

Abstract
Mitochondrial DNA (mtDNA) is a novel danger-associated molecular pattern that on its release into the extracellular milieu acts via toll-like receptor-9, a pattern recognition receptor of the immune system. We hypothesized that plasma mtDNA concentrations will be elevated in septic children, and these elevations are associated with an increase in the severity of illness. In a separate set of in vitro experiments, we test the hypothesis that exposing peripheral blood mononuclear cells (PBMC) to mtDNA activates the immune response and induces tumor necrosis factor (TNF) release. Children with sepsis/systemic inflammatory response syndrome or control groups were enrolled within 24 h of admission to the pediatric intensive care unit. Mitochondrial gene cytochrome c oxidase 1 (COX1) concentrations were measured by realtime quantitative PCR in the DNA extracted from plasma. PBMCs were treated with mtDNA (10μg/mL) and supernatant TNF levels were measured. The median plasma mtDNA concentrations were significantly elevated in the septic patients as compared with the critically ill non-septic and healthy control patients [1.75E+05 (IQR 6.64E+04-3.67E+05) versus 5.73E+03 (IQR 3.90E+03-1.28E+04) and 6.64E+03 (IQR 5.22E+03-1.63E+04) copies/μL respectively]. The median concentrations of plasma mtDNA were significantly greater in patients with MOF as compared with patients without MOF (3.2E+05 (IQR 1.41E+05-1.08E+06) vs. 2.9E+04 (IQR 2.47E+04-5.43E+04) copies/μL). PBMCs treated with mtDNA demonstrated higher supernatant TNF levels as compared with control cells (6.5 ±1.8 vs. 3.5±0.5 pg/mL, P>0.05). Our data suggest that plasma mtDNA is a novel danger-associated molecular pattern in pediatric sepsis and appears to be associated with MOF.
Keywords: Alarmin, DAMP, mitochondrial DNA, pediatric sepsis
Introduction
Since the first description of the “danger hypothesis” by Matzinger, numerous cellular proteins have been classified as “danger-associated molecular patterns” (DAMPs) or “alarmins” (1). During homeostasis, the DAMP proteins are sequestered in different subcellular compartments, e.g., nucleus, cytosol (2, 3) and remain unrecognized by the immune system. However, under stressful conditions, these molecules are actively secreted or passively released into the extracellular milieu and activate the inflammatory cascade via the highly conserved pattern recognition receptors (PRRs). Some recent examples of alarmins include heat shock proteins, high mobility group box-1 (HMGB1), and adenosine triphosphate (1, 4, 5).
The immunomodulating role of extracellular mtDNA was discovered recently, even though the putative bacterial origins of mitochondria have been known for decades. There is abundant mtDNA present in the cell, as a single cell encloses hundreds of mitochondria, and each mitochondrion contains an estimated 2 to 10 copies of its genome (6). Similar to bacteria, mitochondria possess a double-membrane structure and contain 37 genes coding for 2 ribosomal ribonucleic acids (RNAs), 22 transfer RNAs, and 13 polypeptides (7, 8). Furthermore, mtDNA have hypomethylated CpG motifs that resemble bacterial CpG DNA and activate TLR9, a PRR that detects bacterial and viral DNA (9). Thus extracellular mtDNA, due to its similarity to bacterial DNA, can activate signaling pathways and promulgate inflammation. Zhang et al., in a seminal study, demonstrated that patients admitted with trauma had significant elevations of mtDNA concentrations in the plasma and injured tissues (3, 10, 11). The authors validated their results in a rat model of trauma/hemorrhagic shock and noted that plasma mtDNA levels were elevated for 7 days after injury (3). Data regarding the role of plasma mtDNA in adult sepsis are confusing at best. Puskarich et al. (12) did not observe any difference in plasma mtDNA concentrations between septic patients and healthy controls in the emergency department. In contrast, another prospective study on adult patients with severe sepsis and septic shock revealed that a rise of 1 ng/mL plasma mtDNA is associated with an increase in the fatality rate by 0.7% (13). We hypothesized that plasma mtDNA levels would be elevated in children with severe sepsis, and these elevations would be associated with increased severity of illness. In addition, we document that exposing peripheral blood mononuclear cells (PBMC) to mtDNA activates the immune response and induces TNF release.
