

Abstract
The adult skeleton is renewed by remodeling throughout life. Bone remodeling is a process where osteoclasts and osteoblasts work sequentially in the same bone remodeling unit. After the attainment of peak bone mass, bone remodeling is balanced and bone mass is stable for one or two decades until age-related bone loss begins. Age-related bone loss is caused by increases in resorptive activity and reduced bone formation. The relative importance of cortical remodeling increases with age as cancellous bone is lost and remodeling activity in both compartments increases. Bone modeling describes the process whereby bones are shaped or reshaped by the independent action of osteoblast and osteoclasts. The activities of osteoblasts and osteoclasts are not necessarily coupled anatomically or temporally. Bone modeling defines skeletal development and growth but continues throughout life. Modeling-based bone formation contributes to the periosteal expansion, just as remodeling-based resorption is responsible for the medullary expansion seen at the long bones with aging. Existing and upcoming treatments affect remodeling as well as modeling. Teriparatide stimulates bone formation, 70% of which is remodeling based and 20–30% is modeling based. The vast majority of modeling represents overflow from remodeling units rather than de novo modeling. Denosumab inhibits bone remodeling but is permissive for modeling at cortex. Odanacatib inhibits bone resorption by inhibiting cathepsin K activity, whereas modeling-based bone formation is stimulated at periosteal surfaces. Inhibition of sclerostin stimulates bone formation and histomorphometric analysis demonstrated that bone formation is predominantly modeling based. The bone-mass response to some osteoporosis treatments in humans certainly suggests that nonremodeling mechanisms contribute to this response and bone modeling may be such a mechanism. To date, this has only been demonstrated for teriparatide, however, it is clear that rediscovering a phenomenon that was first observed more half a century ago will have an important impact on our understanding of how new antifracture treatments work.
Keywords: bone modeling, bone remodeling, osteoporosis, teriparatide, bisphosphonates, denosumab, romosozumab
Introduction
Osteoporosis is a common condition, affecting one in three postmenopausal women and one in five men, corresponding to 200 million women and men, worldwide [Strom et al. 2011]. Osteoporosis is characterized by low bone mass and deteriorated bone architecture [WHO, 1994]. The immediate clinical consequence of osteoporosis is fracture [Johnell et al. 2005], and osteoporosis-related fractures, vertebral as well as hip, are associated with morbidity and increased mortality [Bliuc et al. 2014; Gerdhem, 2013].
In the course of the past three decades, several drugs have been developed that can prevent fractures; however, although the effect of these treatments on vertebral fractures is impressive, the effect on nonvertebral fractures is less than satisfactory [Black et al. 2007; Cummings et al. 2009]. Moreover significant reduction of vertebral fractures occurs early in the course of therapy, typically within 6 months, whereas reduction of nonvertebral fractures and hip fractures specifically has not been observed before at least 1 year of therapy [Black et al. 1996, 2007; Cummings et al. 2009]. This could be explained by the fact that vertebral fragility is primarily determined by focal areas of erosion creating stress risers on trabeculae [Dempster, 1997], whereas weakness in the peripheral skeleton results from trabecular and cortical bone loss, particularly cortical porosity, that becomes predominant only in older age [Zebaze et al. 2010]. In turn, the elimination of stress risers, which is proportional to the potency of the various antiresorptives, is sufficient to explain the early decrease of vertebral fractures, whereas long-term reversal of the negative bone mineral balance seen in the peripheral skeleton, particularly the progressive restoration of the cortical bone volume, is essential to reduce nonvertebral fractures. As a corollary, spine bone mineral density (BMD) changes have been found to explain less than 50% of vertebral fracture risk reduction [Austin et al. 2012; Cummings et al. 2002; Jacques et al. 2012; Miller et al. 2010; Watts et al. 2004], whereas more recently hip BMD gain with potent parenteral antiresorptives such as zoledronic acid and denosumab has explained up to 60–90% of nonvertebral fracture risk reduction [Austin et al. 2012; Jacques et al. 2012]. Nevertheless relatively large changes at the hip are needed to significantly influence fracture risk, for example, a 6% BMD gain is equivalent to 1% nonvertebral fracture risk reduction with denosumab [Cummings et al. 2009].
Therefore the search for better treatments continues. Improved treatment of osteoporosis may include identification of new treatments, but may also include a better understanding of the mechanisms of action of existing drugs as this could lead to improved use of existing treatments. This review will focus on the importance of the effect of treatments on remodeling and, especially, modeling of bone and how this may affect the outcome of treatments.
Physiology of bone modeling and remodeling
The adult skeleton comprises both cortical and cancellous bone. About 80% of bone is cortical, however, the distribution of cancellous and cortical bone varies between bone sites, for example, cancellous bone comprises 66% and 75% of lumbar and thoracic vertebrae, respectively, whereas only 5% of the bone at the distal radius is cancellous. The femoral neck is in between these extremes with 75% of bone being cortical [Dempster, 2006].
Bone remodeling
The adult skeleton is renewed by remodeling every 10 years. Remodeling persists throughout life. It has been estimated that 3–4 million bone remodeling units (BRUs) are initiated each year and that 1 million BRUs are actively engaged in bone turnover at any time [Manolagas, 2000]. Remodeling is a process characterized by four phases: the activation phase when the osteoclasts are recruited; the resorption phase, when the osteoclasts resorb bone; the reversal phase, where the osteoclasts undergo apoptosis and the osteoblasts are recruited; the formation phase, where the osteoblasts lay down new organic bone matrix that subsequently mineralizes. By definition, bone remodeling is a process where osteoclasts and osteoblasts work sequentially in the same BRU [Dempster, 2002;Eriksen, 1986]. After the attainment of peak bone mass, bone remodeling is balanced and bone mass is stable for a decade or two until age-related bone loss begins. Age-related bone loss is caused by increases in resorptive activity and reduced bone formation [Dempster and Lindsay, 1993]. Abnormalities in bone remodeling cause bone loss or bone gain and are the basis of low and high bone-mass syndromes [Brunkow et al. 2001; Johnson et al. 1997; Motyckova and Fisher, 2002].
