

Here, Ritschka et al. provide insight into new functions for the senescence-associated secretory phenotype (SASP), which can reinforce the arrest and induce senescence in a paracrine manner. They show that SASP promotes a proregenerative response through the induction of cell plasticity and stemness and demonstrate that transient therapeutic delivery of senescent cells could be harnessed to drive tissue regeneration.
Keywords: senescence, plasticity, stem cells, papilloma, CD34, SASP
Abstract
Senescence is a form of cell cycle arrest induced by stress such as DNA damage and oncogenes. However, while arrested, senescent cells secrete a variety of proteins collectively known as the senescence-associated secretory phenotype (SASP), which can reinforce the arrest and induce senescence in a paracrine manner. However, the SASP has also been shown to favor embryonic development, wound healing, and even tumor growth, suggesting more complex physiological roles than currently understood. Here we uncover timely new functions of the SASP in promoting a proregenerative response through the induction of cell plasticity and stemness. We show that primary mouse keratinocytes transiently exposed to the SASP exhibit increased expression of stem cell markers and regenerative capacity in vivo. However, prolonged exposure to the SASP causes a subsequent cell-intrinsic senescence arrest to counter the continued regenerative stimuli. Finally, by inducing senescence in single cells in vivo in the liver, we demonstrate that this activates tissue-specific expression of stem cell markers. Together, this work uncovers a primary and beneficial role for the SASP in promoting cell plasticity and tissue regeneration and introduces the concept that transient therapeutic delivery of senescent cells could be harnessed to drive tissue regeneration.
Cellular senescence can be induced by a variety of stress stimuli. These can include active oncogene signaling, DNA damage, irradiation, or chemotherapy, causing the activation of tumor suppressor networks, including p53, p16 (Cdkn2a), and p21 (Cdkn1a), and cell cycle arrest (Serrano et al. 1997; Schmitt et al. 2002; Coppe et al. 2008; Campisi 2013). Thereby, senescence acts as a cell-intrinsic tumor-suppressive mechanism that prevents the proliferation of damaged cells. However, senescence can also contribute to tumor suppression in a cell-extrinsic manner through senescence-associated secretory phenotype (SASP)-mediated recruitment of immune cells (Kang et al. 2011; Lujambio et al. 2013).
Cellular senescence is also induced upon replicative aging of cells in culture, and senescent cells accumulate in multiple tissues and stem cells during physiological aging (Dimri et al. 1995; Baker et al. 2016). This accumulation of nonproliferating cells contributes to the loss of functional and regenerative capacity in aged tissues, preventing proliferation of both somatic and stem cells in a cell-intrinsic manner (Janzen et al. 2006; Krishnamurthy et al. 2006; Molofsky et al. 2006; Sharpless and DePinho 2007; Sousa-Victor et al. 2014; Baker et al. 2016; Garcia-Prat et al. 2016). Additionally, senescent cells can contribute to loss of tissue function during aging through non-cell-autonomous mechanisms. This can result from SASP proteins disrupting homeostasis or possibly through the induction of paracrine senescence in neighboring cells (Baker et al. 2011; Acosta et al. 2013; Campisi 2013). Therefore, this supports how aging and decreased regeneration may in part result from senescence and tumor-suppressive mechanisms.
Recently, however, beneficial roles for senescence have been emerging, often mediated through the SASP. In the developing embryo, senescent cells are suggested to contribute to tissue development and patterning through both remodeling mediated by senescent cell clearance and tissue growth and patterning through the SASP (Munoz-Espin et al. 2013; Storer et al. 2013). In addition, senescent cells recruit immune cells and limit fibrosis during wound healing and contribute to wound repair through the secretion of growth factors (Krizhanovsky et al. 2008; Jun and Lau 2010; Demaria et al. 2014). However, these same beneficial effects on tissue growth and patterning can have unwanted side effects through promoting tumor growth and progression (Krtolica et al. 2001; Yoshimoto et al. 2013).
Reconciling these beneficial and detrimental aspects of the senescence program has been difficult. Here, we describe how transient exposure to the SASP can promote tissue regeneration. However, prolonged exposure leads to a subsequent cell-intrinsic arrest, likely as a tumor-suppressive response to aberrant regeneration. This work uncovers important mechanistic insights into the biological function of senescence.
Results
To explore the senescence program in epithelial cells, we performed microarray analysis on primary mouse keratinocytes undergoing oncogene-induced senescence (OIS) from the expression of oncogenic HRasV12. As expected, senescent keratinocytes exhibited hallmark features of senescence, including decreased proliferation, a large flat appearance, increased senescence-associated β-galactosidase (SA-β-Gal) activity, and increased expression of p53, p21, and p16 (Supplemental Fig. 1a–d). Analysis of the microarray signature revealed that other known mediators of senescence and the SASP were also increased, including p15 (Cdkn2b), Il1a, and Hmga2 (Fig. 1A). Surprisingly, however, senescent cells also exhibited increased expression of many genes normally associated with somatic and cancer stem cells, including CD34, Lgr6, Prom1, CD44, Ngfr, and Nestin (Fig. 1A). This was further supported by bioinformatics and gene set enrichment analysis (GSEA) using Genomatix software and published signatures of common stem cell populations (Fig. 1B,C; for full gene lists, see Supplemental Table 1; Blanpain et al. 2004). To validate this signature, we performed quantitative PCR (qPCR) analysis for selected skin stem cell genes in senescent cells up to 2 wk after infection (Fig. 1D). As before, we found increased expression of many of these, including CD34, Nestin, Lrig1, and Lgr6 as well as other hair follicle stem cell (HFSC) genes (Supplemental Fig. 1f). Similar observations were also noticed in the primary control cells, which undergo senescence-like changes upon prolonged culture, including adopting a senescent-like morphology and exhibiting increased expression of p16 (Supplemental Fig. 1e,h). Surprisingly, these cells also displayed an increase in the same stem cell genes, although at lower levels than seen in OIS (Supplemental Fig. 1e,g). Supporting the unanticipated link between senescence and stemness, when senescence was induced in additional ways, including treatment of cells with the chemotherapeutic drug etoposide or irradiation, the cells also displayed an increase in the same stem cell genes while undergoing senescence (Fig. 1E–F).
Figure 1.

A stem cell signature is increased in senescent cells. (A) Representative heat map of microarray analysis of HRasV12-induced senescent keratinocytes compared with growing keratinocytes. Example genes are shown to illustrate that stem cell and senescent genes are more highly expressed in senescent cells compared with growing cells. (B) Bioinformatics analysis of genes significantly up-regulated in senescent cells (analysis performed using Genomatix software; full list of up-regulated and down-regulated genes is in Supplemental Table 1). (C) GSEA of stem cell genes (Blanpain et al. 2004) in the gene expression profile of Ras-induced senescent keratinocytes. (NES) Normalized enrichment score. (D) qPCR analysis for skin stem cell and senescence genes in Ras-infected keratinocytes at 4–14 d post-infection (dpi) normalized to vector-infected keratinocytes at 4 dpi. n = 4, except for 14 dpi (n = 3). (E) qPCR analysis of etoposide-treated keratinocytes 3–5 d after treatment normalized to DMSO-treated keratinocytes 3 d after treatment. n = 5, except for day 5 (n = 3). (F) qPCR analysis of irradiated keratinocytes (12 Gy) 5 and 10 d after irradiation normalized to keratinocytes before irradiation. n = 2. (D–F) Error bars indicate mean ± SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001), two-tailed Student's t-test.
