

ABSTRACT
The prostate apoptosis response protein 4 (Par-4) is a tumor-suppressor that has been shown to induce cancer-cell selective apoptosis in a variety of cancers. The regulation of Par-4 expression and activity is a relatively understudied area, and identifying novel regulators of Par-4 may serve as novel therapeutic targets. To identify novel regulators of Par-4, a co-immunoprecipitation was performed in colon cancer cells, and co-precipitated proteins were identified by mass-spectometry. TRIM21 was identified as a novel interacting partner of Par-4, and further shown to interact with Par-4 endogenously and through its PRY-SPRY domain. Additional studies show that TRIM21 downregulates Par-4 levels in response to cisplatin, and that TRIM21 can increase the resistance of colon cancer cells to cisplatin. Furthermore, forced Par-4 expression can sensitize pancreatic cancer cells to cisplatin. Finally, we demonstrate that TRIM21 expression predicts survival in pancreatic cancer patients. Our work highlights a novel mechanism of Par-4 regulation, and identifies a novel prognostic marker and potential therapeutic target for pancreatic cancer.
KEYWORDS: Cisplatin, colon cancer, pancreatic cancer, Par-4, prognosis, proteasome, TRIM21
Abbreviations
- CDDP
cisplatin
- DNA
deoxyribonucleic acid
- NF-κB
nuclear factor kappaB
- Par-4
prostate apoptosis response protein 4
- PARP
poly(ADP-ribose) polymerase
- TRIM21
tripartite motif-containing protein 21
- VEGF
vascular endothelial growth factor
- EGFR
epidermal growth factor receptor
- ISC-4
phenylbutyl isoselenocyanate 4
- Bcl-2
B-cell lymphoma 2
- DR5
death receptor 5
- IRF
interferon regulatory factor
- c-FLIP
flice-like inhibitory protein
- PI3K
Phosphatidylinositol-4,5-bisphosphate 3-kinase
Introduction
While early stage colon cancer has a 5-year survival rate of 60%,1 metastatic colon cancer has a 5-year survival rate of 12.9%.1 The low 5-year survival rate for metastatic colon cancer is due to many factors, including inherent chemoresistance and acquired resistance to treatment.2 The prognosis for pancreatic cancer is even more grim: the 5-year survival rate for early stage pancreatic cancer is 27.1%,1 while metastatic pancreatic cancer has a 5-year survival rate of 2.4%.1 The inherent difficulty in treating pancreatic cancer is related to late diagnosis and the resistance of pancreatic cancer cells to chemotherapy.3 These dismal prognoses underscore an important need for more effective therapeutics and for identifying novel therapeutic targets.
While standard cancer chemotherapies have a broad-based mechanism of action, novel cancer therapies take a more focused approach by targeting proteins that are unique to cancer cells. Thus, targeted therapies are much better tolerated than standard chemotherapy, because patients do not experience the common symptoms that are due to the off-target effects of standard chemotherapy, such as hair loss and immune suppression. Some examples of targeted therapies used in the treatment of colorectal cancer and pancreatic cancer include Avastin and Erlotinib. Avastin is a monoclonal antibody directed against vascular endothelial growth factor (VEGF), thereby inhibiting angiogenesis, and is used in the treatment of colorectal cancer.2 Erlotinib is a small-molecule inhibitor of epidermal growth factor receptor (EGFR), thereby inhibiting cell survival and proliferation, and is used in the treatment of pancreatic cancer.3 While the combination of standard chemotherapy with targeted therapy improves survival compared with standard therapy alone, resistance often develops, which highlights the need for novel drug targets and targeted therapies.2
Prostate apoptosis response protein – 4 (Par-4) is a tumor-suppressor gene that has therapeutic potential as a targeted therapy. Par-4 was originally found to be upregulated in prostate cancer cells undergoing cell death.4 Par-4 is a tumor-suppressor that is activated by PKA and regulates apoptosis in a cell-type-specific manner.5 Par-4 overexpression is sufficient to induce apoptosis in vitro and in vivo in multiple types of cancer, such as breast cancer, lung cancer, and melanoma.6,7 In other cell types, such as Jurkat T lymphocytes,8 Par-4 increases cell susceptibility to pro-apoptotic stimuli. In colon cancer cells, Par-4 overexpression sensitizes cells to apoptosis in response to the chemotherapeutic agent, 5-fluorouracil,9 and Akt inhibitor, ISC-4.10 In pancreatic cancer cells, treatment with inhibitors of NF-κB and Bcl−2 induce Par-4 expression, which in turn sensitizes cells to chemotherapeutic-induced apoptosis.,11,12
Par-4 is able to regulate apoptosis via activation of both the extrinsic13 and intrinsic pathways of apoptosis.8,14–17 Par-4 overexpression can induce apoptosis partly by translocating the death receptors Fas13,18 DR 5,19 and the death ligand, FasL,13 to the cell membrane; this, in turn, results in the cleavage of caspase 813,19 and engagement of the extrinsic pathway of apoptosis. Par-4 overexpression also downregulates the anti-apoptotic protein, Bcl−2,8,14–17 which in turn increases mitochondrial permeability and the release of cytochrome c, caspase-9 cleavage, and engagement of the intrinsic pathway.
While most Par-4 studies have focused on characterizing the function of Par-4, its regulation is a relatively understudied area. Par-4 has been shown to be upregulated in response to treatment with various natural products and small-molecules.20–22 Recently, Par-4 has been shown to be transcriptionally upregulated by FOXO3a in response to treatment with Withaferin A.23 Ubiquitination of target proteins by E3 ligases followed by proteasomal degradation is a common mechanism for downregulating protein levels and activity. The ubiquitination of Par-4 by FBXO45 and its subsequent proteasomal degradation has been demonstrated to regulate cancer cell survival.24
TRIM21, an E3 ligase, was first discovered as an autoantigen associated with autoimmune diseases, such as systemic lupus. Like other E3 ligases, TRIM21 functions by ubiquitination of target substrates. For example, TRIM21 regulates innate immune signaling through ubiquitination of DDX41, an intracellular DNA sensor, thereby inhibiting the innate immune response to intracellular dsDNA.25 In addition, multiple members of the IRF family of proteins, which is a family of transcription factors that are activated and act downstream of toll-like receptors, are substrates of TRIM21.26,27 Thus, TRIM21 negatively regulates the innate immune response to foreign pathogens.
While most studies have examined the role of TRIM21 in regulating innate immune signaling, some studies have implicated TRIM21 in the regulation of other cellular processes. For example, TRIM21 has been shown to positively regulate apoptosis via ubuiqitination of apoptosis inhibitors, such as Bcl−2 and c-FLIP, leading to their degradation.28,29 Furthermore, TRIM21 has been shown to be a negative regulator of B-cell proliferation.30 The above findings suggest a possible tumor-suppressive role of TRIM21. In line with this, 2 recent studies have shown that reduced TRIM21 expression is correlated with poor prognosis in hepatocellular carcinoma and diffuse large B-cell lymphoma.31,32
Identifying novel regulators of Par-4 represents a potential avenue for identifying new drug targets for colon and pancreatic cancer. In this study, we identify TRIM21 as a novel interaction partner of Par-4 in colon cancer cells. Furthermore, we show that TRIM21 can regulate Par-4 levels in response to cisplatin in both pancreatic cancer and colon cancer cell lines. Finally, we show that TRIM21 may represent a potentially novel therapeutic target and biomarker.