Bone remodeling is most prominent on cancellous bone surfaces and it is estimated that 80% of bone remodeling activity takes place in cancellous bone, although cancellous bone only comprises 20% of bone. The relative importance of cortical remodeling increases with age as cancellous bone is lost and the remodeling activity in both compartments increases [Seeman, 2013]. In the cortical bone, remodeling takes place at both the periosteal and endocortical surfaces [Balena et al. 1992; Bliziotes et al. 2006; Dempster et al. 2001; Orwoll, 2003], but it also occurs inside the compact cortical bone. At the cortical surfaces remodeling is a surface-based process similar to the process in cancellous bone, whereas intracortical remodeling is characterized by osteoclasts drilling through the compact bone in the cutting cone followed by osteoblasts filling the cylindrical void in the closing cone [Dempster and Lindsay, 1993]. This is called a Haversian remodeling system [Havers, 1691].
The purposes of remodeling are many including the replacement of old and damaged bone with new bone and calcium homeostasis (long term). By removing old and damaged bone targeted remodeling plays a key role in maintaining the mechanical strength of bone. However, excessive remodeling and repair poses a risk to bone strength as it destabilizes bone and introduces stress concentrators [Dempster, 1997; Einhorn, 1992]. Even targeted remodeling may be harmful according to the following hypothesis. Excessive strain causes regional microdamage, which leads to targeted remodeling removing the damaged bone and a larger volume of the surrounding undamaged bone, this temporary volume deficit increases the strain in neighboring bone and the potential establishment of a vicious cycle between damage and repair [Allen and Burr, 2008; Martin, 1995]. Bone became an important player in calcium homeostasis when our primitive ancestors left the oceans, an environment with a high availability of calcium, and ventured on to dry land where calcium is a scarce resource. There are several examples of bone being a dynamic part of calcium homeostasis, for example, during pregnancy and lactation or when male deer grow antlers, the latter being an extreme example in which sufficient calcium can only be attained by temporarily removing it from the skeleton [Banks et al. 1968a, b]. The potential conflict between preserving bone strength and providing calcium to the rest of the body becomes more obvious with aging when vitamin D production and, thereby calcium absorption, decreases and secondary hyperparathyroidism develops in order to maintain adequate serum calcium levels by increasing bone resorption. Furthermore, the estrogen insufficiency in postmenopausal women also leads to increased remodeling activity. Increased resorptive activity in a young individual is accompanied by complementary increased formation and the balance at each BRU is neutral, therefore the bone loss is merely reflecting an opening of the remodeling space and is therefore reversible. The situation in postmenopausal women and elderly men is very different. The balance between resorption and subsequent formation at each BRU is negative and increased resorptive activity therefore leads to bone loss that is irreversible due to thinning of the trabeculae, loss of trabeculae, and thinning of the cortex.
Bone remodeling also plays a role in the maintenance of acid/base balance, and the release of growth factors embedded in bone. Moreover, it provides a reservoir of labile mineral (short-term homeostasis) and it is the only mechanism by which old, dying, or dead osteocytes can be replaced [Dempster, 2006].
Bone modeling
Bone modeling describes the process whereby bones are shaped or reshaped by the independent action of osteoblasts and osteoclasts. The activities of osteoblasts and osteoclasts are not necessarily coupled anatomically or temporally as is the case in bone remodeling. Bone modeling defines skeletal development and growth and is responsible for the shaping of bones and their movement through space. Even in adults adaptation to permanently changed strain leads to modeling of bone, an example of which is tibial modeling after harvesting fibula for reconstructive surgery [Taddei et al. 2009]. Abnormalities in bone modeling cause skeletal dysplasias or dysmorphias.
Frost and colleagues were the first to describe modeling in bone from adults [Hattner et al. 1965]. The bones investigated were the ribs, femoral heads, iliac crests, humeri, and vertebrae from 75 healthy adults of both sexes. They investigated the shape of the cement line delineating old from newly formed bone at bone-forming sites in cancellous bone and found that the vast majority of these sites had a scalloped morphology, suggesting that bone formation had followed bone resorption; however, 3% of the cement lines were smooth suggesting that formation had taken place on a surface not previously resorbed. The authors deduced that this could represent bone modeling, but it could also represent overflow of formation processes extending beyond the perimeter of the resorption lacunae. There was no effect of age on the prevalence of modeling-based bone formation, and no information was provided about the effect of gender.
This seminal observation was relegated to the library shelves and probably ignored by most researchers for many years. However, 30 years after this first observation, Erben described similar findings in rat bone [Erben, 1996], both at cancellous and endocortical surfaces. Kobayashi and colleagues found modeling in 62% of human iliac crest biopsies. Modeling was found on 2% of the cancellous bone surfaces, but the labeled surface at the modeling sites accounted for 25–50% of the entire labeled surface [Kobayashi et al. 2003].
Bone modeling has been demonstrated in aging humans. Modeling-based bone formation contributes to the periosteal expansion, just as remodeling-based resorption is responsible for the medullary expansion seen at long bones and ribs with aging [Epker and Frost, 1966; Garn et al. 1967; Ruff and Hayes, 1982].
How is bone modeling controlled? Physical activity can stimulate bone modeling. This is seen for example in tennis players where the arm used for tennis has a higher bone mass than the other arm [Kontulainen et al. 2002]. The modeling-based bone formation at the femoral neck in the nonhuman primate study of denosumab was located at the superior endocortex and the inferior periosteal surface [Ominsky et al. 2015], which is consistent with where the greatest stress has been documented by finite element analysis in sideways fall and stance loading, respectively [Nawathe et al. 2015; Ominsky et al. 2015]. However, it should be kept in mind that only one slice from the femoral neck was available for examination from each animal and modeling-based bone formation could therefore not be examined at the anterior and posterior parts of the femoral neck. A recent study examined femoral neck samples from patients who had undergone hip replacement surgery [Cosman et al. 2013]. Bone formation rate was highest on the inferior periosteum and the superior endocortex, which were exactly the same locations where modeling-based bone formation was seen in the monkeys in the denosumab nonhuman primate study [Ominsky et al. 2015]. Bone modeling is also controlled by other factors as modeling-based bone formation was also seen at the ribs, which are not axially loaded, in the denosumab nonhuman primate study [Ominsky et al. 2015]. It is therefore likely that bone modeling is controlled by genetic factors in combination with environmental factors such as physical strain and probably hormonal factors, as it has been demonstrated that the parathyroid hormone (PTH) and inhibition of sclerostin can stimulate modeling-based bone formation [Lindsay et al. 2006; Ominsky et al. 2014].
