

ABSTRACT
Phase II clinical trials indicate that the combination of cysteamine plus epigallocatechin gallate (EGCG) is effective against cystic fibrosis in patients bearing the most frequent etiological mutation (CFTRΔF508). Here, we investigated the interaction between both agents on cultured respiratory epithelia cells from normal and CFTRΔF508-mutated donors. We observed that the combination of both agents affected metabolic circuits (and in particular the tricarboxylic acid cycle) in a unique way and that cysteamine plus EGCG reduced cytoplasmic protein acetylation more than each of the 2 components alone. In a cell-free system, protein cross-linking activity of EGCG was suppressed by cysteamine. Finally, EGCG was able to enhance the conversion of cysteamine into taurine in metabolic flux experiments. Altogether, these results indicate that multiple pharmacological interactions occur between cysteamine and EGCG, suggesting that they contribute to the unique synergy of both agents in restoring the function of mutated CFTRΔF508.
KEYWORDS: acetylation, cysteamine, cystic fibrosis, EP300, epigallocatechin gallate, metabolic flux, metabolic profiling
Introduction
Cystic fibrosis (CF) is the most frequent genetic disease causing premature death in infants and young adults. CF is caused by mutations in CF transmembrane conductance regulator (CFTR), an ABC transporter-class ion channel protein that conducts chloride and thiocyanate ions across epithelial cell membranes. The most frequent mutation of the CFTR gene, yielding a CFTR protein from which one amino acid (F508) is deleted (CFTRΔF508) due to the absence of the corresponding nucleotide triplet, occurs either in a homozygous form (on both alleles) or as a composite mutation (together with a loss-of-function mutation of CFTR affecting the second allele) in 70–90% of CF patients.1,2
The CFTRΔF508 protein is subjected to inappropriate proteostasis, at 2 levels. First, the protein tends to be degraded shortly after its synthesis before it reaches the plasma membrane. Second, the absence of functional CFTRΔF508 protein at the cell surface creates a negative feedback loop that reduces the stability of the few CFTRΔF508 molecules that reach the plasma membrane, favoring their lysosomal degradation over endosomal recycling. Attempts to improve this defective proteostasis have been launched. One particular strategy to enhance the expression of functional CFTRΔF508 protein at the plasma membrane of respiratory epithelia consists in the combined treatment with cysteamine and epigallocatechin gallate (EGCG). Indeed, combination regimens with both compounds have improved the diagnostic sweat test (in which CF patients demonstrate elevated chloride concentrations) to normal levels and have reduced inflammation-associated clinical biomarkers in several phase II trials.3-6
Cysteamine is approved for the treatment of cystinosis, a lysosome storage disease, in which the amino acid cystine accumulates. Cysteamine concentrates inside lysosomes and reacts with cystine to form cysteine and a cysteine-cysteamine complex, which both can leave the organelle.7 In addition, cysteamine has been shown to inhibit the enzymatic activity of transglutaminase-2, hence affecting proteostasis.8 EGCG is an over-the-counter green tea flavonoid with multiple (and mostly unsubstantiated) modes of action and health-related effects.9,10 Experiments in “humanized” mice bearing a CFTRΔF508 knock-in mutation (which renders them biochemically identical to CF patients with respect to the CFTR mutation) indicate that the combination of cysteamine and EGCG is unable to restore CFTR function in conditions of haploinsufficiency for Beclin 16, a pleiotropic protein that is required for autophagy, yet also participates in other vesicle trafficking pathways.11,12 Similarly, in isolated respiratory epithelial cells, knockdown of Beclin 1 or that of the autophagic substrate STQM1/p62 has been shown to reduce or enhance, respectively, the beneficial effects of cysteamine and EGCG on the restoration of CFTRΔF508 function.3-6 These findings have been interpreted to mean that cysteamine plus EGCG requires the induction of autophagy to be effective in improving the plasma membrane localization and function of CFTRΔF508.