Materials and Methods
The study was approved by the University of Pittsburgh Institutional Review Board (IRB no. PRO09060070), and blood samples were collected after obtaining informed consent from parents or guardians. Pediatric patients were eligible for enrollment when the following criteria were fulfilled: admission to the pediatric intensive care unit (PICU), presence of an indwelling catheter for sample collection, and presence of clinical signs and symptoms of sepsis and/or systemic inflammatory response syndrome (SIRS) (see Supplemental Data, Supplemental Digital Content 1, at http://links.lww.com/SHK/A348). In addition, critically ill nonseptic patients, i.e., patients who fulfilled the first two criteria listed above, but did not exhibit any signs or symptoms of SIRS/sepsis and healthy controls were enrolled. Blood samples were also collected from healthy children who were having blood drawn as part of a preoperative evaluation.
Sample preparation
Blood samples from children in the sepsis/SIRS or control groups were obtained within 24 h of admission to the PICU. Blood samples were collected in tubes containing sodium heparin and were centrifuged immediately at 1914 × g for 10min to separate plasma from the cellular components. Samples were stored at −70°C for batch analysis.
DNA was extracted from 200 μL of plasma using the QIAamp Blood Kit (Qiagen GmbH, Hilden, Germany), and the concentration of mitochondrial gene cytochrome c oxidase 1 (COX1) was measured by SYBR green chemistry real-time quantitative PCR (Life Technologies, Grand Island, NY). We want to make the reader aware that COX1 is also an abbreviation for cyclooxygenase; however, in this manuscript, it is being used as an abbreviation for cytochrome c oxidase 1. We chose the COX1 gene to quantify mtDNA, as plasma levels of mtDNA encoding COX1 have been reported to be elevated in severely injured patients (14). The primer sequences for COX1 and β globin used were forward: GCC TCC GTA GAC CTA ACC ATC TTC; reverse: GTA AGT TAC AAT ATG GGA GAT TAT TCC and forward: TTCACTAGCAACCTCAAACAGACA; reverse: TGTCTCCACATGCCCAGTTTCT, respectively. PCR reaction mixture was prepared using SYBR Green PCR master mix (PE Applied Biosystems, Foster City, CA), using the primers described above. PCR was set up in a reaction volume of 20 μL using 10 μL of 2× SYBR green Master Mix (2×), 1 μL forward primer (1 μM), 1 μL reverse primer (1 μM),3 μL of nuclease-free H2O, and 5 μL of plasma nuclear extract. The following thermocycler conditions were used: 3-min incubation at 95°C followed by 40 cycles of an initial denaturation step at 95°C for 30 s, an annealing step of 54°C for 45 s, and an elongation step of 68°C for 1 min.
Standard curve preparation
The linearity of the quantitative assay was assessed by using a cloned plasmid DNA, which was serially diluted to prepare a series of calibrators with known concentrations as described by Chiu et al. (15). Briefly, using primers described above, we amplified bases between 421 and 781 encoding the mitochondrial gene COXI (Ref Seq: NC_012920). The PCR amplicons were cloned into a pGEM-T-Easy vector (Promega, Madison, WI), and the identity of the cloned insert was confirmed. DNA was extracted, quantified, and used as a calibrator. As previously described, the mtDNA copy number of this calibrator was derived by dividing the total DNA concentration by the weight of each plasmid molecule (15). Calibrators were prepared by serial dilution of the stock solution and contained 1 to 109 mt DNA copies/μL.
Isolation of mitochondria and mtDNA preparation
Mitochondria were isolated from THP-1 using the mitochondria isolation kit (Thermo Scientific/Pierce, Logan, UT) as per the manufacturer's suggestions. The isolated mitochondrial lysates were prepared by resuspending mitochondria (1 mg/mL) in mammalian protein extraction reagent (M-PER) (Thermo Scientific/Pierce, Logan, UT) and incubating at 4°C for 10 min with end-over-end rotation. The lysates were centrifuged at 12,000 × g for 1 h, and the supernatants were collected. The samples were flash frozen in liquid nitrogen and stored at −80°C. Protein concentration was determined using the BCA protein assay kit (Thermo Scientific, Rockford, IL). MtDNA was extracted using DNeasy Blood & Tissue kit (Qiagen GmbH, Hilden, Germany) following the protocol recommended by the manufacturer. The mtDNA concentration was determined by a spectrophotometer and the purity was evaluated by quantitative PCR using mitochondria specific primer for COX1 and genomic specific primer for β-globin as previously described. Nuclear DNA contamination was less than 0.01%.