The effect of osteoporosis treatments on bone modeling
Anabolic treatment
It has been known for almost a century that PTH stimulates bone formation [Bauer et al. 1929; Selye, 1932]. In a prescient observation in 1932, Selye deduced that PTH administered in very small doses stimulates osteoblasts and thereby bone apposition without previous osteoclast formation.
Using quadruple tetracycline labeling, Lindsay and colleagues demonstrated that teriparatide (PTH1-34) was in fact able to stimulate bone modeling at trabecular bone [Lindsay et al. 2006]. As Frost and colleagues had shown decades before, formation was assessed to be modeling based if the underlying cement line was smooth and remodeling based if the underlying cement line was scalloped. In control subjects all formation was remodeling based, whereas in women treated with PTH1-34 70% was remodeling based and 20–30% was modeling based on the cancellous and endocortical surfaces, respectively. It was also noted that the second tetracycline label frequently extended beyond the limits of the scalloped reversal line on to the adjacent, previously unresorbed surface. In fact, 50–64% of the modeling-based formation occurred in these extended remodeling units, suggesting that the vast majority of modeling in response to a short course of PTH1-34 represents overflow from remodeling units rather than de novo modeling on previously quiescent surfaces.
Abaloparatide is an analogue of PTH-related protein and under investigation for the treatment of osteoporosis. No animal data or results from investigation of human biopsies on the effects of abaloparatide on modeling are available.
Antiresorptive treatments
Denosumab
Denosumab is an antibody against the RANK-ligand (RANKL) and by neutralizing RANKL osteoclast recruitment, activity and life span are reduced. Denosumab is therefore a strong antiresorptive agent for the treatment of osteoporosis. The current understanding of the mechanisms underlying the antifracture efficacy of antiresorptive treatments is that by inhibiting recruitment and/or activity of osteoclasts, bone remodeling is reduced and, thereby, the remodeling space is refilled, leading to an early increase in bone mass and reduction in stress concentrators. Increased secondary mineralization of older bone also adds to the increase in bone mass later in the course of treatment. Treatment with denosumab has led to very impressive increases in bone mass, especially at sites with a high content of cortical bone [Cummings et al. 2009]. Furthermore, the increases in bone mass seem to continue despite the fact that bone turnover is continuously suppressed [Bone et al. 2013]. This has led to the hypothesis that these increases, at least in part, may be the result of a remodeling-independent mechanism to accrue bone matrix. In order to test this hypothesis, proximal femur and rib samples from cynomolgus monkeys were re-examined. Kostenuik and colleagues investigated the effect of denosumab or placebo treatment on cancellous bone for 16 months in ovariectomized cynomolgus monkeys [Kostenuik et al. 2011]. Mineralizing surfaces in cancellous bone were significantly reduced, and bone mass and bone strength were improved. Labeling of the mineralizing surface was performed 3 times, after 6, 10, and 16 months in this study. On re-examination of the bones from these animals and particularly when examining the cortical bone, it was seen that multilabeled bone formation was ongoing at the cortical surfaces, especially on the superior endocortex and the inferior periosteal surfaces [Ominsky et al. 2015]. The cement lines were smooth and this phenomenon was seen to the same extent in treated and untreated animals. The ribs of the same animals were also examined and it could be demonstrated that denosumab did not alter the surface extent of modeling-based formation, or the cortical area bound by them, relative to ovariectomized control animals. This was in contrast to the significantly reduced remodeling-based bone formation and eroded surfaces in the treated animals. The authors concluded that in this animal model of postmenopausal bone loss denosumab inhibits remodeling. Furthermore, denosumab does not stimulate modeling, but is permissive for modeling.
These observations led to the following hypothesis. In untreated ovariectomized animals bone balance is negative because remodeling-based bone resorption is increased due to loss of estrogen and this is not fully compensated for by remodeling-based bone formation. The overall bone balance becomes negative and the animals lose bone because modeling cannot compensate fully for the negative remodeling balance. When denosumab is administered and bone resorption is fully inhibited, modeling-based bone formation continues unabated and the net result is a bone gain (Figure 1). Another indirect argument for a potential maintenance of the full bone modeling capacity with denosumab has recently been provided by a clinical trial combining denosumab and intermittent teriparatide [Leder et al. 2014; Tsai et al. 2013]. In that study, the two drugs exerted additional effects on areal BMD (aBMD), while markers of bone resorption remained fully suppressed, suggesting that PTH-stimulated bone modeling was taking place in the absence of bone remodeling. The bone gain seen during denosumab treatment may therefore be due to a combination of ongoing bone modeling, reduction of remodeling and, thereby, filling of the remodeling space and increased secondary mineralization of bone [Ominsky et al. 2015].
Figure 1.
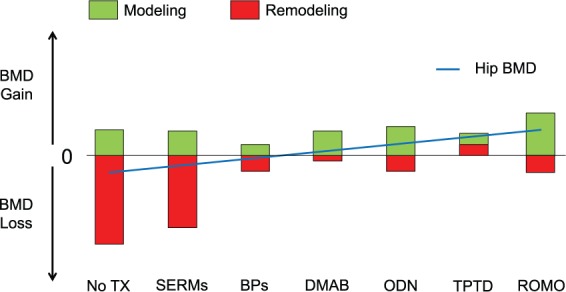
The theoretical contribution of bone remodeling and modeling to the change in hip bone mineral density (BMD) in postmenopausal women with or without existing and upcoming treatments for osteoporosis.
BPs, bisphosphonates [Black et al. 2015; Miller et al. 2012]; Dmab, denosumab [Bone et al. 2013]; No Tx, no treatment; Odn, odanacatib [Langdahl et al. 2012]; Romo, romosozumab [McClung et al. 2014a, b]; SERMs, selective estrogen receptor modulators [Silverman et al. 2012]; TPTD, teriparatide [Neer et al. 2001].
Although bone biopsies have been obtained in the FREEDOM study investigating the effect of denosumab on bone mass and fracture in postmenopausal women with osteoporosis [Reid et al. 2010], it has not yet been reported if any effect on bone modeling was seen.