The clinical trials involving cysteamine and EGCG show that patients bearing CFTRΔF508 or similar class 2 mutations in CFTR respond to the treatment; patients bearing class I CFTR mutations (with no protein rescuable) failed to respond to therapy. The same patients whose primary nasal brushed cells were unable to respond to cysteamine plus EGCG in vitro also demonstrated deficient treatment responses in vivo,5,6 suggesting that cysteamine and EGCG exert their therapeutic efficacy indeed through an indirect effect on misfolded CFTR mutants rather than solely via their possible mucolytic or antibiotic activities.13,14 The synergistic interaction between both agents is particularly strong upon sequential treatment. While cysteamine rapidly increases membrane exposure of CFTRΔF508 in vitro, in cells homozygous for the CFTRΔF508 mutation, this effect is lost upon washout of cysteamine unless EGCG is added to the cell cultures. Indeed, EGCG inhibits the master kinase CK2 that is pivotal in the fragmentation of CFTR and hence stabilizes CFTR mutant at PM after rescue. Conversely, EGCG has no effect on the subcellular localization and function of CFTRΔF508 if added alone to the cells.5,6
Nonetheless, the exact molecular mechanisms that explain the favorable interaction between cysteamine and EGCG remain elusive. Driven by this consideration, we decided to perform an exhaustive functional and metabolomic analysis of drug interactions between cysteamine and EGCG on respiratory epithelial cells. Here we demonstrate that both drugs, if combined, alter the metabolome of cultured cells and that both compounds reduce cellular protein acetylation. We also show that EGCG can favor protein cross-linking that is avoided by cysteamine. Conversely, EGCG accelerates the metabolism of cysteamine to hypotaurine. Hence, cysteamine and EGCG engage in multiple interactions that may contribute to their synergistic capacity to improve the function of the CFTRΔF508 protein.
Results
Metabolic effects of cysteamine and EGCG on respiratory epithelial cells
Two distinct epithelial bronchial cell lines, 16HBE (which is wild type with respect to CFTR gene) and Cfbe41o- (which is homozygously mutated CFTRΔF508) were cultured overnight in the presence of standard doses of cysteamine and EGCG, alone or in combination, followed by metabolomic analyses (Fig. 1). EGCG alone or in combination with cysteamine reduces the intracellular concentration of multiple metabolites, in particular amino acids, amino acid derivatives and nucleotides, in 16HBE cells that are not affected in Cfbe41o- cells (left panel in Fig. 1), pointing to intrinsic differences in the metabolism between cell lines. The combination of cysteamine and EGCG (but neither of the compounds alone) strongly increased the mono-carbohydrates xylitol and ribose/ribulose in 16HBE (but not in Cfbe41o-) cells and oleylcarnitine (C18:1) and the glutathione precursor γ-glutamylcysteine in Cfbe41o- (but not in 16HBE) cells (right panel in Fig. 1). In Cfbe41o- cells, the combination of cysteamine and EGCG profoundly affected core metabolism including glycolysis and the tricarboxylic acid (TCA) cycle, causing an increase in pyruvate/oxaloacetate, lactate, citrate/isocitrate, but a decrease in α-ketoglutarate, fumarate and malate (Fig. 2A and B). The decrease in fumarate and pyruvate/oxaloacetate was more pronounced with the combination of the 2 drugs than with either of them alone (Fig. 2B). In contrast, in 16HBE cells, all identifiable metabolites of the TCA cycle diminished after treatment with the combination of cysteamine and EGCG (Fig. 2B, Supplemental Fig. 1). Accordingly, the ratios between some of the metabolites were affected differentially in 16HBE and Cfbe41o- cells. This applies in particular to the ratio between pyruvate/oxaloacetate and malate or lactate (Supplemental Fig. 2). Altogether, these results point to subtle combination effects of cysteamine and EGCG that are affected by the cellular context (and presumably by the CFTR mutation).
Figure 1.

Heatmap visualization of metabolic profiling in 16HBE and Cfbe41o- cells. 16HBE and Cfbe41o- primary epithelial bronchial cell lines have been cultured overnight in absence or presence of 250 μM cysteamine and 100 μM epigallocatechin gallate (EGCG) alone or in combination. The heatmap depicts average levels of 198 metabolites detected by the combination of 3 analytical methods. Each cell corresponds to the treatment-group average abundancies (4–5 replicates) and both metabolites (rows) and conditions (columns) were clustered by the Ward method on the euclidean distance matrix. Metabolite's fold changes (FC) are shown in log2 scale as compared with their levels in 16HBE cells when cultured in complete medium (CM). Metabolites have been divided in 10 different subgroups as shown in the legend.
Figure 2.

Metabolic changes in tricarboxylic (TCA) cycle in Cfbe41o- cells. (A) Schematic representation of TCA cycle. Colors represent the fold change (FC) of each metabolite in Cfbe41o- cells treated overnight with 250 μM cysteamine (CYS) and 100 μM epigallocatechin gallate (EGCG) compared with its level when the same cell line was cultured in complete medium (CM). Metabolites that are not detected by the 3 metabolic analytical methods are depicted in gray. (B) Boxplot representation of the indicated TCA intermediates according to the metabolic profiling (Fig. 1). Data are shown as log2-normalized area of the relative metabolite peak. Asterisks indicate significant differences (moderate t-test) induced by the combination treatment (cysteamine and EGCG) in with respect to untreated controls for both 16HBE and Cfbe41o- cells. ***p < 0.001. ns: non-significant.