Furthermore, in agreement with the transcript profile, upon OIS induction, we found a significant increase in the number of newborn keratinocytes that were double-positive for CD34 and integrin-α6 proteins, which label a distinct population of HFSCs (Fig. 2A; Supplemental Fig. 2a; Blanpain et al. 2004; Jensen et al. 2010). Interestingly, newborn keratinocytes do not yet express these stem cell markers, which become expressed after establishment of the hair cycle in the adult, suggesting that these markers are being induced de novo during senescence induction (Supplemental Fig. 2b–d). Remarkably, however, even though there was an increase in stemness-associated markers, these cells were still undergoing proliferative arrest, as measured by a progressive decrease in proliferation (Supplemental Fig. 1c) and a loss of clonogenic capacity in the total (Fig. 2B,C) and double-positive CD34/integrin-α6 cells (Fig. 2D; Supplemental Fig. 2e). Indeed, qPCR analysis on the double-positive CD34/integrin-α6 population showed that these cells were not escaping senescence but expressed high levels of the senescence markers p16 and p21 (Fig. 2E; Supplemental Fig. 2f). Together, these results demonstrate that senescent cells, while undergoing senescence arrest and loss of clonogenicity, paradoxically exhibit increased markers normally associated with stemness.
Figure 2.

OIS induces de novo specification of stem cell fate. (A) Representative FACS plots and quantification for α6-integrin+/CD34+ stem cells in Ras- or vector-infected keratinocytes at 7, 10, and 14 dpi. n = 3. (B,C) Representative images of colony-forming assays of vector-infected keratinocytes at 7 dpi and Ras-infected keratinocytes at 7 and 14 dpi (B) and quantification of numbers of colonies of Ras-infected keratinocytes at 7 and 14 dpi normalized to colonies of vector-infected keratinocytes at 7 dpi (C). n = 5, except for 14 dpi (n = 3). (D) Quantification of the numbers of colonies of sorted α6-integrin+/CD34+ cells from Ras-infected keratinocytes at 14 dpi normalized to colonies of sorted α6-integrin+/CD34+ cells from adult epidermis. n = 3. (E) qPCR analysis of sorted α6-integrin+/CD34− and α6-integrin+/CD34+ from Ras-infected keratinocytes at 14 dpi normalized to vector-infected keratinocytes at 4 dpi. n = 2, except for CD34− (n = 3). (A,C,D,E) Error bars indicate mean ± SEM (*) P < 0.05; (**) P < 0.01; (***) P < 0.001, two-tailed Student's t-test.
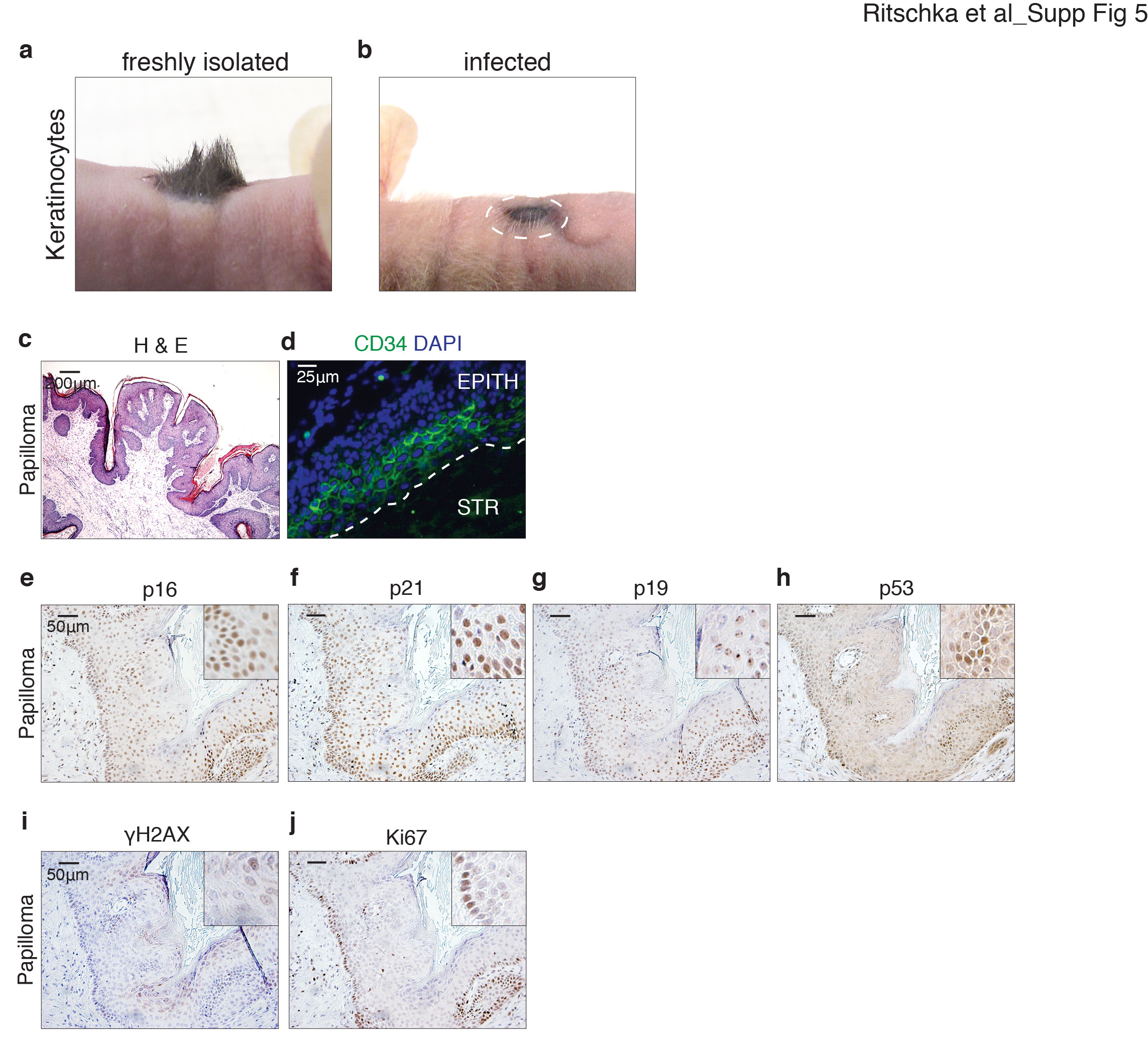
To investigate this unexpected connection between senescence and stem cells in vivo, we explored the incidence and distribution of senescence in papillomas that were induced with DMBA/TPA treatment, an in vivo model of OIS. Upon examination of established papillomas for SA-β-Gal activity, no staining was detectable in the epithelial layers of the tumor, with the strongest reactivity found in the underlying dermal tissue and some regions of the papilloma stroma (Fig. 3A–H). However, a thorough analysis of the expression patterns of known senescence markers—including p16, p21, p19, p53, γH2AX, and Ki67—by immunohistochemistry demonstrated high expression of these proteins in the epithelial cells and the underlying dermis, with a more diffuse expression in the stroma of the benign lesion (Fig. 3I–K, Supplemental Fig. 3a–c). Papilloma arises from the aberrant upward expansion and differentiation of CD34+ stem cells in the basal and early differentiating layers of the epithelial papilloma (Fig. 3L; Malanchi et al. 2008; Lapouge et al. 2012). Detailed quantification of the distribution of the senescence markers revealed that these were most highly expressed in these same stem cell layers (Fig. 3M; Supplemental Fig. 3d). Interestingly, the basal-initiating layer of the papilloma contained cells that were positive for both proliferation markers (such as Ki67 or PCNA) and senescence markers, suggesting a dynamic process of aberrant proliferation and subsequent arrest (Supplemental Fig. 3g–i). Given this result, we extended our analysis to another model of in vivo OIS in the pancreas, where activation of mutant KRasG12D drives senescence and premalignant pancreatic intraepithelial neoplasia (PanIN) lesions (Morton et al. 2010). Detailed histological analysis of these tissues revealed increased expression of senescence markers, including SA-β-Gal, p21, and p53, concomitant with deceased proliferation (Supplemental Fig. 4). Interestingly, here also there was increased activation of the stem cell markers CD44 and Nestin in these senescent lesions. Altogether, this demonstrates that also in vivo in models of premalignant lesions, there is a pronounced association of stem cell markers in senescent cells.