Results
TRIM21 is a novel interacting partner of Par-4
In order to discover new regulators of Par-4, we sought to identify novel binding partners of Par-4. To identify novel binding partners of Par-4, we performed an immunoprecipitation of Par-4 from HCT-116, HT-29, and KM12C colon cancer whole cell lysates that had been transiently transfected with Par-4 plasmid. The purpose of the transfection was to increase the signal. Proteins that co-precipitated with Par-4 were analyzed by mass spectrometry for identification. TRIM21 was identified as an interacting protein in all 3 cell lines tested. A representative list of the top identified proteins in the pull-down and the negative control pull-down is shown in Table 1 To validate the mass-spectrometry result and to confirm that the interaction between Par-4 and TRIM21 was not an artifact of the ectopic Par-4 expression, reciprocal co-immunoprecipitations were performed with endogenous Par-4 in HCT-116, HT-29, and KM12C whole cell lysates. As can be seen in Fig. 1A–C, Par-4 and TRIM21 interact endogenously in colorectal cancer cells.
Table 1.
HCT-116, HT-29, and KM12C cells were grown and either transfected with Par-4 expression plasmid or control plasmid for 48 hrs. Par-4 was immunoprecipitated from whole cell lysates using anti-Par-4 antibody and protein G magnetic beads. An in-solution trypsin digestion was performed on the eluted proteins, and the digests were sent for mass spectrometry analysis. A representative list of the most abundant proteins identified in the digest reveals TRIM21 as a potential novel interacting partner of Par-4.
| IP α HA |
IP α Par-4 |
||
|---|---|---|---|
| Accession | Name | Accesaon | Name |
| gi | 62414289 | vimentin [Homo sapiens] | gi | 55769533 | PRKC apoptosis WT1 regulator protein [Homo sapiens] |
| gi | 58331171 | T-complex protein 1 subunit zeta isoform b [Homo sapiens] | gi | 15208660 | E3 ubiquitin-protein ligase TRIM21 [Homo sapiens] |
| gi | 4502643 | T-complex protein 1 subunit zeta isoform a [Homo sapiens] | gi | 4826998 | splicing factor, proline- and glutamine-rich [Homo sapiens] |
| gi | 31542947 | 60 kDa heat shock protein, mitochondrial [Homo sapiens] | cont | 000394 | Trypa5 | cont | 000394 | Trypa5|P romTArt5 Promega trypsin artifact 5 К to R mods (22391, 2914)(1987, 2003). |
| RRRRRgi | 62422577 | REVERSED neurobeachin isoform 1 [Homo sapiens] | gi | 4506583 | replication protein A 70 kDa DNA-binding subunit [Homo sapiens] |
Figure 1.
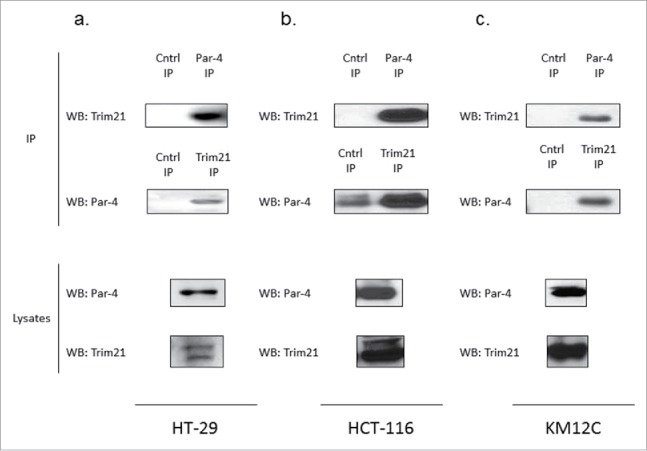
TRIM21 is a novel interacting partner of Par-4. HCT-116, HT-29, and KM12C cells were grown and transfected with either Par-4 expression plasmid or control plasmid for 48 hrs. Co-immunoprecipitations were performed in HT-29, HCT-116, and KM12C cells (a, b, c, respectively) in order to validate the TRIM21/Par-4 interaction.
TRIM21 interacts with Par-4 through its PRY-SPRY domain
Next, we sought to characterize the Par-4/TRIM21 interaction. Specifically, we wanted to identify which domain of TRIM21 is responsible for mediating its interaction with Par-4. TRIM21 is an E3 ligase that is part of the Tripartite motif (TRIM) family. TRIM family members contain a RING finger domain, a B-box domain, and a coiled-coil domain. TRIM21 is unique in that it also contains a C-terminal PRY-SPRY domain, which is a protein-protein interaction domain.33 A schematic of TRIM21 can be seen in Fig. 2A.
Figure 2.
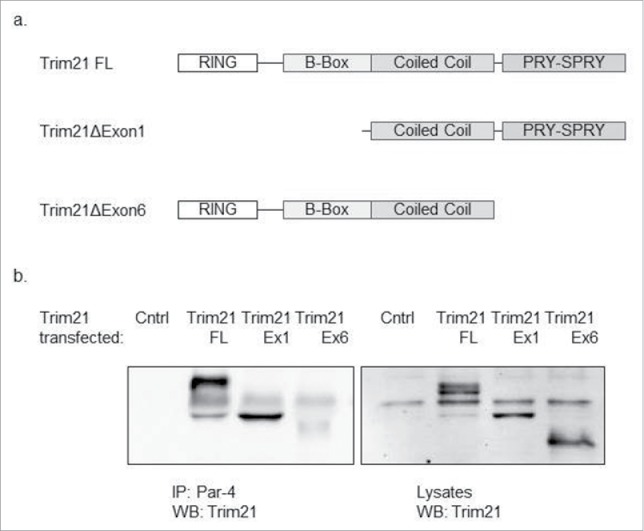
TRIM21 interacts with Par-4 via its PRY-SPRY domain. HCT-116 cells were co-transfected with Par-4 plasmid and different TRIM21 constructs for 48 hrs. Par-4 was immunoprecipitated from whole cell lysates using anti-Par-4 antibody and protein G magnetic beads. The proteins were eluted from the beads and a western blot analysis is performed using anti-TRIM21 antibody. (a) A diagram representing the various TRIM21 constructs that were used (b) Western blots demonstrating that when co-transfecting with the TRIM21 construct that does not contain the PRY-SPRY domain, TRIM21 does not interact with Par-4; in contrast, when co-transfecting with the TRIM21 constructs that contain the PRY-SPRY domain, TRIM21 does interact with Par-4.
To determine which domain of TRIM21 mediates its interaction with Par-4, we obtained 3 TRIM21 constructs: full-length, exon 1 deletion, and exon 6 deletion. The exon 1 deletion does not contain the RING domain nor the B-box domain. These domains are responsible for the E3 ligase activity of TRIM21. The exon 6 deletion does not contain the PRY-SPRY domain. A schematic of these constructs can also be seen in Fig. 2A. To test which domain of TRIM21 is responsible for its interaction with Par-4, we co-transfected HCT-116 cells with combinations of the Par-4 plasmid and each of the 3 TRIM21 constructs. We then performed a co-immunoprecipitation against Par-4, and probed for the presence of TRIM21 by Western Blotting (Fig. 2B). The results show that truncated constructs that contain the PRY-SPRY domain bind to Par-4. However, when the exon containing this motif is deleted, binding is absent, implicating the domain with PRY-SPRY as essential for binding.