Effects of cysteamine and EGCG on protein acetylation and cross-linking
To further explore the metabolic effects of cysteamine and EGCG on respiratory epithelia, we cultured 16HBE and Cfbe41o- cells in the absence or presence of the 2 agents, alone or in combination, and determined the level of cytoplasmic protein acetylation by immunofluorescence.15 Both cysteamine and EGCG reduced the general protein acetylation in Cfbe41o- cells, and the combination of both drugs had a stronger effect than each of them alone (Fig. 3B). These effects were less pronounced on 16HBE cells (Fig. 3A). Neither cysteamine not EGCG reduced the cellular acetyl CoA concentration (Figs. 1 and 4A), and the combination actually enhances the absolute intracellular level of acetyl CoA, reduces CoA and hence elevates the acetyl CoA/CoA ratio (Supplemental Fig. 3). Since acetyl CoA (and the acetyl CoA/CoA ratio) is increased by cysteamine plus EGCG, yet protein acetylation diminishes, we wondered whether the 2 agents might affect the activity of the EP300 acetyltransferase (which already has been shown to be inhibited by EGCG).16 For this, we used a cell-free assay in which a purified recombinant protein corresponding to the enzymatically active fragment of EP300 was mixed with H3 histone, followed by addition of acetyl CoA and measurement of histone H3 acetylation on K56 by immunoblot using an antibody that recognizes acetyl-lysine residues (Fig. 4B). Like the positive control, anacardic acid, EGCG inhibited histone H3 acetylation, and this effect was accompanied by protein polymerization (and hence a global reduction of electrophoretic mobility). Addition of cysteamine was able to reduce this cross-linking effect of EGCG and simultaneously antagonized the inhibition of histone H3 acetylation mediated by EGCG. Hence, EP300 is unlikely to be a direct target of EGCG and cysteamine. However, this result reveals that the protein cross-linking activity of EGCG17,18 can be antagonized by cysteamine.
Figure 3.
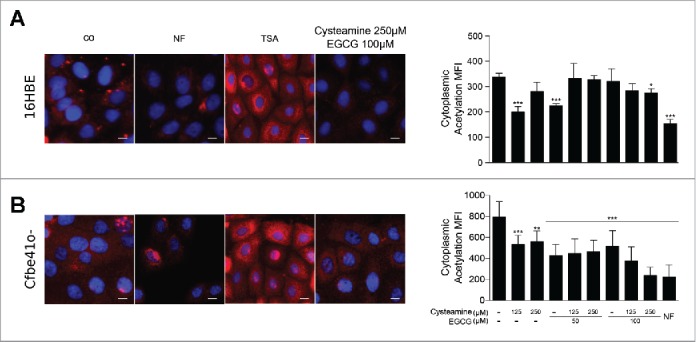
Effects of cysteamine and epigallocatechin gallate (EGCG) on cytoplasmic protein acetylation. 16HBE and Cfbe41o- primary epithelial bronchial cell lines were cultured overnight in absence (co) or presence of 125 μM or 250 μM cysteamine and 50 μM or 100 μM EGCG alone or in combination. Treatment with 10 μM Trichostatin A (TSA) or nutrient free (NF) medium for 5 h was used respectively as negative and positive controls for protein acetylation. Representative pictures and the relative quantifications are shown for 16HBE (A) and Cfbe41o- (B) cells. Data in the graphs are reported as means ± SD of 8 replicates; the experiments were repeated twice yielding similar results. Data were analyzed using Prism (GraphPad Software) or Excel (Microsoft Co., Redmond, WA, USA), and statistical significance was assessed by means of 2-tailed Student's t-test or ANOVA test, as appropriate. *p < 0.05, **p < 0.01, ***p < 0.001. MFI: mean fluorescence intensity. Size bar: 10 μm.
Figure 4.

Effects of cysteamine and epigallocatechin gallate (EGCG) on protein cross-linking in a cell-free system. (A) Boxplot representation of Acetyl CoA according to the metabolic profiling (Fig. 1). Data are shown as log2-normalized area for the relative metabolite peak. Asterisks indicate significant differences (moderate t-test) induced by each treatment with respect to untreated controls for both 16HBE and Cfbe41o- cells. (B) Schematic representation of the in vitro acetylation assay in a cell-free system. Reaction was performed using 1 μg H3 histone, 10 μM Acetyl CoA, 0.25 μg GST-EP300 together with cysteamine and EGCG. (C) Immunoblot for acetyl-lysine residues of the in vitro acetylation assay using the indicated concentrations of cysteamine and EGCG. Anacardic Acid (AA) has been used as control for EP300 activation with consequent reduction of H3 K56 levels. Total H3 immunoblot was performed as loading control. The nitrocellulose membrane was stained with Pierce™ Reversible Protein Stain Kit to assess quality of the tested samples (right side of the panel). The images are representative of one experiment that has been performed at least 3 times yielding similar results. ns: non-significant. **p < 0.01, ***p < 0.001.
