

In this review, Gupte et al. discuss new findings on the diverse roles of PARPs in chromatin regulation, transcription, RNA biology, and DNA repair as well as recent advances that link ADP-ribosylation to stress responses, metabolism, viral infections, and cancer.
Keywords: poly(ADP-ribose) (PAR), mono(ADP-ribose) (MAR), poly(ADP-ribose) polymerase (PARP), gene regulation, DNA repair, RNA biology
Abstract
The discovery of poly(ADP-ribose) >50 years ago opened a new field, leading the way for the discovery of the poly(ADP-ribose) polymerase (PARP) family of enzymes and the ADP-ribosylation reactions that they catalyze. Although the field was initially focused primarily on the biochemistry and molecular biology of PARP-1 in DNA damage detection and repair, the mechanistic and functional understanding of the role of PARPs in different biological processes has grown considerably of late. This has been accompanied by a shift of focus from enzymology to a search for substrates as well as the first attempts to determine the functional consequences of site-specific ADP-ribosylation on those substrates. Supporting these advances is a host of methodological approaches from chemical biology, proteomics, genomics, cell biology, and genetics that have propelled new discoveries in the field. New findings on the diverse roles of PARPs in chromatin regulation, transcription, RNA biology, and DNA repair have been complemented by recent advances that link ADP-ribosylation to stress responses, metabolism, viral infections, and cancer. These studies have begun to reveal the promising ways in which PARPs may be targeted therapeutically for the treatment of disease. In this review, we discuss these topics and relate them to the future directions of the field.
ADP-ribosylation is a reversible post-translational modification (PTM) of proteins resulting in the covalent attachment of a single ADP-ribose unit [i.e., mono(ADP-ribose) (MAR)] or polymers of ADP-ribose units [i.e., poly(ADP-ribose) (PAR)] on a variety of amino acid residues on target proteins (Gibson and Kraus 2012; Daniels et al. 2015a). This modification is mediated by a diverse group of ADP-ribosyl transferase (ADPRT) enzymes that use ADP-ribose units derived from β-NAD+ to catalyze the ADP-ribosylation reaction. These enzymes include bacterial ADPRTs (e.g., cholera toxin and diphtheria toxin) as well as members of three different protein families in yeast and animals: (1) arginine-specific ecto-enzymes (ARTCs), (2) sirtuins, and (3) PAR polymerases (PARPs) (Hottiger et al. 2010). Surprisingly, a recent study showed that the bacterial toxin DarTG can ADP-ribosylate DNA (Jankevicius et al. 2016). How this fits into the broader picture of cellular ADP-ribosylation has yet to be determined.
In this review, we focus on the mono(ADP-ribosyl)ation (MARylation) and poly(ADP-ribosyl)ation (PARylation) of glutamate, aspartate, and lysine residues by PARP family members. While many reviews have been written on PARPs in the past decade, we highlight the current trends and ideas in the field, in particular those discoveries that have been published in the past 2–3 years.
PARPs and friends: writers, readers, erasers, and feeders
PARPs interact physically and functionally with a set of accessory proteins that play key roles in determining the overall outcomes in PARP-dependent pathways. By borrowing from and adding to descriptions used by the histone modification field (Hottiger 2015), PARPs can be thought of as “writers” of ADP-ribose, and the accessory proteins can be thought of as “readers” (ADP-ribose-binding domains [ARBDs]), “erasers” (ADP-ribose and PAR hydrolases), “feeders” (NAD+ synthases), and “consumers” (NAD+ hydrolases) (Fig. 1). These are elaborated on in more detail below.
Figure 1.
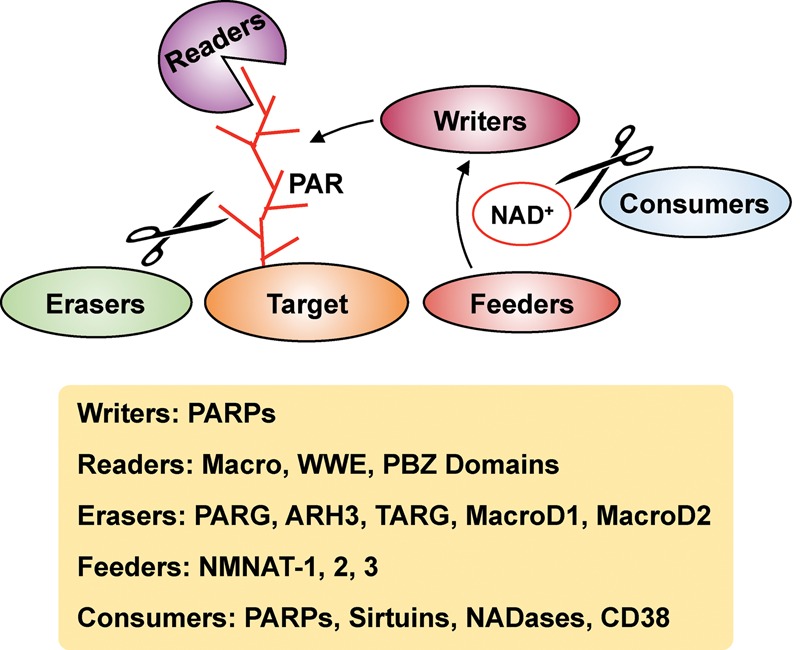
A variety of effectors mediate intracellular ADP-ribosylation dynamics. PARPs act as “writers” that add ADP-ribose moieties to target proteins. The NAD+ required for these PARP-mediated ADP-ribosylation reactions is supplied by nicotinamide mononucleotide adenylyl transferases (NMNATs; “feeders”). The ADP-ribose units on target protein can be recognized by “readers” containing macro, WWE, or PAR-binding zinc finger (PBZ) domains. The removal of ADP-ribose chains is catalyzed by “erasers,” which include PAR glycohydrolase (PARG), ADP-ribosyl hydrolase 3 (ARH3), TARG, and MacroD1/D2. NAD+ levels can be modulated by NAD+ “consumers,” such as PARPs, sirtuins, NADases, and CD38, which hydrolyze NAD+.
The PARP family: ADP-ribose writers
The PARP family consists of 17 members that have distinct structural domains, activities, subcellular localizations, and functions (Gibson and Kraus 2012; Vyas et al. 2013, 2014). PARPs can be thought of as ADP-ribose “writers,” that covalently attach (“write”) ADP-ribose units on substrate proteins (Fig. 1).
Based on their structural domains and functions, the different PARPs can be broadly classified as DNA-dependent PARPs (PARP-1, PARP-2, and PARP-3), Tankyrases (PARP-5a and PARP-5b), Cys–Cys–Cys–His zinc finger (CCCH)-containing and WWE PAR-binding domain-containing PARPs (PARP-7, PARP-12, PARP-13.1, and PARP-13.2), and PAR-binding macrodomain-containing “macro” PARPs (PARP-9, PARP-14, and PARP-15) (Ame et al. 2004; Vyas et al. 2013). The PARP family members can also be categorized according to their catalytic activities: “mono,” “poly,” or inactive. The PARP “monoenzymes” [i.e., mono(ADP-ribosyl) transferases (MARTs); i.e., PARP-3, PARP-4, PARP-6, PARP-10, PARP-14, PARP-15, and PARP-16] catalyze the addition of a single ADP-ribose unit on target proteins through a process called MARylation (Vyas et al. 2014). The PARP “polyenzymes” (i.e., PARP-1, PARP-2, PARP-5a, and PARP-5b) catalyze the polymerization of ADP-ribose units through α(1 → 2) O-glycosidic bonds in linear or branched chains (Gibson and Kraus 2012). In contrast, no enzymatic activity has been described for PARP-9 and PARP-13 (Vyas et al. 2014).
The catalytic domain of many PARPs contains an “H-Y-E” (His–Tyr–Glu) motif. The histidine and tyrosine residues are required for the proper orientation of NAD+, while the glutamate residue is required for catalytic activity. Nonetheless, the H-Y-E motif is not the sole indicator of PARP activity (Vyas et al. 2014). In the mono-PARPs (except for PARP-3 and PARP-4), the glutamate residue is replaced with isoleucine, leucine, or tyrosine, which is associated with the absence of polymerase activity. Although PARP-3 and PARP-4 have the H-Y-E motif, their structurally distinct donor (“D”) loop may explain the lack of polymerase activity by these family members. Strikingly, PARP-9 and PARP-13 do not have the conserved histidine residue in their NAD+-binding pockets, which is likely to account for the lack of catalytic activity (Vyas et al. 2014).
What's in a name? PARPs or ARTDs (ADPRTs, diphtheria toxin-like)?
The PARP family of proteins is defined by the presence of the conserved PARP catalytic domain. Historically, all members of the family were named based on the polymerase activity of the founding member, PARP-1, even though many family members have MART activity. Recent discussions in the PARP field have centered around the need to reform the PARP nomenclature. Hottiger et al. (2010) have proposed using the term ARTD, which represents the basic catalytic activity of the proteins. While some aspects of the new nomenclature are an improvement, the vast majority of the published literature has used the older PARP nomenclature. In addition, the development of PARP inhibitors (PARPis) and their frequent discussion in the popular press may make the “PARP” nomenclature difficult to replace. Furthermore, the use of two different names for each PARP protein may cause confusion. The most important distinction for the field now seems to be between the MARTs and the poly(ADP-ribosyl) transferases. Recently, the terms “monoPARP” and “polyPARP” have gained some traction as jargon in the field, but we prefer the terms “monoenzyme” and “polyenzyme” due to the obvious oxymoron and redundancy, respectively, of the former terms. These terms work equally well with the PARP and ARTD nomenclature (e.g., PARP monoenzyme and ARTD polyenzyme). Clearly, the field needs to adopt a more sensible, consistent, and universal nomenclature. Borrowing from the words of Marshall McLuhan, we must not let the naming of PARPs become a numbing blow from which the field never recovers.
ARBDs: ADP-ribose readers
Studies over the past decade have led to the discovery of motifs, domains, or modules in proteins that can bind to various forms of ADP-ribose on PARP substrate proteins and function as “readers” (Fig. 1). These ARBDs include PAR-binding motifs (PBMs), macrodomains, PAR-binding zinc finger (PBZ) modules, and WWE domains (Table 1; Kalisch et al. 2012; Barkauskaite et al. 2013; Karlberg et al. 2013; Krietsch et al. 2013). Other less well characterized ARBDs include (1) the phosphopeptide-binding Forkhead-associated (FHA) and BRCA1 C-terminal (BRCT) domains, (2) RNA recognition modules (RRMs), and (3) arginine (R)- and glycine (G)-rich motifs (RGGs; also called glycine–arginine-rich [GAR] domains) (Li et al. 2013; Teloni and Altmeyer 2016).
Table 1.
Examples of proteins containing ARBDa

PBMs are short sequences (∼20 amino acids) with a loosely defined consensus ([HKR]1-X2-X3-[AIQVY]4-[KR]5-[KR]6-[AILV]7-[FILPV]8,) (Pleschke et al. 2000; Gagne et al. 2008). Despite being the first PAR reader domains to be defined, the exact nature of their interaction with PAR remains to be elucidated. Macrodomains are larger (∼130- to 190-amino acid) globular domains that can bind to (“read”) an ADP-ribose monomer or the terminal ADP-ribose moiety in a PAR chain (Table 1; Fig. 2A,B; Karras et al. 2005; Feijs et al. 2013; Rack et al. 2016). The PBZs are short modules (∼30 amino acids) with the consensus sequence [K/R]-X-X-C-X-[F/Y]-G-X-X-C-X-[K/R]-[K/R]-X-X-X-X-H-X-X-X-[F/Y]-X-H that bind the ADP-ribose–ADP-ribose junctions of PAR chains as well as ADP-ribose monomers (Table 1; Fig. 2A,B; Ahel et al. 2008). WWE domains, which contain conserved tryptophan (W) and glutamate (E) residues, bind to the iso-ADP-ribose moiety in PAR chains (Table 1; Fig. 2A,B; Wang et al. 2012). Thus, WWE domains bind exclusively to ADP-ribose oligomers or polymers.
Figure 2.
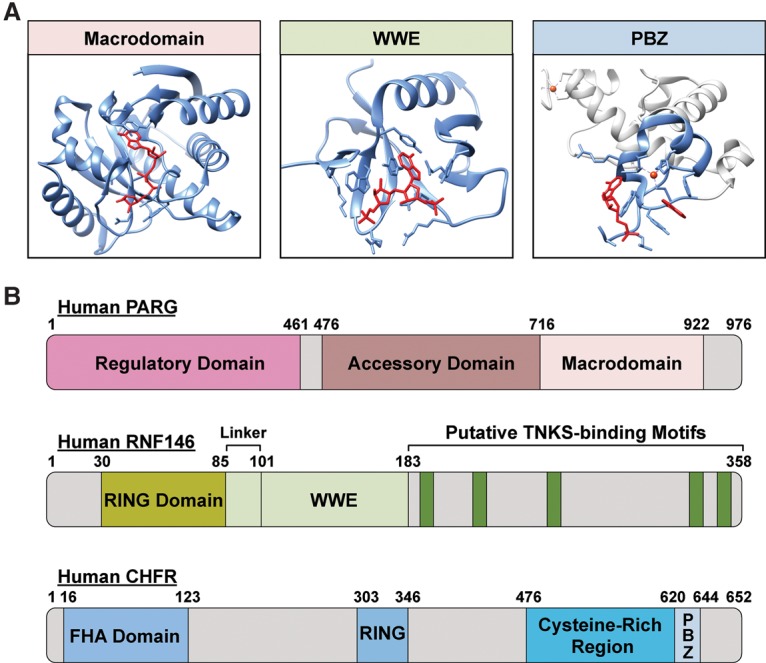
Structures of ARBDs. (A) The ARBDs shown include a macrodomain from Archaeoglobus fulgidus Af1521 (Protein Data Bank [PDB] 2BFQ), a WWE domain from human RNF146 (PDB 3V3L), and a PBZ motif from human CHFR (PDB 2XOY). The ARBDs are shown in blue, and the ADP-ribose ligands are highlighted in red. (B) Schematic showing the structures of example proteins containing ARBDs: human PARG (macrodomain), human RNF146 (WWE domain), and human CHFR (PBZ motif). The different domains in the proteins are indicated.
Various proteins are “functionalized” with these ARBDs, allowing them to provide a functional bridge between ADP-ribosylation and other cellular events. For example, the DNA damage repair protein APLF (aprataxin polynucleotide-kinase-like factor) is recruited to sites of DNA damage through interactions with ADP-ribose via its PBZ motif (Li et al. 2010). Similarly, the RNF146 E3 ubiquitin ligase interacts with its ADP-ribosylated substrates through its WWE domain, allowing it to subsequently ubiquitylate them (Zhang et al. 2011). As discussed below, the discovery of these diverse ARBDs has provided new tools for exploring ADP-ribose-directed or PAR-directed events in cells and biochemical assays.
ADP-ribose and PAR hydrolases: erasers
PAR polymers turn over rapidly in the cell (Tulin and Spradling 2003). Thus, not surprisingly, a number of enzymes have evolved to remove covalently linked ADP-ribose and PAR from proteins (Fig. 1). These “erasers” include ADP-ribosyl hydrolase 3 (ARH3) (Oka et al. 2006), PAR glycohydrolase (PARG) (Slade et al. 2011), TARG/C6orf130 (Sharifi et al. 2013), MacroD1 and MacroD2 (Jankevicius et al. 2013; Rosenthal et al. 2013), and the NUDIX family of hydrolases (Daniels et al. 2015b). Many of these enzymes contain a macrodomain fold, which allows them to interact with ADP-ribosylated substrates. Both PARG and ARH3 catalyze PAR chain degradation through endoglycocidic and exoglycocidic activities, which results in the cleavage of the ribose–ribose bonds but leaves a terminal ADP-ribose moiety attached to the acceptor amino acid residue of the substrate (Oka et al. 2006; Slade et al. 2011; Niere et al. 2012). In contrast, TARG, MacroD1, and MacroD2 can hydrolyze the ester bond between the ribose and acceptor amino acids (aspartates or glutamates), thus facilitating the complete removal of the ADP-ribose moiety (Jankevicius et al. 2013; Rosenthal et al. 2013; Sharifi et al. 2013). The NUDIX family of hydrolases can hydrolyze PAR chains by targeting the phosphodiester bond in the protein-proximal ADP-ribose unit, which results in the formation of a phosphoribose moiety attached to the acceptor amino acid (Daniels et al. 2015b). Not surprisingly, these “erasers” exhibit different specificities and different modes of binding with different targets (Kistemaker et al. 2016). For example, in an in vitro assay using synthetic ADP-ribosylated peptides, MacroD2 binds in a nonspecific manner to its substrates, whereas TARG1 binds in a manner dependent on the sequence of amino acids surrounding the site of ADP-ribosylation (Kistemaker et al. 2013, 2016).
Different “eraser” activities have been shown to contribute to the dynamic turnover of ADP-ribose that is critical for multiple cellular processes. For example, the accumulation of PAR due to loss of PARG activity causes early embryonic lethality as well as increased sensitivity to genotoxic stress (Koh et al. 2004). Interestingly, the “erasers” have different subcellular localizations: MacroD1 and ARH3 localize to the mitochondria (Niere et al. 2012; Jankevicius et al. 2013), whereas MacroD2 and TARG localize predominantly to the nucleus (Jankevicius et al. 2013; Sharifi et al. 2013). This suggests possible localized turnover of PAR, adding another layer of complexity to the regulation of cellular processes by ADP-ribosylation.
Nicotinamide mononucleotide adenylyl transferases (NMNATs): PARP feeders
PARPs are NAD+-dependent enzymes and thus require a source of NAD+ in all of the cellular compartments in which they function (Fig. 1). NMNAT-1, NMNAT-2, and NMNAT-3 comprise a small family of NAD+ synthases that produce NAD+ from nicotinamide mononucleotide and ATP (Emanuelli et al. 2001; Raffaelli et al. 2002; Zhang et al. 2003). Like the PARPs that they “feed,” the NMNATs exhibit distinct subcellular localizations: NMNAT-1 (nucleus), NMNAT-2 (cytoplasm and golgi), and NMNAT-3 (mitochondria) (Berger et al. 2005). NMNAT-1 can be recruited to chromatin by PARP-1, where it directly “feeds” NAD+ to PARP-1, thereby modulating PARP-1 catalytic activity (Zhang et al. 2012). Indeed, expression of a catalytically inactive mutant of NMNAT-1 results in a reduction of PAR accumulation at PARP-1-bound gene promoters and a concomitant attenuation of gene expression (Zhang et al. 2012).