Figure 3.

Senescent epithelial cells display a stem cell signature in vivo. (A) Hematoxylin and eosin (H&E) staining of DMBA/TPA-induced papillomas. Boxed regions E (epithelia), S (stroma), and D (dermis) are representative of areas magnified in C–E. Bars, 50 µm. (B) SA-β-Gal staining of papilloma. Boxed regions E (epithelia), S (stroma), and D (dermis) are representative of areas magnified in F–H. Bars, 50 µm. Immunohistochemistry for the senescence markers p16 and p21 in the epithelia (I), stroma (J), and underlying dermis (K) of chemically induced papillomas. Bars, 25 µm. (L) Immunofluorescence for the epithelial stem cell marker CD34 and DAPI in chemically induced papillomas. (EPITH) Epithelia; (STR) stroma. Bar, 25 µm. (M) Distribution of senescence markers in DMBA/TPA papillomas. All images are representative of at least four biological replicates. (N–Q) Nude mice were grafted with a combination of 4 × 106 dermal fibroblasts and 3 × 106 keratinocytes infected with either vector at 6 dpi (N) or Ras at 6 dpi (O). The number of mice is shown in the bottom right corner. (P) Representative immunohistochemistry for GFP. (Q) Magnification of the boxed region in P.
To investigate whether cells that show increased expression of stem cell markers in culture while undergoing senescence might be escapers from the senescence program or function as stem cells, we used an in vivo assay to assess the properties of these cells. To address this, we performed skin grafting assays, a test of HFSC function in which freshly isolated newborn keratinocytes or purified adult CD34+ HFSCs are grafted along with dermal fibroblasts into full-thickness wounds in the back of nude mice (Blanpain et al. 2004; Jensen et al. 2010). This results in large patches of hair follicles that develop directly from the transplanted cells (Supplemental Fig. 5a), whereas if the keratinocytes are placed in culture prior to transplantation, they lose their regenerative capacity, generating only pigmented scar tissue (Supplemental Fig. 5b). We investigated whether keratinocytes expressing the HRasV12 oncogene and increasing expression of the stem cell markers could function as HFSCs in vivo by transplanting GFP+ HRasV12-infected keratinocytes undergoing senescence. Remarkably, this resulted in the generation of a papilloma that was histologically and molecularly similar to the DMBA/TPA ones, including exhibiting similar patterns of senescence protein expression in the epithelial layers as well as possessing an aberrant layer of CD34+ cells (Supplemental Fig. 5c–j). Indeed, most of the papilloma consisted of GFP+ senescent cells, suggesting that, even in vivo, senescing cells with increased stem cell markers remained senescent. Interestingly, however, while most of the papilloma was GFP+ (likely arising from the proliferation of not-fully senescent cells), it contained small regions that were clearly negative for GFP, suggesting that endogenous keratinocytes were being recruited into a premalignant fate (Fig. 3N–Q).
Given this coexpression of senescence and stem cell genes, we investigated whether these were functionally linked. To address this, we assessed the expression of these same stem cell genes upon disruption of key senescence regulators—including cell cycle inhibitors p53, p16, and p21—and SASP mediators NFKB, CEBPB, IL1A, and p38. As expected, knockdown of the cell cycle inhibitors now blocked the proliferation arrest and allowed senescence bypass/proliferation. Surprisingly, however, loss of these genes had little or no effect on the increased expression of the stem cell genes, which persisted after senescence bypass either transiently (after p53 loss) (Supplemental Fig. 6e) or long-term upon prolonged culture (after p16 or p21 loss) (Fig. 4A; Supplemental Fig. 6c,f). This is clearly seen in cells in which cell cycle inhibitor p16 was knocked down, as these retained high levels of all of the analyzed stem cell genes, including CD34/α6 integrin-positive cells, yet regained colony-forming capacity (Fig. 4B,C; Supplemental Fig. 6a–d).
Figure 4.

Loss of SASP regulator NFκB in senescent cells causes a decrease in stem cell gene expression. (A) qPCR analysis of Ras- and shp16 I-double-infected keratinocytes at 6, 10, and 14 dpi normalized to Ras- and vector-double-infected keratinocytes on the respective days. n = 3. (B,C) Representative colony-forming assay (B) and number of colonies (C) of Ras- and vector-double-infected and Ras- and shp16 II-double-infected keratinocytes at 14 dpi. n = 4. (D) qPCR analysis of Ras- and shRelA II-double-infected keratinocytes at 6, 10, and 14 dpi normalized to Ras- and vector-double-infected keratinocytes on the respective days. n = 3. (E) The number of α6-integrin+/CD34+ cells of Ras, shRelA I, shRelA II, and NFκB superrepressor (SR)-double-infected keratinocytes normalized to Ras- and vector-double-infected keratinocytes at 10 dpi. n = 3. (A,C,D,E) Error bars indicate mean ± SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001, two-tailed Student's t-test.
Next, we investigated whether knockdown of the SASP regulators affected stem cell gene expression. Interestingly, here knockdown of RelA/NFKB caused a decrease in the expression of many of the stem cells genes (Fig. 4D), while loss of CEBPB, IL1A, and p38 had no effect (Supplemental Fig. 7). This was further validated when the NFKB pathway was inhibited with expression of a constitutively activated repressor of the pathway (Fig. 4E). In each instance, inhibition of the NFKB pathway had no effect on the senescence phenotype or proliferative arrest of the cells (Supplemental Fig. 7a). Together, this suggests that the senescence-associated increase in stem cell gene expression is regulated in part by the NFKB-mediated arm of the SASP, while proliferation of cells expressing aberrant activation of stem cell markers is blocked by tumor suppressor genes.