Effects of EGCG on cysteamine metabolism
Cysteamine is metabolized by cysteamine dioxygenase to hypotaurine which then is enzymatically dehydrogenated to taurine. Hypotaurine concentrations increased in cysteamine treated cells by a factor of ∼4, although taurine levels were not affected (Fig. 5A). To investigate a possible drug interaction between cysteamine and EGCG, we cultured cells with non-radioactive isotope-labeled cysteamine (with 4 deuterium atoms in the carbohydrate chain) and followed the metabolism of cysteamine in the cells (Fig. 5B). Metabolic flux measurements indicated that EGCG strongly increased the accumulation of hypotaurine and taurine generated from deuterium-labeled cysteamine over the endogenous molecule (M4/M0 ratio, Fig. 5C). Hence, EGCG modulates the metabolism of cysteamine in cells, pointing to yet another potential interaction between the 2 drugs.
Figure 5.

Fluxomic analysis using isotope labeled cysteamine. (A) Boxplot representation of hypotaurine and taurine according to the metabolic profiling (Fig. 1). Data are shown as log2-normalized area for the relative metabolite peak. Asterisks indicate significant differences (moderate t-test) induced by each treatment with respect to untreated controls for both 16HBE and Cfbe41o- cells. (B) Metabolic conversion of cysteamine in taurine and hypotaurine. Cysteamine was labeled with 4 deuterium atoms corresponding to the hydrogens in the carbon chain. (C) Deuterium enrichment of hypotaurine and taurine after treatment with 250 μM of isotope-labeled cysteamine alone or in combination with 100 μM EGCG. Data are shown as the ratio between the levels of the metabolite enriched with 4 deuterium atoms (M4) and the metabolite enriched with no deuterium atoms (M0). Data are reported as means ± SD of 4 replicates; the experiments were repeated twice yielding similar results. **p < 0.01, ***p < 0.001. ns: non-significant.
Discussion
Our study reveals 3 potential interactions of cysteamine and EGCG at the metabolic level. First, the 2 agents together induce a few changes in the overall metabolome that are distinct from the effects of each of the compounds alone. Some of these cells depend on the cellular environment, as indicated by the comparison of CFTR wild type and CFTRΔF508 mutated cells. The differential effect of the combination regimen on the metabolome may effect the TCA cycle, carbohydrate, lipid and redox metabolism, as exemplified by differential effects on malate, xylitol, oleylcarnitine and γ-glutamylcysteine, respectively. Hence these effects are widespread, suggesting a major metabolic rewiring induced by the combination regimen. Both cysteamine and EGCG reduce cytoplasmic protein acetylation and hence act similar to Caloric restriction mimetics (CRMs). CRMs diminish protein acetylation through an increase in protein deacetylation or a reduction in protein acetylation. This latter effect can be achieved by depletion of acetyl-CoA or by direct inhibition of the enzymatic activity of acetyltransferases. However, neither cysteamine nor EGCG did reduce the intracellular acetyl-CoA level (and actually both together enhanced acetyl-CoA), and the combination was unable to directly inhibit the enzymatic activity of the EP300 acetyltransferase in a cell-free system. Therefore, reduced protein acetylation mediated by cysteamine and EGCG must be attributed to indirect effects on acetyl transferases (or the activation of deacetylases). Interestingly, the known protein cross-linking activity of EGCG17,18 was completely suppressed by cysteamine, pointing to an important functional crosstalk between the 2 agents. On yet another level, we observed that cysteamine metabolism was modulated by EGCG in the sense that the green tea flavonoid stimulated the enzymatic conversion of exogenous cysteamine into hypotaurine and taurine. Since this effect was more pronounced on hypotaurine than in taurine, it might reflect an increase in the activity of cysteamine dioxygenase. However, this effect was also measurable in metabolic flux experiments (in which the ratio of exogenous cysteamine-derived hypotaurine over endogenous cysteine-derived hypotaurine) increased under the influence of EGCG. However, the total cellular levels of hypotaurine did not increase with EGCG more than with cysteamine alone, and neither of the 2 compounds nor their combination was able to raise the concentration of intracellular taurine. This suggests that hypotaurine and taurine are rapidly metabolized or extruded from the cells as they are generated from cysteamine. Irrespective of these open possibilities, there is strong effect of EGCG on cysteamine metabolism that may contribute to the interaction between both agents.
In synthesis, the present study reveals 3 levels of interaction between EGCG and cysteamine with respect to their action on respiratory epithelia. First, both agents affect cellular metabolism through combined effects. Second, EGCG-mediated protein cross-linking can be inhibited by cysteamine. Third, EGCG enhances cysteamine metabolism in the cells. Altogether, these interactions are likely to contribute to the unique synergy of both agents in restoring the function of mutated CFTRΔF508.