Interestingly, ectopic expression of NMNAT-1 in MCF-7 cells increases nuclear NAD(P)H levels (used as an indirect measurement or surrogate for NAD+) without affecting the cytosolic NAD(P)H levels (Zhang et al. 2012). This suggests that cellular NAD+ could be present in distinct pools in different subcellular organelles such that the levels of NAD+ are locally regulated by the availability and activity of the respective NMNATs. Certain PARPs, such as PARP-2, PARP-3, and PARP-7, exhibit differential localization in the nucleus and the cytoplasm during different phases of the cell cycle (Vyas et al. 2013). Moreover, as mentioned in the previous section, the ADP-ribose hydrolases also exhibit distinct subcellular localizations (Niere et al. 2012; Jankevicius et al. 2013; Sharifi et al. 2013). Taken together, these observations suggest that the compartmentalization of the “feeders,” “writers,” and “erasers” could be essential for rapid and coordinated changes in ADP-ribosylation in different subcellular milieus.
NAD+ consumers
Given the dependence of PARP enzymes on NAD+, proteins that limit the cellular NAD+ supply have the potential to affect PARP catalytic activity. Thus, NAD+ consumption (distinguished here from NAD+ conversion to reduced NADH) should also be considered a potential regulator of PARP activity (Fig. 1). Although PARP enzymes, especially PARP-1 under cellular stress conditions, are likely to be the greatest NAD+ consumers, other enzymes also contribute to cellular NAD+ consumption. These include NADases (NAD+ glycohydrolases), CD38, and sirtuins (Lin 2007; Ying 2008). NADases are enzymes that catalyze the hydrolysis of NAD+ to ADP-ribose and nicotinamide. They have been identified in bacteria, fungi, and mammals and may be membrane-anchored (Kim et al. 1988, 1993; Cho et al. 1998; Ghosh et al. 2010). The ADP-ribosyl cyclase CD38, which catalyzes the cyclization of NAD+ to cyclic ADP-ribose, also exhibits significant NADase activity (Zocchi et al. 1993; Aksoy et al. 2006b). In this regard, CD38 knockout mice exhibit significantly higher tissue NAD+ levels than wild-type mice (Aksoy et al. 2006b). CD38 can regulate the NAD+-dependent protein deacetylase activity of the sirtuin Sirt1 by limiting NAD+ (Aksoy et al. 2006a). More broadly, a number of studies have shown that modulation of cellular NAD+ levels can regulate the catalytic activities of sirtuins (Anderson et al. 2002, 2003; Araki et al. 2004; Revollo et al. 2004); presumably, similar mechanisms can regulate the catalytic activities of PARPs as well.
Like the PARP proteins, Sirt1 consumes NAD+ as a consequence of its catalytic activity, which promotes the deacetylation of lysine residues in proteins, generating O-acetyl-ADP-ribose (the acceptor of the acetyl group) and nicotinamide (Haigis and Sinclair 2010; Tong and Denu 2010). Reduction of NAD+ levels by Sirt1 catalytic activity may act to reduce PARP catalytic activity and vice versa. In this regard, PARP-1 and Sirt1 have been shown to function antagonistically, possibly through competition for NAD+ or NMNAT-1 (Rongvaux et al. 2003; Kim et al. 2005; Kolthur-Seetharam et al. 2006; Zhang et al. 2009). Chemical activation of Sirt1 leads to reduced PARP-1 activity, and knockout of Sirt1 increases PARP-1 activity (Kolthur-Seetharam et al. 2006). Furthermore, nicotinamide, a product of the reactions catalyzed by both PARP-1 and Sirt1, can inhibit both of their enzymatic activities (Rongvaux et al. 2003; Kim et al. 2005). However, the functional interplay between PARP-1 and Sirt1 goes beyond competition for NAD+, possibly involving direct interactions between PARP-1 and Sirt1 or coregulation of shared cellular processes (Tulin et al. 2006; El Ramy et al. 2009; Rajamohan et al. 2009; Krishnakumar and Kraus 2010a). It may even involve a role for the Sirt1 catalytic product O-acetyl-ADP-ribose, which binds macroH2A1.1 (Hoff and Wolberger 2005; Kustatscher et al. 2005), a macrodomain-containing histone variant that interacts functionally with PARP-1 (Ouararhni et al. 2006; Nusinow et al. 2007; Timinszky et al. 2009; Hussey et al. 2014). Nonetheless, the studies described here suggest that modulation of NAD+ levels by NAD+ consumers can play a role in regulating the catalytic activity of PARPs.
Recurring themes in PARP biology: DNA repair, transcription, signaling, and beyond, with a focus on PARP-1
PARP family members are expressed in a wide variety of tissues and cell types, some ubiquitously and others in a more restricted manner. Our emerging understanding of the broader PARP family and the biological potential that it represents has led to a growing interest in the contribution of PARPs to molecular, cellular, and physiological outcomes. The field has evolved from its historical focus on the role of PARPs, especially PARP-1 and PARP-2, in DNA damage repair to a much more diverse biology (Fig. 3). In this section, we review the recent advances in PARP biology that highlight the striking functional versatility of PARPs.
Figure 3.
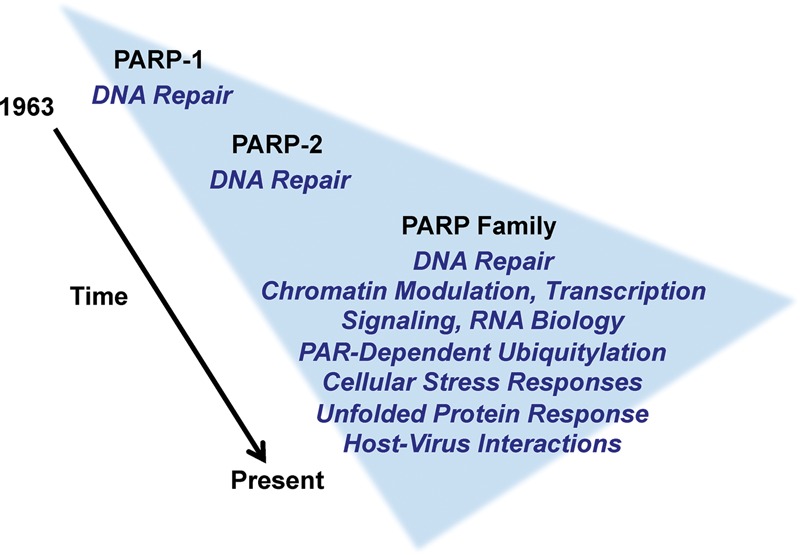
A time line of discoveries for PARPs and ADP-ribosylation. The schematic illustrates our expanding understanding of the functions of PARP family members in different biological processes. Details are discussed in the section “Recurring Themes in PARP Biology: DNA Repair, Transcription, Signaling, and Beyond, with a Focus on PARP-1.”
PARPs in DNA damage repair, cell survival, and cell death
Historically, PARPs, in particular PARP-1, have been studied for their roles in DNA damage repair, including base excision repair (BER) and double-strand break (DSB) repair (De Vos et al. 2012; Beck et al. 2014b). In the case of severe genotoxic stress, PARP-dependent pathways may direct the cell-initiated programmed cell death. Here we describe roles for some of the nuclear PARPs involved in these processes.
PARP-1 and DNA repair
PARP-1 is recruited to nicks and DSBs in genomic DNA in response to DNA damage and is a critical mediator of DNA damage repair (Audebert et al. 2004; Langelier et al. 2012). Indeed, Parp1 knockout mice exhibit heightened sensitivity to DNA-damaging agents (de Murcia et al. 1997). The mechanisms of action by which PARP-1 can promote the repair of damaged DNA have been widely explored, yet some aspects remain unexplained. Activation of PARP-1 at sites of DNA damage results in the production of long PAR chains on PARP-1 itself as well as other proteins associated with the damaged DNA, which in turn recruit PAR-binding proteins. These include (1) XRCC1 (X-ray repair cross-complementing protein 1), a scaffolding protein involved in assembly and activation of the DNA BER machinery (Masson et al. 1998; Okano et al. 2003); (2) CHD4 (chromodomain nucleosome remodeling and histone deacetylase), a part of the repressive nucleosome remodeling and deacetylase (NuRD) complex, which acts to repress transcription and facilitate DNA repair at the break sites (Chou et al. 2010); (3) APLF and CHFR, which have PAR-binding domains that allow APLF recruitment to DNA damage sites and CHFR to regulate antephase checkpoints, respectively (Ahel et al. 2008; Li et al. 2010); and (4) macrodomain-containing proteins, such as ALC1, which is activated in a PAR-dependent manner to enable nucleosome remodeling (Ahel et al. 2009). Moreover, the rapid PAR-dependent recruitment to DNA damage sites of mitotic recombination 11 (MRE11) (Haince et al. 2008) and ataxia telangiectasia-mutated (ATM) (Aguilar-Quesada et al. 2007; Haince et al. 2007), components of the homologous recombination machinery, implicates PARP-1 in homologous recombination as well.
Recent work from a number of laboratories has led to new insights into the role of PARP-1 in DNA damage repair. For example, a recent study by Luijsterburg et al. (2016) explored the contribution of PARP-1 to the nonhomologous end-joining (NHEJ) pathway of DNA repair. In their model, PARP-1 facilitates recruitment of the chromatin remodeler CHD2 to DSBs in a PAR-dependent manner. CHD2 in turn recruits the core components of the NHEJ machinery. Moreover, the presence of CHD2 at the DSB sites leads to chromatin decondensation and the deposition of the histone variant H3.3. Together, CHD2 and H3.3 change the local chromatin structure to a more permissive one for DNA repair by NHEJ, thus facilitating DSB repair (Luijsterburg et al. 2016).
As suggested by the aforementioned observations, a major contribution of PARPs to DSB repair is through the ADP-ribosylation of histones, which potentiates the expansion of compacted chromatin and enables the repair machinery to function competently. Recently, a novel protein, HPF1 (histone PARylation factor 1) or C4orf27, was shown to be a coregulator of PARP-1-dependent histone ADP-ribosylation (Gibbs-Seymour et al. 2016). Loss of HPF1 results in PARP-1 hyperautomodification and a consequent decrease in histone ADP-ribosylation, suggesting that HPF1 restricts PARP-1 automodification and promotes histone ADP-ribosylation. HPF1 is also required for efficient cellular responses to DNA-damaging agents, thus making HPF1 an integral component of genome maintenance by PARP-1 (Gibbs-Seymour et al. 2016).
Furthermore, previous studies of DSB repair have shown that the spatial organization of the repair machinery is important for efficient repair responses (Bekker-Jensen et al. 2006; Misteli and Soutoglou 2009). PAR polymers have been shown recently to potentiate liquid demixing (i.e., separation into distinct phases by forming liquid droplets) (Hyman and Simons 2012) at the sites of DNA damage, which promotes the assembly of intrinsically disordered RNA-binding proteins, such as EWS, FUS, and TAF15 (Altmeyer et al. 2015). This phase separation, which dynamically reorganizes the soluble nuclear space, orchestrates the earliest cellular responses to DNA damage (Altmeyer et al. 2015). These studies highlight some of the recent advances in our understanding of the mechanisms by which PARP-1 contributes to the repair of damaged DNA.
PARP-1: a cellular rheostat?
Importantly, excessive (hyper) PARylation by PARP-1 can direct the cell away from DNA repair pathways toward the activation of cell death pathways. These cell death pathways include parthanatos, a unique form of programmed cell death that occurs independently of caspase and is distinct from necrosis and apoptosis (David et al. 2009). Exposure to N-methyl-N-nitro-N-nitrosoguanidine (MNNG), a DNA-alkylating agent, leads to extensive activation of PARP-1, which triggers the release of apoptosis-inducing factor (AIF) from mitochondria (Yu et al. 2002). AIF then translocates into the nucleus, where it recruits the nuclease MIF (macrophage migration inhibitory factor), which cleaves genomic DNA into large fragments, resulting in chromatinolysis (Wang et al. 2016). Thus, the level of PARP-1 activation can serve as a rheostat: As the strength of the stress stimulus increases, the levels of PARP-1 activity and PAR synthesis increase, leading to different cellular outcomes (i.e., inflammatory responses, DNA repair and cell survival, senescence, or cell death [e.g., apoptosis, necrosis, and parthanatos]) (Luo and Kraus 2012).
PARP-3 and DNA repair
Recent studies have also highlighted an emerging role for another PARP, the PARP monoenzyme PARP-3, in DNA damage repair. PARP-3 is critical for DSB repair, and the loss of PARP-3 results in a delayed response to DSBs as well as increased sensitivity to anti-tumor drugs that cause DSBs (Boehler et al. 2011; Rulten et al. 2011; Beck et al. 2014a). PARP-3 also potentiates NHEJ by enabling the accumulation of APLF at DSBs, which results in the retention of the XRCC4/Lig4 DNA ligation complex required for rapid repair of the DNA breaks (Rulten et al. 2011). Furthermore, PARP-3 is required for phosphorylation of APLF at Ser116, an event that facilitates efficient DSB repair (Fenton et al. 2013). PARP-3 also associates with other DNA repair factors, such as DNA-PKcs, PARP-1, DNA ligase III, DNA ligase IV, Ku70, and Ku80 (Rouleau et al. 2007). Interestingly, the ADP-ribosylation of Ku80 by PARP-3 plays an important role in directing DNA repair toward NHEJ rather than homologous recombination (Beck et al. 2014a). ADP-ribosylation of histone H2B by PARP-3 is also observed near the sites of DNA damage upon the binding of PARP-3 to the nicked DNA (Grundy et al. 2016; Kistemaker et al. 2016). PARP-3 may also function synergistically with PARP-1 to potentiate DNA damage repair, as Parp1 and Parp3 double-knockout mice exhibit decreased survival in response to X-ray irradiation as compared with the individual gene knockouts (Boehler et al. 2011). This suggests that different PARPs might act in concert to ensure efficient DNA damage repair.
Structural analyses of PARPs in DNA repair
Mapping the structural domains and interactions of PARPs with DNA has helped to elucidate the mechanisms by which PARPs can recognize damaged DNA and initiate repair. For PARP-1, the first two zinc finger domains (Zn1 and Zn2) make contacts with the DNA (Langelier et al. 2011), whereas an adjacent third zinc finger domain (Zn3) is required for DNA-dependent PARP-1 catalytic activity as well as chromatin compaction (Langelier et al. 2010). The Zn1, Zn3, and WGR domains are essential for activation of PARP-1 at sites of DNA damage (Langelier et al. 2012). Upon association of PARP-1 with a break site, these protein domains are refolded to promote extensive interdomain contacts that facilitate PARP-1 catalytic activity (Langelier et al. 2012). Moreover, DNA binding induces unfolding of an autoinhibitory helical domain (HD), which then allows for NAD+ binding and activation of PARP-1 (Dawicki-McKenna et al. 2015; Steffen et al. 2016). The recognition of single-stranded breaks by Zn1 and Zn2 promotes the folding of other PARP-1 domains and subsequent allosteric activation (Eustermann et al. 2015).
PARP-2, which lacks the zinc finger domains, has a short N-terminal region that, together with the WGR and catalytic domains, is required for binding to damaged DNA and subsequent activation (Riccio et al. 2016). Moreover, while PARP-2 and PARP-3 share the allosteric activation mechanism with PARP-1, they require 5′ phosphorylation at the DNA breaks for activation, unlike PARP-1 (Langelier et al. 2014). These studies revealed some of the distinguishing features that contribute to the differential activities of different PARPs.
PARPs in gene regulation: a focus on PARP-1
A number of nuclear PARPs have been implicated in gene regulatory outcomes; much of the focus has been on PARP-1. Current models postulate that PARP-1 regulates gene expression through two general mechanisms: (1) by modulating chromatin structure (both local and higher order) and (2) by acting as a transcriptional coregulator (with the transcriptional machinery as well as sequence-specific DNA-binding transcription factors) (Kim et al. 2004; Krishnakumar et al. 2008; Krishnakumar and Kraus 2010b; Ryu et al. 2015).
Regulation of chromatin structure
The regulation of chromatin structure by PARP-1 may involve (1) alterations in nucleosome structure through direct binding or (2) PARylation of histones as well as nonhistone, chromatin-associated proteins. In Drosophila, activation of PARP-1 promotes decondensation of chromatin in response to heat shock or other cellular signaling pathways (Tulin and Spradling 2003; Petesch and Lis 2008). This decondensation could be due to the ADP-ribosylation of histone H1 by PARP-1 or competitive displacement of histone H1 from nucleosomes by PARP-1 (Kim et al. 2004; Krishnakumar et al. 2008). Proinflammatory signaling also induces PARP-1 enzymatic activity and histone ADP-ribosylation at transcriptionally active and accessible chromatin regions (Martinez-Zamudio and Ha 2012). While the actions of PARP-1 on chromatin may occur across broad domains, PARP-1 can also have specific effects at the level of the nucleosome. PARP-1 binds to nucleosomes by recognizing specific structural features (Kim et al. 2004) and can alter nucleosome structure while binding cooperatively with transcription factors, such as the pioneer transcription factor Sox2 (Liu and Kraus 2017). Finally, PARP-1 can also function as a histone chaperone by directly binding or recruiting other factors to facilitate nucleosome assembly (Muthurajan et al. 2014).
Interestingly, a recent study proposed that PARP-1-generated PAR serves as a dynamic source of ATP, which is required for the activity of ATP-dependent chromatin remodeling enzymes during cellular signaling (Wright et al. 2016). In this model, the pyrophosphatase NUDIX5 catalyzes the conversion of ADP-ribose to ATP in the presence of pyrophosphate. This reaction, which was shown to be triggered in response to hormone-dependent signaling, enables the ATP generated to be used by chromatin remodeling enzymes to regulate hormone-dependent gene transcription (Fig. 4A; Wright et al. 2016).
Figure 4.