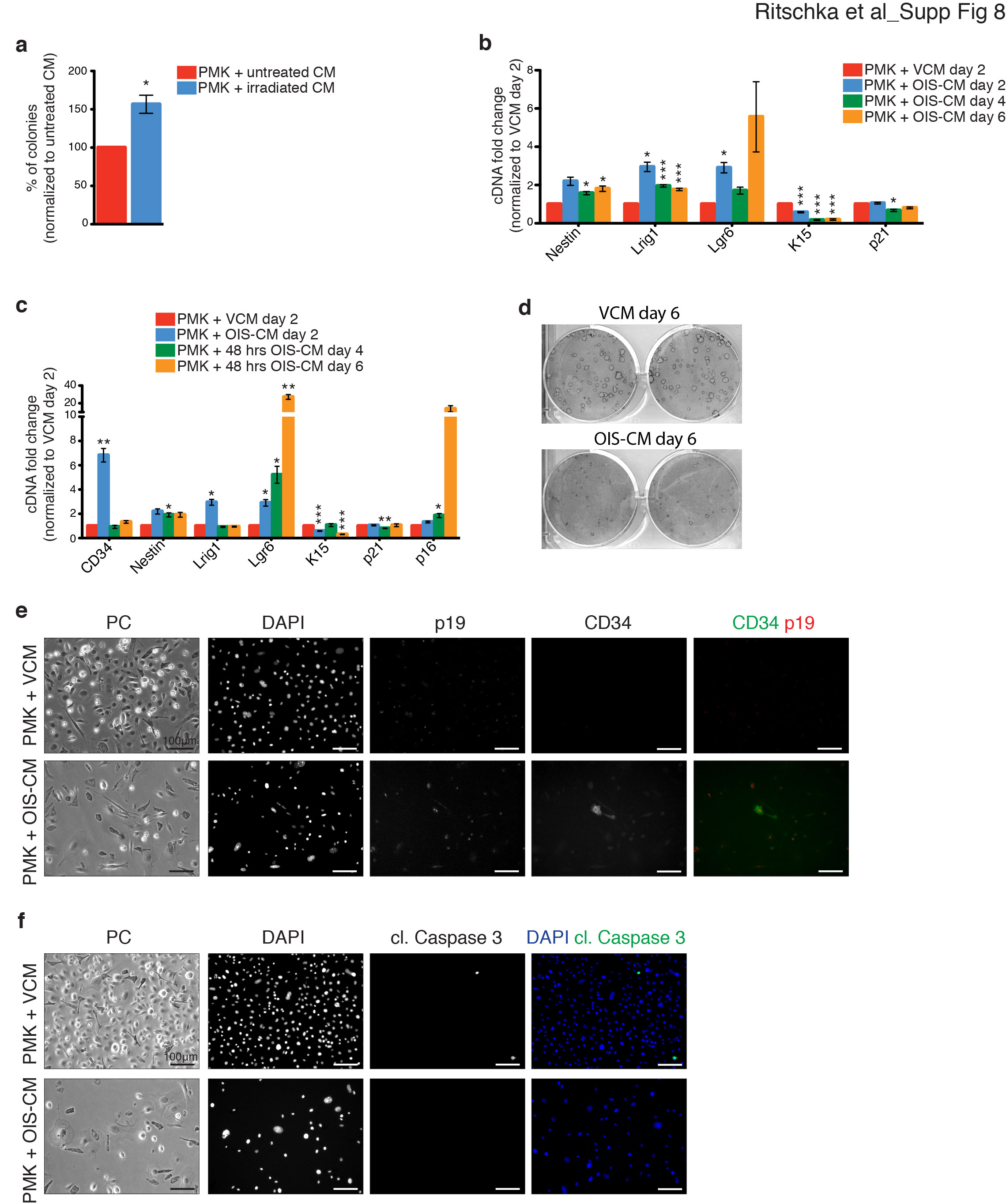
Based on this, we functionally investigated whether the SASP is linked to the increased expression of stem cell genes. To investigate this hypothesis, we took conditioned medium (CM) from vector-infected growing cells (VCM) or OIS keratinocytes (OIS-CM) and added it to proliferating newborn primary mouse keratinocytes (PMKs) during a short-term period (2 d) (Fig. 5A). Remarkably, transient exposure to OIS-CM led to a marked increase in the same stem cell genes as before, demonstrating that the SASP can induce stem cell gene expression (Fig. 5B). Additionally, when tested in colony-forming assays, cells that had been transiently exposed to the SASP exhibited increased clonogenic capacity (Fig. 5C). Importantly, increased stem cell markers and colony formation were also seen in cells that were treated with CM from irradiation-induced senescent keratinocytes (Supplemental Fig. 8a). Next, we assessed whether the SASP-treated cells possessed any functional properties of stem cells in vivo. To examine whether the SASP-treated cells possessed stem cell function, we transplanted GFP+ cells that had been exposed to VCM or OIS-CM into full-thickness grafts. As expected, the grafts containing keratinocytes treated with VCM healed over, presenting mostly with pigmented scar tissue and sparse individual hair follicles (Fig. 5D,E). Remarkably, however, the grafts containing cells that had been transiently treated with OIS-CM developed large thick patches of hair (Fig. 5D,E). Importantly, staining of the grafts for GFP to trace the transplanted cells confirmed that there was a significant increase in the number of hair follicles following exposure to the SASP and that most of the hair follicles were coming from the SASP-treated GFP+ cells (Fig. 5F,G). This demonstrates that the SASP can induce cellular plasticity and stemness, promoting tissue regeneration.
Figure 5.

Transient exposure to the SASP induces stem cell function and tissue regeneration. (A) Scheme representing the strategy used to collect VCM or OIS-CM and subsequent treatment of freshly isolated keratinocytes. (B) qPCR analysis for stem cell and senescence genes on keratinocytes treated for 48 h with VCM or OIS-CM. n = 10. (C) Quantification of the numbers of colonies of keratinocytes treated for 48 h with VCM or OIS-CM. n = 4. (D,E) Representative skin grafts in nude mice that were grafted with a combination of 4 × 106 dermal fibroblasts and 6 × 106 keratinocytes from the β-actin-GFP reporter mouse following 48 h of exposure to VCM or OIS-CM. (F) Representative immunohistochemistry for GFP and DAPI on grafts in D and E. (G) Graph showing the number of hair follicles per field of view. Grafts were analyzed 21–23 d after implantation. n = 6 PMK + OIS-CM; n = 7 PMK + VCM. (H) qPCR analysis for CD34 and p16 on keratinocytes treated for 2–6 d with OIS-CM normalized to VCM at day 2. n = 5, except for OIS-CM day 6 (n = 4) and OIS-CM and VCM day 2 (n = 10). (I) Representative images of keratinocytes treated for 2, 4, and 6 d with VCM or OIS-CM. n = 5. Bars, 100 µm. (J) Quantification of SA-β-Gal-positive cells of keratinocytes treated for 2–6 d with VCM or OIS-CM. n = 4, except for VCM day 6 (n = 3) and VCM and OIS-CM day 4 (n = 2). (B,C,G,H,J) Error bars indicate mean ± SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001, two-tailed Student's t-test.
