. Gallagher, Email: gallagmi@tcd.ie
References
- 1.Ffrench B, Gasch C, O’Leary JJ, Gallagher MF. Developing ovarian cancer stem cell models: laying the pipeline from discovery to clinical intervention. Mol Cancer. 2014;13:262. doi: 10.1186/1476-4598-13-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lajtha LG. Stem cells and their properties. Proc Canadian Cancer Conf. 1967;7:31–39. [PubMed] [Google Scholar]
- 3.Weissman IL, Anderson DJ, Gage F. Stem and progenitor cells: origins, phenotypes, lineage commitments, and transdifferentiations. Annu Rev Cell Dev Biol. 2001;17:387–403. doi: 10.1146/annurev.cellbio.17.1.387. [DOI] [PubMed] [Google Scholar]
- 4.Bajada S, Mazakova I, Richardson JB, Ashammakhi N. Updates on stem cells and their applications in regenerative medicine. J Tissue Eng Regen Med. 2008;2(4):169–183. doi: 10.1002/term.83. [DOI] [PubMed] [Google Scholar]
- 5.He S, Nakada D, Morrison SJ. Mechanisms of stem cell self-renewal. Annu Rev Cell Dev Biol. 2009;25:377–406. doi: 10.1146/annurev.cellbio.042308.113248. [DOI] [PubMed] [Google Scholar]
- 6.Evans M, Kaufman MH. Establishment in culture of pluripotent cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 7.Thomson JA, Itskovitz-Eldor J, Sharipo SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 8.Evans M. Discovering pluripotency: 30 years of mouse embryonic stem cells. Nat Rev Mol Cell Biol. 2011;12:680–686. doi: 10.1038/nrm3190. [DOI] [PubMed] [Google Scholar]
- 9.Kleinsmith L, Pierce B. Multipotency of single embryonal carcinoma cells. Cancer Res. 1964;24:1544–1551. [PubMed] [Google Scholar]
- 10.Metcalf D. The molecular control of cell division, differentiation commitment and maturation in haemopoietic cells. Nature. 1989;339(6219):27–30. doi: 10.1038/339027a0. [DOI] [PubMed] [Google Scholar]
- 11.Dick JE. Normal and leukemic human stem cells assayed in SCID mice. Semin Immunol. 1996;8(4):197–206. doi: 10.1006/smim.1996.0025. [DOI] [PubMed] [Google Scholar]
- 12.Larochelle A, Vormoor J, Hanenberg H, Wang JC, Bhatia M, Lapidot T, Moritz T, Murdoch B, Xiao XL, Williams DA, Dick JE. Identification of primitive human hematopoietic cells capable of repopulating NOD/SCID mouse bone marrow: implications for gene therapy. Nat Med. 1996;2(12):1329–1337. doi: 10.1038/nm1296-1329. [DOI] [PubMed] [Google Scholar]
- 13.Caplin AL. Mesenchymal stem cells. J Orthap Res. 1996;9(5):641–650. doi: 10.1002/jor.1100090504. [DOI] [PubMed] [Google Scholar]
- 14.Friedenstein AJ, Gorskaja JF, Kulagina NN. Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp Haematol. 1976;4(5):267–74. [PubMed] [Google Scholar]
- 15.Andrews PW. From teratocarcinom as to embryonic stem cells. Philos Trans R Soc Lond B. 2002;357(1420):405–417. doi: 10.1098/rstb.2002.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andrews PW, Matin MM, Bahrami AR, Damjanov I, Gokhale P, Draper JS. Embryonic stem (ES) cells and embryonal carcinoma (EC) cells: opposite sides of the same coin. Biochem Soc Trans. 2005;33:1526–30. doi: 10.1042/BST0331526. [DOI] [PubMed] [Google Scholar]
- 17.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 18.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 20.Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121(6):823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 21.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 22.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 23.Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R, et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci U S A. 2006;103(30):11154–11159. doi: 10.1073/pnas.0603672103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65(20):9328–9337. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 25.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 26.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 27.Chen K, Huang Y, Chen J. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34(6):732–740. doi: 10.1038/aps.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez-Losada J, Balmain A. Stem-cell hierarchy in skin cancer. Nat Rev Cancer. 2003;3(6):434–443. doi: 10.1038/nrc1095. [DOI] [PubMed] [Google Scholar]