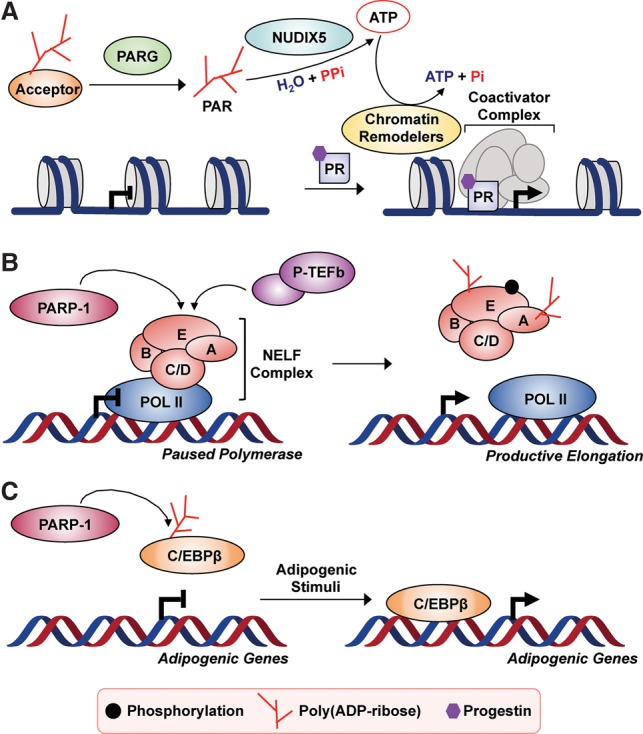
Varied roles of ADP-ribosylation in the regulation of gene regulation. (A) PAR turnover plays a critical role in hormone-dependent gene expression by generating ATP. Free PAR from PARG hydrolysis is further broken down by NUDIX5 to produce ATP. The ATP generated is used by ATP-dependent chromatin remodeling enzymes to modulate nucleosome occupancy at progesterone receptor (PR) target genes to stimulate transcription (Wright et al. 2016). (B) PARP-1 regulates the release of promoter-proximally paused RNA polymerase II into productive transcriptional elongation through ADP-ribosylation of the negative elongation factor (NELF) complex. Phosphorylation of NELF by the P-TEFb (positive-transcription elongation factor b) complex and subsequent ADP-ribosylation by PARP-1 results in the dissociation of NELF from RNA polymerase II, and the resulting release of pausing triggers productive elongation (Gibson et al. 2016). (C) ADP-ribosylation of C/EBPβ regulates the adipogenic transcriptional program. ADP-ribosylation by PARP-1 inhibits the binding of C/EBPβ to DNA. Upon exposure to adipogenic stimuli, there is loss of C/EBPβ PARylation and subsequent DNA binding. This turns on the expression of C/EBPβ-dependent proadipogenic genes (Luo et al. 2017).
PARP-1 can also indirectly alter chromatin architecture by modulating the activity of histone-modifying enzymes and chromatin remodelers. PARylation of KDM5B, a histone lysine demethylase that acts on histone H3 Lys4 trimethyl (H3K4me3), by PARP-1 inhibits the binding of KDM5B to chromatin and its demethylase activity at specific sites across the genome (Krishnakumar and Kraus 2010b). This leads to an increase in the levels of H3K4me3 at the promoters of PARP-1-regulated genes and enhanced expression (Krishnakumar and Kraus 2010b). ADP-ribosylation of Drosophila ISWI, an ATP-dependent nucleosome remodeler, by dPARP inhibits nucleosome binding, ATPase, and chromatin condensation activities of ISWI at heat-shock loci (Sala et al. 2008). ALC1, another ATP-dependent nucleosome remodeler, can bind to PARP-1 in a PAR-dependent manner through its ADP-ribose-binding macrodomain (Ahel et al. 2009). This interaction enhances the nucleosome remodeling activity of ALC1 as well as its recruitment to specific genomic loci (Gottschalk et al. 2009). These examples represent a few of the ways in which PARP-1 can directly or indirectly alter chromatin structure.
Modulation of insulator function
Arrangement of genomic DNA in the three-dimensional space of the nucleus may play an important role in regulating gene transcription (Wu 1997). Among the genomic regulatory elements that direct higher-order chromatin interactions are insulators, which segregate transcriptionally active and inactive chromatin domains in order to maintain stable gene expression patterns (Gaszner and Felsenfeld 2006). The chromatin insulator-binding protein CCCTC-binding factor (CTCF) has been shown to be PARylated by PARP-1. Although PARylation of CTCF does not affect the DNA-binding affinity of CTCF, it is required for optimal function in transcriptional activation and insulation (Yu et al. 2004; Farrar et al. 2010). In this regard, Ong et al. (2013) have shown that ADP-ribosylation of CP190, a CTCF-interacting protein, promotes its interaction with insulator proteins, association with the nuclear lamina, and insulator activity in vivo. Loss of dPARP results in the disruption of the nuclear clustering of CP190 complexes and reduces insulator activity (Ong et al. 2013). Additional studies have shown that PARP-1 can interact with CTCF in a signal-dependent manner. For example, Zhao et al. (2015) demonstrated that PARP-1–CTCF interactions are regulated in response to circadian rhythms, which trigger oscillations in the recruitment of circadian loci to the lamina, causing the silencing of these loci through the acquisition of repressive H3K9me2 histone modifications. Importantly, inhibition of PARP-1 activity by the PARPi olaparib abrogates the recruitment of circadian loci to the nuclear envelope (Zhao et al. 2015). Together, these data suggest that ADP-ribosylation by PARP-1 is integral to the function of insulators in organizing the three-dimensional architecture of the genome.
Alterations of the transcriptional machinery
In addition to its role as a modulator of chromatin structure, PARP-1 has also been shown to function as a coregulator that can alter the function of components of the transcriptional machinery. As a coregulator, PARP-1 interacts with the basal transcription machinery as well as sequence-specific DNA-binding transcription factors, such as NF-κB, HES1, Elk1, Sox2, C/EBPβ, and nuclear hormone receptors (Kraus and Lis 2003; Ryu et al. 2015). These interactions, which may occur as the endpoint of cellular signaling pathways, may lead to recruitment or ADP-ribosylation of various components of the transcriptional machinery. For example, PARP-1 can modulate transcription factor activity by promoting the recruitment of coregulators, such as the lysine acetyltransferase p300 and arginine methyltransferase CARM1, to NF-κB to support its transcriptional activity (Hassa et al. 2003, 2005, 2008). Moreover, PARP-1 may facilitate the exchange of coregulators at promoters, such as inactive Cdk8-positive Mediator for active Cdk8-negative Mediator during retinoic acid-regulated activation (Pavri et al. 2005) or the estrogen-dependent recruitment of the TopoIIβ-containing activation complex in exchange for corepressor complexes present at the promoters of estrogen-responsive genes (Ju et al. 2006).
ADP-ribosylation of transcriptional regulators by PARP-1, which alters or modulates their activities, may also play a critical role in gene regulation. For example, PARP-1 ADP-ribosylates NELF-A and NELF-E of negative elongation factor (NELF), a regulatory complex that promotes promoter-proximal pausing of RNA polymerase II (Adelman and Lis 2012). ADP-ribosylation of NELF by PARP-1 promotes its release from paused RNA polymerase II, allowing productive elongation and RNA production (Fig. 4B; Gibson et al. 2016). Interestingly, ADP-ribosylation of NELF-E is dependent on prior phosphorylation by P-TEFb (positive-transcription elongation factor b), a kinase complex that promotes transcriptional elongation (Adelman and Lis 2012). PARP-1 also ADP-ribosylates C/EBPβ, an adipogenic transcription factor, which inhibits its DNA-binding activity (Fig. 4C; Luo et al. 2017). Adipogenic signals reduce PARP-1-dependent ADP-ribosylation of C/EBPβ, restoring its DNA-binding activity and allowing the adipogenic transcriptional program to proceed, ultimately leading to differentiation into mature adipocytes. These examples illustrate how the actions of PARP-1 are ultimately reflected in modified cellular signaling and alterations in physiological processes, such as stress responses, immune responses, circadian rhythms, and metabolism. While the examples here focus on the inhibitory effects of ADP-ribosylation, positive or stimulatory outcomes are possible as well.
PARPs in RNA biology
An emerging aspect of PARP biology is the roles that PARPs play in RNA biogenesis, processing, and trafficking. Key studies from Chang and colleagues (Leung et al. 2011) brought this new aspect of PARP biology to the forefront. They showed that, during heat shock and other stress conditions, stalled translation complexes aggregate to form RNA-rich cytoplasmic stress granules, which have been implicated in the regulation of mRNA translation and stability. The cytoplasmic stress granules contain PAR as well as six PARPs (PARP-5a, PARP-12, PARP-13.1, PARP-13.2, PARP-14, and PARP-15). Protein components of the stress granules, including Argonautes 1–4 (Ago 1–4, proteins that bind various classes of small noncoding RNAs), are ADP-ribosylated in response to cellular stress. The regulation of stress granules by ADP-ribosylation is required for the relief of microRNA-mediated translational repression and microRNA-directed mRNA cleavage (Leung et al. 2011). A recent study in Drosophila has shown that dPARP-16 and its catalytic activity are required for the formation of Sec body stress assemblies and cell survival in response to amino acid starvation (Aguilera-Gomez et al. 2016). Together, these results implicate cytoplasmic PARPs in the post-transcriptional regulation of gene expression.
PARPs have been implicated in other aspects of RNA biology as well, such as ribosomal RNA synthesis. In this regard, PARP-1 binds to and acts in concert with noncoding pRNAs (promoter-associated RNAs) as well as TIP5 (part of the chromatin remodeling complex NoRC) to suppress expression of ribosomal RNA (rRNA) genes (rDNA) by promoting formation of repressive heterochromatin (Guetg et al. 2012). This requires ADP-ribosylation of TIP5 as well as components of newly synthesized rDNA chromatin (Guetg et al. 2012). In Drosophila, nearly half of nuclear dPARP and PAR are localized in nucleoli, where rRNA transcription and processing occur (Boamah et al. 2012). dPARP plays an essential role in ribosomal biogenesis by maintaining nucleolar structural integrity through PAR synthesis (Boamah et al. 2012). Together, these studies demonstrate that PARP proteins bind to RNAs to modulate RNA biology and maintain cellular integrity.
Cross-talk between ADP-ribosylation and ubiquitylation
Another emerging aspect of PARP biology is the functional interplay between ADP-ribosylation and ubiquitylation. An example of this is PAR-dependent ubiquitylation, a process in which PARylation of a protein serves as a signal for its subsequent ubiquitylation, which may then lead to ubiquitin-dependent degradation of the protein by the proteasome (Pellegrino and Altmeyer 2016). In this regard, RNF146, a ubiquitin E3 ligase, has been shown to bind PAR through its WWE domain (Kang et al. 2011; Zhang et al. 2011). This allows the recruitment of RNF146 to PARylated target proteins, such as those PARylated by Tankyrase (PARP-5), and subsequent ubiquitylation of the target proteins using the RING E3 ligase domain of RNF146 (Zhang et al. 2011). Similarly, the ubiquitin ligase CHFR can bind autoPARylated PARP-1 through its PBZ domain and promote subsequent ubiquitylation and degradation of PARP-1 (Kashima et al. 2012). This results in decreased PARP-1 levels under mitotic stress conditions, causing cell cycle arrest (Kashima et al. 2012). PAR-dependent ubiquitylation is likely to be a general mechanism controlling the stability and degradation of many PARP substrate proteins.
Another interesting aspect of cross-talk between ADP-ribosylation and ubiquitylation comes from the bacterial pathogen Legionella pneumophila. Qiu et al. (2016) recently discovered a mechanism for ubiquitylation independent of E1 and E2 enzymes. Instead of relying on E1 and E2 enzymes, L. pneumophila uses SidE effectors to ubiquitylate multiple endoplasmic reticulum (ER)-associated Rab GTPases in host cells. These SidE effectors, such as SdeA, contain a putative MART motif, which is essential for the function of the effectors and the pathogenicity of L. pneumophila. Strikingly, a fragment from SdeA is capable of ADP-ribosylating ubiquitin (Qiu et al. 2016). Whether the ADP-ribosylated ubiquitin is directly used to modify target proteins is unclear, but this observation indicates a potential alternative mechanism for cross-talk between PARylation and ubiquitylation. Perhaps some currently undiscovered eukaryotic ubiquitin targeting PARP monoenzymes may promote ubiquitylation independent of E1 and E2 enzymes in eukaryotic cells.
The emerging biology of PARP monoenzymes and catalytically inactive PARPs
Although the historical focus of the PARP field has been on PARP polyenzymes, such as PARP-1, PARP-2, and Tankyrase, the focus has shifted to the PARP monoenzymes (i.e., MARTs). In fact, the PARP family comprises more monoenzymes than polyenzymes. These include PARP-3, PARP-4, PARP-6, PARP-7, PARP-8, PARP-10, PARP-11, PARP-12, PARP-14, PARP-15, and PARP-16. In addition, PARP-9 and PARP-13 are catalytically inactive (Ame et al. 2004; Vyas et al. 2014; Hottiger 2015).
Work by Vyas et al. (2013, 2014) has been instrumental in systematically characterizing the PARP family, especially the MARTs, on the basis of their cellular localization as well as phenotypes associated with loss of function. They found that the majority of PARP family members localizes to the cytoplasm, with some having a more specific localization to distinct organelles. They also observed that a number of PARPs exhibited cell cycle-dependent shuttling between the nucleus and cytoplasm (Vyas et al. 2013). Of the MARTs, loss of PARP-7 resulted in a mitotic defect, while the loss of macrodomain-containing PARP-9, PARP-14, and PARP-15 caused an actin cytoskeletal defect. Depletion of PARP-14, in particular, caused a striking morphological phenotype, leading to the elucidation of PARP-14 as a component of focal adhesion complexes and a major player in focal adhesion and cell motility (Vyas et al. 2013). These and other observations have led to increased interest in the biology of PARP monoenzymes as well as the catalytically inactive PARPs.
PARP-16, PARP-14, PARP-10, and PARP-6: counting down the biology of PARP monoenzymes
In this section, we provide a few examples of the emerging biology of PARP monoenzymes.
PARP-16
In a recent study, Jwa and Chang (2012) identified a surprising link between PARP-16 and the unfolded protein response (UPR) (Fig. 5A). They found that PARP-16 is a tail-anchored ER protein, which is inserted into the ER membrane through a C-terminal transmembrane domain. The N terminus of PARP-16, which contains the catalytic domain, faces toward cytoplasm. Interestingly, both PARP-16 and its catalytic activity are required for ER stress responses by regulating the UPR signaling pathway. PARP-16 interacts with PERK and IRE1α, two key components of the UPR pathway, and ADP-ribosylates both proteins under ER stress conditions (Jwa and Chang 2012). ADP-ribosylation of PERK and IRE1α by PARP-16 increases their kinase activities as well as the endonuclease activity of IRE1α. Strikingly, the C-terminal luminal tail of PARP-16 plays an essential role in mediating ER stress response. Although this study highlights a clear role for PARP-16 catalytic activity in mediating the UPR signaling pathway, further elucidation is necessary to understand the mechanisms by which PARP-16 senses luminal ER stress and transduces the signal to its catalytic domain.
Figure 5.
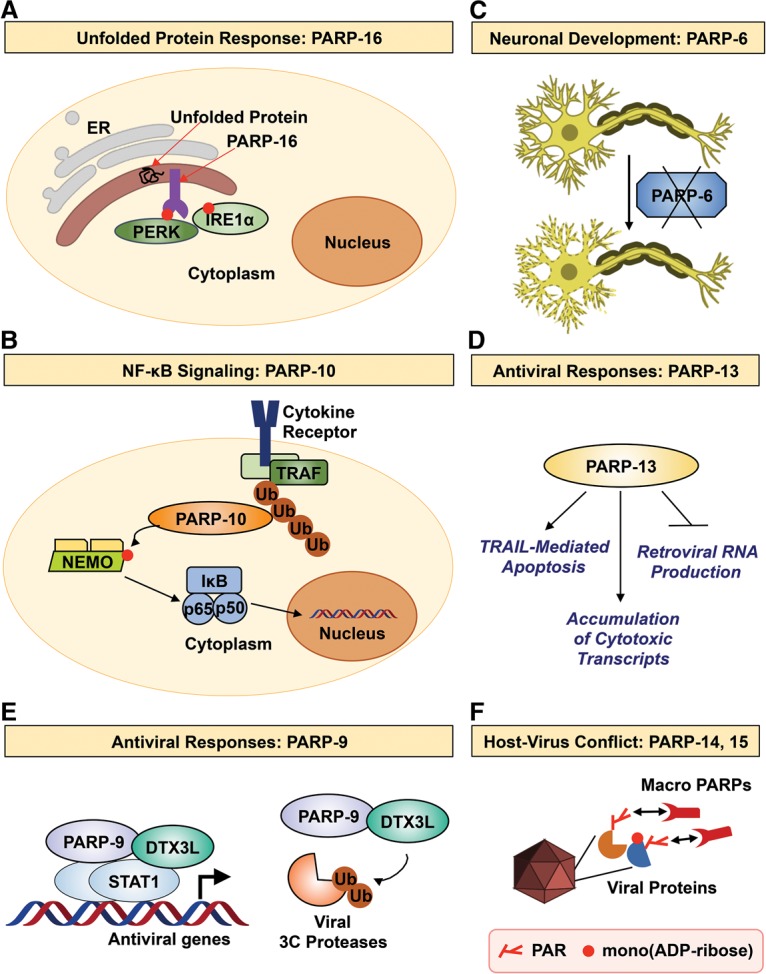
PARP monoenzymes and catalytically inactive PARPs participate in diverse biological processes. (A) PARP-16 regulates the UPR by modulating PERK and IRE1α activity. (B) PARP-10 attenuates NF-κB signaling by inhibiting NEMO. (C) PARP-6 is required for neurogenesis in the hippocampus. (D) PARP-13 inhibits viral pathogens by promoting TRAIL-mediated apoptosis, accumulation of cytotoxic transcripts, and inhibition of retroviral mRNA production. (E) PARP-9 associates with deltex E3 ubiquitin ligase 3L (DTX3L). Together, they promote antiviral gene transcription and trigger degradation of viral 3C proteases. (F) The macroPARPs PARP-14 and PARP-15 are involved in host–virus conflicts via their rapidly evolving macrodomains. See the text for details.