Materials and Methods
Cell culture
Human lung bronchial epithelial cells 16HBE and Cfbe41o- (F508del/F508del-CFTR) were kindly provided by Dr. DC Gruenert (California Pacific Medical Center Research Institute, San Francisco, CA, USA) and cultured as recommended by American Type Culture Collection in Minimum Essential Medium Earle's salt (200 mM L-glutamine, 10% fetal bovine serum and 100 units/mL penicillin G sodium and 100 μg/mL streptomycin sulfate). Twenty-four h post seeding, cells were treated with 250 μM Cysteamine (M9768; Sigma-Aldrich, Saint Louis, MO, USA), 250 μM D4-Cysteamine (D-6501; CDN Isotopes), 100 μM epigallocatechin gallate (EGCG, E4143; Sigma-Aldrich) or their combination for 18 h before analysis.
Immunofluorescence
16HBE and Cfbe41o- cells were seeded in 384-well imaging plates (BD Falcon, Sparks, MD, USA) 24 h before stimulation. Cells were treated for 18 h with cysteamine, EGCG alone or in combination. In parallel, Nutrient free (NF) medium (EBSS, Earle's Balanced Salt Solution; E2888; Sigma-Aldrich) and 10 μM Trichostatin A (TSA; T8552; Sigma-Aldrich) were used respectively as negative and positive controls for protein acetylation. Then, the cells were fixed with 4% paraformaldeyde (PFA, w:v in PBS) for 15 min at room temperature. Non-specific binding sites were blocked with 5% bovine serum albumin in PBS (v:v) followed by overnight incubation at 4°C with acetyl-lysine primary antibody (623401; BioLegend) and the appropriate Alexa Fluor 568 conjugated secondary antibody (Molecular Probes-Invitrogen, Eugene, OR, USA). In the case of cytoplasmic acetyl-lysine staining, an additional step of blocking using anti acetylated-tubulin antibody was performed (#5335; Cell Signaling Technologies, Danvers, MA, USA). Nuclei were stained with 10 μM Hoechst 33342 (Molecular Probes-Invitrogen). Images were acquired using a BD pathway 855 automated microscope (BD Imaging Systems, San Jose, USA) equipped with a 40x objective (Olympus, Center Valley, PA, USA). For quantitative analyses of protein acetylation, cell surfaces were segmented into cytoplasmic and nucleic regions, and average staining intensity of each individual cell was measured for statistical analysis. Data are reported as means ± SD of 8 replicates and experiments were repeated twice yielding similar results. Data were analyzed using Prism (GraphPad Software, Inc., La Jolla, CA, USA) or Excel (Microsoft Co., Redmond, WA, USA), and statistical significance was assessed by means of 2-tailed Student's t-test or ANOVA tests, as appropriate.
In vitro acetylation assay
Recombinant GST-EP300 fusion protein, corresponding to amino acids 1066–1707 (14–418; Millipore, Billerica, MA, USA) was assessed for its acetyltransferase activity on the EP300 natural substrate recombinant histone H3 protein (M2503S; New England Biolabs, Ipswich, MA, USA). Briefly, 0.25 μg of EP300 HAT domain was incubated in the presence of an HAT assay buffer (250 mM Tris-HCl, pH 8.0, 50% glycerol, 0.5 mM EDTA and 5 mM dithiothreitol), 1 μg of histone H3 protein, 10 μM of acetyl-CoA (A2056; Sigma-Aldrich) as well as cysteamine and EGCG (alone or in combination) at the indicated concentration for 1 h at 30°C. Fifty μM Anacardic acid (A7236; Sigma-Aldrich) was used as internal positive control of the experiment. The reaction was stopped by adding 4x SDS buffer and boiling the samples. Acetylation of substrate proteins was measured by immunoblotting using specific antibodies against H3K56 (#4243; Cell Signaling Technologies) or H3 (#4499; Cell Signaling Technologies). Experiments were repeated at least 3 times yielding similar results.