TRIM21 is not sufficient to downregulate Par-4 levels
Given that TRIM21 is an E3 ligase and interacts with Par-4, we hypothesized that TRIM21 may downregulate Par-4 protein levels via ubiquitination and subsequent proteasomal degradation. The endogenous expression patterns of Par-4 and TRIM21 across a panel of colorectal cancer cell lines appears to show an inverse relationship (Fig. 3A). This inverse relationship suggests that TRIM21 could downregulate Par-4 levels. To test this hypothesis, we ectopically expressed the 3 TRIM21 constructs in SW480 cells by transient transfection, and probed for the expression of Par-4 by Western blotting. The results demonstrate that TRIM21 overexpression is not sufficient to downregulate Par-4 levels (Fig. 3B). These results were corroborated by repeating the experiment using HCT-116 cells (data not shown). This conclusion is further supported by the observation that ectopic TRIM21 expression and TRIM21 knock-down do not regulate the stabilization of Par-4.24
Figure 3.
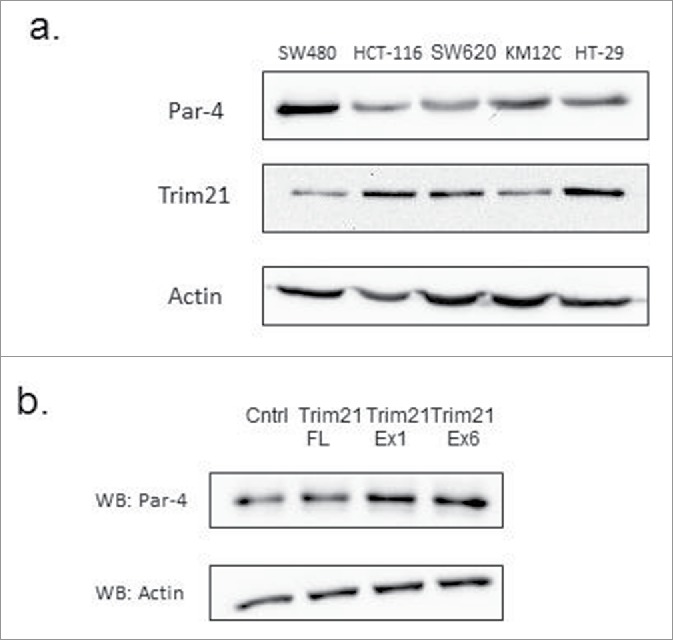
TRIM21 is not sufficient to downregulate Par-4 protein levels. (a) Western blots of endogenous expression levels of Par-4 and TRIM21 in colon cancer cell lines show an inverse correlation in expression. (b) SW480 cells were transfected with either a control plasmid or various TRIM21 constructs for 48 hours. Whole cell lysates were collected and Par-4 protein levels are analyzed by Western blot with actin as a loading control.
Ectopic expression of TRIM21 downregulates Par-4 in the presence of cisplatin
Given that TRIM21 was not sufficient to downregulate Par-4 levels, we hypothesized that TRIM21 may regulate Par-4 levels in response to a stimulus. Cisplatin (CDDP) is one such stimulus that has been shown to affect Par-4 activity via promoting its cleavage by caspase 3.34 Furthermore, Par-4 downregulation has been demonstrated to induce cisplatin resistance in pancreatic cancer cells via a PI3K/Akt-dependent EMT pathway.35
To test whether TRIM21 modulates Par-4 levels in response to cisplatin treatment, we transfected HCT-116 and HT-29 cells with TRIM21 full-length plasmid or control plasmid, and treated the transfected cells with increasing concentrations of cisplatin for 24 hrs. We probed for changes in Par-4 levels by Western blotting. As can be seen in Figs. 4A and 4B, in the presence of TRIM21 overexpression, cisplatin downregulates Par-4 levels relative to control transfected cells. In HT-29 cells, this downregulation is evident even at the lowest cisplatin dose tested, and this downregulation occurs at all doses up to 15 μg/ml (Fig. 4A). In HCT-116 cells, this downregulation occurs at all doses at or above 3.75 μg/ml with the exception of the 7.5 μg/ml dose (Fig. 4B).
Figure 4.
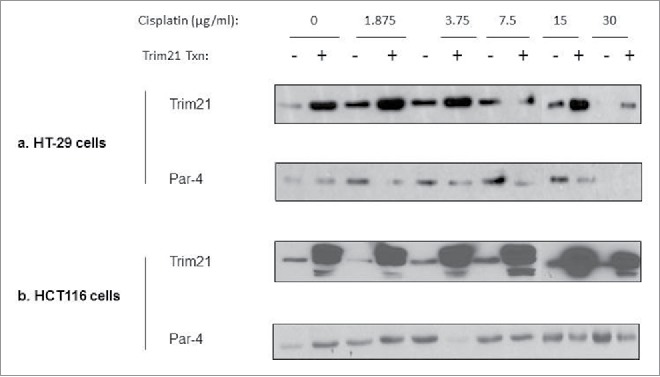
Ectopic expression of TRIM21 downregulates Par-4 in the presence of cisplatin in colon cancer cells. Colon cancer cell lines, transfected with or without TRIM21 expression plasmid for 48 hrs, were treated with increasing doses of cisplatin for 24 hrs. The cisplatin concentrations are in units of μg/ml. Western blots of Par-4 and TRIM21 levels are shown with actin shown as a loading control (a) HT-29 (b) HCT-116.
Cisplatin downregulates Par-4 in a dose- and proteaseome-dependent manner
Given our observations that cisplatin downregulates Par-4 at multiple doses, we asked whether cisplatin downregulates Par-4 in a dose-dependent manner. To determine this, we treated TRIM21-transfected HCT-116 cells with increasing doses of cisplatin and performed a time-course. Par-4 levels from whole cell lysates were then examined via Western blotting. The data show that cisplatin-induced Par-4 downregulation occurs in a dose-dependent manner: Par-4 levels are reduced more quickly at higher doses (Fig. 5A).
Figure 5.
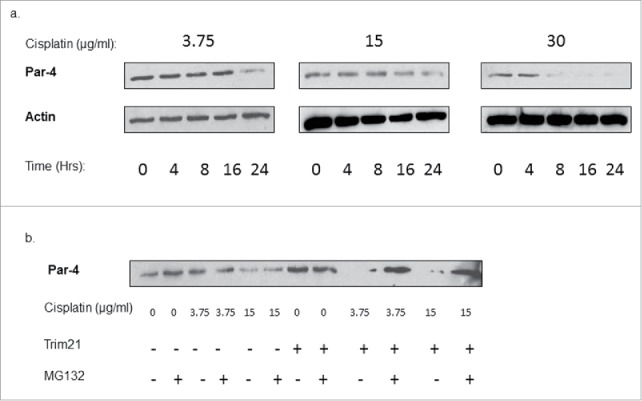
Cisplatin downregulates Par-4 in a dose- and proteasome-dependent manner. (a) Western blots showing Par-4 expression levels in TRIM21-transfected HCT-116 cells over time at different doses of cisplatin. Cisplatin doses are in units of μg/ml. Time is in units of hours. Actin is shown as a loading control. (b) HCT-116 cells were transfected with or without TRIM21 expression plasmid for 48 hrs, then treated with the indicated doses of cisplatin in μg/ml for 24 hrs, and with or without 10 μM MG132. Blot shows Par-4 expression levels, and actin is shown as a loading control.