PARP-14
Recent studies have linked PARP-14 signaling pathways to the normal functions as well as cancers of immune cells. For example, PARP-14 regulates the class distribution, affinity repertoire, and recall capacity of antibody responses, which require efficient differentiation and interactions among B cells, Th cells, and dendritic cells (Cho et al. 2013). Furthermore, PARP-14 is required for IL-4-dependent enhancement of glycolysis in B cells as well as Myc-induced oncogenesis (Cho et al. 2011). PARP-14 inhibits STAT1 phosphorylation and proinflammatory gene expression in IFNγ-stimulated macrophages while enhancing STAT6 phosphorylation and anti-inflammatory gene expression in IL-4-stimulated macrophages, in part by antagonizing the actions of PARP-9 and by ADP-ribosylating STAT1 (Iwata et al. 2016). In multiple myeloma, a cancer of plasma cells, Jun N-terminal kinase 2 (JNK2) signals for cell survival through PARP-14, which binds to and inhibits JNK1 to promote the survival of myeloma cells and reduce their sensitization to anti-myeloma agents (Barbarulo et al. 2013).
PARP-10
A number of PARP family members play key roles in cellular stress responses controlled by the transcription factor NF-κB. Verheugd et al. (2013) observed a role for PARP-10 as a regulator of NF-κB signaling, a critical cellular pathway activated in response to a variety of pathogens (Fig. 5B). They found that PARP-10 negatively regulates the induction of NF-κB-dependent genes encoding cytokines. This regulation of NF-κB signaling requires both the catalytic activity and two unique ubiquitin interaction motifs in PARP-10. The ubiquitin interaction motifs recognize K63-linked polyubiquitin and can prevent the ubiquitylation of the upstream NF-κB activator NEMO (a subunit of the IκB kinase complex) (Verheugd et al. 2013). Furthermore, PARP-10 directly ADP-ribosylates NEMO. These events lead to the inhibition of nuclear localization of the p65 subunit of NF-κB and subsequent attenuation of NF-κB-dependent gene expression (Verheugd et al. 2013). This example with PARP-10 shows again how PARP family members function at the crossroads of NF-κB-dependent cellular stress responses and ubiquitin-dependent cellular processes.
PARP-6 and PARP-3
Recently, Huang et al. (2016a) described a role for PARP-6 in the development of hippocampal neurons (Fig. 5C). This work is based on the formation of the neural circuits during neurodevelopment, which is largely dependent on hippocampal dendrite morphogenesis (Dotti et al. 1988) and is characterized by widespread dendritic growth and branching. Huang et al. (2016a) observed increased expression of PARP-6 during dendrite morphogenesis. Moreover, they observed that loss of PARP-6 at this stage leads to a reduction in dendritic complexity in primary rat hippocampal neurons. Importantly, the requirement for PARP-6 was found to be directly linked to its catalytic activity as well as its N-terminal cysteine-rich domain (CRD). Both catalytically dead and CRD deletion mutants have defective branching and growth of the hippocampal neurons (Huang et al. 2016a). However, the exact targets of ADP-ribosylation by PARP-6 have not yet been elucidated.
Interestingly, PARP-3 has also been found to play a key role in neural development. PARP-3 is essential for ectodermal specification and neural crest development in zebrafish, possibly through transcriptional regulation (Rouleau et al. 2011). Together, these studies illustrate how the catalytic activity of a PARP monoenzyme can play key roles during development.
Role of PARP monoenzymes and catalytically inactive PARPs in immune and antiviral responses
As noted above, many members of the PARP family play key roles in cellular stress responses, including proinflammatory and pathogen responses. Early studies showed that Parp1-null mice are resistant to septic shock due to decreased serum levels of proinflammatory cytokines, such as TNFα and IL6, likely due to effects on cytokine gene expression (Oliver et al. 1999). PARP-1 has been implicated in inflammation and innate immune responses by modulating NF-κB activity (Hassa and Hottiger 2002). However, the broader role of PARP family members in different types of immune stresses requires further exploration. In this section, we describe some of the recent studies that have examined this question, with a focus on the role of PARP monoenzymes and catalytically inactive PARPs in antiviral responses.
Role of PARPs in antiviral responses
Antiviral activity for PARP family members was first discovered for PARP-13, also called ZAP (zinc finger antiviral protein), a catalytically inactive PARP family member (Fig. 5D). PARP-13 can inhibit the production of retroviral RNA (Gao et al. 2002) and inhibit endogenous retrotransposition by long interspersed nuclear elements (LINEs) and Alu elements (Goodier et al. 2015; Moldovan and Moran 2015). Furthermore, PARP-13 can directly target cellular transcripts for degradation, such as TRAILR4 mRNA, which encodes a decoy receptor for the cytotoxic ligand TRAIL (Todorova et al. 2014). As such, PARP-13 promotes TRAIL-mediated apoptosis, a well-known antiviral defense strategy (Todorova et al. 2014). PARP-13 also induces the expression of genes that attenuate the RISC-mediated transcript silencing, which promotes the accumulation of cytotoxic transcripts that promote the innate antiviral defense (Seo et al. 2013).
Recently, another catalytically inactive PARP, PARP-9, was shown to associate with deltex E3 ubiquitin ligase 3L (DTX3L) to mediate immune responses to viral pathogens (Fig. 5E; Zhang et al. 2015). Interferon stimulation of cells leads to activation of its downstream transcription factor, STAT1, and subsequent binding of STAT1 to genomic loci to induce proinflammatory gene expression. Zhang et al. (2015) showed that the phenotypic outcomes of a STAT1 mutant that is hyperresponsive to interferon signaling depend on the PARP-9–DTX3L complex. Multiple domains of PARP-9 and DTX3L are required for the interaction with STAT1. This interaction induced the E3 ubiquitin ligase activity of the complex toward host histone H2BJ to promote interferon-stimulated gene expression. Activation of the PARP-9–DTX3L complex's ubiquitin ligase activity also stimulated the degradation of the viral 3C proteases via the immunoproteasome (Zhang et al. 2015). Thus, PARP-9–DTX3L acts on both host and viral proteins to mediate efficient immune responses to viral pathogens.
Another PAR family member, PARP-12, a catalytically active monoenzyme, has also been shown to be involved in antiviral responses. In this regard, intracellular PARP-12 expression is up-regulated upon stimulation by type II interferons (Welsby et al. 2014). Furthermore, ectopic expression of PARP-12 leads to increased NF-κB signaling, implicating PARP-12 in cellular immune responses (Welsby et al. 2014).
In contrast to the antiviral roles of PARPs noted above, ADP-ribosylation has also been linked to proviral responses. Viral protein-mediated degradation of PARG during herpes simplex virus 1 (HSV-1) replication suggests that increases in PARylation could promote viral infection (Grady et al. 2012). Furthermore, ADP-ribosylation also promotes replication and genome integration for certain types of viruses (Kuny and Sullivan 2016). Collectively, these examples point to key roles for PARPs and ADP-ribosylation in viral responses but also illustrate the complex nature of the biology. Moreover, these studies emphasize that the roles of PARPs in host–virus interactions should be assessed in a context-specific manner.
PARPs and host–virus conflicts
An important aspect of host defense against viral infection is the ability to combat constantly evolving viruses. The rapid evolution of viral protein sequences to adapt to host defenses and the consequent changes in host-derived proteins to overcome this represent an interesting aspect of evolutionary biology (Adhya and Basu 2010; Daugherty and Malik 2012; Salomon and Orth 2013). This aspect of evolution can be traced to specific domains in host proteins that mediate competition with viral proteins. Recent work by Daugherty et al. (2014) explored the role of PARPs in the evolution of host–virus defenses (Fig. 5F). They showed that PARP-9, PARP-13, PARP-14, and PARP-15 in primates have diversified through positive selection. Specifically, the macrodomains from PARP-9, PARP-14, and PARP-15 exhibit characteristics suggestive of multiple rounds of positive selection. These studies predict a connection between the antiviral response and the catalytic activity of PARP monoenzymes. Furthermore, they suggest that PARP-14 and PARP-15 may have gone through multiple cycles of gene birth and loss, supporting the theory that they are evolving rapidly in response to viral conflicts (Daugherty et al. 2014).
Interestingly, macrodomain-containing proteins have also been identified in a variety of virus families, including the Coronaviridae, Togaviridae, and Hepeviridae (Table 1; Egloff et al. 2006; Malet et al. 2009; Li et al. 2016). A macrodomain-containing protein from the hepatitis E virus, a member of the Hepeviridae family, can catalyze the removal of MAR and PAR from modified proteins. Moreover, this catalytic activity is required for efficient viral replication (Li et al. 2016). The presence of functional macrodomains in viruses points toward novel mechanisms of pathogenesis and may provide a deeper insight into the evolutionary pressures on the rapidly evolving mammalian macroPARPs.
From enzymes to modifications: new technologies to detect and study of ADP-ribosylation
The recent development of new methods, reagents, and other tools has greatly facilitated the detection and study of ADP-ribosylation. This has been particularly important as the field has moved beyond the study of PARP proteins to the study of ADP-ribosylation. The new methods, reagents, and tools include (1) reagents to detect MAR, oligo-(ADP-ribose), and PAR in cells and biochemical assays; (2) mass spectrometry (MS)-based proteomic approaches to identify ADP-ribosylated proteins and the specific sites of ADP-ribosylation; (3) chemical biology approaches to assign ADP-ribosylation events to specific PARP family members; (4) genomic assays to determine the sites of ADP-ribosylation across the genome; and (5) tools to assay the levels of NAD+ in various cellular compartments (Fig. 6). These will be introduced and discussed in the sections below.
Figure 6.
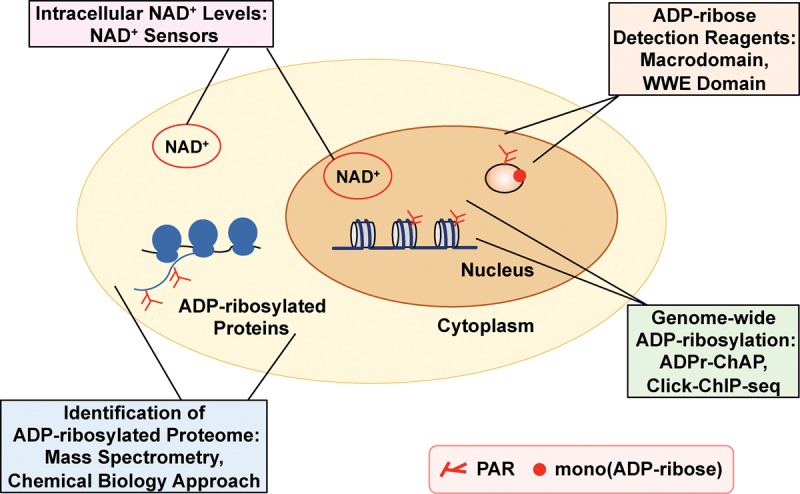
New technologies to detect and study cellular ADP-ribosylation. The schematic illustrates different technologies that are discussed in detail in the text.
ADP-ribose detection reagents
A large part of the recent progress in the PARP field, especially as it relates to the catalytic activity of the enzymes, can be attributed to the development of novel ADP-ribose-detecting reagents. For decades, the field has relied on the anti-PAR monoclonal antibody 10H, which binds to PAR chains >10 ADP-ribose units in length (Kawamitsu et al. 1984). Although useful, this antibody has left the field blind to MARylation and oligo(ADP-ribosyl)ation events. Other studies have reported the production of anti-ADP-ribose antibodies, but their specificity and utility have not been assessed broadly (Meyer and Hilz 1986).
The discovery of a diverse set of naturally occurring ARBDs, as noted above, has provided new tools for exploring ADP-ribose and PAR in cells through the use of ARBD-GFP fusions, which allow real-time tracking of localized ADP-ribose and PAR synthesis in cells (Timinszky et al. 2009; Murawska et al. 2011; Aguilera-Gomez et al. 2016). The macrodomain from the archaebacterial protein Af1521, which recognizes MAR and the terminal ADP-ribose of PAR, has been fused with a GST tag and used to enrich for ADP-ribosylated targets in genomic and proteomic screens (Bartolomei et al. 2016; Martello et al. 2016). Similarly, the WWE domain from mammalian RNF146 has been fused with a GST tag and used to determine the genomic localization of PAR (Bartolomei et al. 2016). ARBDs, including the aforementioned macrodomain and WWE domain, have also been fused to the Fc region of rabbit immunoglobulin to create recombinant antibody-like fusion proteins that bind ADP-ribose or PAR and function much like monoclonal antibodies (Gibson et al. 2016; Luo et al. 2017). These Fc-fused reagents can be used in a broad range of assays, including immunoblotting, immunofluorescence, chromatin immunoprecipitation (ChIP), and ELISA (Gibson et al. 2016; Luo et al. 2017). The use of these ARBD-based reagents in detecting dynamic changes in ADP-ribosylation in various biological systems highlights their importance and the potential for future discoveries that they provide (Gibson et al. 2016; Teloni and Altmeyer 2016).
In addition to ARBD-based reagents, approaches from chemical biology have also facilitated the detection of ADP-ribosylated proteins in cells. For example, Westcott et al. (2017) have shown that the alkyne–adenosine analog N6-propargyl adenosine (N6pA) can be metabolically incorporated into ADP-ribose in mammalian cells. In combination with click chemistry approaches using the alkyne moeity, N6pA allows the fluorescence detection and proteomic analysis of ADP-ribosylated proteins.
New techniques in PARP proteomics and MS
Compared with other types of PTMs, the identification of sites of ADP-ribosylation on a proteome-wide scale has lagged behind other PTMs until recently. Although previous studies have demonstrated clear effects of ADP-ribosylation of target proteins, the sites of PARylation were not mapped in most cases. For example, PARylation of KDM5B, a histone H3 Lys4 demethylase, by PARP-1 at unspecified sites prevents its binding to chromatin and blocks its enzymatic activity (Krishnakumar and Kraus 2010b). Additional studies have shown that a number of DNA-binding transcription factors (e.g., Sox and YY1) are PARylated by PARP-1 (Oei and Shi 2001; Gao et al. 2009), but, again, those sites were not mapped. Examples where the sites of ADP-ribosylation on target proteins have been mapped are NFAT (ADP-ribosylation increases DNA binding) (Olabisi et al. 2008), p53 (ADP-ribosylation inhibits nuclear export) (Kanai et al. 2007), CTFC (ADP-ribosylation is required for optimal function in transcriptional activation and insulator function) (Farrar et al. 2010), STAT1 (ADP-ribosylation inhibits signal-regulated phosphorylation) (Iwata et al. 2016), NELF-E (ADP-ribosylation inhibits RNA binding and NELF-dependent promoter-proximal pausing by RNA polymerase II) (Fig. 4B; Gibson et al. 2016), and C/EBPβ (ADP-ribosylation inhibits DNA binding) (Fig. 4C; Luo et al. 2017).
The nature of ADP-ribosylation has made it difficult to study: (1) It is labile, (2) it is heterogeneous (i.e., mono, oligo, and poly), (3) and its polymers are bulky and charged. Moreover, due to the presence of multiple active PARPs in a cell at any given time and their compensatory nature, determining the contribution made by one specific PARP to the ADP-ribosyl proteome has proven to be difficult. Recently, a number of novel MS-based techniques have been developed to identify proteins that are ADP-ribosylated (protein or peptide identification) as well as the specific sites of ADP-ribosylation (Daniels et al. 2015a). These methods rely on approaches to (1) degrade PAR chains down to a single ADP-ribosyl moiety and (2) cleave the ADP-ribosyl moiety to produce an identifiable mass shift in the mass spectrometer. The different methods discussed below use different approaches, both enzymatic and chemical, to achieve this.
Chemical cleavage
Zhang et al. (2013) recently reported a method to enrich for the universe of ADP-ribosylated peptides using boronate affinity chromatography, in which the ADP-ribose unit forms an ester linkage with boronate beads. Subsequently, the bead-bound peptides can be eluted using hydroxylamine (NH2OH), which cleaves the ester bond between a protein-proximal ADP-ribose and the side chain carboxyl group of an aspartic acid or glutamic acid residue (Fig. 7A,B). This results in the generation of aspartic acid or glutamic acid residues modified with hydroxamic acid, adding an additional mass of 15.0109 Da, which is detectable as a mass shift during MS (Table 2). This method yields both protein and site identifications.
Figure 7.
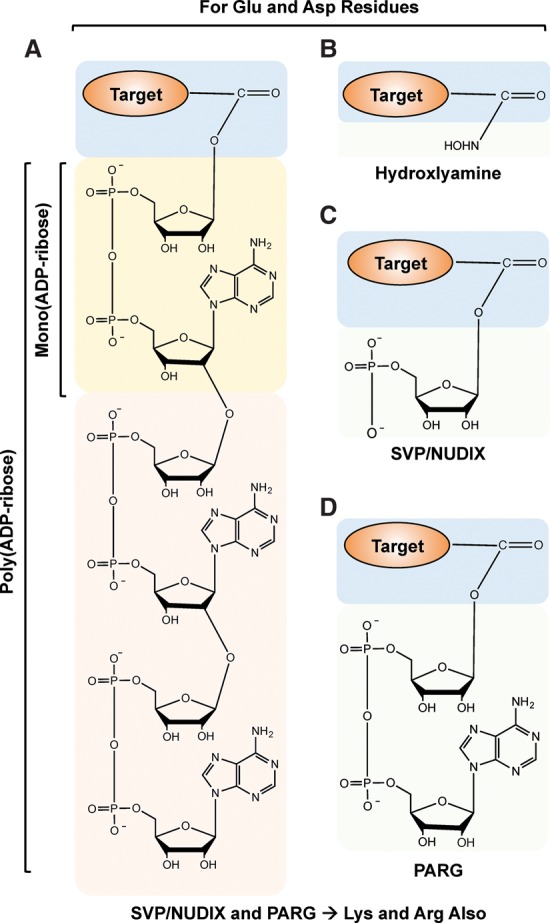
Chemistry of PAR cleavage for use in MS methods. (A) Chemical structure of a PAR covalently linked to an amino acid in a target protein. (B–D) Structure of terminal moieties attached to an amino acid in the target protein after ADP-ribose cleavage using hydroxylamine (B), snake venom phosphodiesterase (SVP) or NUDIX (C), or PARG (D). The chemical structures shown in A–D are for Glu and Asp residues. Note that SVP, NUDIX, and PARG can also hydrolyze ADP-ribose linked to Lys and Arg residues (not shown). The exact nature of the chemical structures for SVP-, NUDIX-, and PARG-cleaved Lys-ADP-ribose and Arg-ADP-ribose have not been determined experimentally (Daniels et al. 2015a).
Table 2.