As transient exposure to the SASP induces stemness and regeneration, we asked whether longer exposure to the SASP would amplify these properties. Accordingly, PMK were treated with VCM or OIS-CM for longer time periods. We found that the increased expression of HFSC genes after a transient exposure to the SASP for 2 d was increased further upon repeated exposure to the same SASP for 6 d (Fig. 5H; Supplemental Fig. 8b). Interestingly, this increased expression seemed to be dependent on the SASP and not the length of time in culture, as removal of the OIS-CM after 2 d resulted in restoration of baseline levels of the stem cell genes (Supplemental Fig. 8c). Surprisingly, however, with repeated exposure, the recipient cells now exhibited intrinsic features of senescence, including proliferative decline and increased expression of p16 and SA-β-Gal (Fig. 5H–J), similar to paracrine senescence induced upon long-term exposure to the SASP (Acosta et al. 2013). Additional analysis demonstrated that the CD34-positive cells now expressed the cell cycle inhibitor p19 and that the entire population was not apoptotic and had lost the ability to form colonies in clonogenic assays (Supplemental Fig. 8d–f). This suggests that the intrinsic cell cycle arrest of senescence may be activated as a tumor-suppressive mechanism to block proliferation of cells with enhanced plasticity.
Together, these data demonstrate that transient exposure to the SASP can be beneficial in inducing cell plasticity and regeneration, whereas prolonged exposure could lead to paracrine and cell-intrinsic senescence arrest. To find support for this in an in vivo setting, we examined a previously described model of transposon-mediated intrahepatic gene transfer to stably deliver oncogenic Ras coexpressing GFP, which induces senescence in single adult hepatocytes in murine livers (Kang et al. 2011). These senescent cells are subsequently cleared by the immune system, while delivery of an inactive effector loop mutant form of Ras that is incapable of signaling to downstream pathways fails to induce senescence and circumvents the clearance. As described, delivery of oncogenic NRasG12V caused induction of senescence markers in individual hepatocytes (Fig. 6A; Supplemental Fig. 9a). When we examined the surrounding tissue for previously described markers of stem and cancer stem cells in the liver, we found increased expression of CD44 in cells immediately adjacent to senescent cells, which was not seen in the inactive Ras-expressing hepatocytes (Fig. 6B; Supplemental Fig. 9b). While CD44 is a well-known marker of cancer stem cells, here we cannot exclude that it may also be labeling macrophages or other immune cells. In addition, expression of oncogenic Ras led to a dramatic increase in expression of Nestin in the surrounding tissue, which was also absent from the inactive control (Fig. 6C; Supplemental Fig. 9c). As Nestin is also a well-described mediator of stemness and plasticity in the liver (Tschaharganeh et al. 2014), this further supports that the SASP can induce stem cell properties in surrounding tissue.
Figure 6.
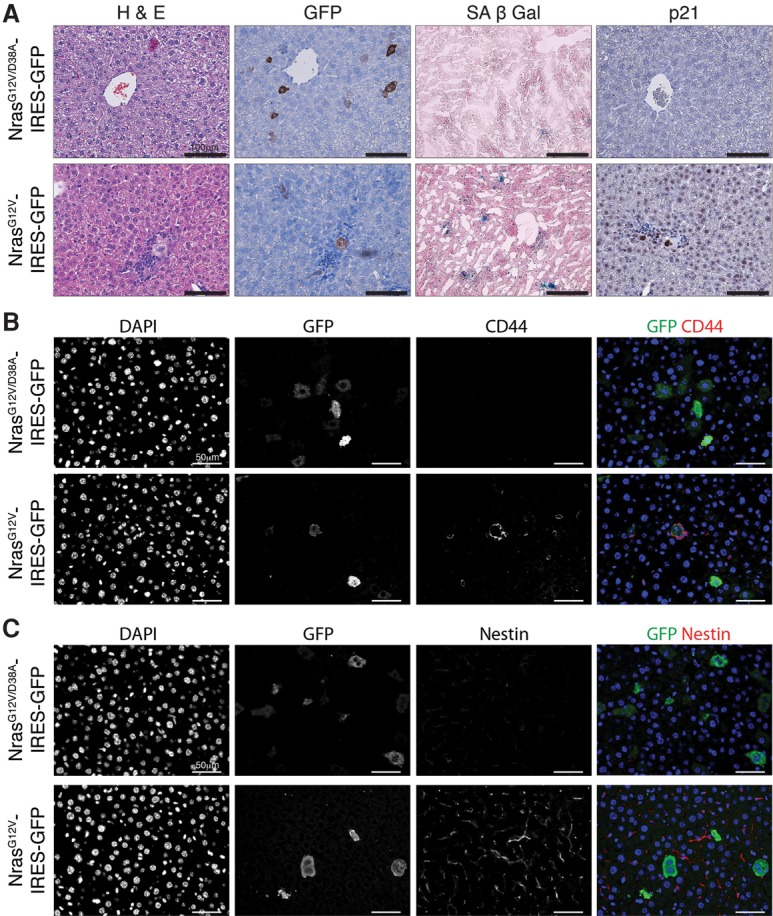
Induction of senescence in single cells in livers induces an increase in stem cell markers in the surrounding tissue of the liver. Representative histology of liver sections 6 d after delivery of control (NrasG12V/D38A-IRES-GFP) or mutant (NrasG12V-IRES-GFP) transposon constructs. (A) H&E staining, immunohistochemistry for GFP and p21, and SA-β-Gal staining. Bars, 100 µm. (B,C) Immunofluorescence for CD44, GFP, and DAPI (B) and Nestin, GFP, and DAPI (C). Bars, 50 µm. All images are representative of at least three biological replicates.
Discussion
Collectively, our study uncovers a novel and fundamental biological function of senescence and the SASP. We identified how transient exposure to the SASP can provide regenerative signals that induce cell plasticity and stemness, which are beneficial for tissue regeneration (Fig. 7). However, prolonged or aberrant exposure to the SASP subsequently provokes a cell-intrinsic senescence block to these growth-promoting signals, resulting in paracrine senescence responses and decreased regenerative capacity (Fig. 7).
Figure 7.
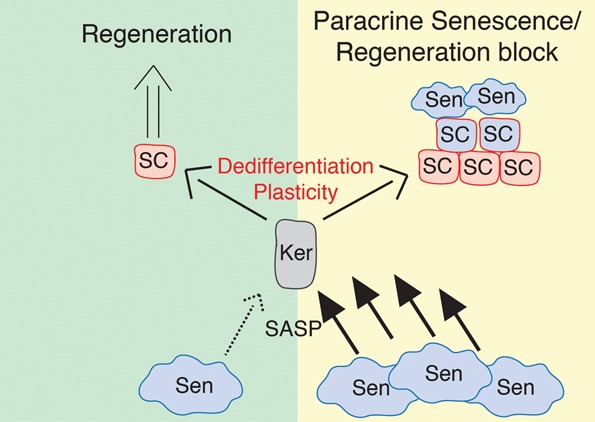
Schematic diagram illustrating transient and chronic exposure to SASP. Transient or low-level exposure to the SASP (dashed arrows) is beneficial, inducing plasticity and regeneration. However, prolonged or chronic exposure to the SASP (solid arrows) induces increased stemness, which is blocked by paracrine senescence arrest.