Other antiresorptives
The same increase in bone mass at predominantly cortical sites has not been seen with other antiresorptives including potent bisphosphonates [Yang et al. 2013]. This has yet to be investigated. However, there could potentially be reasons why bisphosphonates would not have the same effect on cortical bone as denosumab. It has been demonstrated that osteoblasts take up bisphosphonates [Coxon et al. 2008], and animal studies have shown that bisphosphonates suppress bone formation by lining cells, that is, bone modeling [Gasser et al. 2000; Gasser and Green, 2006]. Furthermore, if bisphosphonates are co-administered with PTH the effect of PTH is blunted, more so if bisphosphonates are administered frequently [Cosman et al. 2011; Finkelstein et al. 2006]. These findings may together suggest that bisphosphonates inhibit osteoblasts directly and therefore also potentially inhibit modeling-based bone formation. Depending on their affinity for the bone matrix, bisphosphonates have also been shown to reach the osteocytes lacunae [Roelofs et al. 2010], and could therefore exert some negative effects on these cells and their role as mechanostatic censors and PTH-responsive cells, although others have shown anti-apoptotic effects of bisphosphonates on osteocytes in cellular and mouse models [Bonnet et al. 2013; Plotkin et al. 2006]. Finally, bisphosphonates attach to bone and therefore are likely to be preferentially sequestered in cancellous bone and the accessibility of bisphosphonates to cortical bone is less than that of denosumab, therefore bisphosphonates may not inhibit cortical bone remodeling to the same extent as denosumab [Roelofs et al. 2012].
Bone formation-sparing antiresorptive treatment
Odanacatib
Resorbing osteoclasts adhere very tightly to the bone surface, seal off the resorption lacunae, and generate an acidic environment in the resorption lacunae by secreting protons. Bone mineral is dissolved by the acidic environment and the collagen and other noncollagenous proteins are degraded by proteases such as metalloproteinases and cathepsin K [Duong, 2012].
Odanacatib is an inhibitor of cathepsin K. Treatment with odanacatib therefore has a different mechanism of action compared with denosumab as treatment with odanacatib leaves the osteoclasts alive and unaffected, but inhibits bone resorption by inhibiting cathepsin K activity [Duong, 2012].
The effects of odanacatib on bone have been investigated in adult rhesus monkeys. Treatment with odanacatib resulted in increased BMD and bone strength at the lumbar spine and the hip [Cusick et al. 2012; Masarachia et al. 2012]. Histomorphometric analyses of vertebrae, proximal femur and transiliac bone biopsies demonstrated that odanacatib reduced cancellous bone remodeling in the lumbar vertebrae and hip, and decreased intracortical remodeling at several femoral sites in monkeys. However, treatment with odanacatib preserved or enhanced endocortical bone formation and dosedependently stimulated modeling-based bone formation at the periosteal surfaces [Cusick et al. 2012; Masarachia et al. 2012]. The effect of odanacatib on cortical bone was also investigated at the central femur. Treatment with odanacatib stimulated bone formation both at the periosteal surface and at the endocortex. At the endocortex bone modeling was stimulated whereas bone remodeling was reduced. The intracortical remodeling was also reduced. These changes led to increased cortical thickness and volume [Pennypacker et al. 2014]. Whether a similar increase of modeling-based bone formation with odanacatib occurs in humans, particularly in estrogen-deprived and older individuals in whom the viability and/or activity of lining cells could be reduced, remains to be demonstrated. An interaction between mechanical loading and cathepsin K inhibition on bone modeling has been postulated, which if true, could explain some differences in bone-mass gain observed with odanacatib at loaded (i.e. hip) compared with less loaded (i.e. radius) sites. The mechanisms by which cathepsin K inhibition, which primarily occurs at remodeling sites, can increase bone modeling, particularly at the periosteal surface, also remains to be elucidated.
Combined anabolic and antiresorptive treatment
Osteocytes are terminally differentiated osteoblasts which become embedded in newly formed bone matrix and produce sclerostin. Sclerostin binds to lipoprotein-related peptide (LRP) 5/6 and thereby inhibits LRP5/6 from binding to the frizzled receptor and activating the Wnt pathway [Poole et al. 2005]. Activation of the Wnt canonical pathway induces translocation of β-catenin to the nucleus of the osteoblasts and subsequently gene transcription that stimulates bone formation through stimulation of osteoblast differentiation, proliferation, and survival [Baron and Rawadi, 2007]. Osteocytes control bone formation by the release of sclerostin as sclerostin inhibits osteoblastic bone formation. Individuals who produce reduced amounts of sclerostin have a high bone mass and reduced fracture risk [Brunkow et al. 2001; Hamersma et al. 2003], and therefore inhibition of sclerostin by antibodies is being investigated as a potential new anabolic treatment of osteoporosis. Inhibition of sclerostin by romosozumab, a sclerostin antibody, has been investigated in cynomolgus monkeys [Ominsky et al. 2010]. BMD and strength increased dose dependently. Histomorphometric analyses of bone samples revealed increased bone formation on trabecular, periosteal, endocortical, and intracortical surfaces despite decreased resorptive activity. The study also demonstrated that inhibition of sclerostin by romosozumab predominantly stimulates modeling-based bone formation at both cancellous and endocortical surfaces [Ominsky et al. 2014].
Implications of remodeling and modeling on the long-term effects of osteoporosis drugs on bone mass and strength
Bone mass, as evaluated by aBMD, remains the most important determinant of bone strength, explaining up to 80% of the failure load [Zysset et al. 2013]. Hence greater gains in aBMD, and thereby higher aBMD values, have been associated with lesser fracture risk, both in the presence and absence of osteoporosis therapy [Cosman et al. 2014; Schwartz et al. 2010]. However, large differences in BMD gain, particularly at sites of predominantly cortical bone such as the hip, have been noted between osteoporosis drugs, and even among antiresorptives. Hence relatively weak antiresorptives such as selective estrogen receptor modulators induce a small (1–2%) initial gain of hip BMD, pertaining to the partial refilling of the remodeling space, but later do not prevent the loss of hip aBMD [Silverman et al. 2012], because new BRUs continue to be activated and remodeling-based bone loss continues, particularly intracortically, which is not fully compensated for by the amount of modeling-based bone formation (Figure 1). With more potent bisphosphonates, greater inhibition of bone remodeling allows greater gains in aBMD initially but long-term clinical trials have consistently shown a plateauing effect after 2–3 years at the hip [Black et al. 2006, 2015; Miller et al. 2012]. This phenomenon could be explained by a new equilibrium reached between the amount of bone removed by the residual bone remodeling and the amount of new bone deposited by modeling-based bone formation, even though the latter may be somewhat negatively affected by bisphosphonates [Gasser et al. 2000] (Figure 1). However, with a complete suppression of bone remodeling, as achieved with denosumab, and provided bone modeling is sustained, as suggested by the studies on monkeys [Ominsky et al. 2015], then a positive bone accrual could be maintained long term, thereby potentially explaining the continuous BMD increase observed with this drug for up to 10 years [Papapoulos et al. 2015]. Eventually, with new compounds such as odanacatib and particularly romosozumab, that both inhibit bone remodeling while promoting bone modeling, even if transiently, an even greater gain of aBMD could be observed (Figure 1).