Methodology for the MS-based detection of protein ADP-ribosylation

An advantage of using hydroxylamine is that the resulting mass shift on the amino acids is the same irrespective of the nature of the ADP-ribose chain (poly, oligo, or mono). Moreover, given that this is a chemical cleavage, there is no inherent bias attached to this methodology, as there might be in an enzymatic reaction. However, the hydroxylamine chemistry is relevant only in the context of aspartic acid and glutamic acid residues; it cannot be used to screen for ADP-ribosylation of other amino acids, such as lysine or arginine, which are also ADP-ribosylated. Using this approach, Zhang et al. (2013) identified >1000 ADP-ribosylation sites on 340 proteins, thus generating an extensive list of both known and previously unknown PARP targets. A similar approach using hydroxylamine was also reported by Gagne et al. (2015) to determine the sites of automodification on PARP-1, PARP-2, and PARP-3. This versatile method has yielded new insights into the biology of ADP-ribosylation and PARP targets.
Enzymatic cleavage
Enzymatic cleavage of PAR and ADP-ribose to reduce complexity using enzymes, such as snake venom phosphodiesterase (SVP), ARH3, PARG, or NUDIX hydrolases, is becoming a popular approach as a prelude to MS (Messner et al. 2010; Chapman et al. 2013; Sharifi et al. 2013; Daniels et al. 2014, 2015b; Martello et al. 2016). A number of groups have used SVP, a pyrophosphatase that cleaves ADP-ribose subunits to phosphoribose (also known as ribose-5′-phosphate), which remains covalently attached to the amino acid in the ADP-ribosylated protein, and 5′-AMP, which is released (Chapman et al. 2013; Daniels et al. 2014). The phosphoribose moiety remaining at the site of modification can be identified by MS as a shift of 212.01 Da (Fig. 7A,C). Phosphoribosylated peptides can be enriched using techniques developed previously for phosphopeptides, such as immobilized metal affinity chromatography (IMAC) (Chapman et al. 2013; Daniels et al. 2014). This approach is not restricted to acidic residues, unlike hydroxylamine treatment, providing a major advantage for this approach (Table 2). Moreover, the coenrichment of phosphorylated amino acids allows for the study of the dynamics between phosphorylation and ADP-ribosylation (Daniels et al. 2014). This approach has been used to identify sites of ADP-ribosylation on automodified PARP-1, including glutamate, aspartate, and lysine residues (Chapman et al. 2013), as well as endogenous ADP-ribosylation sites from whole cells (Daniels et al. 2014).
One of the caveats of using SVP is that it is difficult to obtain commercially and is typically purified in small batches, which may affect reproducibility. To address this issue, Palazzo et al. (2015) and Daniels et al. (2015b) explored the use of NUDIX hydrolase enzymes instead of SVP. Enzymes in the NUDIX superfamily of hydrolases catalyze the hydrolysis of nucleoside diphosphates linked to other moieties, including ADP-ribose (Fig. 7A,C). The investigators showed that different NUDIX enzymes, including the RNA 5′ pyrophosphohydrolase (RppH) from Escherichia coli (EcRppH) and the human NUDIX enzyme HsNudT16, can be used for digestion of PAR chains for MS (Table 2). However, in a direct comparison with SVP, only two of the 14 phosphoribosylated peptides detected from automodified PARP-1 by Daniels et al. (2015b) were found in all three enzyme-treated samples (SVP vs. EcRppH and HsNudT16). Palazzo et al. (2015) detected only two distinct sites of ADP-ribosylation on PARP-1, but these sites overlapped those identified by Daniels et al. (2015b). HsNudT16 has also been used to identify serine residues on histones as novel acceptors of ADP-ribosylation (Leidecker et al. 2016). While the use of NUDIX enzymes has some advantages, these enzymes appear to have an inherent bias for features of the peptide to which the ADP-ribose is attached. The simultaneous use of multiple enzymes could overcome this bias, but such an approach needs to be explored further. Moreover, the use of other types of enzymes should be explored as an alternative to the NUDIX enzymes. For example, the recently characterized phosphodiesterase ENPP1 has been used to identify the sites of automodification in PARP-1 and PARP-10 (Palazzo et al. 2016).
Nielsen and colleagues (Martello et al. 2016) have used PARG to cleave PAR chains to ADP-ribose monomers to reduce the complexity of PAR prior to MS (Fig. 7A, D). In addition, they have combined this approach with an ADP-ribose enrichment step using the Af1521 macrodomain (Martello et al. 2016) based on a prior affinity enrichment approach performed without PARG cleavage (Jungmichel et al. 2013). This allows for the selection of peptides that carry a single ADP-ribose unit, the site of which can be identified using MS (Table 2). Using this approach, they were able to profile >900 ADP-ribosylation sites in HeLa cells as well as in the mouse liver. Notably, they observed a striking difference in the amino acids that were ADP-ribosylated in HeLa cells versus liver tissue. In HeLa cells, sites of glutamate, lysine, arginine, and aspartate ADP-ribosylation were comparably enriched, while, in liver tissue, sites of arginine ADP-ribosylation were markedly enriched (Martello et al. 2016). Although this technique allows for the identification of all amino acids that are ADP-ribosylated, bias for the macrodomain against ADP-ribosylated glutamic acid could lead to the loss of a number of targets (Daniels et al. 2014).
MS methods for the detection of ADP-ribosylation
Some of the approaches discussed above use collision-induced dissociation (CID) or higher-energy collisional dissociation (HCD) to fragment the ions during MS (Zhang et al. 2013; Daniels et al. 2014). The use of these MS techniques may lead to a loss of the labile ADP-ribose units from amino acid residues, resulting in a reduced number of sites identified. The use of electron transfer dissociation (ETD) has been suggested as an alternative that allows the ADP-ribose chains to remain intact, thus providing wider coverage (Rosenthal et al. 2015). Although the use of ETD alone yielded fewer peptides carrying an ADP-ribose moiety compared with HCD, the ETD fragmentation did allow for the detection of certain unique peptides not present in the HCD subset (Rosenthal et al. 2015). Moreover, a combinatorial approach using the two techniques together was more robust in the identification of ADP-ribosylated peptides (Rosenthal et al. 2015). The benefits and drawbacks of each of the approaches mentioned here must be considered when designing experiments to detect ADP-ribosylated proteins.
PARP chemical biology and NAD+ analog sensitivity
Although our ability to identify ADP-ribosylated proteins in cells has improved dramatically, as noted above, determining which PARP family member is responsible for the ADP-ribosylation has been elusive. Many PARPs that share cellular compartments have overlapping protein substrates, which include other PARPs. Thus, traditional methods to identify the PARP proteins responsible for a specific ADP-ribosylation event, such as genetic or RNAi-mediated depletion of a particular family member, may be inadequate or yield confounding results (e.g., depletion of a PARP family member may alter ADP-ribosylation of a target protein indirectly by affecting the catalytic activity of another PARP family member).
Recent studies have described chemical biology approaches for PARPs that allow for identification of PARP-specific ADP-ribosylation events. These methods make use of unnatural NAD+ analogs that can be used only by PARPs that have appropriate mutations in their catalytic domain. In general, the mutations create a “hole” in the PARP catalytic domain that can be filled by an extra chemical moiety (“bump”) on the NAD+ analog, conceptually similar to an approach developed by Specht and Shokat (2002) to identify targets of specific cellular kinases. The addition of a “clickable” moiety (e.g., alkyne) on the NAD+ analog has expanded the utility of this approach, enabling the identification of novel target proteins and the sites of ADP-ribosylation catalyzed by different PARP family members.
Targeting the nicotinamide moiety of NAD+ for PARP analog sensitivity
Carter-O'Connell et al. (2014) have developed an NAD+ analog-sensitive PARP (asPARP) approach in which an ethyl group is added at the C-5 position of the nicotinamide moiety. They also added an alkyne at the N-6 position of the adenosine moiety, generating 5-Et-6-a-NAD+, to allow for copper-catalyzed “click” chemistry. In initial experiments, they used this NAD+ analog with a PARP-1 mutant in which Lys903 (K903) was mutated to alanine (K903A). K903, which is semiconserved in the PARP family, is present in the NAD+-binding pocket. The mutation of the lysine to alanine at this site causes a change in the specificity of PARP-1, allowing it to catalyze MARylation, but not PARylation (a limitation of this particular approach), with 5-Et-6-a-NAD+ (Fig. 8A,B; Carter-O'Connell et al. 2014). Using the asPARP-1 mutant with the NAD+ analog coupled to click chemistry and MS allowed the isolation and enrichment of PARP-1-modified proteins. A corresponding mutation in PARP-2 also conferred analog sensitivity with 5-Et-6-a-NAD+ (Carter-O'Connell et al. 2014). Small modifications to the approach conferred analog sensitivity to the PARP monoenzymes PARP-10 and PARP-11 (Carter-O'Connell et al. 2016). These studies highlight the utility of the asPARP approach for identifying the target substrates of specific PARP family members.
Figure 8.
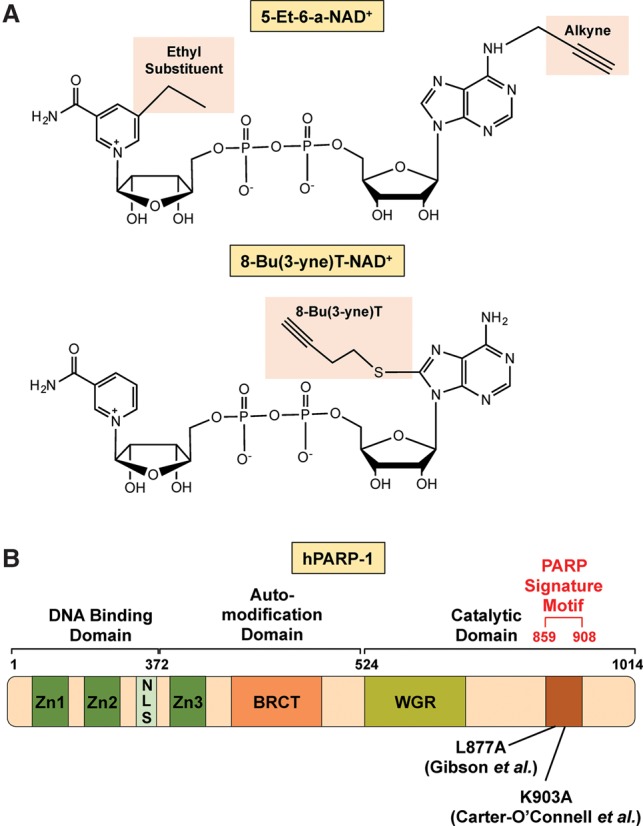
Chemical biology approaches to identify PARP-specific ADP-ribosylation events. (A) Molecular structures of the NAD+ analogs used by Carter-O'Connell et al. (2014) (top) and Gibson et al. (2016) (bottom) for their analog-sensitive PARP approaches. The substituents for NAD+, each analog as well as the “clickable” groups, are highlighted in pink. (B) Schematic of human PARP-1 showing the different protein domains. The highlighted amino acids in human PARP-1 were mutated by Carter-O'Connell et al. (2014) and Gibson et al. (2016) to confer analog sensitivity.
Targeting the adenosine moiety of NAD+ for PARP analog sensitivity
Gibson et al. (2016) have developed an NAD+ asPARP approach using a single, bifunctional alkyne-containing R group at the C-8 position of the adenosine moiety, generating 8-Bu(3-yne)T-NAD+, which both confers analog sensitivity and allows for copper-catalyzed “click” chemistry. They used this NAD+ analog with a PARP-1 mutant in which Leu877 (L877) was mutated to alanine (L877A), conferring analog sensitivity (Fig. 8A,B). In addition, using mutation of a homologous residue, the approach was extended to PARP-2 and PARP-3 (with L443A and L394A mutations, respectively) (Gibson et al. 2016). Importantly, the approach preserves the polymerase catalytic activity of both PARP-1 and PARP-2.
Using this asPARP approach, Gibson et al. (2016) were able to identify the universe of targets (both protein IDs and site IDs) for PARP-1, PARP-2, and PARP-3 in HeLa nuclear extracts. They observed a significant overlap in the targets of PARP-1, PARP-2, and PARP-3, although there was still a distinct population of targets for each individual PARP. Moreover, the three PARPs exhibit a degree of sequence preference at their ADP-ribosylation sites. (Gibson et al. 2016). The PARP-1, PARP-2, and PARP-3 ADP-ribosylated proteomes identified in this study showed an enrichment for proteins involved in transcription and RNA biology, thus supporting a role for these PARPs beyond DNA damage repair (Gibson et al. 2016). Given the conservation of the gatekeeper residues identified by Carter-O'Connell et al. (2014) and Gibson et al. (2016), this approach should be broadly applicable across the PARP family.
Cataloging the ADP-ribosylated proteome
The proteomics technologies described in this and the previous section have led to a rapidly expanding database of PARP targets that require further exploration to determine the specific roles of site-specific ADP-ribosylation in biological outcomes. Nevertheless, these proteomics studies have provided a glimpse into the role of different PARPs in various aspects of cell biology. Further development of these approaches can be achieved by methodological improvements as well as by combining one or more of the technologies described above for improved detection and identification of targets in different cellular contexts. Recently, the Leung group (Vivelo et al. 2016) created a database (http://ADPriboDB.leunglab.org) that provides a comprehensive list of ADP-ribosylated proteins derived from >400 publications. Currently, the database contains information regarding ADP-ribosylation of 2389 unique protein targets (Vivelo et al. 2016). This archive will be a valuable resource for the field, especially if the information that it contains on ADP-ribosylation can be leveraged with new technologies for the synthesis of site-specific ADP-ribosylated peptides (Moyle and Muir 2010; van der Heden van Noort et al. 2010; Kistemaker et al. 2013, 2016), which can be used for binding assays with ARBDs or as antigens for antibody generation.
Genomic analyses of ADP-ribosylation
Genomic approaches have revolutionized our understanding of how PTMs, largely modifications of histone proteins, affect genome function. The role of PARPs in DNA repair and gene regulation has been widely studied, as described above. How ADP-ribosylation of chromatin-associated proteins affects these biological outcomes is a burgeoning area of study. However, until recently, methods to detect ADP-ribosylation of chromatin-associated proteins were elusive. Recent advances in the detection of ADP-ribosylation, as described in the previous section, have allowed the development of technologies for genomic analysis of ADP-ribosylation. Two such approaches are described below.
ADP-ribose chromatin affinity precipitation (ADPr-ChAP)
Recently, Bartolomei et al. (2016) described a protocol for the affinity purification of chromatin using two well-characterized ARBDs to enrich for ADP-ribosylated chromatin, which they call ADPr-ChAP. In the ADPr-ChAP-seq protocol, mammalian cells were fixed using 4% formaldehyde followed by lysis and chromatin shearing. Genomic DNA cross-linked to ADP-ribosylated chromatin-associated proteins was enriched by affinity precipitation with PAR-binding Af1521 and WWE domains. The genomic DNA in the precipitated material was then sequenced and mapped to the human genome to identify regions enriched for ADP-ribosylation (Fig. 9A). The investigators determined genome-wide changes in ADP-ribosylation upon short-term induction of DNA damage with H2O2 (Bartolomei et al. 2016). They found that, upon H2O2-induced DNA damage, ADP-ribose is enriched in heterochromatin (which is marked with histone H3K9me3) and depleted at active promoters. Moreover, ADP-ribosylation was found to correlate with higher levels of total histone H3. In locus-specific ADPr-ChAP assays, they observed increased ADP-ribosylation at specific genomic loci upon other stimuli, such as the induction of adipogenesis (Bartolomei et al. 2016). As illustrated in this study, ADPr-ChAP can be used to explore dynamic changes in ADP-ribosylation that occur at a genomic level in response to various cellular perturbations. However, since many PARPs that share cellular compartments have overlapping protein substrates, including other PARPs, the combination of ADPr-ChAP with genetic or RNAi-mediated depletion of a particular PARP family member may yield ambiguous results regarding the specific PARPs responsible for the ADP-ribosylation events identified.
Figure 9.
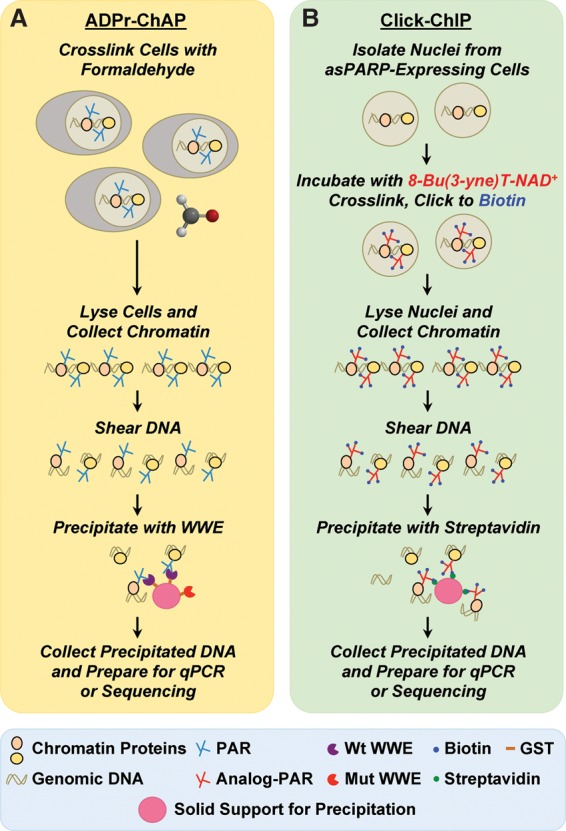
Comparison of techniques used to map genome-wide ADP-ribosylation. (A) ADPr-ChAP by Bartolomei et al. (2016). (B) Click-ChIP-seq (click chemistry-based chromatin isolation and precipitation with deep sequencing) by Gibson et al. (2016). Details of the protocols are provided in the text.