Primary human monocytes (PBMC) isolation and stimulation
PBMC were isolated from buffy coats obtained from plasma of volunteer donors by the Ficoll-Hypaque procedure as described previously (16). Isolated PBMC were maintained in culture in RPMI-1640 medium containing 10% fetal bovine serum, 1% penicillin/streptomycin, 0.35% β-mercaptoethanol, and 2% glutamine, 10mmol/L 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (pH 7.2). Where indicated, stimulation was performed with 10 μg/mL mtDNA and recombinant 1 μg/mL HMGB1 (Sigma Chemical Co., St. Louis, MO) for 4h.
ELISA
Tumor necrosis factor (TNF) concentrations were measured in culture supernatants from treated cells using a commercially available sandwich ELISA (Biosource, Camarillo, CA). All procedures were performed as recommended by the manufacturer.
Statistical analysis
Clinical data including patient age, sex, C-reactive protein (CRP), pediatric risk of mortality index (PRISM) are expressed as mean ± standard error of the mean (SEM), or median [interquartile range (IQR), i.e., 25th–75th percentile], as appropriate. To compare the correlation between plasma mtDNA and CRP, we used Spearman correlation test. Statistical calculations were performed with SigmaPlot 11.0 (Systat Software Inc, San Jose, CA).
Results
There were 28 septic and 10 critically ill non-septic patients enrolled in this study. In addition, 20 healthy controls were also recruited for this study. The median age for the septic patients was 4.0 [0.12–20] y, and 54% were male (Table 1). The median age for healthy control and critically ill non-septic patients was 8.8 [2–16] and 6.5 [2–12] y respectively. A primary infection was present in almost 70% of the cohort (bacterial 45%, viral 19.3%, fungal 6.4%, and 25% culture-negative). Seventy percent of the patients had underlying chronic medical problems at the time of enrollment. MOF was present in 75% of patients, and there were two deaths in this cohort, thereby the overall mortality rate in this cohort was 7.1%. The overall PICU mortality for all patients during the same period was 1.7%.
Table 1. Demographic data of the study subjects.
| Healthy control patients | |
| Number | 20 |
| Age in years [range] | 8.8 [2–16] |
| Males (%) | 12 (60) |
| Critically ill non-septic patients | |
| Number | 10 |
| Age in years [range] | 6.50 [2–12] |
| Males (%) | 4 (44) |
| Sepsis/SIRS patients | |
| Number | 28 |
| Age in years [range] | 4.0 [0.12–20] |
| Males (%) | 15 (53.6) |
| Primary infection found (%) | 20 (71.4) |
| Bacterial (%) | 13 (45) |
| Viral (%) | 6 (19.3) |
| Fungal (%) | 2 (6.4) |
| History of chronic illness (%) | 20 (71.4) |
| Mortality (%) | 2 (7.1) |
| CRP (range) | 3.5 [0.04–35.80] |
| PRISM | 9.5 [1–32] |
| Septic shock (%) | 20 (71.4) |
| Multiple organ failure (%) | 21 (75) |
Median [range] presented for age, CRP, and PRISM.
CRP indicates C-reactive protein; PRISM, pediatric risk of mortality index; SIRS, systemic inflammatory response syndrome.
Within the first 24 h after admission to the PICU, the median plasma mtDNA concentrations were significantly elevated in the septic patients as compared with the critically ill non-septic and healthy control patients (1.75E+05 (IQR 6.64E+04−3.67E+05) versus 5.73E+03 (IQR 3.90E+03-1.28E+04) and 6.64E+03 (IQR 5.22E+03-1.63E+04) copies/μL respectively; P = 0.001, Fig. 1). There was no difference between plasma mtDNA concentrations in the critically ill non-septic and healthy control patients (P = 1.0).
Fig. 1. Box and whisker plots of plasma mtDNA levels in healthy controls, non-septic and septic children within 24 h after admission to the PICU (*, P<0.05, vs. control patients).
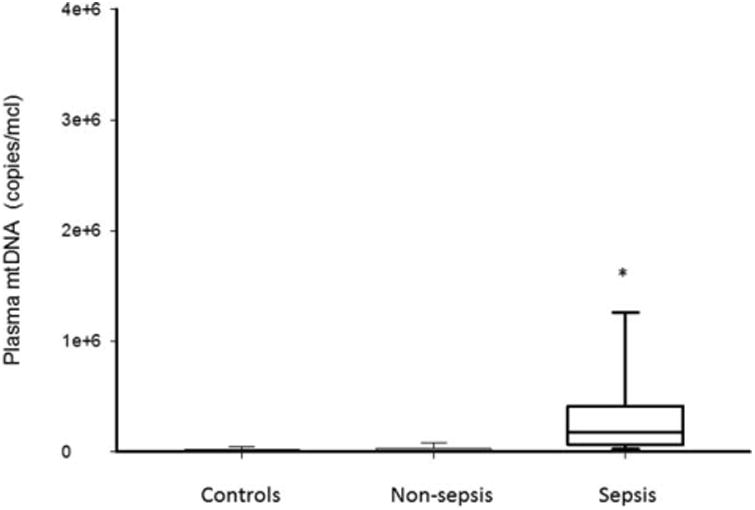
The line within the box denotes the median and the box spans the interquartile range (25–75th percentiles). The whiskers mark the minimum and maximum plasma mtDNA concentrations. mtDNA indicates mitochondrial DNA; PICU, pediatric intensive care unit.