Given that TRIM21 is an E3 ligase, we asked whether the downregulation of Par-4 occurs through proteasomal degradation. To test this hypothesis, we transfected HCT-116 cells with control plasmid or TRIM21 plasmid, treated cells with 2 different doses of cisplatin, and treated cells either with or without 10 μM MG132, a proteasome inhibitor. The effects on Par-4 levels were determined by Western blotting. The data show that the Par-4 downregulation in reponse to TRIM21/cisplatin is abrogated by co-treatment with MG132 (Fig. 5B). This suggests that the downregulation of Par-4 in response to cisplatin occurs at least partially through the proteasome pathway.
Cisplatin downregulates Par-4 in both the cytoplasmic and nuclear compartments
To determine an intracellular location of the Par-4 downregulation in response to cisplatin, we transfected HCT-116 cells with or without TRIM21 and treated with 3.75 μg/ml of cisplatin or carrier, then performed a nuclear-cytoplasmic fractionation to determine whether the Par-4 downregulation was localized to a specific subcellular compartment. Par-4 protein levels were examined by Western blotting. As can be seen in,Fig. 6 the Par-4 downregulation occurs in both the cytoplasmic and nuclear compartments, with the downregulation being slightly more pronounced in the nuclear compartment. Lamin A/B and β-tubulin were probed to confirm the integrity of the subcellular fractionation and to ensure that there was no cross-contamination between compartments during the fractionation.
Figure 6.
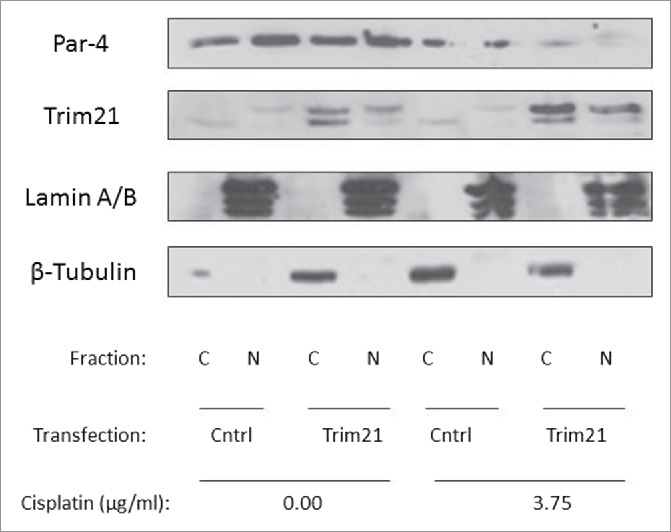
Cisplatin downregulates Par-4 in both the cytoplasmic and nuclear compartments. HCT-116 cells were either transfected with plasmid encoding TRIM21 or control plasmid for 48 hrs and treated with or without 3.75 μg/ml of cisplatin. Then, nuclear-cytoplasmic fractionation was performed and Par-4 and TRIM21 levels were examined by Western blotting, with Lamin A/B and β-tubulin serving as markers validating the integrity of the fractionation.
Cisplatin downregulates Par-4 in pancreatic cancer cells
Given that Par-4 was downregulated in response to cisplatin in the presence of overexpressed TRIM21 in colon cancer cells, we wanted to examine whether we could extend this observation to a different type of cancer. To test that hypothesis, we transfected AsPc-1, BxPc-3, and MiaPaca-2 cells, which are all pancreatic cancer cell lines, with empty vector (control) or with TRIM21 for 48 hrs, and treated the cells with increasing doses of cisplatin for 24 hours. We probed whole cell lystates for Par-4 expression via Western blotting. We observed an enhanced dose-dependent downregulation of Par-4 in response to cisplatin treatment in TRIM21-transfected cells compare with control cells (Fig. 7A–C).
Figure 7.

Cisplatin downregulates Par-4 in pancreatic cancer cells. Pancreatic cancer cell lines were either transfected with TRIM21 plasmid or control plasmid for 48 hrs, and then treated with increasing concentrations of cisplatin for 24 hrs. Cisplatin doses are in units of μg/ml. Western blots showing Par-4 expression are shown with actin as a loading control. Western blots validating TRIM21 overexpression are also included. (a) BxPc-3 (b) MiaPaca-2 (c) AsPc-1.
TRIM21 is a potential therapeutic target in colon and pancreatic cancer
Given the effect of TRIM21 on Par-4 in response to cisplatin, we hypothesized that TRIM21 could contribute to cisplatin resistance. To address this question, we performed an MTT viability assay on HCT-116 cells that had either been transfected with TRIM21 plasmid or control plasmid, followed by treatment with increasing concentrations of cisplatin. The data show that TRIM21 decreases the sensitivity of HCT-116 cells to cisplatin (Fig. 8A). Specifically, HCT-116 cells had statistically significant increases in viability at cisplatin doses between 0.5–10 μg/ml, with a right shift in IC50 from approximately 5 μg/ml to 10 μg/ml between the control and TRIM21 transfected cells, respectively. Likewise, we performed an analogous experiment in Panc-1 cells, a pancreatic cancer cell line. To our surprise, ectopic TRIM21expression in Panc-1 cells had no effect on Panc-1 cell viability in response to cisplatin (data not shown). Despite that finding, we show in Fig. 8B that ectopic Par-4 expression sensitizes Panc-1 cells to cisplatin-induced apoptosis as indicated by PARP cleavage, specifically at the 15 and 30 μg/ml doses. PARP cleavage was examined by Western blotting and the percentage of the cleavage product was determined using densitometry. Densitometric analysis revealed that Par-4 overexpression increased the levels of apoptosis in Panc-1 cells by 2- to 3-fold relative to the control transfected cells at the 15 and 30 μg/ml doses of cisplatin (Fig. 8B).
Figure 8.

TRIM21 is a potential therapeutic target in colon and pancreatic cancer. (a) HCT-116 cells were either transfected with a plasmid encoding TRIM21 or control plasmid for 48 hrs, and then treated with increasing doses of cisplatin for 24 hrs, and viability was assessed by MTT assay. Viability is plotted as a percentage of control samples. Asterisks indicate statistically significant differences in viability between TRIM21 and control transfected cells. (b) Panc-1 cells were either transfected with a plasmid encoding Par-4 or control plasmid for 48 hrs, and then treated with increasing doses of cisplatin. Cisplatin doses are shown in units of μg/ml. Levels of PARP and Par-4 levels were examined by Western blotting. Percentage of cleaved PARP, as determined by densitometric analysis, is shown below the blots. Kaplan-Meier survival curves showing overall survival, Fig. 8c, and progression-free survival, Fig. 8d, of a cohort of pancreatic cancer patients stratified by TRIM21 mRNA expression levels. Data was obtained.