Click-ChIP (click chemistry-based chromatin isolation and precipitation)
Gibson et al. (2016) repurposed their asPARP technology, described above, to determine the genomic localization of PARP-specific ADP-ribosylation, with a focus on PARP-1. In the genomic version of the method, which they call “Click-ChIP-seq” (Click-ChIP with deep sequencing), they used the asPARP-1 approach in nuclei collected from mouse embryonic fibroblasts ectopically expressing an asPARP-1 mutant. The nuclei expressing asPARP-1 were incubated with the clickable NAD+ analog 8-Bu(3-yne)T-NAD+ and then cross-linked with 1% formaldehyde followed by clicking the analog to biotin. The chromatin was sheared, PARP-1 ADP-ribosylated chromatin was precipitated using streptavidin beads, and the enriched genomic DNA was subjected to deep sequencing (Fig. 9B; Gibson et al. 2016). Click-ChIP-seq showed that PARP-1-mediated ADP-ribosylation events are enriched at transcriptionally active promoters and correspond with the enrichment of histone H3K4me3 (a marker for active promoters). Genome-wide correlations were also observed between PARP-1-mediated ADP-ribosylation events, chromatin-bound PARP-1, and NELF-B, a subunit of the NELF. An inverse correlation was observed with RNA polymerase II, supporting the role for ADP-ribosylation of NELF in the release of promoter-proximal paused RNA polymerase II into productive elongation (Fig. 4B; Gibson et al. 2016).
Cellular NAD+ sensors
PARPs are dependent on NAD+ for their catalytic activity. Although NAD+ has historically been thought to pass freely from one cellular compartment to another, the results from recent studies (e.g., Zhang et al. 2012; Cambronne et al. 2016) and the fact that the three NMNATs have discreet and distinct subcellular localizations have challenged this view. Moreover, NAD+ and PAR levels fluctuate during key biological processes, exemplified by a precipitous drop in both during the transition from mitotic cell growth to cellular differentiation during adipogenesis studies (Pekala et al. 1981; Janssen and Hilz 1989; Luo et al. 2017). As such, the dynamic NAD+ synthesis and its subcellular localization are important considerations for PARP function.
The ability to measure intracellular NAD+ levels was limited by the tools available until recently, with the advent of cellular sensors that can assay rapid and dynamic changes in NAD+ in intact cells. The Peredox sensor, which combines a circularized GFP T-Sapphire component with the NADH-binding Rex protein, is a redox sensor that can be used for ratiometric measurement of NADH/NAD+ levels (Hung et al. 2011). However, Peredox cannot be used for the direct measurement of NAD+. The SoNar sensor has a circularly permuted (cp) YFP linked to a Rex protein variant that can bind to either NADH or NAD+ (Zhao et al. 2015). It also has increased fluorescence in response to the binding of both substrates at distinct excitation wavelengths. This allows for ratiometric determinations as well as independent detection of either NADH or NAD+. Moreover, SoNar is able to detect cellular changes in NAD+/NADH over a greater dynamic range than Peredox (Zhao et al. 2015). More recently, Cambronne et al. (2016) reported a sensor that solely detects NAD+. This sensor has a bipartite NAD+-binding domain (which can reversibly bind NAD+) and cpVenus as the fluorescence component. Significantly, the addition of nuclear, cytoplasmic, and mitochondrial localization sequences to the sensors has enabled the study of physiological NAD+ levels in each cellular compartment separately (Cambronne et al. 2016).
From the bench to the clinic: advances in PARP-related therapeutics
No review on PARPs would be complete without at least a mention of the vast clinical potential represented by this family of proteins. In fact, the associated readers, erasers, and feeders noted above also hold great clinical potential. As highlighted at ClinicalTrials.gov, there are >200 clinical trials aimed at evaluating the therapeutic potential of a new generation of PARPis. While the vast majority is focused on cancer, others are aimed at cardiovascular disease, inflammation, infections, and other common ailments.
The use of PARPis in cancer treatment has received considerable attention. Inhibition of PARP-1 results in accumulation of DNA DSBs in BRCA1/2-defective cancer cells and induces cell death via a phenomenon known as synthetic lethality (Bryant et al. 2005; Farmer et al. 2005). The discovery that cancers with BRCA1/2 mutations are sensitive to PARPis has sparked great interest in developing these drugs to treat ovarian and breast cancer (Curtin and Szabo 2013). In fact, in 2014, the U.S. Food and Drug Administration approved the use of AstraZeneca's Lynparza (olaparib) for the treatment of women with advanced ovarian cancer associated with BRCA1/2 mutations. Undoubtedly, applications for treating cancers without BRCA1/2 mutations will also be found, since PARPs intersect with many different cellular stress and genome integrity pathways. Furthermore, given the important role of nuclear PARPs in gene regulation, PARPis may be useful in targeting transcriptionally addicted cancers (Franco and Kraus 2015; Wang et al. 2015).
Molecular strategies for targeting PARPs
The majority of currently available PARPis has a relatively broad spectrum across the PARP family (Wahlberg et al. 2012). However, as is becoming increasingly clear, different PARPs have very different functional roles. Thus, inhibiting multiple PARPs may have pleiotropic effects and unintended consequences. Profiling an array of PARPis revealed that most of them target the NAD+-binding pocket, which results in relatively nonspecific inhibition across the different family members (Wahlberg et al. 2012). However, compounds such as olaparib and veliparib showed increased selectivity for PARP-1 through PARP-4, suggesting that these and other structurally similar PARPis may have an appropriate level of specificity for clinical use (Wahlberg et al. 2012).
In a recent screen with different PARPis, the Schuler group (Thorsell et al. 2016) tested the capability of each inhibitor to specifically attenuate the in vitro catalytic activity of a panel of PARPs. Furthermore, they used X-ray crystallography to elucidate the mechanisms of action for the different PARPis. Using this strategy, they assayed 11 members of the PARP family, including PARP-1, PARP-2, and PARP-3, and the Tankyrases. They were able to determine that inhibitors such as veliparib and niraparib have the highest selectivity for PARP-1 and PARP-2, whereas other inhibitors, such as olaparib, have a high potency but relatively lower selectivity for PARP-1 and PARP-2 (Thorsell et al. 2016). This study not only provides critical information regarding selectivity and potency of different clinically used PARPis but also establishes assays that can be used in the future to test new PARPis.
A recent study explored a new strategy that targets allosteric regulation as a selective way of inhibiting PARP-1 (Steffen et al. 2014). Historically, most PARPis are nicotinamide mimics that target the PARP NAD+-binding pocket (Steffen et al. 2013), making them broadly effective against many PARPs. The genetic disruption of PARP-1 interdomain contacts prevents DNA damage-dependent catalytic activation and shows increased sensitivity to platinum-based anti-cancer agents while having no effect on PARP-1 recruitment to sites of DNA damage or transcriptional regulation (Steffen et al. 2014). These results highlight the potential of synergistic drug combinations using allosteric PARPis with DNA-damaging agents.
Recent progress in the development of drug target engagement assays to evaluate the binding of drugs to their targets in cells (e.g., cellular thermal shift assay [CETSA]) may be helpful in evaluating PARPis before going into the clinic. For example, the CETSA was able to distinguish between olaparib, a well-characterized PARP-1 inhibitor that binds directly to PARP-1, and iniparib, a proposed PARP-1 inhibitor that reached phase III clinical trials but showed no efficacy (Martinez Molina et al. 2013). Similarly, CETSA was used to assess the inhibitory potential of the Tankyrase inhibitor XAV939 toward PARP-1 (Thorsell et al. 2016). The structural differences revealed for different PARPs—for example, PARP-1 and PARP-3—have also been used to assess the selectivity of different inhibitors (Lehtio et al. 2009). The development of these and other molecular strategies will help in optimizing PARPis with respect to specificity and potency.
PARPis in the clinic
PARPis are making their way into clinical trials and the clinic. As noted above, olaparib is approved for use in BRCA1/2-deficient ovarian cancer. It may also have efficacy in breast cancer and castration-resistant prostate cancer (Fong et al. 2009; Mateo et al. 2015). Olaparib was shown to be highly effective in castration-resistant prostate cancer patients who have mutations in DNA repair genes. Out of 16 olaparib-treated patients with mutations in DNA repairs genes, such as BRCA1/2, ATM, CHK2, 14 showed a reduction in tumor biomarkers, such as prostate-specific antigen (PSA), as well as the levels and the number of circulating tumor cells (Mateo et al. 2015). Much of the effect of PARP inhibition in prostate cancer is thought to come from the dual role of PARP-1, which has been shown to regulate not only DNA damage-related pathways in prostate cancer cells but also the function of the androgen receptor as a transcriptional regulator (Schiewer et al. 2012). These studies suggest that targeting PARP-1 in hormone-dependent cancers may have beneficial effects due to effects beyond DNA damage.
Recently, Drew et al. (2016) examined the efficacy of another PARPi, rucaparib, in advanced breast and ovarian cancers. Rucaparib broadly targets PARP-1 and PARP-2 and has also been shown to have some inhibitory effect on the activity of Tankyrase1 and Tankyrase2 (PARP-5a and PARP-5b) (Thomas et al. 2007; Haikarainen et al. 2014). Rucaparib administered in continuous doses was shown to be effective against germline BRCA mutant cancers in stage II clinical trials (Drew and Calvert 2008; Brown et al. 2016; Drew et al. 2016).
PARPis are also being tested against small cell lung cancer (SCLC). SCLC is a highly aggressive cancer with few good treatment options currently available. Interestingly, PARP-1 is overexpressed in SCLC (Byers et al. 2012). Moreover, targeting PARP-1 with talazoparib reduces the growth of SCLC cell lines and tumor growth in preclinical models (Cardnell et al. 2013). Currently, talazoparib is being tested in clinical trials for SCLC. Although PARPis have limited effect in SCLC tumors with activated mTOR pathways, a recent study by Cardnell et al. (2016) showed that a combined therapy of PARPis with the mTOR inhibitor BKM-120 can have an additive effect on the inhibition of tumor progression. However, the efficacy of this therapeutic regime in patients needs to be determined.
Although PARPis have been shown to have a high efficacy in solid tumors, such as breast cancer and SCLC, these inhibitors also target normal cells, which can have adverse effects. In a recent study, a bioactivatable drug targeting NAD(P)H:quinone oxidoreductase 1 (NQO1), β-lapachone, was used in conjunction with a PARPi to specifically target NQO1-overexpressing tumor cells (Huang et al. 2016b). The synergistic effect of PARP inhibition in combination with β-lapachone-induced DNA damage specifically promoted tumor cell death and showed efficacy even in cancers with wild-type BRCA1/2 (Huang et al. 2016b).
The results with PARPis described here hint at just a few of the many promising ways in which PARPis may be used to treat cancers and other diseases.
Conclusions and perspectives
The rapidly evolving nature of the PARP field is reflected in the new and exciting studies discussed here. Our understanding of the contribution of PARPs to a diverse array of biological processes has been greatly enhanced by recent studies that have ventured beyond the traditional roles of PARPs. This has led to many new discoveries, especially about the roles of PARP monoenzymes and catalytically inactive PARPs. Moreover, the development of new technologies and tools for the detection of ADP-ribosylation has led to a shift in the focus of the field from PARP proteins to their catalytic products. The anticipated use of these tools and methodologies holds great promise for the future of the PARP field.
At the same time, a number of aspects of the biology of PARPs and related proteins remain to be explored. For example, we still know very little about the broad spectrum of biology dependent on the PARP family of proteins. In addition, our understanding of the different catalytic-dependent and catalytic-independent functions of PARPs is limited. Furthermore, while numerous examples of ADP-ribose “reader” domains exist in nature, a comprehensive understanding of the functions of the proteins that contain these domains has been elusive.
With respect to ADP-ribosylation, the factors that drive selectivity and specificity for different substrates by different PARPs have been incompletely elucidated. Additionally, determining the repertoire of targets of distinct PARPs and their sites of ADP-ribosylation in different tissues is in its infancy. Likewise, the broader spectrum of amino acids that function as acceptors of ADP-ribose is still being defined (e.g., serine and cysteine) (Leidecker et al. 2016; Westcott et al. 2017). Such information would provide new insights into the biological roles of PARP across tissues and in disease states. One of the greatest needs and most significant challenges in the field, however, is moving beyond the identification of sites of ADP-ribosylation toward the determination of the functional relevance of ADP-ribosylation at those sites, which will reveal the definitive biological consequences of ADP-ribosylation. Finally, the field has not fully explored the therapeutic potential of PARPis. In conclusion, even after five decades of research on PARPs and ADP-ribosylation, much work remains to be done.
Competing interest statement
W.L.K. is a founder and consultant for Ribon Therapeutics, Inc.
Acknowledgments
We thank Dae-Seok Kim, Keun Woo Ryu, and Balaji Parameswaran for critical reading of the manuscript and intellectual input. The PARP-related work in the Kraus laboratory is funded by the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK069710), the Cancer Prevention and Research Institute of Texas (RP160319), and the Cecil H. and Ida Green Center for Reproductive Biology Sciences Endowment.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.291518.116.
References
- Adelman K, Lis JT. 2012. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13: 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhya D, Basu A. 2010. Epigenetic modulation of host: new insights into immune evasion by viruses. J Biosci 35: 647–663. [DOI] [PubMed] [Google Scholar]
- Aguilar-Quesada R, Munoz-Gamez JA, Martin-Oliva D, Peralta A, Valenzuela MT, Matinez-Romero R, Quiles-Perez R, Menissier-de Murcia J, de Murcia G, Ruiz de Almodovar M, et al. 2007. Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol Biol 8: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera-Gomez A, van Oorschot MM, Veenendaal T, Rabouille C. 2016. In vivo vizualisation of mono-ADP-ribosylation by dPARP16 upon amino-acid starvation. Elife 5: e21475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahel I, Ahel D, Matsusaka T, Clark AJ, Pines J, Boulton SJ, West SC. 2008. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 451: 81–85. [DOI] [PubMed] [Google Scholar]
- Ahel D, Horejsi Z, Wiechens N, Polo SE, Garcia-Wilson E, Ahel I, Flynn H, Skehel M, West SC, Jackson SP, et al. 2009. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325: 1240–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy P, Escande C, White TA, Thompson M, Soares S, Benech JC, Chini EN. 2006a. Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochem Biophys Res Commun 349: 353–359. [DOI] [PubMed] [Google Scholar]
- Aksoy P, White TA, Thompson M, Chini EN. 2006b. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun 345: 1386–1392. [DOI] [PubMed] [Google Scholar]
- Altmeyer M, Neelsen KJ, Teloni F, Pozdnyakova I, Pellegrino S, Grofte M, Rask MB, Streicher W, Jungmichel S, Nielsen ML, et al. 2015. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat Commun 6: 8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ame JC, Spenlehauer C, de Murcia G. 2004. The PARP superfamily. Bioessays 26: 882–893. [DOI] [PubMed] [Google Scholar]
- Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Cohen H, Lin SS, Manchester JK, Gordon JI, Sinclair DA. 2002. Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. J Biol Chem 277: 18881–18890. [DOI] [PubMed] [Google Scholar]
- Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. 2003. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423: 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. 2004. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305: 1010–1013. [DOI] [PubMed] [Google Scholar]
- Audebert M, Salles B, Calsou P. 2004. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem 279: 55117–55126. [DOI] [PubMed] [Google Scholar]
- Barbarulo A, Iansante V, Chaidos A, Naresh K, Rahemtulla A, Franzoso G, Karadimitris A, Haskard DO, Papa S, Bubici C. 2013. Poly(ADP-ribose) polymerase family member 14 (PARP14) is a novel effector of the JNK2-dependent pro-survival signal in multiple myeloma. Oncogene 32: 4231–4242. [DOI] [PubMed] [Google Scholar]
- Barkauskaite E, Jankevicius G, Ladurner AG, Ahel I, Timinszky G. 2013. The recognition and removal of cellular poly(ADP-ribose) signals. FEBS J 280: 3491–3507. [DOI] [PubMed] [Google Scholar]
- Bartolomei G, Leutert M, Manzo M, Baubec T, Hottiger MO. 2016. Analysis of chromatin ADP-ribosylation at the genome-wide level and at specific loci by ADPr-ChAP. Mol Cell 61: 474–485. [DOI] [PubMed] [Google Scholar]
- Beck C, Boehler C, Guirouilh Barbat J, Bonnet ME, Illuzzi G, Ronde P, Gauthier LR, Magroun N, Rajendran A, Lopez BS, et al. 2014a. PARP3 affects the relative contribution of homologous recombination and nonhomologous end-joining pathways. Nucleic Acids Res 42: 5616–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck C, Robert I, Reina-San-Martin B, Schreiber V, Dantzer F. 2014b. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res 329: 18–25. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J. 2006. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol 173: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, Ziegler M. 2005. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 280: 36334–36341. [DOI] [PubMed] [Google Scholar]
- Boamah EK, Kotova E, Garabedian M, Jarnik M, Tulin AV. 2012. Poly(ADP-Ribose) polymerase 1 (PARP-1) regulates ribosomal biogenesis in Drosophila nucleoli. PLoS Genet 8: e1002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehler C, Gauthier LR, Mortusewicz O, Biard DS, Saliou JM, Bresson A, Sanglier-Cianferani S, Smith S, Schreiber V, Boussin F, et al. 2011. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc Natl Acad Sci 108: 2783–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JS, Kaye SB, Yap TA. 2016. PARP inhibitors: the race is on. Br J Cancer 114: 713–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917. [DOI] [PubMed] [Google Scholar]
- Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, Giri U, Peyton M, Fan YH, Diao L, et al. 2012. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov 2: 798–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, Cohen MS, Goodman RH. 2016. Biosensor reveals multiple sources for mitochondrial NAD+. Science 352: 1474–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardnell RJ, Feng Y, Diao L, Fan YH, Masrorpour F, Wang J, Shen Y, Mills GB, Minna JD, Heymach JV, et al. 2013. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res 19: 6322–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardnell RJ, Feng Y, Mukherjee S, Diao L, Tong P, Stewart CA, Masrorpour F, Fan Y, Nilsson M, Shen Y, et al. 2016. Activation of the PI3K/mTOR pathway following PARP inhibition in small cell lung cancer. PLoS One 11: e0152584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-O'Connell I, Jin H, Morgan RK, David LL, Cohen MS. 2014. Engineering the substrate specificity of ADP-ribosyltransferases for identifying direct protein targets. J Am Chem Soc 136: 5201–5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-O'Connell I, Jin H, Morgan RK, Zaja R, David LL, Ahel I, Cohen MS. 2016. Identifying family-member-specific targets of mono-ARTDs by using a chemical genetics approach. Cell Rep 14: 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JD, Gagne JP, Poirier GG, Goodlett DR. 2013. Mapping PARP-1 auto-ADP-ribosylation sites by liquid chromatography-tandem mass spectrometry. J Proteome Res 12: 1868–1880. [DOI] [PubMed] [Google Scholar]
- Cho YS, Han MK, Kwark OS, Phoe MS, Cha YS, An NH, Kim UH. 1998. Auto-ADP-ribosylation of NAD glycohydrolase from Neurospora crassa. Comp Biochem Physiol B Biochem Mol Biol 120: 175–181. [DOI] [PubMed] [Google Scholar]
- Cho SH, Ahn AK, Bhargava P, Lee CH, Eischen CM, McGuinness O, Boothby M. 2011. Glycolytic rate and lymphomagenesis depend on PARP14, an ADP ribosyltransferase of the B aggressive lymphoma (BAL) family. Proc Natl Acad Sci 108: 15972–15977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SH, Raybuck A, Wei M, Erickson J, Nam KT, Cox RG, Trochtenberg A, Thomas JW, Williams J, Boothby M. 2013. B cell-intrinsic and -extrinsic regulation of antibody responses by PARP14, an intracellular (ADP-ribosyl)transferase. J Immunol 191: 3169–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiacovo MP, Elledge SJ. 2010. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci 107: 18475–18480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin NJ, Szabo C. 2013. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol Aspects Med 34: 1217–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels CM, Ong SE, Leung AK. 2014. Phosphoproteomic approach to characterize protein mono- and poly(ADP-ribosyl)ation sites from cells. J Proteome Res 13: 3510–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels CM, Ong SE, Leung AK. 2015a. The promise of proteomics for the study of ADP-ribosylation. Mol Cell 58: 911–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels CM, Thirawatananond P, Ong SE, Gabelli SB, Leung AK. 2015b. Nudix hydrolases degrade protein-conjugated ADP-ribose. Sci Rep 5: 18271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Malik HS. 2012. Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet 46: 677–700. [DOI] [PubMed] [Google Scholar]
- Daugherty MD, Young JM, Kerns JA, Malik HS. 2014. Rapid evolution of PARP genes suggests a broad role for ADP-ribosylation in host-virus conflicts. PLoS Genet 10: e1004403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David KK, Andrabi SA, Dawson TM, Dawson VL. 2009. Parthanatos, a messenger of death. Front Biosci (Landmark Ed) 14: 1116–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawicki-McKenna JM, Langelier MF, DeNizio JE, Riccio AA, Cao CD, Karch KR, McCauley M, Steffen JD, Black BE, Pascal JM. 2015. PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol Cell 60: 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, et al. 1997. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci 94: 7303–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos M, Schreiber V, Dantzer F. 2012. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol 84: 137–146. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. 1988. The establishment of polarity by hippocampal neurons in culture. J Neurosci 8: 1454–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew Y, Calvert H. 2008. The potential of PARP inhibitors in genetic breast and ovarian cancers. Ann N Y Acad Sci 1138: 136–145. [DOI] [PubMed] [Google Scholar]
- Drew Y, Ledermann J, Hall G, Rea D, Glasspool R, Highley M, Jayson G, Sludden J, Murray J, Jamieson D, et al. 2016. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br J Cancer 114: 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff MP, Malet H, Putics A, Heinonen M, Dutartre H, Frangeul A, Gruez A, Campanacci V, Cambillau C, Ziebuhr J, et al. 2006. Structural and functional basis for ADP-ribose and poly(ADP-ribose) binding by viral macro domains. J Virol 80: 8493–8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ramy R, Magroun N, Messadecq N, Gauthier LR, Boussin FD, Kolthur-Seetharam U, Schreiber V, McBurney MW, Sassone-Corsi P, Dantzer F. 2009. Functional interplay between Parp-1 and SirT1 in genome integrity and chromatin-based processes. Cell Mol Life Sci 66: 3219–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelli M, Carnevali F, Saccucci F, Pierella F, Amici A, Raffaelli N, Magni G. 2001. Molecular cloning, chromosomal localization, tissue mRNA levels, bacterial expression, and enzymatic properties of human NMN adenylyltransferase. J Biol Chem 276: 406–412. [DOI] [PubMed] [Google Scholar]
- Eustermann S, Wu WF, Langelier MF, Yang JC, Easton LE, Riccio AA, Pascal JM, Neuhaus D. 2015. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol Cell 60: 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921. [DOI] [PubMed] [Google Scholar]
- Farrar D, Rai S, Chernukhin I, Jagodic M, Ito Y, Yammine S, Ohlsson R, Murrell A, Klenova E. 2010. Mutational analysis of the poly(ADP-ribosyl)ation sites of the transcription factor CTCF provides an insight into the mechanism of its regulation by poly(ADP-ribosyl)ation. Mol Cell Biol 30: 1199–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feijs KL, Forst AH, Verheugd P, Luscher B. 2013. Macrodomain-containing proteins: regulating new intracellular functions of mono(ADP-ribosyl)ation. Nat Rev Mol Cell Biol 14: 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AL, Shirodkar P, Macrae CJ, Meng L, Koch CA. 2013. The PARP3- and ATM-dependent phosphorylation of APLF facilitates DNA double-strand break repair. Nucleic Acids Res 41: 4080–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et al. 2009. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361: 123–134. [DOI] [PubMed] [Google Scholar]
- Franco HL, Kraus WL. 2015. No driver behind the wheel? Targeting transcription in cancer. Cell 163: 28–30. [DOI] [PubMed] [Google Scholar]
- Gagne JP, Isabelle M, Lo KS, Bourassa S, Hendzel MJ, Dawson VL, Dawson TM, Poirier GG. 2008. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res 36: 6959–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagne JP, Ethier C, Defoy D, Bourassa S, Langelier MF, Riccio AA, Pascal JM, Moon KM, Foster LJ, Ning Z, et al. 2015. Quantitative site-specific ADP-ribosylation profiling of DNA-dependent PARPs. DNA Repair (Amst) 30: 68–79. [DOI] [PubMed] [Google Scholar]
- Gao G, Guo X, Goff SP. 2002. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science 297: 1703–1706. [DOI] [PubMed] [Google Scholar]
- Gao F, Kwon SW, Zhao Y, Jin Y. 2009. PARP1 poly(ADP-ribosyl)ates Sox2 to control Sox2 protein levels and FGF4 expression during embryonic stem cell differentiation. J Biol Chem 284: 22263–22273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaszner M, Felsenfeld G. 2006. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet 7: 703–713. [DOI] [PubMed] [Google Scholar]
- Ghosh J, Anderson PJ, Chandrasekaran S, Caparon MG. 2010. Characterization of Streptococcus pyogenes β-NAD+ glycohydrolase: re-evaluation of enzymatic properties associated with pathogenesis. J Biol Chem 285: 5683–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs-Seymour I, Fontana P, Rack JG, Ahel I. 2016. HPF1/C4orf27 is a PARP-1-interacting protein that regulates PARP-1 ADP-ribosylation activity. Mol Cell 62: 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Kraus WL. 2012. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 13: 411–424. [DOI] [PubMed] [Google Scholar]
- Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H, Schwede F, Yu Y, Kraus WL. 2016. Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 353: 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL, Pereira GC, Cheung LE, Rose RJ, Kazazian HH Jr. 2015. The broad-spectrum antiviral protein ZAP restricts human retrotransposition. PLoS Genet 11: e1005252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai Y, Swanson SK, Washburn MP, Florens L, Ladurner AG, Conaway JW, et al. 2009. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc Natl Acad Sci 106: 13770–13774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SL, Hwang J, Vastag L, Rabinowitz JD, Shenk T. 2012. Herpes simplex virus 1 infection activates poly(ADP-ribose) polymerase and triggers the degradation of poly(ADP-ribose) glycohydrolase. J Virol 86: 8259–8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy GJ, Polo LM, Zeng Z, Rulten SL, Hoch NC, Paomephan P, Xu Y, Sweet SM, Thorne AW, Oliver AW, et al. 2016. PARP3 is a sensor of nicked nucleosomes and monoribosylates histone H2B(Glu2). Nat Commun 7: 12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guetg C, Scheifele F, Rosenthal F, Hottiger MO, Santoro R. 2012. Inheritance of silent rDNA chromatin is mediated by PARP1 via noncoding RNA. Mol Cell 45: 790–800. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. 2010. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5: 253–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haikarainen T, Narwal M, Joensuu P, Lehtio L. 2014. Evaluation and structural basis for the inhibition of tankyrases by PARP inhibitors. ACS Med Chem Lett 5: 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haince JF, Kozlov S, Dawson VL, Dawson TM, Hendzel MJ, Lavin MF, Poirier GG. 2007. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J Biol Chem 282: 16441–16453. [DOI] [PubMed] [Google Scholar]
- Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, Poirier GG. 2008. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem 283: 1197–1208. [DOI] [PubMed] [Google Scholar]
- Hassa PO, Hottiger MO. 2002. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-κB in inflammatory disorders. Cell Mol Life Sci 59: 1534–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassa PO, Buerki C, Lombardi C, Imhof R, Hottiger MO. 2003. Transcriptional coactivation of nuclear factor-κB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. J Biol Chem 278: 45145–45153. [DOI] [PubMed] [Google Scholar]
- Hassa PO, Haenni SS, Buerki C, Meier NI, Lane WS, Owen H, Gersbach M, Imhof R, Hottiger MO. 2005. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-κB-dependent transcription. J Biol Chem 280: 40450–40464. [DOI] [PubMed] [Google Scholar]
- Hassa PO, Covic M, Bedford MT, Hottiger MO. 2008. Protein arginine methyltransferase 1 coactivates NF-κB-dependent gene expression synergistically with CARM1 and PARP1. J Mol Biol 377: 668–678. [DOI] [PubMed] [Google Scholar]
- Hoff KG, Wolberger C. 2005. Getting a grip on O-acetyl-ADP-ribose. Nat Struct Mol Biol 12: 560–561. [DOI] [PubMed] [Google Scholar]
- Hottiger MO. 2015. SnapShot: ADP-ribosylation signaling. Mol Cell 58: 1134–1134 e1131. [DOI] [PubMed] [Google Scholar]
- Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. 2010. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci 35: 208–219. [DOI] [PubMed] [Google Scholar]
- Huang JY, Wang K, Vermehren-Schmaedick A, Adelman JP, Cohen MS. 2016a. PARP6 is a regulator of hippocampal dendritic morphogenesis. Sci Rep 6: 18512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Motea EA, Moore ZR, Yao J, Dong Y, Chakrabarti G, Kilgore JA, Silvers MA, Patidar PL, Cholka A, et al. 2016b. Leveraging an NQO1 bioactivatable drug for tumor-selective use of poly(ADP-ribose) polymerase inhibitors. Cancer Cell 30: 940–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung YP, Albeck JG, Tantama M, Yellen G. 2011. Imaging cytosolic NADH–NAD+ redox state with a genetically encoded fluorescent biosensor. Cell Metab 14: 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussey KM, Chen H, Yang C, Park E, Hah N, Erdjument-Bromage H, Tempst P, Gamble MJ, Kraus WL. 2014. The histone variant macroH2A1 regulates target gene expression in part by recruiting the transcriptional coregulator PELP1. Mol Cell Biol 34: 2437–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Simons K. 2012. Cell biology. Beyond oil and water—phase transitions in cells. Science 337: 1047–1049. [DOI] [PubMed] [Google Scholar]
- Iwata H, Goettsch C, Sharma A, Ricchiuto P, Goh WW, Halu A, Yamada I, Yoshida H, Hara T, Wei M, et al. 2016. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADPribosylation. Nat Commun 7: 12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankevicius G, Hassler M, Golia B, Rybin V, Zacharias M, Timinszky G, Ladurner AG. 2013. A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat Struct Mol Biol 20: 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankevicius G, Ariza A, Ahel M, Ahel I. 2016. The toxin-antitoxin system DarTG catalyzes reversible ADP-ribosylation of DNA. Mol Cell 64: 1109–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen OE, Hilz H. 1989. Differentiation of 3T3-L1 pre-adipocytes induced by inhibitors of poly(ADP-ribose) polymerase and by related noninhibitory acids. Eur J Biochem 180: 595–602. [DOI] [PubMed] [Google Scholar]
- Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. 2006. A topoisomerase IIβ-mediated dsDNA break required for regulated transcription. Science 312: 1798–1802. [DOI] [PubMed] [Google Scholar]
- Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML. 2013. Proteome-wide identification of poly(ADP-ribosyl)ation targets in different genotoxic stress responses. Mol Cell 52: 272–285. [DOI] [PubMed] [Google Scholar]
- Jwa M, Chang P. 2012. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1α-mediated unfolded protein response. Nat Cell Biol 14: 1223–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisch T, Ame JC, Dantzer F, Schreiber V. 2012. New readers and interpretations of poly(ADP-ribosyl)ation. Trends Biochem Sci 37: 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai M, Hanashiro K, Kim SH, Hanai S, Boulares AH, Miwa M, Fukasawa K. 2007. Inhibition of Crm1–p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat Cell Biol 9: 1175–1183. [DOI] [PubMed] [Google Scholar]
- Kang HC, Lee YI, Shin JH, Andrabi SA, Chi Z, Gagne JP, Lee Y, Ko HS, Lee BD, Poirier GG, et al. 2011. Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proc Natl Acad Sci 108: 14103–14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlberg T, Langelier MF, Pascal JM, Schuler H. 2013. Structural biology of the writers, readers, and erasers in mono- and poly(ADP-ribose) mediated signaling. Mol Aspects Med 34: 1088–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karras GI, Kustatscher G, Buhecha HR, Allen MD, Pugieux C, Sait F, Bycroft M, Ladurner AG. 2005. The macro domain is an ADP-ribose binding module. EMBO J 24: 1911–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima L, Idogawa M, Mita H, Shitashige M, Yamada T, Ogi K, Suzuki H, Toyota M, Ariga H, Sasaki Y, et al. 2012. CHFR protein regulates mitotic checkpoint by targeting PARP-1 protein for ubiquitination and degradation. J Biol Chem 287: 12975–12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamitsu H, Hoshino H, Okada H, Miwa M, Momoi H, Sugimura T. 1984. Monoclonal antibodies to poly(adenosine diphosphate ribose) recognize different structures. Biochemistry 23: 3771–3777. [DOI] [PubMed] [Google Scholar]
- Kim UH, Rockwood SF, Kim HR, Daynes RA. 1988. Membrane-associated NAD+ glycohydrolase from rabbit erythrocytes is solubilized by phosphatidylinositol-specific phospholipase C. Biochim Biophys Acta 965: 76–81. [DOI] [PubMed] [Google Scholar]
- Kim UH, Kim MK, Kim JS, Han MK, Park BH, Kim HR. 1993. Purification and characterization of NAD glycohydrolase from rabbit erythrocytes. Arch Biochem Biophys 305: 147–152. [DOI] [PubMed] [Google Scholar]
- Kim MY, Mauro S, Gevry N, Lis JT, Kraus WL. 2004. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 119: 803–814. [DOI] [PubMed] [Google Scholar]
- Kim MY, Zhang T, Kraus WL. 2005. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev 19: 1951–1967. [DOI] [PubMed] [Google Scholar]
- Kistemaker HA, van Noort GJ, Overkleeft HS, van der Marel GA, Filippov DV. 2013. Stereoselective ribosylation of amino acids. Org Lett 15: 2306–2309. [DOI] [PubMed] [Google Scholar]
- Kistemaker HA, Nardozza AP, Overkleeft HS, van der Marel GA, Ladurner AG, Filippov DV. 2016. Synthesis and macrodomain binding of mono-ADP-ribosylated peptides. Angew Chem Int Ed Engl 55: 10634–10638. [DOI] [PubMed] [Google Scholar]
- Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM. 2004. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci 101: 17699–17704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolthur-Seetharam U, Dantzer F, McBurney MW, de Murcia G, Sassone-Corsi P. 2006. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle 5: 873–877. [DOI] [PubMed] [Google Scholar]
- Kraus WL, Lis JT. 2003. PARP goes transcription. Cell 113: 677–683. [DOI] [PubMed] [Google Scholar]
- Krietsch J, Rouleau M, Pic E, Ethier C, Dawson TM, Dawson VL, Masson JY, Poirier GG, Gagne JP. 2013. Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Mol Aspects Med 34: 1066–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar R, Kraus WL. 2010a. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 39: 8–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar R, Kraus WL. 2010b. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell 39: 736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar R, Gamble MJ, Frizzell KM, Berrocal JG, Kininis M, Kraus WL. 2008. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 319: 819–821. [DOI] [PubMed] [Google Scholar]
- Kuny CV, Sullivan CS. 2016. Virus–host interactions and the ARTD/PARP family of enzymes. PLoS Pathog 12: e1005453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustatscher G, Hothorn M, Pugieux C, Scheffzek K, Ladurner AG. 2005. Splicing regulates NAD metabolite binding to histone macroH2A. Nat Struct Mol Biol 12: 624–625. [DOI] [PubMed] [Google Scholar]
- Langelier MF, Ruhl DD, Planck JL, Kraus WL, Pascal JM. 2010. The Zn3 domain of human poly(ADP-ribose) polymerase-1 (PARP-1) functions in both DNA-dependent poly(ADP-ribose) synthesis activity and chromatin compaction. J Biol Chem 285: 18877–18887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier MF, Planck JL, Roy S, Pascal JM. 2011. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: structural and functional insights into DNA-dependent PARP-1 activity. J Biol Chem 286: 10690–10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier MF, Planck JL, Roy S, Pascal JM. 2012. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 336: 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier MF, Riccio AA, Pascal JM. 2014. PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res 42: 7762–7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtio L, Jemth AS, Collins R, Loseva O, Johansson A, Markova N, Hammarstrom M, Flores A, Holmberg-Schiavone L, Weigelt J, et al. 2009. Structural basis for inhibitor specificity in human poly(ADP-ribose) polymerase-3. J Med Chem 52: 3108–3111. [DOI] [PubMed] [Google Scholar]