Discussion
Modeling-based bone formation in the adult skeleton has largely been ignored although it was demonstrated in human bone samples more than 50 years ago [Hattner et al. 1965]. Under normal circumstances modeling-based bone formation in cancellous bone represents a tiny fraction of total bone formation. This may be different at other surfaces and skeletal sites, however, and needs to be explored.
There is probably a limit to how much bone mass can be attained and how efficiently fractures can be prevented by inhibiting bone resorption. At least in theory, modeling-based bone formation seems a more efficient way to increase bone mass. It is also more rapid as no bone is removed prior to new bone deposition. This may be important if new concepts in osteoporosis treatment, such as ‘treat to target’, are to gain interest [Lewiecki et al. 2013]. However, one caveat should be considered with regard to bone modeling. Modeling-based formation does not replace older bone, which is presumably less biomechanically competent. It also does not replace old, dying, or dead osteocytes, which we now know play a crucial role not only in bone metabolism but also in the systemic regulation of phosphate and energy metabolism [Dallas et al. 2013]. However, on the positive side it does provide the skeleton with a new pool of young, viable osteocytes with a projected life span of decades.
Potent antiresorptive agents such as denosumab may be permissive to modeling-based bone formation and this in association with a low rate of remodeling may contribute to prolonged gains in bone mass with such agents [Bone et al. 2013]. Anabolic agents, such as teriparatide and romozosumab, and bone formation-sparing antiresorptives, such as odanacatib, stimulate modeling-based bone formation in both cancellous and cortical bone. The outcome of these treatments may depend on how effectively they also inhibit bone resorption.
However, it should be remembered that modeling-based bone formation depends mainly on mechanical forces and the ability of lining cells to form new bone upon such stimulation. Hence what happens in monkeys and rodents is not necessarily true in older women, but nevertheless suggests a possible interaction between mechanical stimulation of modeling and drug-induced inhibition of remodeling on the net bone mineral balance in a given region of interest. Therefore, the long-term BMD changes are expected to be variable depending on the strain imparted to the load-bearing bones, including the level of physical activity, age, and geometry of the hip.
The bone-mass response to some osteoporosis treatments in humans certainly suggests that nonremodeling mechanisms contribute to this response and bone modeling may be such a mechanism. To date, this has only been demonstrated by bone histomorphometry for teriparatide (PTH1-34) [Lindsay et al. 2006]. However, it is clear that rekindled interest in a phenomenon that was first observed more half a century ago will have an important impact on our understanding of how new antifracture agents work.
Footnotes
Funding: Bente Langdahl, has received research funding from Eli Lilly and Novo Nordisk, and serves on advisory boards for and has received speaker fee from Eli Lilly, Amgen, UCB, and Merck
Serge Ferrari has received research funding from, serves on advisory boards for and has received speaker fee from Merck, Amgen, UCB, Eli Lilly, Roche, and Agnovos.
David W. Dempster has done consulting for and received speaker fee from Amgen, Eli Lilly, Radius Health, Merck, Mereo Biopharmaceuticals, and Ultragenyx.
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Bente Langdahl, Medical Department of Endocrinology, Aarhus University Hospital, Tage-Hansensgade 2, Aarhus, DK-8000, Denmark.
Serge Ferrari, Department of Geriatric Medicine, Geneva University Hospital, Geneva, Switzerland.
David W. Dempster, Department of Clinical Pathology and Cell Biology, College of Physicians and Surgeons of Columbia University, and Regional Bone Center, Helen Hayes Hospital, New York State Department of Health, West Haverstraw, NY, USA
References
- Allen M., Burr D. (2008) Skeletal microdamage: less about biomechanics and more about remodeling. Clin Rev Bone Miner Metabol 6: 24–30. [Google Scholar]
- Austin M., Yang Y., Vittinghoff E., Adami S., Boonen S., Bauer D., et al. (2012) Relationship between bone mineral density changes with denosumab treatment and risk reduction for vertebral and nonvertebral fractures. J Bone Miner Res 27: 687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balena R., Shih M., Parfitt A. (1992) Bone resorption and formation on the periosteal envelope of the ilium: a histomorphometric study in healthy women. J Bone Miner Res 7: 1475–1482. [DOI] [PubMed] [Google Scholar]
- Banks W., Jr, Epling G., Kainer R., Davis R. (1968a) Antler growth and osteoporosis. I. Morphological and morphometric changes in the costal compacta during the antler growth cycle. Anat Rec 162: 387–398. [DOI] [PubMed] [Google Scholar]
- Banks W., Jr, Epling G., Kainer R., Davis R. (1968b) Antler growth and osteoporosis. II. Gravimetric and chemical changes in the costal compacta during the antler growth cycle. Anat Rec 162: 399–406. [DOI] [PubMed] [Google Scholar]
- Baron R., Rawadi G. (2007) Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology 148: 2635–2643. [DOI] [PubMed] [Google Scholar]
- Bauer W., Aub J., Albright F. (1929) Studies of calcium and phosphorus metabolism: V. A study of the bone trabeculae as a readily available reserve supply of calcium. J Exp Med 49: 145–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black D., Cummings S., Karpf D., Cauley J., Thompson D., Nevitt M., et al. (1996) Randomised trial of effect of alendronate on risk of fracture in women with existing vertebral fractures. Fracture Intervention Trial Research Group. Lancet 348: 1535–1541. [DOI] [PubMed] [Google Scholar]