Altogether, these findings help to reconcile previously described beneficial and detrimental attributes of senescent cells. This study adds to the growing body of work demonstrating beneficial roles for the SASP, including during embryonic development (Munoz-Espin et al. 2013; Storer et al. 2013) and wound repair (Krizhanovsky et al. 2008; Jun and Lau 2010; Demaria et al. 2014) through a previously undescribed plasticity-inducing action. Indeed, we propose a model in which senescent cells act as niche-like signaling centers in which the damaged cell prevents its own continued proliferation and produces a SASP that activates the immune system to promote its own clearance (Xue et al. 2007; Acosta et al. 2008; Kuilman et al. 2008; Kang et al. 2011). In addition, the senescent cell produces extracellular matrix (ECM) and growth factors to facilitate repair and growth (Coppe et al. 2006; Krizhanovsky et al. 2008; Demaria et al. 2014), while, as we now describe, the SASP also induces plasticity and stemness in neighboring cells, which can replace the cleared senescent cell and promote tissue regeneration. Therefore, in an unperturbed setting, this would act beneficially to restore homeostasis and regeneration while eliminating damaged or aged cells.
However, when this process is perturbed with excessive accumulation or impaired clearance of senescent cells, as is seen during aging and premalignant lesion formation, the consequences of aberrant senescence induction become obvious and likely depend on the duration or strength of the SASP exposure and the status of the target cell. Indeed, when the clearance of senescent cells is perturbed, pronounced SASP exposure leads to tumor formation and progression (Krtolica et al. 2001; Kang et al. 2011; Rodier and Campisi 2011; Yoshimoto et al. 2013) or paracrine senescence and aging (Baker et al. 2011; Acosta et al. 2013).
Our study also suggests that paracrine senescence induction by the SASP (Acosta et al. 2013) might include a cell-intrinsic arrest to counter abnormal or prolonged regenerative stimuli. In this case, the recipient cells likely identify this extended regeneration as protumorigenic and activate cell-intrinsic tumor-suppressive mechanisms as a counteractive protection. This further supports the theory that both the loss of the ability in mammals to regenerate tissues and organs and the decline in tissue function seen during aging might be the result of cell-intrinsic senescence and tumor suppression (Janzen et al. 2006; Krishnamurthy et al. 2006; Molofsky et al. 2006; Sharpless and DePinho 2007; Sousa-Victor et al. 2014; Garcia-Prat et al. 2016).
The SASP has also been shown to promote tumor formation in a number of settings (Krtolica et al. 2001; Kang et al. 2011; Rodier and Campisi 2011; Yoshimoto et al. 2013). Our data develop these findings, suggesting that NFKB-mediated SASP exposure might contribute to plasticity during tumor formation through dedifferentiation and amplification of the stem cell of origin, as has been demonstrated previously in colon cancer models (Myant et al. 2013; Schwitalla et al. 2013). In addition, SASP effects could result in an expansion of stem cell numbers, thereby increasing the likelihood that these cells could acquire mutations or favor the induction of cells to reside in a more plastic undefined state that might be more susceptible to transformation (Tschaharganeh et al. 2014; Varga et al. 2014). This could help explain how senescence can favor tumor formation during aging, and it will be interesting to determine whether age-associated in vivo senescence has similar effects on plasticity induction.
During tumor formation, dysregulation of tissue-specific stem cells often contributes to cancer stem cell properties. Our data support the notion that regenerative attempts might promote de novo instruction of stem cells, which could then progress to cancer stem cells upon deregulation or mutation. For example, while CD34-positive cells are primary regulators of HFSCs in the skin, they are also the cells of origin and tumor-propagating cells in squamous cell carcinoma upon loss of p53-mediated arrest (Lapouge et al. 2012). Additionally, Nestin is a marker of stem cells in many tissues, including the brain and skin, yet aberrant activation of Nestin in liver tumors drives cell plasticity and malignancy (Li et al. 2003; Toma et al. 2005; Tschaharganeh et al. 2014; Neradil and Veselska 2015). Furthermore, while NFKB is a potent mediator of age- and tumor-associated inflammation and dedifferentiation, it also contributes to HFSC regeneration upon skin wounding (Chen et al. 2015) and is a key driver of hair follicle initiation and development (Schmidt-Ullrich et al. 2001; Zhang et al. 2009), further supporting how NFKB-mediated processes regulating normal tissue development and regeneration can contribute to tumor formation and cancer stem cell fate when misregulated (Myant et al. 2013; Schwitalla et al. 2013). It is also interesting here that cells that bypass senescence arrest through knockdown of p16 still retain expression of the stem cell signature and now exhibit increased clonal growth capacity, suggesting that escape from senescence may lead to more aggressive cancer stem cell properties mediated by NFKB and a retained SASP. Indeed, this is supported by previous studies demonstrating that cells that escape from chemotherapy-induced senescence can acquire cancer stem cell properties (Achuthan et al. 2011).
This study also contributes to our understanding of the role of senescence in papilloma and presents a tractable model to study papilloma development independently of the longer two-stage DMBA/TPA skin carcinogenesis protocol (Balmain et al. 1984; Kemp 2005; Abel et al. 2009). Interestingly, the senescence-causing HRasV12 mutation is induced in vivo with the DMBA/TPA procedure, which develops from an aberrant expansion of CD34+ tissue stem cells (Morris 2004; Malanchi et al. 2008; Abel et al. 2009; Lapouge et al. 2012). However, what causes the accumulation of stem cells and papilloma induction as well as the potential contribution of senescent cells remain unclear. Our results suggest that papilloma formation in the DMBA-TPA model arises through multiple events. Keratinocytes harboring the mutation can clonally amplify and produce a SASP that enhances stemness in neighboring keratinocytes while progressing to become terminally differentiated and senescent. However, simultaneously, there is a pronounced induction of senescence in the dermal fibroblasts, suggesting that the SASP from these cells might also contribute to the dedifferentiation and amplification of the overlying keratinocyte stem cells. While papilloma has been described as an in vivo model of senescence, it was unexpected that the epithelial keratinocytes would not stain positive for SA-β-Gal in vivo, yet again raising interesting discussions regarding the identification of senescent cells. However, given that these cells express many additional senescence markers (including p16, p19, p21, and p53) and have lost proliferation markers as well as the fact that it is possible to undergo senescence without expression of SA-β-Gal (Lee et al. 2006), it seems reasonable that they can still be described as senescent.
Overall, however, our discovery that timely controlled exposure to the SASP can directly promote tissue regeneration identifies important new biological underpinnings for the senescence program, supporting the idea that senescence is primarily a beneficial and regenerative process. However, when perturbed, this can have detrimental effects seen during tumor formation and aging. This also suggests that a better understanding of the underlying mechanisms might be harnessed for functional regeneration of tissues and organs.
Materials and methods
Animal use
Wild-type C57Bl6/J, p53–/–, p21–/–, FVB/N, β-Actin GFP reporter, and Swiss Nude mice were housed in accordance with the Ethical Committee for Animal Experimentation (CEEA) of the Government of Catalonia. Papillomas were generated as described previously (Abel et al. 2009). Briefly, shaved dorsal back skin of seven to nine w FVB/N female mice was treated with a single dose of 100 nM DMBA (Sigma-Aldrich) and subsequent twice-weekly applications of 6.8 nM TPA (Sigma-Aldrich) for a period of up to 20 wk. Hair reconstitution assays were performed as described previously (Jensen et al. 2010). PanIn lesions were obtained from Pdx1-Cre; LSL-KrasG12D/+ mice as described in Morton et al. (2010). To induce senescence in single cells in the liver, transposable constructs of oncogenic Nras (NrasG12V-IRES-GFP) or an inactive effector loop mutant of Nras (NrasG12V/D38A-IRES-GFP) were injected into livers as described in Kang et al. (2011).