- 29.Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123(5):1911–1918. doi: 10.1172/JCI66024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17(4):362–375. doi: 10.1016/j.ccr.2009.12.049. [DOI] [PubMed] [Google Scholar]
- 31.Oishi N, Wang XW. Novel therapeutic strategies for targeting liver cancer stem cells. Int J Biol Sci. 2011;2011(7):517–535. doi: 10.7150/ijbs.7.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Landen CN, Goodman B, Katre AA, Steg AD, Nick AM, Stone RL, et al. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mole Cancer Ther. 2010;9(12):3186–3199. doi: 10.1158/1535-7163.MCT-10-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silva IA, Bai S, McLean K, Yang K, Griffith K, Thomas D, et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011;71(11):3991–4001. doi: 10.1158/0008-5472.CAN-10-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng E, Long B, Sullivan P, McClellan S, Finan MA, Reed E, et al. CD44+/CD24- ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin Exp Metastasis. 2012;29(8):939–948. doi: 10.1007/s10585-012-9482-4. [DOI] [PubMed] [Google Scholar]
- 35.Chen J, Wang J, Chen D, Yang J, Yang C, Zhang Y, et al. Evaluation of characteristics of CD44 + CD117+ ovarian cancer stem cells in three dimensional basement membrane extract scaffold versus two dimensional monocultures. BMC Cell Biol. 2013;14:7. doi: 10.1186/1471-2121-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cioffi M, D’Alterio C, Camerlingo R, Tirino V, Consales C, Riccio A, et al. Identification of a distinct population of CD133(+)CXCR4(+) cancer stem cells in ovarian cancer. Sci Rep. 2015;5:10357. doi: 10.1038/srep10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burgos-Ojeda D, Rueda BR, Buckanovich RJ. Ovarian cancer stem cell markers: prognostic and therapeutic implications. Cancer Lett. 2012;322(1):1–7. doi: 10.1016/j.canlet.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi Y, Ingram PN, Yang K, Coffman L, Iyengar M, Bai S, et al. Identifying an ovarian cancer cell hierarchy regulated by bone morphogenetic protein 2. Proc Natl Acad Sci U S A. 2015;112(50):E6882–8. doi: 10.1073/pnas.1507899112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang F, Qiu Q, Khanna A, Todd NW, Deepak J, Xing L, et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol Cancer Res. 2009;7(3):330–338. doi: 10.1158/1541-7786.MCR-08-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bradshaw A, Wickremesekera A, Brasch HD, Chibnall AM, Davis PF, Tan ST, Itinteang T. Cancer stem cell in glioblastoma multiofrme. Front Surg. 2016;3:48. doi: 10.3389/fsurg.2016.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cabrera MC, Hollingsworth RE, Hurt EM. Cancer stem cell plasticity and tumor hierarchy. World J Stem Cells. 2015;7(1):27–36. doi: 10.4252/wjsc.v7.i1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pardal R, Molofsky AV, He S, Morrison SJ. Stem cell self-renewal and cancer cell proliferation are regulated by common networks that balance the activation of proto-oncogenes and tumor suppressors. Cold Spring Harb Symp Quant Biol. 2005;2005(70):177–185. doi: 10.1101/sqb.2005.70.057. [DOI] [PubMed] [Google Scholar]
- 43.Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan C, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5(2):100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore N, Lyle S. Quiescent, slow-cycling stem cell populations in cancer: a review of the evidence and discussion of significance. J Oncol. 2011 doi: 10.1155/2011/396076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chefetz I, Alvero AB, Holmbery JC, Lebowitz N, Craveiro V, Yang-Hartwich Y, Yin G, Squillace L, Gurrea Soteras M, Aldo P, Mor G. TLR2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle. 2013;12(3):511–21. doi: 10.4161/cc.23406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yonghyun K, Kyeung MJ, Juyoun J, Do-Hyun N. Cancer stem cells and their mechanism of chemo-radiation resistance. Int J Stem Cells. 2009;2(2):109–114. doi: 10.15283/ijsc.2009.2.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li L, Bhatia R. Stem cell quiescence. Clin Cancer Res. 2011;17(15):4936–4941. doi: 10.1158/1078-0432.CCR-10-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28(3):275–280. doi: 10.1038/nbt.