To investigate if changes in plasma mtDNA correlate with severity of illness, we examined plasma mtDNA levels in the presence or absence of shock. Patients who were admitted with shock demonstrated significantly higher median plasma mtDNA concentrations as compared with patients without shock (1.77E+05 (IQR 9.50E+04-4.27+05) vs. 6.89E+04 (IQR 4.96E+04-8.56+04) copies/μL respectively; P = 0.023, Fig. 2). However, plasma mtDNA concentration did not correlate to a commonly used acute phase reactant, CRP, (ρ 0.08, P = 0.71) (data not shown).
Fig. 2. Box and whisker plots of plasma mtDNA levels in septic children who were admitted with or without shock to the PICU (*, P<0.05, vs. septic children without shock).
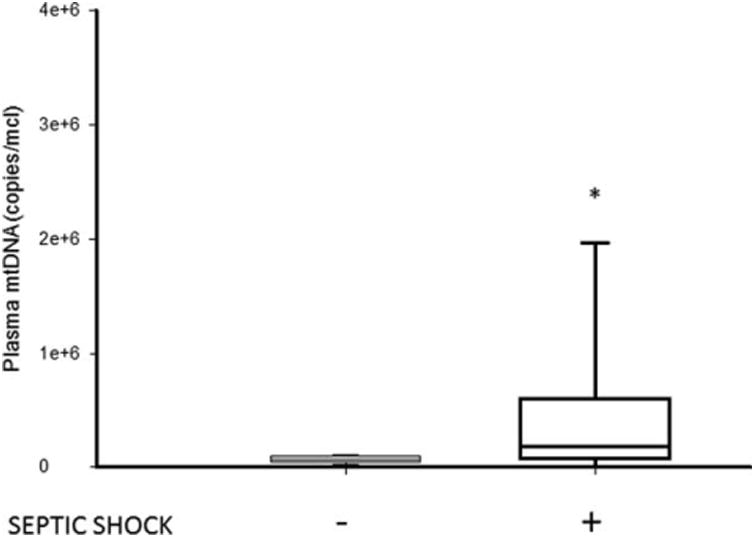
The line within the box denotes the median and the box spans the interquartile range (25–75th percentiles). The whiskers mark the minimum and maximum plasma mtDNA concentrations. mtDNA indicates mitochondrial DNA; PICU, pediatric intensive care unit.
To determine the specificity of plasma mtDNA release following sepsis, we measured the plasma concentrations of β globin, a gene present in all nucleated cells of the body. The median plasma mtDNA levels in septic patients were significantly higher than β globin concentrations—1.39E+05 versus 5.09E+02copies/μL, P < 0.001 respectively. The median concentrations of plasma mtDNA were significantly greater in patients with MOF as compared with patients without MOF (3.2E+05 (IQR 1.41E+05-1.08E+06) vs. 2.9E+04 (IQR 2.47E+04-5.43E+04) copies/μL) (Fig. 3). However, the presence or absence of MOF did not correlate with median plasma β globin concentrations (8.75E+02 (IQR 1.14E+02-1.89E+03) vs. 4.19E+02 (IQR 1.34E+02-5.98E+02) copies/μL) (Fig. 3). The median plasma β globin concentrations were not significantly different in the septic patients as compared with the critically ill non-septic and healthy control patients (2.81E+02 (IQR 7.16E+01-1.29E+03) vs. 2.22E+02 (IQR 1.79E+02-8.92E+02) and 3.66E+02 (IQR 2.53E+02-1.80E+03) copies/μL respectively; P > 0.05). Since there was minimal overlap in the plasma mtDNA concentrations in septic patients with and without organ failure, a receiver-operating characteristic curve was not performed. Taken together, these data support the notion that plasma mtDNA concentrations are increased in pediatric septic patients and correlate with the severity of illness.