Given the effects of TRIM21 on Par-4, and in light of the recent literature suggesting that TRIM21 is a favorable prognostic marker in cohorts of hepatocellular carcinoma and diffuse large B-cell lymphoma patients,31,32 we asked whether TRIM21 has prognostic significance in either colorectal cancer or pancreatic cancer. To answer this question, we downloaded data from the TCGA database, stratified by TRIM21 expression: TRIM21 high (Z-score > 2), TRIM21 low (Z-score < −2), and TRIM21 intermediate (−2 < Z-score < 2). Then, we plotted Kaplain Meier survival curves of these 3 TRIM21 expression groups in the colorectal and pancreatic cancer cohorts. There was no statistically significant difference in overall survival or disease-free survival in the colorectal cancer cohort between the 3 TRIM21 expression groups (data not shown). In contrast, in pancreatic cancer there was a dramatic increase in overall survival in the TRIM21 low cohort relative to the intermediate and high cohorts (p = 0.0043), as can be seen in Fig. 8C. Though the difference in overall survival was not significantly different between the TRIM21 high and intermediate groups (p = 0.2121), the TRIM21 high group seems to be trending toward reduced overall survival relative to the intermediate group (Fig. 8C). Likewise, the trends in disease-free survival follow the same patterns as the trends in overall survival in the pancreatic cancer cohort. Specifically, low TRIM21 expression correlates with increased disease-free survival, whereas high TRIM21 expression correlates with reduced disease-free survival relative to the TRIM21 intermediate cohort, as can be seen in Fig. 8D. In the disease-free survival curve, the TRIM21 high, intermediate, and low expression groups were all significantly different from each other (p < 0.01).
Discussion
In this study, we identified TRIM21 as a novel interaction partner of Par-4, and demonstrate that they interact endogenously. We show that the binding occurs through the PRY-SPRY domain of TRIM21. Though TRIM21 is not sufficient to downregulate Par-4 levels, we show that in response to cisplatin TRIM21 downregulates Par-4 in a dose- and proteasome-dependent manner in both the nucleus and cytoplasm. Furthermore, we show that TRIM21 can increase the resistance of cancer cells to cisplatin and that by overexpressing Par-4, cancer cells can be sensitized to cisplatin-induced apoptosis. Finally, we demonstrate that TRIM21 expression can predict overall and disease-free survival in a cohort of pancreatic cancer patients.
Par-4 is an important tumor-suppressor whose expression is downregulated in several cancers.36–38 In addition, its ability to selectively induce and sensitize cancer cells toward apoptosis underscores the tremendous therapeutic potential of Par-4.6,18,19,39 Therefore, understanding the mechanisms of Par-4 regulation is an important, yet understudied, area of research, and our work represents an advance in our understanding of how Par-4 is regulated. Chen et al. showed that Par-4 levels could be post-translationally regulated by the proteasome through targeting by Fbxo45.24 Brasseur et al. showed that cleaved Par-4 is also regulated by the proteasome.40 We now report a stimulus-dependent regulation of Par-4 by the proteasome. That stimulus is cisplatin. This observation is significant, since cisplatin and its analogs are used in the treatment of colon and pancreatic cancer. Additionally, the subcellular localization of Par-4 is important, because its nuclear localization is correlated with its apoptotic function.41 Thus, our finding that its downregulation is present in the nucleus, along with in the cytoplasm, is also significant.
By identifying a novel mechanism of regulation of Par-4, we in turn discovered a potentially new therapeutic target: TRIM21. This is especially important for pancreatic cancer, where most patients ultimately succumb to their disease,1 and are therefore in need of novel, effective therapies. Here, we show that low TRIM21 levels correlate with prolonged overall and disease-free survival in pancreatic cancer patients. Conversely, we showed that high TRIM21 levels correlate with lower disease-free survival. This intriguing finding is the opposite of the protective effects of TRIM21 in hepatcellular carcinoma and diffuse large B-cell lymphoma.31,32 This illustrates the complexity of disease biology, and the prognostic differences of TRIM21 may reflect differences in downstream TRIM21 targets, such as Par-4, in different types of cancer. Further study of the role of TRIM21 in pancreatic cancer is needed.
Our work has given rise to the concept that, in the presence of high TRIM21 levels, cisplatin downregulates Par-4, at least partially through the proteasome. Other work has been done on the relationship between cisplatin and Par-4. For example, cisplatin has been shown to activate Par-4 via cleavage by caspase 3.34 In this instance, the cleaved Par-4 had more apoptotic activity and a greater nuclear localization than full-length Par-4. In another study, Par-4 downregulation was shown to confer resistance to cisplatin through the PI3K/Akt pathway.35 In this example, Par-4 overexpression reversed the resistance phenotype. Our work adds to the complex relationship between cisplatin and Par-4, and gives rise to interesting questions. For example, understanding the mechanisms immediately downstream of cisplatin-induced DNA damage that determine whether Par-4 is cleaved to a more active form versus downregulated would be an important area of study. Finding ways to switch from cisplatin-induced Par4 downregulation to cisplatin-induced Par-4 cleavage could be beneficial. In addition, it would be interesting to examine whether Par-4 downregulation occurs solely in response to the DNA-crosslinking that occurs during cisplatin treatment,42 or whether this response extends to other types of DNA damage, such as double-strand breaks.
In conclusion, our data show that TRIM21 is a novel interaction partner of Par-4 and a novel regulator of Par-4 in response to cisplatin treatment. Our model suggests that TRIM21 can influence sensitivity of cancer cells to cisplatin. Targeting TRIM21 may potentially be a way to enhance the effectiveness of cisplatin treatment, and may represent an important therapeutic target, especially in pancreatic cancer.
Materials/methods
Cell culture, transfection, plasmids, reagents, and antibodies
HCT-116, SW480, and HT-29 colorectal cancer cells were obtained from ATCC and maintained in RPMI (Cellgro, Manassas, VA) + 10% fetal bovine serum (FBS). Panc-1 cells were a kind gift from Dr. Arun Sharma and were maintained in DMEM (Cellgro, Manassas, VA) + 10% FBS. FBS was obtained from Atlanta Biologicals (Norcross, GA). Cell lines were grown at 37°C and 5% CO2. For experiments, cells were seeded in 6-well plates at a seeding density of 300,000 cells/well. Transient transfections were performed 24 hours post-seeding. Drug treatments were performed 48 hours post-seeding. The cells were transiently transfected using PolyJet DNA Transfection Reagent (SignaGen Laboratories, Rockville, MD), according to the manufacturer's instructions. The Par-4 plasmid and anti-TRIM21 shRNA plasmids were obtained from Origene Technologies (Rockville, MD). The plasmids encoding for full-length TRIM21 and deletion mutant TRIM21 were kind gifts from Dr. Caroline Jefferies at the Royal College of Surgeons in Ireland. The anti-TRIM21, anti-Lamin A/C, and anti-Par-4 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-actin and anti-β-tubulin antibodies were obtained from Sigma-Aldrich (Saint Louis, MO). The anti-PARP antibody was obtained from Cell-Signaling (Danvers, MA). Cisplatin was obtained from Acros Organics (Thermo Fisher, Waltham, MA). MG132 was obtained from Fisher (Thermo Fisher, Waltham, MA).
Western blot analyses
To examine changes in protein expression, cells were washed twice with PBS and were lysed into lysis buffer (50mm HEPES, 100 mm NaCl, 10 mm EDTA, 0.5% NP40, 10% glycerol, supplemented with 0.0001% Tween20, 0.1 mM PMSF, 0.1 mM NaVO4, 0.5 mM NaF, 5 μg/ml leupeptin, 0.1 mm DTT). The proteins were quantified according to the BCA Assay (Thermo Scientific Inc., Rockford, IL) and loaded equally onto 10% SDS-polyacrylamide gels. Proteins were electrophoresed at 150 V and transferred to nitrocellulose membranes using a Trans-Blot SD Semi-Dry Transfer Cell (BioRad, Hercules, CA). Membranes were blocked with 5% non-fat dry milk for 1 h and incubated with primary antibody overnight. The blots were washed 3X in TBS with 0.1% Tween20 and incubated for 1 hr in appropriate HRP-conjugated secondary antibodies (Amersham, Piscataway, NJ). Blots were washed 3X and chemiluminescent detection was performed using Amersham ECL Prime Western Blotting Detection Reagent (Thermo Scientific Inc., Rockford, IL). The blots were either exposed to autoradiography film (GE Healthcare Life Sciences, Pittsburgh, PA) and scanned or imaged using the Molecular Imager Gel Doc XR System (Bio-Rad, Hercules, CA).