Cultured cells preparation for metabolomics analysis
Cells were cultured in 6-well plates being at an approximate 80% of confluence the day of the experiment. After the corresponding treatment, wells plates are placed upon ice under chemical hood and processed as follows: wells are softly and quickly (< 2 s) rinsed with cold milliQ water (+4°C), 500 µL of cold methanol/water (9:1, v:v, −20°C, with internal standards) is added and finally scrapped for 30 s. Two wells from the same treatment were pooled in microcentrifuge tubes before adding cold chloroform (100 µL, −20°C). Solution is vortexed for 30 s and centrifuged at 15 000 rpm for 10 min at +4°C. The supernatant was collected and evaporated in microcentrifuge tubes at 40°C in a pneumatically-assisted concentrator (Techne DB3). Three hundred µL of methanol were added on dried extract and split in 2 parts of 150 µL: the first one was used for the GC-MS experiment, the second one was used for the LC-MS experimentation. Concerning the GC-MS aliquots, methanol-solubilized aliquots were transferred in a glass tube and evaporated again. Fifty µL of methoxyamine (20 mg/mL in pyridine) is added on dried extracts and then stored at room temperature in the dark for 16 h. The day after, 80 µL of trimethylsilyltrifluoroacetamide (MSTFA) is added and final derivatization occurs during 30 min at 40°C. Samples are then transferred in vials and directly injected into GC-MS. After the second evaporation of the LC-MS aliquots, the LC-MS dried extracts are solubilized with 300 µL of MilliQ water, centrifugate (10 min at 15000 g, +4°C) and aliquoted in 3 microcentrifuge tubes (100 µL). Aliquots are transferred in UHPLC vials and injected into UHPLC/MS or kept at −80°C until injection.19
Untargeted analysis of intracellular metabolites by ultra-high performance liquid chromatography (UHPLC) coupled to a quadrupole- time of flight (QTOF) mass spectrometer
Profiling of intracellular metabolites was performed on a RRLC 1260 system (Agilent Technologies, Waldbronn, Germany) coupled to a QTOF 6520 (Agilent Technologies) equipped with an electrospray source operating in both positive and negative mode and full scan mode from 50 to 1000 Da. The gas temperature was set at 350°C with a gas flow of 12 L/min. The capillary voltage was set at 3.5 kV, and the fragmentor at 120 V. Two reference masses were used to maintain the mass accuracy during analysis: m/z 121.050873 and m/z 922.009798 in positive mode and m/z 112.985587 and m/z 980.016375 in negative mode. Ten μL of sample were injected on a SB-Aq column (100 mm × 2.1 mm particle size 1.8 μm) from Agilent Technologies, protected by a guard column XDB-C18 (5 mm × 2.1 mm particle size 1.8 μm) and heated at 40°C. The gradient mobile phase consisted of water with 0.2% of acetic acid (A) and acetonitrile (B). The flow rate was set to 0.3 mL/min. Initial condition is 98% phase A and 2% phase B. Molecules are then eluted using a gradient from 2% to 95% phase B in 7 min. The column was washed using 95% mobile phase B for 3 min and equilibrated using 2% mobile phase B for 3 min. The autosampler was kept at +4°C.19
Targeted analysis of intracellular metabolites by ultra-high performance liquid chromatography (UHPLC) coupled to a Triple Quadrupole (QQQ) mass spectrometer
Targeted analysis was performed on a RRLC 1260 system (Agilent Technologies) coupled to a Triple Quadrupole 6410 (Agilent Technologies) equipped with an electrospray source operating in positive mode. The gas temperature was set at 350°C with a gas flow of 12 L/min. The capillary voltage was set at 3.5 kV. Ten μL of sample were injected on a Column Zorbax Eclipse plus C18 (100 mm × 2.1 mm particle size 1.8 µm) from Agilent technologies, protected by a guard column XDB-C18 (5 mm × 2.1 mm particle size 1.8 μm) and heated at 40°C. The gradient mobile phase consisted of 2 mM of dibutylamine acetate (73345; Sigma-Aldrich) in water (A) and acetonitrile (B). The flow rate was set to 0.2 ml/min, with gradient as follow: initial condition is 90% phase A and 10% phase B, maintained during 4 min. Molecules are then eluted using a gradient from 10% to 95% phase B over 3 min. The column was washed using 95% mobile phase B for 3 min and equilibrated using 10% mobile phase B for 3 min. The autosampler was kept at +4°C. The scan mode used is the MRM for biological samples. Peak detection and integration of 22 analytes were performed using the Agilent Mass Hunter quantitative software.19 Fifteen analytes were used for reporting and interpretation.
Widely-targeted analysis of intracellular metabolites gas chromatography (GC) coupled to a triple quadrupole (QQQ) mass spectrometer
The GC-MS/MS method was performed on a 7890A gas chromatography (Agilent Technologies) coupled to a triple quadrupole 7000C (Agilent Technologies) equipped with an electronic impact source (EI) operating in positive mode. The injection was performed in split less mode with a front inlet temperature set at 250°C. The transfer line and the ion-source temperature were respectively at 250°C and 230°C. The septum purge flow was fixed at 3 mL/min. The purge flow set to split vent and operated at 80 mL/min during 1 min. Gas saver mode was set at 15 mL/min after 5 min. The helium gas flowed at 1 mL/min through the column (J & W Scientific HP-5MS, 30 m × 0.25 mm, i.d. 0.25 mm, d.f., Agilent Technologies). Column temperature was held at 60°C for 1 min, then raised to 210°C (10°C/min), followed by a step to 230°C (5°C/min) and reached 325°C (15°C/min), and be hold at this temperature for 5 min. The collision gas was nitrogen. The scan mode used is the MRM for biological samples. Peak detection and integration of the 122 analytes were performed using the Agilent Mass Hunter quantitative software.19 Ninety-one were used for reporting and interpretation.