Fig. 3. Box and whisker plots of plasma mtDNA and β globin concentrations in children with sepsis with or without MOF who were admitted to the PICU (*, P<0.05, vs. mtDNA MOF-).
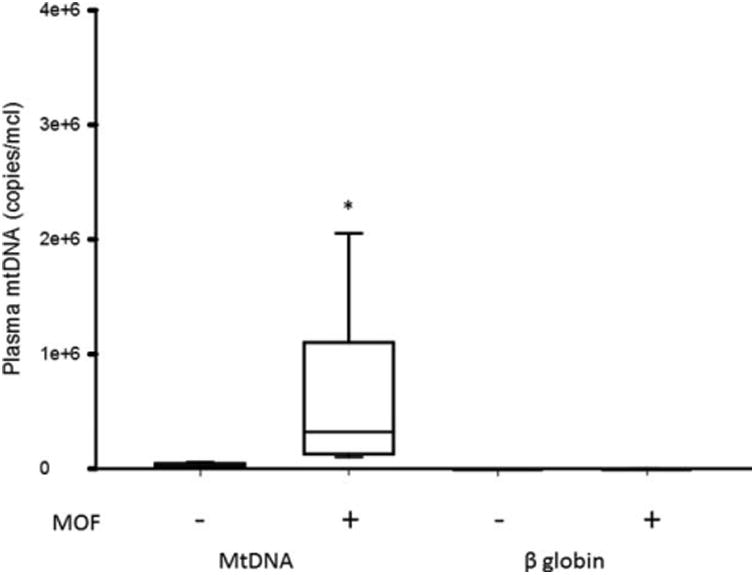
The line within the box denotes the median and the box spans the interquartile range (25–75th percentiles). The whiskers mark the minimum and maximum plasma mtDNA concentrations.
To understand the biological relevance of these findings, we investigated the role of mtDNA in activating the innate immune response. PBMCs were exposed to mtDNA and TNF levels were measured in culture supernatants. As shown in Fig. 4, PBMCs treated with mtDNA demonstrated higher supernatant TNF levels as compared with control cells (6.5 ± 1.8 vs. 3.5±0.5 pg/mL, P>0.05; Fig. 4). Recent data suggest that HMGB1, a nuclear DNA-binding protein, released from necrotic cells is an essential component of DNA-containing immune complexes that stimulate cytokine production via a TLR9-MYD88 pathway (17). Addition of HMGB1 (1 μg/mL) significantly amplified mtDNA-induced TNF secretion as compared with control cells (8.4 ±1.6 vs. 3.5 ±0.5 pg/mL, P < 0.05; Fig. 4). In contrast, treatment with denatured HMGB1 (boiled for 10min) and mtDNA negated the augmentation of TNF secretion previously noted with non-boiled HMGB1. An additional control included exposure of PBMC cells to mitochondrial lysates—supernatant TNF levels in mitochondrial lysate stimulated cells were 15.3 ± 2.9 pg/mL. These data demonstrate that extracellular mtDNA induces TNF secretion, and the additional presence of HMGB1 makes this response more robust.
Fig. 4. ELISA results demonstrating the effects of mtDNA treatment on TNF levels in the supernatants of PBMCs.
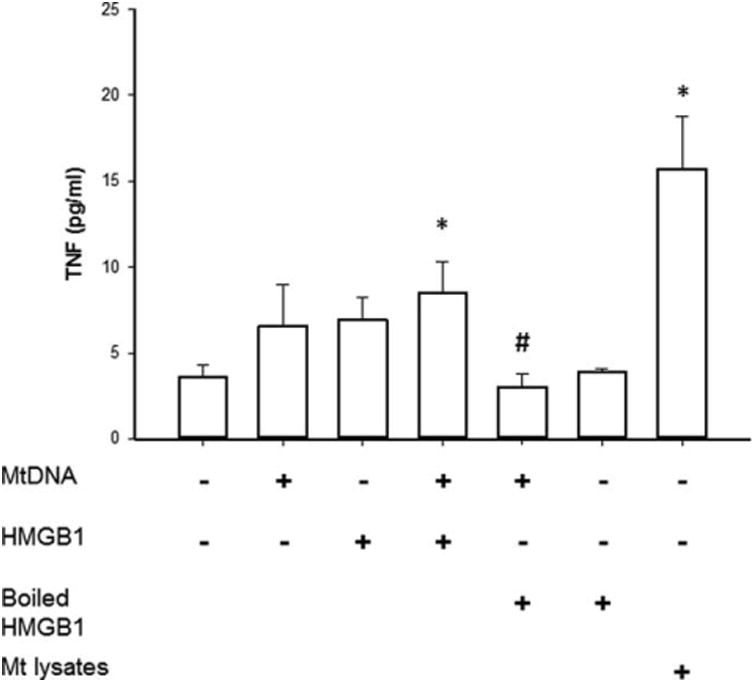
Control cells were maintained in a basal growth medium. Cells were preconditioned with mtDNA, HMGB1, or boiled HMGB1 (1 μg/mL) for 6h. Data represent the mean±sem of three separate experiments with each condition performed in triplicate (*, P<0.05, vs. control cells alone #, P<0.05, vs. cells treated with mtDNA and HMGB1).