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. 2013;14(6):329–340. doi: 10.1038/nrm3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, Elf SE, Miyata Y, Sashida G, Huang G, Di Giandomenico S, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4(1):37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng T. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287(5459):1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 52.Matsumoto A, Takeishi S, Kanie T, Susaki E, Onoyama I, Tateishi Y, et al. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2011;9(3):262–271. doi: 10.1016/j.stem.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 53.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81(3):323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 54.Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature. 2014;510(7505):393–396. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell. 2014;15(1):37–50. doi: 10.1016/j.stem.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao MQ, Choi YP, Kang S, Youn JH, Cho NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. 2010;29(18):2672–2680. doi: 10.1038/onc.2010.35. [DOI] [PubMed] [Google Scholar]
- 57.Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008;22(4):436–448. doi: 10.1101/gad.1627008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10(2):R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen D, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64(3):706–721. doi: 10.1124/pr.111.005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michalet S, Dijoux-Franca M-G. ABC transporters and resistance to antibiotics. In: Ahcne B, Jean B, Jacques R, editors. ABC transporters and multidrug resistance. Hoboken, NJ, USA: Wiley; 2009. pp. 177–193. [Google Scholar]
- 61.Boumendjel A, Boutonnat J, Robert J. ABC transporters and multidrug resistance. Hoboken, NJ, USA: Wiley; 2009. [Google Scholar]
- 62.Januchowski R, Zawierucha P, Andrzejewska M, Ruciński M, Zabel M. Microarray-based detection and expression analysis of ABC and SLC transporters in drug-resistant ovarian cancer cell lines. Biomed Pharmacother. 2013;67(3):240–245. doi: 10.1016/j.biopha.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 63.Eyre R, Harvey I, Stemke-Hale K, Lennard TWJ, Tyson-Capper A, Meeson AP. Reversing paclitaxel resistance in ovarian cancer cells via inhibition of the ABCB1 expressing side population. Tumour Biol. 2014;35(10):9879–9892. doi: 10.1007/s13277-014-2277-2. [DOI] [PubMed] [Google Scholar]
- 64.Zhang Q, Dou H, Xu P, Zhuang S, Liu P. Tumor recurrence and drug resistance properties of side population cells in high grade ovary cancer. Drug Res. 2015;65(3):153–157. doi: 10.1055/s-0034-1375609. [DOI] [PubMed] [Google Scholar]
- 65.Sun M, Yang C, Zheng J, Wang M, Chen M, Le DQS, et al. Enhanced efficacy of chemotherapy for breast cancer stem cells by simultaneous suppression of multidrug resistance and antiapoptotic cellular defense. Acta Biomater. 2015;28:171–182. doi: 10.1016/j.actbio.2015.09.029. [DOI] [PubMed] [Google Scholar]
- 66.To KW, Poon DC, Wei Y, Wang F, Lin G, Fu L. Vatalanib sensitizes ABCB1 and ABCG2-overexpressing multidrug resistant colon cancer cells to chemotherapy under hypoxia. Biochem pharmacol. 2015;97(1);27–37. doi:10.1016/j.bcp.2015.06.034. [DOI] [PubMed]
- 67.Hashida S, Yamamoto H, Shien K, Miyoshi Y, Ohtsuka T, Suzawa K, et al. Acquisition of cancer stem cell-like properties in non-small cell lung cancer with acquired resistance to afatinib. Cancer Sci. 2015;106(10):1377–1384. doi: 10.1111/cas.12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Y, Bieber MM, Teng NNH. Hedgehog signaling regulates drug sensitivity by targeting ABC transporters ABCB1 and ABCG2 in epithelial ovarian cancer. Mol Carcinog. 2014;53(8):625–634. doi: 10.1002/mc.22015. [DOI] [PubMed] [Google Scholar]
- 69.Tamaki A, Ierano C, Szakacs G, Robey RW, Bates SE. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011;50(1):209–232. doi: 10.1042/bse0500209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu M, Ocana A, Tannock IF. Reversal of ATP-binding cassette drug transporter activity to modulate chemoresistance: why has it failed to provide clinical benefit? Cancer Metastasis Rev. 2013;32(1–2):211–227. doi: 10.1007/s10555-012-9402-8. [DOI] [PubMed] [Google Scholar]