- Leidecker O, Bonfiglio JJ, Colby T, Zhang Q, Atanassov I, Zaja R, Palazzo L, Stockum A, Ahel I, Matic I. 2016. Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat Chem Biol 12: 998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Vyas S, Rood JE, Bhutkar A, Sharp PA, Chang P. 2011. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol Cell 42: 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GY, McCulloch RD, Fenton AL, Cheung M, Meng L, Ikura M, Koch CA. 2010. Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. Proc Natl Acad Sci 107: 9129–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Lu LY, Yang CY, Wang S, Yu X. 2013. The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev 27: 1752–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Debing Y, Jankevicius G, Neyts J, Ahel I, Coutard B, Canard B. 2016. Viral macro domains reverse protein ADP-ribosylation. J Virol 90: 8478–8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H. 2007. Nicotinamide adenine dinucleotide: beyond a redox coenzyme. Org Biomol Chem 5: 2541–2554. [DOI] [PubMed] [Google Scholar]
- Liu Z, Kraus WL. 2017. Catalytic-independent functions of PARP-1 determine Sox2 pioneer activity at intractable genomic loci. Mol Cell (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, de Krijger I, Wiegant WW, Shah RG, Smeenk G, de Groot AJ, Pines A, Vertegaal AC, Jacobs JJ, Shah GM, et al. 2016. PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Mol Cell 61: 547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Kraus WL. 2012. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev 26: 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Ryu KW, Kim D, Nandu T, Medina CJ, Gupte R, Gibson BA, Soccio RE, Yu Y, Gupta RK, et al. 2017. PARP-1 controls the adipogenic transcriptional program by PARylating C/EBPβ and modulating its transcriptional activity. Mol Cell 65: 260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malet H, Coutard B, Jamal S, Dutartre H, Papageorgiou N, Neuvonen M, Ahola T, Forrester N, Gould EA, Lafitte D, et al. 2009. The crystal structures of Chikungunya and Venezuelan equine encephalitis virus nsP3 macro domains define a conserved adenosine binding pocket. J Virol 83: 6534–6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martello R, Leutert M, Jungmichel S, Bilan V, Larsen SC, Young C, Hottiger MO, Nielsen ML. 2016. Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat Commun 7: 12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. 2013. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341: 84–87. [DOI] [PubMed] [Google Scholar]
- Martinez-Zamudio R, Ha HC. 2012. Histone ADP-ribosylation facilitates gene transcription by directly remodeling nucleosomes. Mol Cell Biol 32: 2490–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, de Murcia G. 1998. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol 18: 3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, et al. 2015. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med 373: 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner S, Altmeyer M, Zhao H, Pozivil A, Roschitzki B, Gehrig P, Rutishauser D, Huang D, Caflisch A, Hottiger MO. 2010. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res 38: 6350–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T, Hilz H. 1986. Production of anti-(ADP-ribose) antibodies with the aid of a dinucleotide-pyrophosphatase-resistant hapten and their application for the detection of mono(ADP-ribosyl)ated polypeptides. Eur J Biochem 155: 157–165. [DOI] [PubMed] [Google Scholar]
- Misteli T, Soutoglou E. 2009. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat Rev Mol Cell Biol 10: 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan JB, Moran JV. 2015. The zinc-finger antiviral protein ZAP inhibits LINE and Alu retrotransposition. PLoS Genet 11: e1005121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle PM, Muir TW. 2010. Method for the synthesis of mono-ADP-ribose conjugated peptides. J Am Chem Soc 132: 15878–15880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murawska M, Hassler M, Renkawitz-Pohl R, Ladurner A, Brehm A. 2011. Stress-induced PARP activation mediates recruitment of Drosophila Mi-2 to promote heat shock gene expression. PLoS Genet 7: e1002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthurajan UM, Hepler MR, Hieb AR, Clark NJ, Kramer M, Yao T, Luger K. 2014. Automodification switches PARP-1 function from chromatin architectural protein to histone chaperone. Proc Natl Acad Sci 111: 12752–12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niere M, Mashimo M, Agledal L, Dolle C, Kasamatsu A, Kato J, Moss J, Ziegler M. 2012. ADP-ribosylhydrolase 3 (ARH3), not poly(ADP-ribose) glycohydrolase (PARG) isoforms, is responsible for degradation of mitochondrial matrix-associated poly(ADP-ribose). J Biol Chem 287: 16088–16102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusinow DA, Hernandez-Munoz I, Fazzio TG, Shah GM, Kraus WL, Panning B. 2007. Poly(ADP-ribose) polymerase 1 is inhibited by a histone H2A variant, MacroH2A, and contributes to silencing of the inactive X chromosome. J Biol Chem 282: 12851–12859. [DOI] [PubMed] [Google Scholar]
- Oei SL, Shi Y. 2001. Poly(ADP-ribosyl)ation of transcription factor Yin Yang 1 under conditions of DNA damage. Biochem Biophys Res Commun 285: 27–31. [DOI] [PubMed] [Google Scholar]
- Oka S, Kato J, Moss J. 2006. Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J Biol Chem 281: 705–713. [DOI] [PubMed] [Google Scholar]
- Okano S, Lan L, Caldecott KW, Mori T, Yasui A. 2003. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol Cell Biol 23: 3974–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olabisi OA, Soto-Nieves N, Nieves E, Yang TT, Yang X, Yu RY, Suk HY, Macian F, Chow CW. 2008. Regulation of transcription factor NFAT by ADP-ribosylation. Mol Cell Biol 28: 2860–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver FJ, Menissier-de Murcia J, Nacci C, Decker P, Andriantsitohaina R, Muller S, de la Rubia G, Stoclet JC, de Murcia G. 1999. Resistance to endotoxic shock as a consequence of defective NF-κB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J 18: 4446–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, Van Bortle K, Ramos E, Corces VG. 2013. Poly(ADP-ribosyl)ation regulates insulator function and intrachromosomal interactions in Drosophila. Cell 155: 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouararhni K, Hadj-Slimane R, Ait-Si-Ali S, Robin P, Mietton F, Harel-Bellan A, Dimitrov S, Hamiche A. 2006. The histone variant mH2A1.1 interferes with transcription by down-regulating PARP-1 enzymatic activity. Genes Dev 20: 3324–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo L, Thomas B, Jemth AS, Colby T, Leidecker O, Feijs KL, Zaja R, Loseva O, Puigvert JC, Matic I, et al. 2015. Processing of protein ADP-ribosylation by Nudix hydrolases. Biochem J 468: 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo L, Daniels CM, Nettleship JE, Rahman N, McPherson RL, Ong SE, Kato K, Nureki O, Leung AK, Ahel I. 2016. ENPP1 processes protein ADP-ribosylation in vitro. FEBS J 283: 3371–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R, Lewis B, Kim TK, Dilworth FJ, Erdjument-Bromage H, Tempst P, de Murcia G, Evans R, Chambon P, Reinberg D. 2005. PARP-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol Cell 18: 83–96. [DOI] [PubMed] [Google Scholar]
- Pekala PH, Lane MD, Watkins PA, Moss J. 1981. On the mechanism of preadipocyte differentiation. Masking of poly(ADP-ribose) synthetase activity during differentiation of 3T3-L1 preadipocytes. J Biol Chem 256: 4871–4876. [PubMed] [Google Scholar]
- Pellegrino S, Altmeyer M. 2016. Interplay between ubiquitin, SUMO, and poly(ADP-ribose) in the cellular response to genotoxic stress. Front Genet 7: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petesch SJ, Lis JT. 2008. Rapid, transcription-independent loss of nucleosomes over a large chromatin domain at Hsp70 loci. Cell 134: 74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleschke JM, Kleczkowska HE, Strohm M, Althaus FR. 2000. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem 275: 40974–40980. [DOI] [PubMed] [Google Scholar]
- Qiu J, Sheedlo MJ, Yu K, Tan Y, Nakayasu ES, Das C, Liu X, Luo ZQ. 2016. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 533: 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rack JG, Perina D, Ahel I. 2016. Macrodomains: structure, function, evolution, and catalytic activities. Annu Rev Biochem 85: 431–454. [DOI] [PubMed] [Google Scholar]
- Raffaelli N, Sorci L, Amici A, Emanuelli M, Mazzola F, Magni G. 2002. Identification of a novel human nicotinamide mononucleotide adenylyltransferase. Biochem Biophys Res Commun 297: 835–840. [DOI] [PubMed] [Google Scholar]
- Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, Gupta MP. 2009. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol 29: 4116–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S. 2004. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem 279: 50754–50763. [DOI] [PubMed] [Google Scholar]
- Riccio AA, Cingolani G, Pascal JM. 2016. PARP-2 domain requirements for DNA damage-dependent activation and localization to sites of DNA damage. Nucleic Acids Res 44: 1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Andris F, Van Gool F, Leo O. 2003. Reconstructing eukaryotic NAD metabolism. Bioessays 25: 683–690. [DOI] [PubMed] [Google Scholar]
- Rosenthal F, Feijs KL, Frugier E, Bonalli M, Forst AH, Imhof R, Winkler HC, Fischer D, Caflisch A, Hassa PO, et al. 2013. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat Struct Mol Biol 20: 502–507. [DOI] [PubMed] [Google Scholar]
- Rosenthal F, Nanni P, Barkow-Oesterreicher S, Hottiger MO. 2015. Optimization of LTQ-Orbitrap mass spectrometer parameters for the identification of ADP-ribosylation sites. J Proteome Res 14: 4072–4079. [DOI] [PubMed] [Google Scholar]
- Rouleau M, McDonald D, Gagne P, Ouellet ME, Droit A, Hunter JM, Dutertre S, Prigent C, Hendzel MJ, Poirier GG. 2007. PARP-3 associates with polycomb group bodies and with components of the DNA damage repair machinery. J Cell Biochem 100: 385–401. [DOI] [PubMed] [Google Scholar]
- Rouleau M, Saxena V, Rodrigue A, Paquet ER, Gagnon A, Hendzel MJ, Masson JY, Ekker M, Poirier GG. 2011. A key role for poly(ADP-ribose) polymerase 3 in ectodermal specification and neural crest development. PLoS One 6: e15834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulten SL, Fisher AE, Robert I, Zuma MC, Rouleau M, Ju L, Poirier G, Reina-San-Martin B, Caldecott KW. 2011. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol Cell 41: 33–45. [DOI] [PubMed] [Google Scholar]
- Ryu KW, Kim DS, Kraus WL. 2015. New facets in the regulation of gene expression by ADP-ribosylation and poly(ADP-ribose) polymerases. Chem Rev 115: 2453–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala A, La Rocca G, Burgio G, Kotova E, Di Gesu D, Collesano M, Ingrassia AM, Tulin AV, Corona DF. 2008. The nucleosome-remodeling ATPase ISWI is regulated by poly-ADP-ribosylation. PLoS Biol 6: e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon D, Orth K. 2013. What pathogens have taught us about posttranslational modifications. Cell Host Microbe 14: 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, Liu F, Planck JL, Ravindranathan P, Chinnaiyan AM, et al. 2012. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov 2: 1134–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo GJ, Kincaid RP, Phanaksri T, Burke JM, Pare JM, Cox JE, Hsiang TY, Krug RM, Sullivan CS. 2013. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe 14: 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi R, Morra R, Appel CD, Tallis M, Chioza B, Jankevicius G, Simpson MA, Matic I, Ozkan E, Golia B, et al. 2013. Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J 32: 1225–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, Ahel M, Leys D, Ahel I. 2011. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 477: 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht KM, Shokat KM. 2002. The emerging power of chemical genetics. Curr Opin Cell Biol 14: 155–159. [DOI] [PubMed] [Google Scholar]
- Steffen JD, Brody JR, Armen RS, Pascal JM. 2013. Structural implications for selective targeting of PARPs. Front Oncol 3: 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen JD, Tholey RM, Langelier MF, Planck JL, Schiewer MJ, Lal S, Bildzukewicz NA, Yeo CJ, Knudsen KE, Brody JR, et al. 2014. Targeting PARP-1 allosteric regulation offers therapeutic potential against cancer. Cancer Res 74: 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen JD, McCauley MM, Pascal JM. 2016. Fluorescent sensors of PARP-1 structural dynamics and allosteric regulation in response to DNA damage. Nucleic Acids Res 44: 9771–9783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teloni F, Altmeyer M. 2016. Readers of poly(ADP-ribose): designed to be fit for purpose. Nucleic Acids Res 44: 993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas HD, Calabrese CR, Batey MA, Canan S, Hostomsky Z, Kyle S, Maegley KA, Newell DR, Skalitzky D, Wang LZ, et al. 2007. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol Cancer Ther 6: 945–956. [DOI] [PubMed] [Google Scholar]
- Thorsell AG, Ekblad T, Karlberg T, Low M, Pinto AF, Tresaugues L, Moche M, Cohen MS, Schuler H. 2016. Structural basis for potency and promiscuity in poly(ADP-ribose) polymerase (PARP) and tankyrase inhibitors. J Med Chem 10.1021/acs.jmedchem.6b00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timinszky G, Till S, Hassa PO, Hothorn M, Kustatscher G, Nijmeijer B, Colombelli J, Altmeyer M, Stelzer EH, Scheffzek K, et al. 2009. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat Struct Mol Biol 16: 923–929. [DOI] [PubMed] [Google Scholar]
- Todorova T, Bock FJ, Chang P. 2014. PARP13 regulates cellular mRNA post-transcriptionally and functions as a pro-apoptotic factor by destabilizing TRAILR4 transcript. Nat Commun 5: 5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L, Denu JM. 2010. Function and metabolism of sirtuin metabolite O-acetyl-ADP-ribose. Biochim Biophys Acta 1804: 1617–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulin A, Spradling A. 2003. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science 299: 560–562. [DOI] [PubMed] [Google Scholar]
- Tulin A, Naumova NM, Menon AK, Spradling AC. 2006. Drosophila poly(ADP-ribose) glycohydrolase mediates chromatin structure and SIR2-dependent silencing. Genetics 172: 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heden van Noort GJ, van der Horst MG, Overkleeft HS, van der Marel GA, Filippov DV. 2010. Synthesis of mono-ADP-ribosylated oligopeptides using ribosylated amino acid building blocks. J Am Chem Soc 132: 5236–5240. [DOI] [PubMed] [Google Scholar]
- Verheugd P, Forst AH, Milke L, Herzog N, Feijs KL, Kremmer E, Kleine H, Luscher B. 2013. Regulation of NF-κB signalling by the mono-ADP-ribosyltransferase ARTD10. Nat Commun 4: 1683. [DOI] [PubMed] [Google Scholar]
- Vivelo CA, Wat R, Agrawal C, Tee HY, Leung AK. 2016. ADPriboDB: the database of ADP-ribosylated proteins. Nucleic Acids Res 45: D204–D209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas S, Chesarone-Cataldo M, Todorova T, Huang YH, Chang P. 2013. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat Commun 4: 2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT, Ahel I, Chang P. 2014. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat Commun 5: 4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlberg E, Karlberg T, Kouznetsova E, Markova N, Macchiarulo A, Thorsell AG, Pol E, Frostell A, Ekblad T, Oncu D, et al. 2012. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotechnol 30: 283–288. [DOI] [PubMed] [Google Scholar]
- Wang Z, Michaud GA, Cheng Z, Zhang Y, Hinds TR, Fan E, Cong F, Xu W. 2012. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev 26: 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, Yuzugullu H, Von T, Li H, Lin Z, et al. 2015. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 163: 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, Kim B, Bao L, Harraz MM, Chang C, et al. 2016. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 354: aad6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsby I, Hutin D, Gueydan C, Kruys V, Rongvaux A, Leo O. 2014. PARP12, an interferon-stimulated gene involved in the control of protein translation and inflammation. J Biol Chem 289: 26642–26657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott NP, Fernandez JP, Molina H, Hang HC. 2017. Chemical proteomics reveals ADPribosylation of small GTPases during oxidative stress. Nat Chem Biol doi: 10.1038/nchembio.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright RH, Lioutas A, Le Dily F, Soronellas D, Pohl A, Bonet J, Nacht AS, Samino S, Font-Mateu J, Vicent GP, et al. 2016. ADP-ribose-derived nuclear ATP synthesis by NUDIX5 is required for chromatin remodeling. Science 352: 1221–1225. [DOI] [PubMed] [Google Scholar]
- Wu C. 1997. Chromatin remodeling and the control of gene expression. J Biol Chem 272: 28171–28174. [DOI] [PubMed] [Google Scholar]
- Ying W. 2008. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10: 179–206. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. 2002. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297: 259–263. [DOI] [PubMed] [Google Scholar]
- Yu W, Ginjala V, Pant V, Chernukhin I, Whitehead J, Docquier F, Farrar D, Tavoosidana G, Mukhopadhyay R, Kanduri C, et al. 2004. Poly(ADP-ribosyl)ation regulates CTCF-dependent chromatin insulation. Nat Genet 36: 1105–1110. [DOI] [PubMed] [Google Scholar]
- Zhang X, Kurnasov OV, Karthikeyan S, Grishin NV, Osterman AL, Zhang H. 2003. Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem 278: 13503–13511. [DOI] [PubMed] [Google Scholar]
- Zhang T, Berrocal JG, Frizzell KM, Gamble MJ, DuMond ME, Krishnakumar R, Yang T, Sauve AA, Kraus WL. 2009. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J Biol Chem 284: 20408–20417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA, Schirle M, Shi X, Hild M, Bauer A, et al. 2011. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol 13: 623–629. [DOI] [PubMed] [Google Scholar]
- Zhang T, Berrocal JG, Yao J, DuMond ME, Krishnakumar R, Ruhl DD, Ryu KW, Gamble MJ, Kraus WL. 2012. Regulation of poly(ADP-ribose) polymerase-1-dependent gene expression through promoter-directed recruitment of a nuclear NAD+ synthase. J Biol Chem 287: 12405–12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang J, Ding M, Yu Y. 2013. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat Methods 10: 981–984. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Mao D, Roswit WT, Jin X, Patel AC, Patel DA, Agapov E, Wang Z, Tidwell RM, Atkinson JJ, et al. 2015. PARP9-DTX3L ubiquitin ligase targets host histone H2BJ and viral 3C protease to enhance interferon signaling and control viral infection. Nat Immunol 16: 1215–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sifakis EG, Sumida N, Millan-Arino L, Scholz BA, Svensson JP, Chen X, Ronnegren AL, Mallet de Lima CD, Varnoosfaderani FS, et al. 2015. PARP1- and CTCF-mediated interactions between active and repressed chromatin at the lamina promote oscillating transcription. Mol Cell 59: 984–997. [DOI] [PubMed] [Google Scholar]
- Zocchi E, Franco L, Guida L, Benatti U, Bargellesi A, Malavasi F, Lee HC, De Flora A. 1993. A single protein immunologically identified as CD38 displays NAD+ glycohydrolase, ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities at the outer surface of human erythrocytes. Biochem Biophys Res Commun 196: 1459–1465. [DOI] [PubMed] [Google Scholar]