- Black D., Delmas P., Eastell R., Reid I., Boonen S., Cauley J., et al. (2007) Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 356: 1809–1822. [DOI] [PubMed] [Google Scholar]
- Black D., Reid I., Cauley J., Cosman F., Leung P., Lakatos P., et al. (2015) The effect of 6 versus 9 years of zoledronic acid treatment in osteoporosis: a randomized second extension to the HORIZON-Pivotal Fracture Trial (PFT). J Bone Miner Res 30: 934–944. [DOI] [PubMed] [Google Scholar]
- Black D., Schwartz A., Ensrud K., Cauley J., Levis S., Quandt S., et al. (2006) Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA 296: 2927–2938. [DOI] [PubMed] [Google Scholar]
- Bliuc D., Nguyen T., Eisman J., Center J. (2014) The impact of nonhip nonvertebral fractures in elderly women and men. J Clin Endocrinol Metab 99: 415–423. [DOI] [PubMed] [Google Scholar]
- Bliziotes M., Sibonga J., Turner R., Orwoll E. (2006) Periosteal remodeling at the femoral neck in nonhuman primates. J Bone Miner Res 21: 1060–1067. [DOI] [PubMed] [Google Scholar]
- Bone H., Chapurlat R., Brandi M., Brown J., Czerwinski E., Krieg M., et al. (2013) The effect of three or six years of denosumab exposure in women with postmenopausal osteoporosis: results from the FREEDOM extension. J Clin Endocrinol Metab 98: 4483–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet N., Lesclous P., Saffar J., Ferrari S. (2013) Zoledronate effects on systemic and jaw osteopenias in ovariectomized periostin-deficient mice. PLoS One 8: e58726. doi: 10.1371/journal.pone.0058726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunkow M., Gardner J., Van Ness J., Paeper B., Kovacevich B., Proll S., et al. (2001) Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet 68: 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosman F., Cauley J., Eastell R., Boonen S., Palermo L., Reid I., et al. (2014) Reassessment of fracture risk in women after 3 years of treatment with zoledronic acid: when is it reasonable to discontinue treatment? J Clin Endocrinol Metab 99: 4546–4554. [DOI] [PubMed] [Google Scholar]
- Cosman F., Dempster D., Nieves J., Zhou H., Roimisher C., Houle Y., et al. (2013) Bone remodeling and structure in the proximal femur. J Bone Miner Res 28(Suppl. 1): FR037. [Google Scholar]
- Cosman F., Eriksen E., Recknor C., Miller P., Guanabens N., Kasperk C., et al. (2011) Effects of intravenous zoledronic acid plus subcutaneous teriparatide [rhPTH(1-34)] in postmenopausal osteoporosis. J Bone Miner Res 26: 503–511. [DOI] [PubMed] [Google Scholar]
- Coxon F., Thompson K., Roelofs A., Ebetino F., Rogers M. (2008) Visualizing mineral binding and uptake of bisphosphonate by osteoclasts and non-resorbing cells. Bone 42: 848–860. [DOI] [PubMed] [Google Scholar]
- Cummings S., Karpf D., Harris F., Genant H., Ensrud K., LaCroix A., et al. (2002) Improvement in spine bone density and reduction in risk of vertebral fractures during treatment with antiresorptive drugs. Am J Med 112: 281–289. [DOI] [PubMed] [Google Scholar]
- Cummings S., San Martin J., McClung M., Siris E., Eastell R., Reid I., et al. (2009) Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 361: 756–765. [DOI] [PubMed] [Google Scholar]
- Cusick T., Chen C., Pennypacker B., Pickarski M., Kimmel D., Scott B., et al. (2012) Odanacatib treatment increases hip bone mass and cortical thickness by preserving endocortical bone formation and stimulating periosteal bone formation in the ovariectomized adult rhesus monkey. J Bone Miner Res 27: 524–537. [DOI] [PubMed] [Google Scholar]
- Dallas S., Prideaux M., Bonewald L. (2013) The osteocyte: an endocrine cell … and more. Endocr Rev 34: 658–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster D. (2006) Anatomy and functions of the adult skeleton. In: Favus M. (ed), Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 6th edn. Washington, DC: American Society for Bone and Mineral Research, pp. 7–11. [Google Scholar]
- Dempster D. (2002) Bone remodeling. In: Coe F., Favus M. (eds) Disorders of Bone and Mineral Metabolism. Baltimore, MD: Lippincott, Williams and Wilkins, pp. 315–343. [Google Scholar]
- Dempster D. (1997) Exploiting and bypassing the bone remodeling cycle to optimize the treatment of osteoporosis. J Bone Miner Res 12: 1152–1154. [DOI] [PubMed] [Google Scholar]
- Dempster D., Cosman F., Kurland E., Zhou H., Nieves J., Woelfert L., et al. (2001) Effects of daily treatment with parathyroid hormone on bone microarchitecture and turnover in patients with osteoporosis: a paired biopsy study. J Bone Miner Res 16: 1846–1853. [DOI] [PubMed] [Google Scholar]
- Dempster D., Lindsay R. (1993) Pathogenesis of osteoporosis. Lancet 341: 797–801. [DOI] [PubMed] [Google Scholar]
- Duong L. (2012) Therapeutic inhibition of cathepsin K-reducing bone resorption while maintaining bone formation. BoneKey Rep 1: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einhorn T. (1992) Bone strength: the bottom line. Calcif Tissue Int 51: 333–339. [DOI] [PubMed] [Google Scholar]
- Epker B., Frost H. (1966) Periosteal appositional bone growth from age two to age seventy in man. A tetracycline evaluation. Anat Rec 154: 573–577. [DOI] [PubMed] [Google Scholar]
- Erben R. (1996) Trabecular and endocortical bone surfaces in the rat: modeling or remodeling? Anat Rec 246: 39–46. [DOI] [PubMed] [Google Scholar]
- Eriksen E. (1986) Normal and pathological remodeling of human trabecular bone: three dimensional reconstruction of the remodeling sequence in normals and in metabolic bone disease. Endocr Rev 7: 379–408. [DOI] [PubMed] [Google Scholar]
- Finkelstein J., Leder B., Burnett S., Wyland J., Lee H., de la Paz A., et al. (2006) Effects of teriparatide, alendronate, or both on bone turnover in osteoporotic men. J Clin Endocrinol Metab 91: 2882–2887. [DOI] [PubMed] [Google Scholar]