- 71.McGrogan BT, Gilmartin B, Carney DN, McCann A. Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys Acta. 2008;1785:96–132. doi: 10.1016/j.bbcan.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 72.Ceccaldi R, Sarangi P, D’Andrea A. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17:337–349. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- 73.Mou K, D’Andrea A. Crosstalk between the nucleotide excision repair and fanconi anaemia/BRCA pathways. DNA Repair. 2014;19:130–134. doi: 10.1016/j.dnarep.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 74.Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev. 2007;107(5):1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 75.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22(47):7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 76.Ferry KV, Hamilton TC, Johnson SW. ERCC1 contributes to Cisplatin resistance. Biochem Pharmacol. 2000;60(9):1305–1313. doi: 10.1016/S0006-2952(00)00441-X. [DOI] [PubMed] [Google Scholar]
- 77.Rosell R, Taron M, Barnadas A, Scagliotti G, Sarries C, Roig B. Nucleotide excision repair pathways involved in cisplatin resistance in non-small-cell lung cancer. Cancer Control. 2003;10(4):297–305. doi: 10.1177/107327480301000404. [DOI] [PubMed] [Google Scholar]
- 78.Welsh C, Day R, McGurk C, Masters JR, Wood RD, Köberle B. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int J Cancer. 2004;110(3):352–361. doi: 10.1002/ijc.20134. [DOI] [PubMed] [Google Scholar]
- 79.Wang Q. DNA damage responses in cancer stem cells: Implications for cancer therapeutic strategies. World J Biol Chem. 2015;6(3):57–64. doi: 10.4331/wjbc.v6.i3.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsai L, Yu C, Lo J, Sung W, Lee H, Chen S, Chou M. Enhanced cisplatin resistance in oral-cancer stem-like cells is correlated with upregulation of excision-repair cross-complementation group 1. J Dent Sci. 2012;7(2):111–117. doi: 10.1016/j.jds.2012.03.006. [DOI] [Google Scholar]
- 81.Wang Q, Milum K, Han C, Huang Y, Wani G, Thomale J, Wani AA. Differential contributory roles of nucleotide excision and homologous recombination repair for enhancing cisplatin sensitivity in human ovarian cancer cells. Mol Cancer. 2011;10:24. doi: 10.1186/1476-4598-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Joenje H, Patel KJ. The emerging genetic and molecular basis of fanconi anaemia. Nat Rev Genet. 2001;2(6):446–457. doi: 10.1038/35076590. [DOI] [PubMed] [Google Scholar]
- 83.Grompe M, D’Andrea A. Fanconi anemia and DNA repair. Hum Mol Genet. 2001;10(20):2253–2259. doi: 10.1093/hmg/10.20.2253. [DOI] [PubMed] [Google Scholar]
- 84.Poll EH, Arwert F, Joenje H, Wanamarta AH. Differential sensitivity of fanconi anaemia lymphocytes to the clastogenic action of cis-diamminedichloroplatinum (II) and trans-diamminedichloroplatinum (II) Hum Genet. 1985;1(3):206–210. doi: 10.1007/BF00284574. [DOI] [PubMed] [Google Scholar]
- 85.Kennedy RD, D’Andrea AD. The fanconi anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19(24):2925–2940. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 86.Yarde DN, Oliveira V, Mathews L, Wang X, Villagra A, Boulware D, et al. Targeting the fanconi anemia/BRCA pathway circumvents drug resistance in multiple myeloma. Cancer Res. 2009;69(24):9367–9375. doi: 10.1158/0008-5472.CAN-09-2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297(5588):1837–1848. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- 88.Yang H, Haijuan L, Qiubai F, Fan J, Holloman WK, Pavletich NP. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature. 2005;433(7026):653–657. doi: 10.1038/nature03234. [DOI] [PubMed] [Google Scholar]