Discussion
Considerable attention has been focused on mitochondrial dysfunction in sepsis (10,18–21); however, only recently has the role of extracellular mtDNA as a DAMP molecule piqued the interest of the research community. TLR9 is a pattern recognition receptor with specific affinity for unmethylated cytosine and guanine nucleotides separated by a phosphate backbone (CpG). Due to its putative bacterial origins, mitochondria have abundant unmethylated CpG dinucleotide repeats that can stimulate TLR9 to generate an immune response. TLR9 activation involves signaling via the myeloid differentiation primary response gene 88, interleukin (IL)-1 receptor-activated kinase, and tumor necrosis factor receptor (TNFR)-associated factor 6 leading to downstream activation of several pro-inflammatory transcription factors (11, 22). Collins et al. (23) isolated nuclear and mtDNA from murine liver and noted that intra-articular injection of only purified mtDNA caused inflammation and arthritis. Similarly, rats injected with hepatic mtDNA demonstrated signs of inflammation (reduced activity, ruffled fur, and shivering) within 6 h after injection. Furthermore, rats injected with mtDNA demonstrated significantly elevated cytokine (TNF, IL-6) concentrations along with evidence of lung injury as compared with control rats and nuclear DNA injected rats (24). In patients with trauma and hemorrhagic shock, plasma mtDNA levels were noted to be several thousand folds higher as compared with healthy volunteers. It is notable that the bony specimens in these trauma patients demonstrated significantly higher mtDNA levels as compared with plasma (3).
In support of the DAMP hypothesis, in vitro experiments demonstrated that neutrophils exposed to mtDNA induced p38 mitogen-activated protein kinase activation as well as the release of matrix metalloproteinase 8 and 9 (3, 11). Taken together, these findings suggest that activation of the innate immune response by circulating mtDNA provides a mechanistic link between shock and inflammation.
In this study, we demonstrate that critically ill children admitted to the PICU with severe sepsis and septic shock had elevated plasma mtDNA concentrations. Significantly higher plasma mtDNA levels were observed in septic children with MOF as compared with children with no MOF We acknowledge that there is a significant preponderance of mtDNA (multiple copies/cell) as compared with β globin (two copies/cell), and this may explain the substantial (∼178 fold) difference between plasma β globin and mtDNA concentrations in septic children with MOF. Our findings were consistent with the results by Yamanouchi et al. who noted elevated plasma mtDNA concentrations in adult septic patients for 5 days after admission to the ICU. However, in addition to validating the role of plasma mtDNA in pediatric sepsis, we have advanced the field by determining the potential utility of plasma mtDNA as a biomarker for MOF. Furthermore, large-scale cohort studies are warranted to confirm our results and elucidate the underlying mechanisms.
It is noteworthy that we were unable to find a significant association between plasma mtDNA and serum CRP. Multiple studies have highlighted that CRP lacks the specificity to discriminate between bacterial, viral, and noninfectious inflammatory conditions. In addition to this criticism of CRP, there are conflicting data regarding the role of CRP as a biomarker to predict major infection or sepsis (25, 26). We chose to compare plasma mtDNA with CRP as CRP continues to be commonly used in clinical practice to detect bacterial infections. Of note, we did not compare the plasma mtDNA levels with peak CRP levels as the concentration of CRP peaks at 36 to 50 h after inflammation ensues (27). We were unable to perform this comparison due to lack of serial measurement of mtDNA and CRP levels. Along with the obvious limitation of age-matched plasma controls, this study was also not powered to identify an association of plasma mtDNA with demographic parameters, i.e., age and sex of the patient. Therefore, further validation of these data is required in a larger cohort of pediatric septic patients.