- Garn S., Rohmann C., Wagner B., Ascoli W. (1967) Continuing bone growth throughout life: a general phenomenon. Am J Phys Anthropol 26: 313–317. [DOI] [PubMed] [Google Scholar]
- Gasser J., Green J. (2006) Chronic subcutaneous, but not single intravenous, dosing of rats with bisphosphonates results in reduced anabolic response to PTH. J Bone Miner Res (Suppl. 1): F386. [Google Scholar]
- Gasser J., Kneissel M., Thomsen J., Mosekilde L. (2000) PTH and interactions with bisphosphonates. J Musculoskelet Neuronal Interact 1: 53–56. [PubMed] [Google Scholar]
- Gerdhem P. (2013) Osteoporosis and fragility fractures: vertebral fractures. Best Pract Res Clin Rheumatol 27: 743–755. [DOI] [PubMed] [Google Scholar]
- Hamersma H., Gardner J., Beighton P. (2003) The natural history of sclerosteosis. Clin Genet 63: 192–197. [DOI] [PubMed] [Google Scholar]
- Hattner R., Epker B., Frost H. (1965) Suggested sequential mode of control of changes in cell behaviour in adult bone remodelling. Nature 206: 489–490. [DOI] [PubMed] [Google Scholar]
- Havers C. (1691) Osteologia Nova. [Google Scholar]
- Jacques R., Boonen S., Cosman F., Reid I., Bauer D., Black D., et al. (2012) Relationship of changes in total hip bone mineral density to vertebral and nonvertebral fracture risk in women with postmenopausal osteoporosis treated with once-yearly zoledronic acid 5 mg: the HORIZON-Pivotal Fracture Trial (PFT). J Bone Miner Res 27: 1627–1634. [DOI] [PubMed] [Google Scholar]
- Johnell O., Kanis J., Oden A., Johansson H., De Laet C., Delmas P., et al. (2005) Predictive value of BMD for hip and other fractures. J Bone Miner Res 20: 1185–1194. [DOI] [PubMed] [Google Scholar]
- Johnson M., Gong G., Kimberling W., Recker S., Kimmel D., Recker R. (1997) Linkage of a gene causing high bone mass to human chromosome 11 (11q12-13). Am J Hum Genet 60: 1326–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S., Takahashi H., Ito A., Saito N., Nawata M., Horiuchi H., et al. (2003) Trabecular minimodeling in human iliac bone. Bone 32: 163–169. [DOI] [PubMed] [Google Scholar]
- Kontulainen S., Sievanen H., Kannus P., Pasanen M., Vuori I. (2002) Effect of long-term impact-loading on mass, size, and estimated strength of humerus and radius of female racquet-sports players: a peripheral quantitative computed tomography study between young and old starters and controls. J Bone Miner Res 17: 2281–2289. [DOI] [PubMed] [Google Scholar]
- Kostenuik P., Smith S., Jolette J., Schroeder J., Pyrah I., Ominsky M. (2011) Decreased bone remodeling and porosity are associated with improved bone strength in ovariectomized cynomolgus monkeys treated with denosumab, a fully human RANKL antibody. Bone 49: 151–161. [DOI] [PubMed] [Google Scholar]
- Langdahl B., Binkley N., Bone H., Gilchrist N., Resch H., Rodriguez P., et al. (2012) Odanacatib in the treatment of postmenopausal women with low bone mineral density: five years of continued therapy in a phase II study. J Bone Miner Res 27: 2251–2258. [DOI] [PubMed] [Google Scholar]
- Leder B., Tsai J., Uihlein A., Burnett-Bowie S., Zhu Y., Foley K., et al. (2014) Two years of Denosumab and teriparatide administration in postmenopausal women with osteoporosis (The DATA Extension Study): a randomized controlled trial. J Clin Endocrinol Metab 99: 1694–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewiecki E., Cummings S., Cosman F. (2013) Treat-to-target for osteoporosis: is now the time? J Clin Endocrinol Metab 98: 946–953. [DOI] [PubMed] [Google Scholar]
- Lindsay R., Cosman F., Zhou H., Bostrom M., Shen V., Cruz J., et al. (2006) A novel tetracycline labeling schedule for longitudinal evaluation of the short-term effects of anabolic therapy with a single iliac crest bone biopsy: early actions of teriparatide. J Bone Miner Res 21: 366–373. [DOI] [PubMed] [Google Scholar]
- Manolagas S. (2000) Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev 21: 115–137. [DOI] [PubMed] [Google Scholar]
- Martin B. (1995) Mathematical model for repair of fatigue damage and stress fracture in osteonal bone. J Orthop Res 13: 309–316. [DOI] [PubMed] [Google Scholar]
- Masarachia P., Pennypacker B., Pickarski M., Scott K., Wesolowski G., Smith S., et al. (2012) Odanacatib reduces bone turnover and increases bone mass in the lumbar spine of skeletally mature ovariectomized rhesus monkeys. J Bone Miner Res 27: 509–523. [DOI] [PubMed] [Google Scholar]
- McClung M., Chines A., Brown J., Diez-Perez A., Resch H., Caminis J., et al. (2014a) Effects of 2 years of treatment with Romosozumab followed by 1 year of Denosumab or placebo in postmenopausal women with low bone mineral density. J Bone Miner Res (Suppl. 1): 1152. [DOI] [PubMed] [Google Scholar]
- McClung M., Grauer A., Boonen S., Bolognese M., Brown J., Diez-Perez A., et al. (2014b) Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med 370: 412–420. [DOI] [PubMed] [Google Scholar]
- Miller P., Delmas P., Huss H., Patel K., Schimmer R., Adami S., et al. (2010) Increases in hip and spine bone mineral density are predictive for vertebral antifracture efficacy with ibandronate. Calcif Tissue Int 87: 305–313. [DOI] [PubMed] [Google Scholar]
- Miller P., Recker R., Reginster J., Riis B., Czerwinski E., Masanauskaite D., et al. (2012) Efficacy of monthly oral ibandronate is sustained over 5 years: the MOBILE long-term extension study. Osteoporos Int 23: 1747–1756. [DOI] [PubMed] [Google Scholar]
- Motyckova G., Fisher D. (2002) Pycnodysostosis: role and regulation of cathepsin K in osteoclast function and human disease. Curr Mol Med 2: 407–421. [DOI] [PubMed] [Google Scholar]
- Nawathe S., Nguyen B., Barzanian N., Akhlaghpour H., Bouxsein M., Keaveny T. (2015) Cortical and trabecular load sharing in the human femoral neck. J Biomech 48: 816–822. [DOI] [PubMed] [Google Scholar]
- Neer R., Arnaud C., Zanchetta J., Prince R., Gaich G., Reginster J., et al. (2001) Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 344: 1434–1441. [DOI] [PubMed] [Google Scholar]