- 89.Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ. The fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell. 2004;15(4):607–620. doi: 10.1016/j.molcel.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 90.Rothfuss A, Grompe M. Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the fanconi anemia/BRCA pathway. Mol Cell Biol. 2004;24(1):123–134. doi: 10.1128/MCB.24.1.123-134.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen Q, Van der Sluis PC, Boulware D, Hazlehurst LA, Dalton WS. The FA/BRCA pathway is involved in melphalan-induced DNA interstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood. 2005;106(2):698–705. doi: 10.1182/blood-2004-11-4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen CC, Taniguchi T, D’Andrea A. The fanconi anemia (FA) pathway confers glioma resistance to DNA alkylating agents. J Mol Med. 2007;85(4):497–509. doi: 10.1007/s00109-006-0153-2. [DOI] [PubMed] [Google Scholar]
- 93.Balacescu O, Balacescu L, Tudoran O, Todor N, Rus M, Buiga R, et al. Gene expression profiling reveals activation of the FA/BRCA pathway in advanced squamous cervical cancer with intrinsic resistance and therapy failure. BMC Cancer. 2014;14:246. doi: 10.1186/1471-2407-14-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, et al. Disruption of the fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003;9(5):568–574. doi: 10.1038/nm852. [DOI] [PubMed] [Google Scholar]
- 95.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;2012(12):68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Santarpia M, Altavilla G, Margeli M, Cirauqui B, Mesiti M, Cavallari V, Ramirez J, Sanchez-Ronco M, Santarpia L, Taron M, Rosell R. PIK3CA mutations and BRCA expression in breast cancer: potential biomarkers for chemoresistance. Cancer Investig. 2008;26(10):1044–1051. doi: 10.1080/07357900802112701. [DOI] [PubMed] [Google Scholar]
- 97.Norquist B, Wurz K, Pennil C, Garcia R, Gross J, Sakai W, Karlan B, Taniguchi T, Swisher E. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29(22):3008–3015. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sakai W, Swisher E, Karlan B, Aqarwal M, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia D, Couch F, Urban N, Taniguchi T. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rytelewski M, Tong JG, Buensuceso A, Leong HS, Maleki Vareki S, Figueredo R, et al. BRCA2 inhibition enhances cisplatin-mediated alterations in tumor cell proliferation, metabolism, and metastasis. Mol Oncol. 2014;8(8):1429–1440. doi: 10.1016/j.molonc.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ma MH, Yang HH, Parker K, Manyak S, Friedman JM, Altamirano C, et al. The proteasome inhibitor PS-341 markedly enhances sensitivity of multiple myeloma tumor cells to chemotherapeutic agents. Clin Cancer Res. 2003;9(3):1136–1144. [PubMed] [Google Scholar]
- 101.Mitsiades N, Mitsiades CS, Richardson PG, Poulaki V, Tai Y, Chauhan D, et al. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood. 2003;101(6):2377–2380. doi: 10.1182/blood-2002-06-1768. [DOI] [PubMed] [Google Scholar]
- 102.Eckstein N. Platinum resistance in breast and ovarian cancer cell lines. J Exp Clin Cancer Res. 2011;30:91. doi: 10.1186/1756-9966-30-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tapia G, Diaz-Padill I. Molecular Mechanisms of Platinum Resistance in Ovarian Cancer. In Ivan Diaz-Padilla (Ed.): Ovarian Cancer - A Clinical and Translational Update: InTech. 2013.
- 104.Sprick MR, Walczak H. The interplay between the Bcl-2 family and death receptor-mediated apoptosis. Biochim Biophys Acta. 2004;1644(2–3):125–132. doi: 10.1016/j.bbamcr.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 105.Duprez L, Wirawan E, Vanden Berghe T, Vandenabeele P. Major cell death pathways at a glance. Microbes Infect. 2009;11(13):1050–1062. doi: 10.1016/j.micinf.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 106.Mansouri A, Zhang Q, Ridgway LD, Tian L, Claret FX. Cisplatin resistance in an ovarian carcinoma is associated with a defect in programmed cell death control through XIAP regulation. Oncol Res. 2003;13:399–404. doi: 10.3727/096504003108748410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fan Y, Wang L, Han X, Liu X, Ma H, Ding Y. Upregulation of Fas in epithelial ovarian cancer reverses the development of resistance to cisplatin. BMB Rep. 2015;48(1):30–35. doi: 10.5483/BMBRep.2015.48.1.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Peter ME, Hadji A, Murmann AE, Brockway S, Putzbach W, Pattanayak A, Ceppi P. The role of CD95 and CD95 ligand in cancer. Cell Death Differ. 2015;22(4):549–559. doi: 10.1038/cdd.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gherman C, Braicu OL, Zanoaga OA, Pileczki V, Maralani M. Caffeic acid phenethyl ester activates pro-apoptotic and epithelial-mesenchymal transition-related genes in ovarian cancer cells A2780 and A2780cis. Mol Cell Biochem. 2016;413(1–2):189–198. doi: 10.1007/s11010-015-2652-3. [DOI] [PubMed] [Google Scholar]