PBMCs treated with extracellular mtDNA demonstrated higher TNF levels as compared with control cells. Our results were consistent with another study that proved that treatment with extracellular mtDNA can activate neutrophils, with subsequent activation of downstream signaling pathways, i.e., phosphorylation of p38 and p44/42 (11). It was noteworthy that simultaneous presence of HMGB1 amplified the mtDNA-induced TNF secretory response. Besides its pivotal role as a DAMP, the presence of HMGB1 in the extracellular compartment serves many other functions, e.g., migration of monocytes (28) and induces the proliferation of smooth muscle cells and mesoangioblasts (29, 30). Furthermore, HMGB1 contributes to dendritic cell maturation and leads to induction of immune responses (31–33). For example, it has been suggested that HMGB1 binds to substances derived from microbes and injured tissues, creating complexes that modulate the innate immune response by mediating pro-inflammatory cytokine release. In our study, the addition of extracellular HMGB1 amplified the mtDNA-induced TNF response. Our experiments using boiled HMGB1 affirm that HMGB1 is an adjuvant in the mtDNA-induced TNF response. Taken together, our observations suggest that plasma mtDNA contributes to the innate inflammatory response and sepsis-induced mtDNA release may play a role in organ failure. Although not ready for prime-time, the use of mtDNA concentrations can assist in patient stratification for clinical research, and opens up additional lines of investigation as a potential therapeutic target.
The precise mechanism that leads to mtDNA release into the extracellular milieu is unknown at present. In an elegant study, Yousefi et al. (34) demonstrated that lipopolysaccharide from gram-negative bacteria activates IL-5 or interferon-γ-primed eosinophils to release mitochondrial DNA in a rapid fashion—a process that is independent of cell death. The various cell types and tissues that contribute to active mtDNA release are unclear at present.
In summary, our data suggest that plasma mtDNA may act as a novel DAMP in pediatric sepsis, and appears to be associated with MOF. Our in vitro data demonstrate that extracellular mtDNA induces TNF secretion, and simultaneous addition of HMGB1 amplifies this response. Further inquiry into the release of mtDNA and its function in the extracellular milieu may lead to new strategies for preventing septic shock and MOF.
Acknowledgments
Sources of support: NIH-R01 GM098474 (RKA), R01 GM108618 (JAC).
Footnotes
The authors report no conflicts of interest.
Supplemental digital content is available for this article. Direct URL citation appears in the printed text and is provided in the HTML and PDF versions of this article on the journal's Web site (www.shockjournal.com).
References
- 1.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 2.Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta. 2010;1805(1):53–71. doi: 10.1016/j.bbcan.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1768–1773. doi: 10.1164/ajrccm.164.10.2106117. [DOI] [PubMed] [Google Scholar]
- 5.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81(1):1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 6.Wiesner RJ, Ruegg JC, Morano I. Counting target molecules by exponential polymerase chain-reaction - copy number of mitochondrial-DNA in rat-tissues. Biochem Biophys Res Commun. 1992;183(2):553–559. doi: 10.1016/0006-291x(92)90517-o. [DOI] [PubMed] [Google Scholar]
- 7.Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410(2):103–123. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- 8.Andersson SG, Karlberg O, Canback B, Kurland CG. On the origin of mitochondria: a genomics perspective. Philos Trans R Soc Lond B Biol Sci. 2003;358(1429):165–177. doi: 10.1098/rstb.2002.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol. 2006;22:409–437. doi: 10.1146/annurev.cellbio.21.122303.115827. [DOI] [PubMed] [Google Scholar]
- 10.West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11(6):389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock. 2010;34(1):55–59. doi: 10.1097/SHK.0b013e3181cd8c08. [DOI] [PubMed] [Google Scholar]