Tissue culture
Primary mouse keratinocytes and dermal fibroblast cultures were established from 1- to 2-d-old mice. Retrovirus was produced by transiently transfecting the Phoenix packaging cell line (G. Nolan, Stanford University, Stanford, CA) with MSCV or HRasV12, and cells were infected with virus twice for 2 h, drug-selected in 1 µg/mL puromycin for 48 h, and maintained for up to another 4–14 d prior to analysis. For double infections, keratinocytes were infected three times for 2 h with retrovirus (produced by transient transfection with HRasV12, MLP, shp16 I, shp16 II, shRelA I, shRelA II, NFκB superrepressor, shCEBPβ, shIl1α, and shp38α), drug-selected in 1 µg/mL puromycin for 48 h followed by a second drug selection in 25 µg/mL hygromycin for 48 h, and maintained for 6–14 d prior to analysis. Keratinocytes were treated with 100 nM etoposide (Sigma) for 48 h and then kept in normal medium or exposed to 12 Gy of irradiation. For proliferation, BrdU was added to cultured keratinocytes for 16 h, and detection was performed using a BrdU in situ detection kit (BD Pharmingen) as per the manufacturer's protocol. SA-β-Gal activity was detected in cultured cells as described previously (Dimri et al. 1995). Colony-forming assays were performed as described previously (Jensen et al. 2010). The following sequences of shRNAs were taken from Fellmann et al. (2013): shp16 I (TGCTGTTGACAGTGAGCGCTGGCATGAGAAACTGAGCGAATAGTGAAGCCACAGATGTATTCGCTCAGTTTCTCATGCCATTGCCTACTGCCTCGGA), shp16 II (TGCTGTTGACAGTGAGCGCAGCAGCTCTTCTGCTCAACTATAGTGAAGCCACAGATGTATAGTTGAGCAGAAGAGCTGCTATGCCTACTGCCTCGGA), shCEBPβ (TGCTGTTGACAGTGAGCGCCCGTTTCGAGCATTAAAGTGATAGTGAAGCCACAGATGTATCACTTTAATGCTCGAAACGGATGCCTACTGCCTCGGA), and shIl1α (TGCTGTTGACAGTGAGCGCCAGGATGTGGACAAACACTATTAGTGAAGCCACAGATGTAATAGTGTTTGTCCACATCCTGATGCCTACTGCCTCGGA). Sequences for shRelA I and shRelA II were obtained from Mirimus, Inc.
Immunohistochemistry and immunofluorescence
Papillomas were fixed in 4% PFA either overnight or for 10 min at room temperature and washed in PBS, and then tissues were processed for paraffin or OCT embedding and hematoxylin and eosin (H&E) staining. Skin grafts were fixed in 4% PFA for 12 min at room temperature, washed in PBS, and embedded in OCT, and 60-µm sections were cut. Immunohistochemistry and immunofluorescence analysis was performed using standard procedures. Antigen retrieval, where necessary, was performed by boiling deparaffinized sections for 20 min in 0.01 M citric acid buffer (pH 6.0). Primary antibodies were incubated overnight at 4°C, and secondary antibodies were incubated for 1 h at room temperature in 1% serum and 0.1% Triton-X in PBS. Sections were treated with streptavidin–biotin–peroxidase (VectaStain Elite, Vector Laboratories), and immune complexes were visualized with diaminobenzidine (Vector Laboratories) as per the manufacturer's protocol. The following primary antibodies were used at the indicated dilutions: anti-p16 (1:200; Santa Cruz Biotechnology, M-156), anti-p16 (1:200; Abcam, 2D9A12), anti-p21 (1:200; BD Pharmingen, SXM30), anti-p19ARF (1:200; Abcam, ab80), anti-Ki67 (1:200; Abcam, ab15580), anti-PCNA (1:200; Sigma-Aldrich, P8824), anti-MCM2 (1:100; Cell Signaling), anti-p53 (1:200; Leica Microsystems, CM5), anti-P-Histone H2AX (1:200; Cell Signaling, Ser139), anti-CD34 (1:100; eBioscience, RAM34), anti-CD44 (1:200; Abcam, ab157107), anti-CD44 (1:100; BD Biosciences, 550538), anti-Nestin (1:100; Millipore, MAB353), anti-GFP (1:200; Santa Cruz Biotechnology, B-2), anti-GFP (1:200; Invitrogen, A11122), and cleaved Caspase-3 (1:400; Cell Signaling, 9661L). Biotin or Alexa-conjugated secondary antibodies (VectaStain Elite or Molecular Probes) were used at a dilution of 1:2000–1:500. Representative pictures were acquired with a Leica DMI 6000B microscope. For SA-β-Gal staining in solid tissue, premalignant lesions whole-mount-stained with SA-β-Gal were fixed in 4% PFA overnight at 4°C, and 10-µm paraffin sections were cut.
CM
CM was collected and filtered from MSCV- or HrasV12-infected primary mouse keratinocytes 7 d post-infection (dpi) or from untreated or irradiated keratinocytes 7 d after irradiation and added to primary mouse keratinocytes every 24 h for up to 6 d. For hair reconstitution assay, the CM was collected, filtered, and pooled from primary mouse keratinocytes infected with either MSCV or HrasV12 at 6–8 dpi and normalized with fresh medium according to cell number.
Flow cytometry
Epidermal cells were isolated from the dorsal back skin of adult C57Bl6/J mice or 1- to 2-d-old mice. Cultured keratinocytes were isolated through incubation with 0.05% trypsin for 10 min at 37°C followed by inactivation of trypsin by the addition of DMEM containing 10% serum and centrifugation at 1000 rpm for 7 min. Cell suspensions were incubated for 60 min on ice with the following antibodies at the indicated dilutions: APC-conjugated or FITC-conjugated anti-CD34 (1:200; BD Pharmingen, clone RAM34), FITC-conjugated anti-α6-integrin (1:200; Serotec, CD49f clone NKI-GoH3), and APC-conjugated lineage antibody cocktail (1:200; BD Pharmingen). Keratinocytes were sorted on the basis of single cellularity, viability (DAPI), and/or CD34/CD49f status. FACS purification was performed on a FACS Aria system equipped with FACS DiVa software (BD Biosciences). FACS analysis was performed using LSRII FACS analyzers (BD Biosciences) or LSR Fortessa (BD Biosciences) and analyzed using Flowjo software.
Microarray analysis
Total RNA was isolated from four separate technical replicates of primary mouse keratinocytes infected with MSCV and HRasV12 at 6 dpi using an RNAEasy mini RNA extraction kit (Qiagen). RNA was subsequently labeled and hybridized to Agilent 8x60 one-color gene expression arrays. Bioinformatics analysis was performed using Genomatix software (Genomatix Software GmbH). The microarray data have been uploaded to the Gene Expression Omnibus database with accession number GSE93564.