- 110.Sussman RT, Ricci MS, Hart LS, Sun SY, El-Deiry WS. Chemotherapy-resistant side-population of colon cancer cells has a higher sensitivity to TRAIL than the non-SP, a higher expression of c-Myc and TRAIL-receptor DR4. Cancer Biol Ther. 2007;6(9):1490–1495. doi: 10.4161/cbt.6.9.4905. [DOI] [PubMed] [Google Scholar]
- 111.Rogalska A, Marczak A, Gajek A, Szwed M, Śliwińska A, Drzewoski J, Jóźwiak Z. Induction of apoptosis in human ovarian cancer cells by new anticancer compounds, epothilone A and B. Toxicol in Vitro. 2013;27(1):239–249. doi: 10.1016/j.tiv.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 112.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 113.Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9(6):447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- 114.Anderson NS, Turner L, Livingston S, Chen R, Nicosia SV, Kruk PA. Bcl-2 expression is altered with ovarian tumor progression: an immunohistochemical evaluation. J Ovarian Res. 2009;2:16. doi: 10.1186/1757-2215-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kirkin V, Joos S, Zörnig M. The role of Bcl-2 family members in tumorigenesis. Biochim Biophys Acta. 2004;1644(2–3):229–249. doi: 10.1016/j.bbamcr.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 116.Madjd Z, Mehrjerdi AZ, Sharifi AM, Molanaei S, Shahzadi SZ, Asad-Lari M. CD44+ cancer cells express higher levels of the anti-apoptotic protein Bcl-2 in breast tumours. Cancer Immun. 2009;9:4. [PMC free article] [PubMed] [Google Scholar]
- 117.Ma S, Lee T, Zheng B, Chan KW, Guan X. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27(12):1749–1758. doi: 10.1038/sj.onc.1210811. [DOI] [PubMed] [Google Scholar]
- 118.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22(56):9030–9040. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 119.Kuribayashi K, El-Deiry WS. Regulation of programmed cell death by the p53 pathway. Adv Exp Med Biol. 2008;615:201–221. doi: 10.1007/978-1-4020-6554-5_10. [DOI] [PubMed] [Google Scholar]
- 120.Bragado P, Armesilla A, Silva A, Porras A. Apoptosis by cisplatin requires p53 mediated p38alpha MAPK activation through ROS generation. Apoptosis. 2007;12(9):1733–1742. doi: 10.1007/s10495-007-0082-8. [DOI] [PubMed] [Google Scholar]
- 121.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14(5):359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Meek, David W. Tumour suppression by p53: a role for the DNA damage response? Nat Rev. Cancer 9 (10), pp. 714–723. DOI: 10.1038/nrc2716. [DOI] [PubMed]
- 123.Miao W, Liu X, Wang H, Fan Y, Lian S, Yang X, et al. p53 upregulated modulator of apoptosis sensitizes drug-resistant U251 glioblastoma stem cells to temozolomide through enhanced apoptosis. Mol Med Rep. 2015;11(6):4165–4173. doi: 10.3892/mmr.2015.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting notch, hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12(8):445–464. doi: 10.1038/nrclinonc.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim Y, Kahn M. The role of the Wnt signaling pathway in cancer stem cells: prospects for drug development. Res Rep Biochem. 2014;4:1–12. doi: 10.2147/RRBC.S53823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Boone JD, Arend RC, Johnston BE, Cooper SJ, Gilchrist SA, Oelschlager DK, et al. Targeting the Wnt/β-catenin pathway in primary ovarian cancer with the porcupine inhibitor WNT974. Lab Investig. 2016;96(2):249–259. doi: 10.1038/labinvest.2015.150. [DOI] [PubMed] [Google Scholar]
- 127.Zhang Z, Wu J, Luo Q, Liu Q, Wu Q, Ye G, et al. Pygo2 activates MDR1 expression and mediates chemoresistance in breast cancer via the Wnt/β-catenin pathway. Oncogene. 2016;35(36):4787–4797. doi: 10.1038/onc.2016.10. [DOI] [PubMed] [Google Scholar]