- 12.Puskarich MA, Shapiro NI, Trzeciak S, Kline JA, Jones AE. Plasma levels of mitochondrial DNA in patients presenting to the emergency department with sepsis. Shock. 2012;38(4):337–340. doi: 10.1097/SHK.0b013e318266a169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kung CT, Hsiao SY, Tsai TC, Su CM, Chang WN, Huang CR, Wang HC, Lin WC, Chang HW, Lin YJ, et al. Plasma nuclear and mitochondrial DNA levels as predictors of outcome in severe sepsis patients in the emergency room. J Transl Med. 2012;10:130. doi: 10.1186/1479-5876-10-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simmons JD, Lee YL, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, Gillespie MN, Richards WO. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg. 2013;258(4):591–596. doi: 10.1097/SLA.0b013e3182a4ea46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiu RW, Chan LY, Lam NY, Tsui NB, Ng EK, Rainer TH, Lo YM. Quantitative analysis of circulating mitochondrial DNA in plasma. Clin Chem. 2003;49(5):719–726. doi: 10.1373/49.5.719. [DOI] [PubMed] [Google Scholar]
- 16.Janciauskiene S, Wright HT, Lindgren S. Atherogenic properties of human monocytes induced by the carboxyl terminal proteolytic fragment of alpha-1-antitrypsin. Atherosclerosis. 1999;147(2):263–275. doi: 10.1016/s0021-9150(99)00194-x. [DOI] [PubMed] [Google Scholar]
- 17.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 18.Singer M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence. 2014;5(1):66–72. doi: 10.4161/viru.26907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singer P. From mitochondrial disturbances to energy requirements. World Rev Nutr Diet. 2013;105:1–11. doi: 10.1159/000341247. [DOI] [PubMed] [Google Scholar]
- 20.Carre JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P, et al. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med. 2010;182(6):745–751. doi: 10.1164/rccm.201003-0326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singer M. Mitochondrial function in sepsis: acute phase versus multiple organ failure. Crit Care Med. 2007;35(9 Suppl):S441–S448. doi: 10.1097/01.CCM.0000278049.48333.78. [DOI] [PubMed] [Google Scholar]
- 22.Klinman DM. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol. 2004;4(4):249–258. doi: 10.1038/nri1329. [DOI] [PubMed] [Google Scholar]
- 23.Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. 2004;75(6):995–1000. doi: 10.1189/jlb.0703328. [DOI] [PubMed] [Google Scholar]
- 24.Zhang JZ, Liu Z, Liu J, Ren JX, Sun TS. Mitochondrial DNA induces inflammation and increases TLR9/NF-kappaB expression in lung tissue. Int J Mol Med. 2014;33(4):817–824. doi: 10.3892/ijmm.2014.1650. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Attia MW. Review: C-reactive protein has moderate diagnostic accuracy for serious bacterial infection in children with fever. Evid Based Med. 2009;14(2):56. doi: 10.1136/ebm.14.2.56. [DOI] [PubMed] [Google Scholar]
- 26.Sanders S, Barnett A, Correa-Velez I, Coulthard M, Doust J. Systematic review of the diagnostic accuracy of C-reactive protein to detect bacterial infection in nonhospitalized infants and children with fever. J Pediatr. 2008;153(4):570–574. doi: 10.1016/j.jpeds.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 27.Standage SW, Wong HR. Biomarkers for pediatric sepsis and septic shock. Expert Rev Anti Infect Ther. 2011;9(1):71–79. doi: 10.1586/eri.10.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, Lepantalo M, Carpen O, Parkkinen J, Rauvala H. Regulation of monocyte migration by amphoterin (HMGB1) Blood. 2004;104(4):1174–1182. doi: 10.1182/blood-2003-10-3536. [DOI] [PubMed] [Google Scholar]
- 29.Degryse B, Bonaldi T, Scaffidi P, Muller S, Resnati M, Sanvito F, Arrigoni G, Bianchi ME. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152(6):1197–1206. doi: 10.1083/jcb.152.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palumbo R, Sampaolesi M, De Marchis F, Tonlorenzi R, Colombetti S, Mondino A, Cossu G, Bianchi ME. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol. 2004;164(3):441–449. doi: 10.1083/jcb.200304135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bondanza A, Zimmermann VS, Dell'Antonio G, Cin ED, Balestrieri G, Tincani A, Amoura Z, Piette JC, Sabbadini MG, Rovere-Querini P, et al. Requirement of dying cells and environmental adjuvants for the induction of autoimmunity. Arthritis Rheum. 2004;50(5):1549–1560. doi: 10.1002/art.20187. [DOI] [PubMed] [Google Scholar]
- 32.Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, Arnold B, Bianchi ME, Manfredi AA, Rovere-Querini P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005;174(12):7506–7515. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 33.Huegli RW, Staedele H, Messmer P, Regazzoni P, Steinbrich W, Gross T. Displaced anterior column acetabular fracture: closed reduction and percutaneous CT-navigated fixation. Acta Radiol. 2004;45(6):618–621. doi: 10.1080/02841850410008199. [DOI] [PubMed] [Google Scholar]
- 34.Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, Schmid I, Straumann A, Reichenbach J, Gleich GJ, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14(9):949–953. doi: 10.1038/nm.1855. [DOI] [PubMed] [Google Scholar]