Co-IP/Mass-Spec
To identify novel binding partners of Par-4, HCT-116, HT-29, and SW480 cells were cultured in 2 10-cm dishes and transfected with 5 μg of Par-4 plasmid per dish using SignaGen Polyjet transfection reagent, according to the manufacturer's instructions. After 48 hours of transfection, cells were lysed in lysis buffer (50mm HEPES, 100 mm NaCl, 10 mm EDTA, 0.5% NP40, 10% glycerol, supplemented with 0.0001% Tween20, 0.1 mM PMSF, 0.1 mM NaVO4, 0.5 mM NaF, 5 μg/ml leupeptin, 0.1 mm DTT), and the total protein concentration was determined as above.
To perform the co-immunoprecipitation, approximately 3,000 μg of total protein from the lysate was mixed with 2 μg of anti-Par-4 antibody. As a negative control, the same amount of total protein from the lysate was mixed with anti-HA antibody. The antibody-lysate mixtures were allowed to mix overnight at 4° C. The next day the antibody-antigen complexes were pulled down using Dynabeads Protein G (Thermo Fisher Scientific, Leesport, PA) and washed with lysis buffer, according to the manufacturer's instructions. Proteins are eluted from the beads by boiling at 100°C for 10 mins in digestion buffer (6 M urea, 50 mM Tris-HCl, pH = 8.0). Samples are then cooled on ice and eluate removed from beads.
To perform the in-solution digestion, 5 μl of 200 mM DTT/50 mM Tris-HCl/pH = 8.0 was added to solution, mixed, and allowed to incubate for 1 hr at room temperature. After 1 hr, 20 μl of 200 mM Iodoacetamide/50 mM Tris-HCl/pH = 8.0 is added, mixed, and allowed to incubate at room temperature in the dark for 1 hr. After 1 hr, 775 μl of 50 mM Tris-HCl/1 mM CaCl2/pH = 7.6. 2 microliters of trypsin (0.2 μg/ml) is added, mixed, and allowed to incubate at 37°C overnight. Formic acid is added to bring pH down to 3–4. Solutions are snap frozen in liquid nitrogen and spun to dryness under vacuum. Residue is resuspended in milliQ water, snap frozen in liquid nitrogen, and dried using a speed vac. This process is repeated 2 more times. The final residue is submitted for protein identification via MALDI-TOF mass spectrometry. Proteins are identified from mass spectra using Paragon Algorithm.
MTT assay
To examine changes in cell viability, cells were seeded at a density of 6 × 103 cells/well in a 96-well culture plate. After 24 hours, the cells were transfected with the appropriate plasmid. Twenty-four hours post-transfection, the medium was replaced with medium containing different concentrations of cisplatin. Twenty-four hours post-cisplatin treatment, MTT reagent was added (Calbiochem) and the cells were incubated for 3.5 hrs at 37° C. After incubation, the media was aspirated off, crystals were dissolved in MTT solvent (4 mM HCl, 0.1% Nonidet P-40, in isopropanol), and viability was assessed by measuring the absorbance at 570 nm with 630 nm absorbance as the reference. Percent viability is expressed as the absorbance normalized to the DMSO control.
Immunofluoresence
Cells are grown to approximately 50% confluence on 22 × 22 mm coverslips (#1.5). Media is removed and cells are washed with PBS, followed by fixation with 4% paraformaldehyde in PBS for 15 mins at room temperature. Cells are washed in PBS, then permeabilized 3 × 5 mins with 0.1% Triton X-100 in PBS. Cells are washed with PBS, then blocked with 10% BSA in PBS for 1 hr at room temperature. After one hour, the cells are washed with PBS, a mixture of anti-Par-4 antibody (1:50) and anti-TRIM21 antibody (1:50) in 4% BSA/PBS for 2 hours at room temperature. After the incubation in primary antibody, the cells are washed 4X with PBS for 10 mins each at room temperature with gentle shaking. After washing, the cells are then incubated in a mixture of secondary antibody in 4% BSA/PBS: anti-rabbit FITC (1:100) and anti-mouse Ro (1:100). The secondary antibody incubation is performed in the dark for 2 hours at room temperature. After incubation in secondary antibody, the cells are washed 4X with PBS for 10 mins each at room temperature with gentle shaking. After washing, the cells are incubated with DAPI (0.5 μg/ml in PBS) for 5 mins at room temperature in the dark. After incubation with DAPI, the cells are washed 4X with PBS for 10 mins each at roomp temperatue with gentle shaking. Finally, the coverslips are mounted on glass slides using ProLong Diamond Antifade mountant (Thermo Fisher, Leesport, PA). Slides are imaged using DeltaVision Elite Inverted Microscope. Six random fields are imaged to ensure representativeness.
Nuclear-cytoplasmic fractionation
To perform the nuclear-cytoplasmic fractionation, the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher, Leesport, PA) was used, and the manufacturer's instructions were followed. The resultant extracts were quantified and loaded equally onto a 10% acrylamide gel, and proteins probed by Western blotting.
Statistical analyses
The statistical analyses were performed using GraphPad Prism software, version 6.04 (GraphPad Software, Inc., San Diego, CA, USA). All plots were created using GraphPad Prism. All experiments were repeated 3 times with representative experiments shown in the results. For the MTT assay, 2-way ANOVA was used to determine differences between transfectants and controls, using the Sidak correction to correct for multiple comparisons. For the Kaplan-Meier survival curves, the log-rank test was used to determine differences between groups. The pancreatic cancer TCGA data was accessed and downloaded from www.cbioportal.org. TRIM21 high expression was considered to be those that had a Z-score greater than 2. TRIM21 low expression was considered to be those than had a Z-score lower than −2. Patients with intermediate TRIM21 expression had Z-scores between +/− 2. Densitometric analysis was performed using ImageJ.43 The threshold for statistical significance is P < 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The Penn State Hershey Imaging Core is acknowledged for their help in obtaining images.
Funding
This work was funded by start-up funds granted to Rosalyn B. Irby.