Real-time qPCR and analysis
Total RNA from cultured or FACS-sorted cells was purified using Trizol or an RNAEasy microRNA extraction kit (Qiagen) and reverse-transcribed using qScript supermix (Quanta Biosciences). Real-time qPCR was performed using gene-specific primers (Supplemental Table 2) and a LightCycler 480 (Roche). qPCR analysis for vector time-course keratinocytes (Supplemental Fig. 1e) was performed on the following biological replicates: 6 dpi (n = 12), 8 dpi (n = 10), 10 dpi (n = 9), 12 dpi (n = 8), and 14 dpi (n = 3).
Statistical analysis
Results are presented as mean ± SEM. Statistical significance was determined by the two-tailed unpaired Student's t-test.
Supplementary Material
Acknowledgments
We thank Ryan Driskell and Fiona Watt for technical advice, Guillaume Filion and Panagiotis Papasaikas for advice on statistics, Pura Muñoz-Cánoves for β-actin GFP mice, the Centre for Genomic Regulation Bioinformatics unit for assistance, and Pura Muñoz-Cánoves and Juan Valcarcel for critical reading of the manuscript. M.S. and B.R. were supported with fellowships from the “La Caixa” foundation. This work was funded by grants SAF2010-18829 and SAF2013-49082-P (to W.M.K.) from the Spanish Ministry for Economy and Competitiveness, the Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) from the Generalitat de Catalunya, and Centre for Genomic Regulation core funding.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.290635.116.
Freely available online through the Genes & Development Open Access option.
References
- Abel EL, Angel JM, Kiguchi K, DiGiovanni J. 2009. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc 4: 1350–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achuthan S, Santhoshkumar TR, Prabhakar J, Nair SA, Pillai MR. 2011. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J Biol Chem 286: 37813–37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. 2008. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006–1018. [DOI] [PubMed] [Google Scholar]
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, et al. 2013. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15: 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. 2011. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. 2016. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530: 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmain A, Ramsden M, Bowden GT, Smith J. 1984. Activation of the mouse cellular Harvey-ras gene in chemically induced benign skin papillomas. Nature 307: 658–660. [DOI] [PubMed] [Google Scholar]
- Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E. 2004. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell 118: 635–648. [DOI] [PubMed] [Google Scholar]
- Campisi J. 2013. Aging, cellular senescence, and cancer. Annu Rev Physiol 75: 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Wang L, Plikus MV, Jiang TX, Murray PJ, Ramos R, Guerrero-Juarez CF, Hughes MW, Lee OK, Shi S, et al. 2015. Organ-level quorum sensing directs regeneration in hair stem cell populations. Cell 161: 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Kauser K, Campisi J, Beausejour CM. 2006. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem 281: 29568–29574. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME, et al. 2014. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31: 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci 92: 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan Y, et al. 2013. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep 5: 1704–1713. [DOI] [PubMed] [Google Scholar]
- Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, et al. 2016. Autophagy maintains stemness by preventing senescence. Nature 529: 37–42. [DOI] [PubMed] [Google Scholar]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. 2006. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443: 421–426. [DOI] [PubMed] [Google Scholar]
- Jensen KB, Driskell RR, Watt FM. 2010. Assaying proliferation and differentiation capacity of stem cells using disaggregated adult mouse epidermis. Nat Protoc 5: 898–911. [DOI] [PubMed] [Google Scholar]
- Jun JI, Lau LF. 2010. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 12: 676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. 2011. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479: 547–551. [DOI] [PubMed] [Google Scholar]
- Kemp CJ. 2005. Multistep skin cancer in mice as a model to study the evolution of cancer cells. Semin Cancer Biol 15: 460–473. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. 2006. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 443: 453–457. [DOI] [PubMed] [Google Scholar]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. 2008. Senescence of activated stellate cells limits liver fibrosis. Cell 134: 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. 2001. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci 98: 12072–12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. 2008. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133: 1019–1031. [DOI] [PubMed] [Google Scholar]
- Lapouge G, Beck B, Nassar D, Dubois C, Dekoninck S, Blanpain C. 2012. Skin squamous cell carcinoma propagating cells increase with tumour progression and invasiveness. EMBO J 31: 4563–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D, Hwang ES. 2006. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 5: 187–195. [DOI] [PubMed] [Google Scholar]
- Li L, Mignone J, Yang M, Matic M, Penman S, Enikolopov G, Hoffman RM. 2003. Nestin expression in hair follicle sheath progenitor cells. Proc Natl Acad Sci 100: 9958–9961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, et al. 2013. Non-cell-autonomous tumor suppression by p53. Cell 153: 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, Huber M, Hohl D, Cano A, Birchmeier W, et al. 2008. Cutaneous cancer stem cell maintenance is dependent on β-catenin signalling. Nature 452: 650–653. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. 2006. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443: 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RJ. 2004. A perspective on keratinocyte stem cells as targets for skin carcinogenesis. Differentiation 72: 381–386. [DOI] [PubMed] [Google Scholar]
- Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, Jamieson NB, Oien KA, Lowy AM, Brunton VG, et al. 2010. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci 107: 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, et al. 2013. Programmed cell senescence during mammalian embryonic development. Cell 155: 1104–1118. [DOI] [PubMed] [Google Scholar]
- Myant KB, Scopelliti A, Haque S, Vidal M, Sansom OJ, Cordero JB. 2013. Rac1 drives intestinal stem cell proliferation and regeneration. Cell Cycle 12: 2973–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neradil J, Veselska R. 2015. Nestin as a marker of cancer stem cells. Cancer Sci 106: 803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Campisi J. 2011. Four faces of cellular senescence. J Cell Biol 192: 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Ullrich R, Aebischer T, Hulsken J, Birchmeier W, Klemm U, Scheidereit C. 2001. Requirement of NF-κB/Rel for the development of hair follicles and other epidermal appendices. Development 128: 3843–3853. [DOI] [PubMed] [Google Scholar]
- Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW. 2002. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109: 335–346. [DOI] [PubMed] [Google Scholar]
- Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, et al. 2013. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152: 25–38. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. 2007. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol 8: 703–713. [DOI] [PubMed] [Google Scholar]
- Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, et al. 2014. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506: 316–321. [DOI] [PubMed] [Google Scholar]
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al. 2013. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155: 1119–1130. [DOI] [PubMed] [Google Scholar]
- Toma JG, McKenzie IA, Bagli D, Miller FD. 2005. Isolation and characterization of multipotent skin-derived precursors from human skin. Stem Cells 23: 727–737. [DOI] [PubMed] [Google Scholar]
- Tschaharganeh DF, Xue W, Calvisi DF, Evert M, Michurina TV, Dow LE, Banito A, Katz SF, Kastenhuber ER, Weissmueller S, et al. 2014. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 158: 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga J, De Oliveira T, Greten FR. 2014. The architect who never sleeps: tumor-induced plasticity. FEBS Lett 588: 2422–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. 2007. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445: 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. 2013. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499: 97–101. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Tomann P, Andl T, Gallant NM, Huelsken J, Jerchow B, Birchmeier W, Paus R, Piccolo S, Mikkola ML, et al. 2009. Reciprocal requirements for EDA/EDAR/NF-κB and Wnt/β-catenin signaling pathways in hair follicle induction. Dev Cell 17: 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}