- 128.Wu X, Luo F, Li J, Zhong X, Liu K. Tankyrase 1 inhibitior XAV939 increases chemosensitivity in colon cancer cell lines via inhibition of the Wnt signaling pathway. Int J Oncol. 2016;48(4):1333–1340. doi: 10.3892/ijo.2016.3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Barghout SH, Zepeda N, Xu Z, Steed H, Lee C, Fu Y. Elevated β-catenin activity contributes to carboplatin resistance in A2780cp ovarian cancer cells. Biochem Biophys Res Commun. 2015;468(1–2):173–178. doi: 10.1016/j.bbrc.2015.10.138. [DOI] [PubMed] [Google Scholar]
- 130.Yang W, Yan H-X, Chen L, Liu Q, He Y-Q, Yu L-X, et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008;68(11):4287–4295. doi: 10.1158/0008-5472.CAN-07-6691. [DOI] [PubMed] [Google Scholar]
- 131.Chau WK, Ip CK, Mak ASC, Lai H-C, Wong AST. c-Kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/β-catenin-ATP-binding cassette G2 signaling. Oncogene. 2013;32(22):2767–81. doi: 10.1038/onc.2012.290. [DOI] [PubMed] [Google Scholar]
- 132.McAuliffe SM, Morgan SL, Wyant GA, Tran LT, Muto KW, Chen YS. et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. In Proceedings of the National Academy of Sciences of the United States of America. 2012;109(43):E2939-48. [DOI] [PMC free article] [PubMed]
- 133.Yabuuchi S, Pai SG, Campbell NR, de Wilde RF, Oliveira E, Korangath P, et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett. 2013;335(1):41–51. doi: 10.1016/j.canlet.2013.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kast RE. Potential for all-trans retinoic acid (tretinoin) to enhance interferon-alpha treatment response in chronic myelogenous leukemia, melanoma, myeloma and renal cell carcinoma. Cancer Biol Ther. 2008;7(10):1515–9. doi: 10.4161/cbt.7.10.6573. [DOI] [PubMed] [Google Scholar]
- 135.Loaiza-Bonilla A, Gore SD, Carraway HE. Novel approaches for myelodysplastic syndromes: beyond hypomethylating agents. Curr Opin Hematol. 2010;17(2):104–9. doi: 10.1097/MOH.0b013e3283366bb8. [DOI] [PubMed] [Google Scholar]
- 136.Haque A, Banik NL, Ray SK. Emerging role of combination of all-trans retinoic acid and interferon-gamma as chemoimmunotherapy in the management of human glioblastoma. Neurochem Res. 2007;32(12):2203–9. doi: 10.1007/s11064-007-9420-z. [DOI] [PubMed] [Google Scholar]
- 137.Pasquali D, Rossi V, Bellastella G, Bellastella A, Sinisi AA. Natural and synthetic retinoids in prostate cancer. Curr Pharm Design. 2006;12(15):1923–9. doi: 10.2174/138161206776873554. [DOI] [PubMed] [Google Scholar]
- 138.Brtko J. Role of retinoids and their cognate nuclear receptors in breast cancer chemoprevention. Cent Eur J Public Health. 2007;15(1):3–6. doi: 10.1007/s10389-006-0055-3. [DOI] [PubMed] [Google Scholar]
- 139.Fenaux P, Wang ZZ, Degos L. Treatment of acute promyelocytic leukemia by retinoids. Curr Top Microbiol Immunol. 2007;313:101–28. doi: 10.1007/978-3-540-34594-7_7. [DOI] [PubMed] [Google Scholar]
- 140.Connolly RM, Nguyen K, Sukumar S. Molecular pathways: current role and future directions of the retinoic acid pathway in cancer prevention and treatment. Clin Cancer Res. 2013;19(7):1651–1659. doi: 10.1158/1078-0432.CCR-12-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chiesa MD, Passalacqua R, Michiara M, Franciosi V, Di Costanzo F, Bisagni G, et al. Tamoxifen vs tamoxifen plus 13-cis-retinoic acid vs tamoxifen plus interferon alpha-2a as first-line endocrine treatments in advanced breast cancer: updated results of a phase II, prospective, randomised multicentre trial. Acta Biomed. 2007;78(3):204–9. [PubMed] [Google Scholar]
- 142.Sutton LM, Warmuth MA, Petros WP, Winer EP. Pharmacokinetics and clinical impact of all-trans retinoic acid in metastatic breast cancer: a phase II trial. Cancer Chemother Pharmacol. 1997;40(4):335–341. doi: 10.1007/s002800050666. [DOI] [PubMed] [Google Scholar]
- 143.Sellerio AL, Cuisani E, Ben-Moshe NB, Coco S, Piccinini A, Myers CR, Sethna JP, Gianmpetro C, Zapperi A, La Porta CAM. Overshoot during phenotypic switching of cancer cell populations. Sci Rep. 2015;5:15464. doi: 10.1038/srep15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.