References
- 1.Fast Stats: An interactive tool for access to SEER cancer statistics. Surveillance, Epidemiology, and End Results (SEER) Program National Cancer Institute. [Google Scholar]
- 2.Pai SG, Fuloria J. Novel therapeutic agents in the treatment of metastatic colorectal cancer. World J Gastrointest Oncol 2016; 8:99-104; PMID:26798440; http://dx.doi.org/ 10.4251/wjgo.v8.i1.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kourie HR, Gharios J, Elkarak F, Antoun J, Ghosn M. Is metastatic pancreatic cancer an untargetable malignancy? World J Gastrointest Oncol 2016; 8:297-304; PMID:26989465; http://dx.doi.org/ 10.4251/wjgo.v8.i3.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sells SF, Wood DP Jr., Joshi-Barve SS, Muthukumar S, Jacob RJ, Crist SA, Humphreys S, Rangnekar VM. Commonality of the gene programs induced by effectors of apoptosis in androgen-dependent and -independent prostate cells. Cell Growth Differ 1994; 5:457-66; PMID: 8043520 [PubMed] [Google Scholar]
- 5.Gurumurthy S, Goswami A, Vasudevan KM, Rangnekar VM. Phosphorylation of Par-4 by protein kinase A is critical for apoptosis. Mol Cell Biol 2005; 25:1146-61; PMID:15657440; http://dx.doi.org/ 10.1128/MCB.25.3.1146-1161.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irby RB, Kline CL. Par-4 as a potential target for cancer threapy. Exp Opin Therap Targets 2013; 1:77-87; PMID:23062118; http://dx.doi.org/11479426 10.1517/14728222.2013.731047 [DOI] [PubMed] [Google Scholar]
- 7.Lucas T, Pratscher B, Krishnan S, Fink D, Gunsberg P, Wolschek M, Wacheck V, Muster T, Romirer I, Wolff K, et al.. Differential expression levels of Par-4 in melanoma. Melanoma Res 2001; 11:379-83; PMID:11479426; http://dx.doi.org/ 10.1097/00008390-200108000-00008 [DOI] [PubMed] [Google Scholar]
- 8.Boehrer S, Chow KU, Beske F, Kukoc-Zivojnov N, Puccetti E, Ruthardt M, Baum C, Rangnekar VM, Hoelzer D, Mitrou PS, et al.. In lymphatic cells par-4 sensitizes to apoptosis by down-regulating bcl-2 and promoting disruption of mitochondrial membrane potential and caspase activation. Cancer Res 2002; 62:1768-75; PMID:11912153 [PubMed] [Google Scholar]
- 9.Kline CL, Shanmugavelandy SS, Kester M, Irby RB. Delivery of PAR-4 plasmid in vivo via nanoliposomes sensitizes colon tumor cells subcutaneously implanted into nude mice to 5-FU. Cancer Biol Ther 2009; 8:1831-7; PMID:19729995; http://dx.doi.org/ 10.4161/cbt.8.19.9592 [DOI] [PubMed] [Google Scholar]
- 10.Sharma AK, Kline CL, Berg A, Amin S, Irby RB. The Akt inhibitor ISC-4 activates prostate apoptosis response protein-4 and reduces colon tumor growth in a nude mouse model. Clin Cancer Res 2011; 17:4474-83; PMID:21555373; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Azmi AS, Ahmad A, Banerjee S, Rangnekar VM, Mohammad RM, Sarkar FH. Chemoprevention of pancreatic cancer: characterization of Par-4 and its modulation by 3,3′ diindolylmethane (DIM). Pharm Res 2008; 25:2117-24; PMID:18427961; http://dx.doi.org/ 10.1007/s11095-008-9581-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Azmi AS, Wang Z, Burikhanov R, Rangnekar VM, Wang G, Chen J, Wang S, Sarkar FH, Mohammad RM. Critical role of prostate apoptosis response-4 in determining the sensitivity of pancreatic cancer cells to small-molecule inhibitor-induced apoptosis. Mol Cancer Ther 2008; 7:2884-93; PMID:18790769; http://dx.doi.org/ 10.1158/1535-7163.MCT-08-0438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chakraborty M, Qiu SG, Vasudevan KM, Rangnekar VM. Par-4 drives trafficking and activation of Fas and Fasl to induce prostate cancer cell apoptosis and tumor regression. Cancer Res 2001; 61:7255-63; PMID:11585763. [PubMed] [Google Scholar]
- 14.Camandola S, Mattson MP. Pro-apoptotic action of PAR-4 involves inhibition of NF-kappaB activity and suppression of BCL-2 expression. J Neurosci Res 2000; 61:134-9; PMID:10878585; http://dx.doi.org/ 10.1002/1097-4547(20000715)61:2%3c134::AID-JNR3%3e3.0.CO;2-P [DOI] [PubMed] [Google Scholar]
- 15.Cheema SK, Mishra SK, Rangnekar VM, Tari AM, Kumar R, Lopez-Berestein G. Par-4 transcriptionally regulates Bcl-2 through a WT1-binding site on the bcl-2 promoter. J Biol Chem 2003; 278:19995-20005; PMID:12644474; http://dx.doi.org/ 10.1074/jbc.M205865200 [DOI] [PubMed] [Google Scholar]
- 16.Chendil D, Das A, Dey S, Mohiuddin M, Ahmed MM. Par-4, a pro-apoptotic gene, inhibits radiation-induced NF kappa B activity and Bcl-2 expression leading to induction of radiosensitivity in human prostate cancer cells PC-3. Cancer Biol Ther 2002; 1:152-60; PMID:12170775; http://dx.doi.org/ 10.4161/cbt.61 [DOI] [PubMed] [Google Scholar]
- 17.Qiu G, Ahmed M, Sells SF, Mohiuddin M, Weinstein MH, Rangnekar VM. Mutually exclusive expression patterns of Bcl-2 and Par-4 in human prostate tumors consistent with down-regulation of Bcl-2 by Par-4. Oncogene 1999; 18:623-31; PMID:9989812; http://dx.doi.org/ 10.1038/sj.onc.1202344 [DOI] [PubMed] [Google Scholar]
- 18.Bergmann M, Kukoc-Zivojnov N, Chow KU, Trepohl B, Hoelzer D, Weidmann E, Mitrou PS, Boehrer S. Prostate apoptosis response gene-4 sensitizes neoplastic lymphocytes to CD95-induced apoptosis. Annals of hematology 2004; 83:646-53; PMID:15316756; http//dx.doi.org/ 10.1007/s00277-004-0922-3 [DOI] [PubMed] [Google Scholar]
- 19.Lee TJ, Jang JH, Noh HJ, Park EJ, Choi KS, Kwon TK. Overexpression of Par-4 sensitizes TRAIL-induced apoptosis via inactivation of NF-kappaB and Akt signaling pathways in renal cancer cells. J Cell Biochem 2010; 109:885-95; PMID:20127709; http://dx.doi.org/ 10.1002/jcb.22504 [DOI] [PubMed] [Google Scholar]
- 20.Thayyullathil F, Rahman A, Pallichankandy S, Patel M, Galadari S. ROS-dependent prostate apoptosis response-4 (Par-4) up-regulation and ceramide generation are the prime signaling events associated with curcumin-induced autophagic cell death in human malignant glioma. FEBS open bio 2014; 4:763-76; PMID:25349781; http://dx.doi.org/ 10.1016/j.fob.2014.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z, DuBois RN. Par-4, a proapoptotic gene, is regulated by NSAIDs in human colon carcinoma cells. Gastroenterology 2000; 118:1012-7; PMID:10833474; http://dx.doi.org/ 10.1016/S0016-5085(00)70352-0 [DOI] [PubMed] [Google Scholar]
- 22.Srinivasan S, Ranga RS, Burikhanov R, Han SS, Chendil D. Par-4-dependent apoptosis by the dietary compound withaferin A in prostate cancer cells. Cancer Res 2007; 67:246-53; PMID:17185378; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2430 [DOI] [PubMed] [Google Scholar]