- Ominsky M., Libanati C., Niu Q., Boyce R., Kostenuik P., Wagman R., et al. (2015) Sustained modeling-based bone formation during adulthood in cynomolgus monkeys May contribute to continuous BMD gains with denosumab. J Bone Miner Res 30: 1280–1289. [DOI] [PubMed] [Google Scholar]
- Ominsky M., Niu Q., Li C., Li X., Ke H. (2014) Tissue-level mechanisms responsible for the increase in bone formation and bone volume by sclerostin antibody. J Bone Miner Res 29: 1424–1430. [DOI] [PubMed] [Google Scholar]
- Ominsky M., Vlasseros F., Jolette J., Smith S., Stouch B., Doellgast G., et al. (2010) Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res 25: 948–959. [DOI] [PubMed] [Google Scholar]
- Orwoll E. (2003) Toward an expanded understanding of the role of the periosteum in skeletal health. J Bone Miner Res 18: 949–954. [DOI] [PubMed] [Google Scholar]
- Papapoulos S., Lippuner K., Roux C., Lin C., Kendler D., Lewiecki E., et al. (2015) The effect of 8 or 5 years of denosumab treatment in postmenopausal women with osteoporosis: results from the FREEDOM Extension study. Osteoporos Int 26: 2773–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennypacker B., Chen C., Zheng H., Shih M., Belfast M., Samadfam R., et al. (2014) Inhibition of cathepsin K increases modeling-based bone formation, and improves cortical dimension and strength in adult ovariectomized monkeys. J Bone Miner Res 29: 1847–1858. [DOI] [PubMed] [Google Scholar]
- Plotkin L., Manolagas S., Bellido T. (2006) Dissociation of the pro-apoptotic effects of bisphosphonates on osteoclasts from their anti-apoptotic effects on osteoblasts/osteocytes with novel analogs. Bone 39: 443–452. [DOI] [PubMed] [Google Scholar]
- Poole K., Van Bezooijen R., Loveridge N., Hamersma H., Papapoulos S., Lowik C., et al. (2005) Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J 19: 1842–1844. [DOI] [PubMed] [Google Scholar]
- Reid I., Miller P., Brown J., Kendler D., Fahrleitner-Pammer A., Valter I., et al. (2010) Effects of denosumab on bone histomorphometry: the FREEDOM and STAND studies. J Bone Miner Res 25: 2256–2265. [DOI] [PubMed] [Google Scholar]
- Roelofs A., Coxon F., Ebetino F., Lundy M., Henneman Z., Nancollas G., et al. (2010) Fluorescent risedronate analogues reveal bisphosphonate uptake by bone marrow monocytes and localization around osteocytes in vivo. J Bone Miner Res 25: 606–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelofs A., Stewart C., Sun S., Blazewska K., Kashemirov B., McKenna C., et al. (2012) Influence of bone affinity on the skeletal distribution of fluorescently labeled bisphosphonates in vivo. J Bone Miner Res 27: 835–847. [DOI] [PubMed] [Google Scholar]
- Ruff C., Hayes W. (1982) Subperiosteal expansion and cortical remodeling of the human femur and tibia with aging. Science 217: 945–948. [DOI] [PubMed] [Google Scholar]
- Schwartz A., Bauer D., Cummings S., Cauley J., Ensrud K., Palermo L., et al. (2010) Efficacy of continued Alendronate for fractures in women with and without prevalent vertebral fracture: the FLEX trial. J Bone Miner Res 25: 976–982. [DOI] [PubMed] [Google Scholar]
- Seeman E. (2013) Age- and menopause-related bone loss compromise cortical and trabecular microstructure. J Gerontol A Biol Sci Med Sci 68: 1218–1225. [DOI] [PubMed] [Google Scholar]
- Selye H. (1932) On the stimulation of new bone formation with parathyroid extract and irradiated ergosterol. Endocrinology 16: 547–558. [Google Scholar]
- Silverman S., Chines A., Kendler D., Kung A., Teglbjaerg C., Felsenberg D., et al. (2012) Sustained efficacy and safety of bazedoxifene in preventing fractures in postmenopausal women with osteoporosis: results of a 5-year, randomized, placebo-controlled study. Osteoporos Int 23: 351–363. [DOI] [PubMed] [Google Scholar]
- Strom O., Borgstrom F., Kanis J., Compston J., Cooper C., McCloskey E., et al. (2011) Osteoporosis: burden, health care provision and opportunities in the EU: a report prepared in collaboration with the International Osteoporosis Foundation (IOF) and the European Federation of Pharmaceutical Industry Associations (EFPIA). Arch Osteoporos 6: 59–155. [DOI] [PubMed] [Google Scholar]
- Taddei F., Balestri M., Rimondi E., Viceconti M., Manfrini M. (2009) Tibia adaptation after fibula harvesting: an in vivo quantitative study. Clin Orthop Relat Res 467: 2149–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J., Uihlein A., Lee H., Kumbhani R., Siwila-Sackman E., McKay E., et al. (2013) Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet 382: 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts N., Cooper C., Lindsay R., Eastell R., Manhart M., Barton I., et al. (2004) Relationship between changes in bone mineral density and vertebral fracture risk associated with risedronate: greater increases in bone mineral density do not relate to greater decreases in fracture risk. J Clin Densitom 7: 255–261. [DOI] [PubMed] [Google Scholar]
- WHO (1994) Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO Study Group. World Health Organ Tech Rep Ser 843: 1–129. [PubMed] [Google Scholar]
- Yang L., Sycheva A., Black D., Eastell R. (2013) Site-specific differential effects of once-yearly zoledronic acid on the hip assessed with quantitative computed tomography: results from the HORIZON Pivotal Fracture Trial. Osteoporos Int 24: 329–338. [DOI] [PubMed] [Google Scholar]
- Zebaze R., Ghasem-Zadeh A., Bohte A., Iuliano-Burns S., Mirams M., Price R., et al. (2010) Intracortical remodelling and porosity in the distal radius and post-mortem femurs of women: a cross-sectional study. Lancet 375: 1729–1736. [DOI] [PubMed] [Google Scholar]
- Zysset P., Dall’ara E., Varga P., Pahr D. (2013) Finite element analysis for prediction of bone strength. Bonekey Rep 2: 386. [DOI] [PMC free article] [PubMed] [Google Scholar]