Fluxomic analysis
The same extraction protocol as above applies excepted that the extraction solvent did not contain any labeled internal standards. Derivatization of GC aliquot was handle in glass tubes. Fifty µL of O-ethylhydroxylamine hydrochloride (20 mg/mL in pyridine, Sigma-Aldrich) was added to the dried extracts, stored at room temperature in the dark for 16 h. Silylation was carried out with 80 µL of N-tert-butyldimethylsilyl-N-methyltrifluoroacetamid (MSTBFA, Sigma-Aldrich) at 40°C for 60 min. The GC-MS/MS method was performed on a 7890A gas chromatography (Agilent Technologies) coupled to a triple quadrupole 7000C (Agilent Technologies) equipped with an EI operating in positive mode. The front inlet temperature was 250°C, the injection was performed in split less mode. The transfer line and the ion-source temperature were 250°C and 230°C, respectively. The septum purge flow was fixed at 3 mL/min, the purge flow to split vent operated at 80 mL/min during 1 min and gas saver mode was set to 15 mL/min after 5 min. The helium gas flowed through the column (J & W Scientific HP-5MS, 30 m × 0.25 mm, i.d. 0.25 mm, d.f., Agilent Technologies) at 1 mL/min. Column temperature was held at 70°C for 1 min, then raised to 120°C (15°C/min), followed by a step of 1 min at this temperature, and a second step to reach 325°C (10°C/min), holding this temperature for 3 min. The collision gas was nitrogen. The scan mode used is the MRM for biological samples with the Q1 corresponding to the maximally derivatized molecule and with Q3 corresponding to a typical fragment of silyl group (73 or 147). Peak detection and integration of the isotopomers of 37 analytes were performed using the Agilent Mass Hunter quantitative software (B.07.01). After manual verification and QC protocols, 31 isotopomer distributions entered data analysis and interpretation.
Statistical analysis
Statistical analyses and representation of the metabolomics data were performed on the log2 transformed peak areas centered on the untreated 16HBE samples. Data are reported as such without back-transformation. For differential analysis, moderated statistics were computed from the original design with the explicit p-values reported for each comparison of interest.20
Supplementary Material
Disclosure of potential conflicts of interest
V.R., L.M. and G.K. are listed as inventors on a patent application describing the use of cysteamine for the treatment of CF.
Funding
GK is supported by the Ligue contre le Cancer (équipes labelisées); Agence Nationale de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Fondation Bettencourt-Schueller; Fondation de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); the Swiss Bridge Foundation, ISREC and the Paris Alliance of Cancer Research Institutes (PACRI). LM and VR are sustained by IERFC non-profit foundation, Regional Cystic Fibrosis Associations of Campania, Sicilia, Lazio, Puglia and E-Rare (Rescue CFTR preclinic).
References
- [1].Drumm ML, Ziady AG, Davis PB. Genetic variation and clinical heterogeneity in cystic fibrosis. Annu Rev Pathol 2012; 7:267-82; PMID:22017581; http://dx.doi.org/ 10.1146/annurev-pathol-011811-120900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet 2015; 16:45-56; PMID:25404111; http://dx.doi.org/ 10.1038/nrg3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Luciani A, Villella VR, Esposito S, Gavina M, Russo I, Silano M, Guido S, Pettoello-Mantovani M, Carnuccio R, Scholte B, et al.. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on DeltaF508 cystic fibrosis transmembrane conductance regulator. Autophagy 2012; 8:1657-72; PMID:22874563; http://dx.doi.org/ 10.4161/auto.21483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Villella VR, Esposito S, Bruscia EM, Vicinanza M, Cenci S, Guido S, Pettoello-Mantovani M, Carnuccio R, De Matteis MA, Luini A, et al.. Disease-relevant proteostasis regulation of cystic fibrosis transmembrane conductance regulator. Cell Death Differ 2013; 20:1101-15; PMID:23686137; http://dx.doi.org/ 10.1038/cdd.2013.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].De Stefano D, Villella VR, Esposito S, Tosco A, Sepe A, De Gregorio F, Salvadori L, Grassia R, Leone CA, De Rosa G, et al.. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 2014; 10:2053-74; PMID:25350163; http://dx.doi.org/ 10.4161/15548627.2014.973737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tosco A, De Gregorio F, Esposito S, De Stefano D, Sana I, Ferrari E, Sepe A, Salvadori L, Buonpensiero P, Di Pasqua A, et al.. A novel treatment of cystic fibrosis acting on-target: cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ 2016; 23:1380-93; PMID:27035618; http://dx.doi.org/ 10.1038/cdd.2016.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Besouw M, Masereeuw R, van den Heuvel L, Levtchenko E. Cysteamine: an old drug with new potential. Drug Discov Today 2013; 18:785-92; PMID:23416144; http://dx.doi.org/ 10.1016/j.drudis.2013.02.003 [DOI] [PubMed] [Google Scholar]
- [8].Jeon JH, Lee HJ, Jang GY, Kim CW, Shim DM, Cho SY, Yeo EJ, Park SC, Kim IG. Different inhibition characteristics of intracellular transglutaminase activity by cystamine and cysteamine. Exp Mol Med 2004; 36:576-81; PMID:15675041; http://dx.doi.org/ 10.1038/emm.2004.74 [DOI] [PubMed] [Google Scholar]
- [9].Legeay S, Rodier M, Fillon L, Faure S, Clere N. Epigallocatechin Gallate: A Review of Its Beneficial Properties to Prevent Metabolic Syndrome. Nutrients 2015; 7:5443-68; PMID:26198245; http://dx.doi.org/ 10.3390/nu7075230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chakrawarti L, Agrawal R, Dang S, Gupta S, Gabrani R. Therapeutic effects of EGCG: a patent review. Expert Opin Ther Pat 2016; 26:907-16; PMID:27338088; http://dx.doi.org/ 10.1080/13543776.2016.1203419 [DOI] [PubMed] [Google Scholar]
- [11].Galluzzi L, Kroemer G. Common and divergent functions of Beclin 1 and Beclin 2. Cell Res 2013; 23:1341-2; PMID:24018378; http://dx.doi.org/ 10.1038/cr.2013.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sahni S, Merlot AM, Krishan S, Jansson PJ, Richardson DR. Gene of the month: BECN1. J Clin Pathol 2014; 67:656-60; PMID:24811486; http://dx.doi.org/ 10.1136/jclinpath-2014-202356 [DOI] [PubMed] [Google Scholar]
- [13].Devereux G, Fraser-Pitt D, Robertson J, Devlin E, Mercer D, O'Neil D. Cysteamine as a future intervention in cystic fibrosis against current and emerging pathogens: A patient-based ex vivo study confirming its antimicrobial and mucoactive potential in sputum. EBioMedicine 2015; 2:1507-12; PMID:26629546; http://dx.doi.org/ 10.1016/j.ebiom.2015.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vidigal PG, Musken M, Becker KA, Haussler S, Wingender J, Steinmann E, Kehrmann J, Gulbins E, Buer J, Rath PM, et al.. Effects of green tea compound epigallocatechin-3-gallate against Stenotrophomonas maltophilia infection and biofilm. PLoS One 2014; 9:e92876; PMID:24690894; http://dx.doi.org/ 10.1371/journal.pone.0092876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pietrocola F, Lachkar S, Enot DP, Niso-Santano M, Bravo-San Pedro JM, Sica V, Izzo V, Maiuri MC, Madeo F, Marino G, et al.. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ 2015; 22:509-16; PMID:25526088; http://dx.doi.org/ 10.1038/cdd.2014.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Choi KC, Jung MG, Lee YH, Yoon JC, Kwon SH, Kang HB, Kim MJ, Cha JH, Kim YJ, Jun WJ, et al.. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res 2009; 69:583-92; PMID:19147572; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2442 [DOI] [PubMed] [Google Scholar]
- [17].Chen R, Wang JB, Zhang XQ, Ren J, Zeng CM. Green tea polyphenol epigallocatechin-3-gallate (EGCG) induced intermolecular cross-linking of membrane proteins. Arch Biochem Biophys 2011; 507:343-9; PMID:21211509; http://dx.doi.org/ 10.1016/j.abb.2010.12.033 [DOI] [PubMed] [Google Scholar]
- [18].Chu C, Deng J, Xiang L, Wu Y, Wei X, Qu Y, Man Y. Evaluation of epigallocatechin-3-gallate (EGCG) cross-linked collagen membranes and concerns on osteoblasts. Mater Sci Eng C Mater Biol Appl 2016; 67:386-94; PMID:27287135; http://dx.doi.org/ 10.1016/j.msec.2016.05.021 [DOI] [PubMed] [Google Scholar]
- [19].Enot DP, Niso-Santano M, Durand S, Chery A, Pietrocola F, Vacchelli E, Madeo F, Galluzzi L, Kroemer G. Metabolomic analyses reveal that anti-aging metabolites are depleted by palmitate but increased by oleate in vivo. Cell Cycle 2015; 14:2399-407; PMID:26098646; http://dx.doi.org/ 10.1080/15384101.2015.1064206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43:e47; PMID:25605792; http://dx.doi.org/ 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.