- 23.Das TP, Suman S, Alatassi H, Ankem MK, Damodaran C. Inhibition of AKT promotes FOXO3a-dependent apoptosis in prostate cancer. Cell Death Dis 2016; 7:e2111; PMID:26913603; http://dx.doi.org/ 10.1038/cddis.2015.403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X, Sahasrabuddhe AA, Szankasi P, Chung F, Basrur V, Rangnekar VM, Pagano M, Lim MS, Elenitoba-Johnson KS. Fbxo45-mediated degradation of the tumor-suppressor Par-4 regulates cancer cell survival. Cell Death Differ 2014; 21:1535-45; PMID:24992930; http://dx.doi.org/ 10.1038/cdd.2014.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu YJ. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat Immunol 2013; 14:172-8; PMID:23222971; http://dx.doi.org/ 10.1038/ni.2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lazzari E, Korczeniewska J, Ni Gabhann J, Smith S, Barnes BJ, Jefferies CA. TRIpartite motif 21 (TRIM21) differentially regulates the stability of interferon regulatory factor 5 (IRF5) isoforms. PloS one 2014; 9:e103609; PMID:25084355; http://dx.doi.org/ 10.1371/journal.pone.0103609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higgs R, Ni Gabhann J, Ben Larbi N, Breen EP, Fitzgerald KA, Jefferies CA. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J Immunol 2008; 181:1780-6; PMID:18641315; http://dx.doi.org/ 10.4049/jimmunol.181.3.1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jauharoh SN, Saegusa J, Sugimoto T, Ardianto B, Kasagi S, Sugiyama D, Kurimoto C, Tokuno O, Nakamachi Y, Kumagai S, et al.. SS-A/Ro52 promotes apoptosis by regulating Bcl-2 production. Biochem Biophys Res Commun 2012; 417:582-7; PMID:22178074; http://dx.doi.org/ 10.1016/j.bbrc.2011.12.010 [DOI] [PubMed] [Google Scholar]
- 29.Shibata N, Ohoka N, Sugaki Y, Onodera C, Inoue M, Sakuraba Y, Takakura D, Hashii N, Kawasaki N, Gondo Y, et al.. Degradation of stop codon read-through mutant proteins via the ubiquitin-proteasome system causes hereditary disorders. J Biol Chem 2015; 290:28428-37; PMID:26442586; http://dx.doi.org/ 10.1074/jbc.M115.670901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Espinosa A, Zhou W, Ek M, Hedlund M, Brauner S, Popovic K, Horvath L, Wallerskog T, Oukka M, Nyberg F, et al.. The Sjogren's syndrome-associated autoantigen Ro52 is an E3 ligase that regulates proliferation and cell death. J Immunol 2006; 176:6277-85; PMID:16670339; http://dx.doi.org/ 10.4049/jimmunol.176.10.6277 [DOI] [PubMed] [Google Scholar]
- 31.Ding Q, He D, He K, Zhang Q, Tang M, Dai J, Lv H, Wang X, Xiang G, Yu H. Downregulation of TRIM21 contributes to hepatocellular carcinoma carcinogenesis and indicates poor prognosis of cancers. Tumour Biol 2015; 36:8761-72; PMID:26055142; http://dx.doi.org/ 10.1007/s13277-015-3572-2 [DOI] [PubMed] [Google Scholar]
- 32.Brauner S, Zhou W, Backlin C, Green TM, Folkersen L, Ivanchenko M, Lofstrom B, Xu-Monette ZY, Young KH, Moller Pedersen L, et al.. Reduced expression of TRIM21/Ro52 predicts poor prognosis in diffuse large B cell lymphoma patients with and without rheumatic disease. J Intern Med 2015; 278:323-32; PMID:25880119; http://dx.doi.org/ 10.1111/joim.12375 [DOI] [PubMed] [Google Scholar]
- 33.Oke V, Wahren-Herlenius M. The immunobiology of Ro52 (TRIM21) in autoimmunity: a critical review. J Autoimmun 2012; 39:77-82; PMID:22402340; http://dx.doi.org/ 10.1016/j.jaut.2012.01.014 [DOI] [PubMed] [Google Scholar]
- 34.Chaudhry P, Singh M, Parent S, Asselin E. Prostate apoptosis response 4 (Par-4), a novel substrate of caspase-3 during apoptosis activation. Mol Cell Biol 2012; 32:826-39; PMID:22184067; http://dx.doi.org/ 10.1128/MCB.06321-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan J, You Y, Xu T, Yu P, Wu D, Deng H, Zhang Y, Bie P. Par-4 downregulation confers cisplatin resistance in pancreatic cancer cells via PI3K/Akt pathway-dependent EMT. Toxicol Lett 2013; 224:7-15; PMID:24144893; http://dx.doi.org/ 10.1016/j.toxlet.2013.10.008 [DOI] [PubMed] [Google Scholar]
- 36.Cook J, Krishnan S, Ananth S, Sells SF, Shi Y, Walther MM, Linehan WM, Sukhatme VP, Weinstein MH, Rangnekar VM. Decreased expression of the pro-apoptotic protein Par-4 in renal cell carcinoma. Oncogene 1999; 18:1205-8; PMID:10022126; http://dx.doi.org/ 10.1038/sj.onc.1202416 [DOI] [PubMed] [Google Scholar]
- 37.Joshi J, Fernandez-Marcos PJ, Galvez A, Amanchy R, Linares JF, Duran A, Pathrose P, Leitges M, Canamero M, Collado M, et al.. Par-4 inhibits Akt and suppresses Ras-induced lung tumorigenesis. EMBO J 2008; 27:2181-93; PMID:18650932; http://dx.doi.org/ 10.1038/emboj.2008.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moreno-Bueno G, Fernandez-Marcos PJ, Collado M, Tendero MJ, Rodriguez-Pinilla SM, Garcia-Cao I, Hardisson D, Diaz-Meco MT, Moscat J, Serrano M, et al.. Inactivation of the candidate tumor suppressor par-4 in endometrial cancer. Cancer Res 2007; 67:1927-34; PMID:17332319; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2687 [DOI] [PubMed] [Google Scholar]
- 39.Lee TJ, Lee JT, Kim SH, Choi YH, Song KS, Park JW, Kwon TK. Overexpression of Par-4 enhances thapsigargin-induced apoptosis via down-regulation of XIAP and inactivation of Akt in human renal cancer cells. J Cell Biochem 2008; 103:358-68; PMID:18041764; http://dx.doi.org/ 10.1002/jcb.21642 [DOI] [PubMed] [Google Scholar]
- 40.Brasseur K, Fabi F, Adam P, Parent S, Lessard L, Asselin E. Post-translational regulation of the cleaved fragment of Par-4 in ovarian and endometrial cancer cells. Oncotarget 2016; 7:36971-87; PMID:27175591; http://dx.doi.org/ 10.18632/oncotarget.9235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El-Guendy N, Zhao Y, Gurumurthy S, Burikhanov R, Rangnekar VM. Identification of a unique core domain of par-4 sufficient for selective apoptosis induction in cancer cells. Mol Cell Biol 2003; 23:5516-25; PMID:12897127; http://dx.doi.org/ 10.1128/MCB.23.16.5516-5525.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dilruba S, Kalayda GV. Platinum-based drugs: past, present and future. Cancer Chemother Pharmacol 2016; 77:1103-24; PMID:26886018; http://dx.doi.org/ 10.1007/s00280-016-2976-z [DOI] [PubMed] [Google Scholar]
- 43.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9:671-5; PMID:22930834; http://dx.doi.org/ 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]