

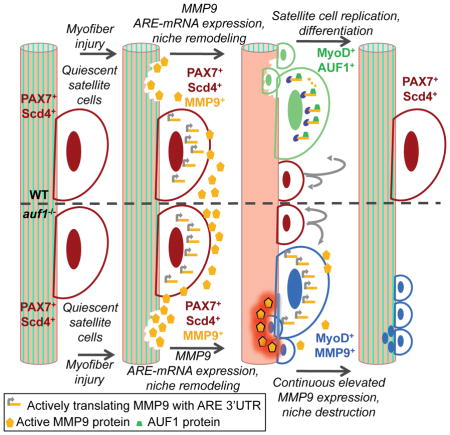
SUMMARY
Following skeletal muscle injury, muscle stem cells (satellite cells) are activated, proliferate, and differentiate to form myofibers. We show that mRNA decay protein AUF1 regulates satellite cell function through targeted degradation of specific mRNAs containing 3′ AU-rich elements (AREs). Auf1−/− mice undergo accelerated skeletal muscle wasting with age and impaired skeletal muscle repair following injury. Satellite cell mRNA analysis and regeneration studies demonstrate that auf1−/− satellite cell self-renewal is impaired due to increased stability and overexpression of ARE-mRNAs, including cell-autonomous overexpression of matrix metalloprotease MMP9. Secreted MMP9 degrades the skeletal muscle matrix, preventing satellite cell-mediated regeneration and return to quiescence. Blocking MMP9 activity in auf1−/− mice restores skeletal muscle repair and maintenance of the satellite cell population. Control of ARE-mRNA decay by AUF1 represents a mechanism for adult stem cell regulation and is implicated in human skeletal muscle wasting diseases.
Keywords: AUF1, AU-rich elements, mRNA decay, satellite cells, muscle stem cells, muscle regeneration
Graphical Abstract
INTRODUCTION
Many key regulatory mRNAs are controlled through post-transcriptional mechanisms, typically the targeted destabilization of the mRNA, its selective translation, or both (Moore et al., 2014). The regulated stability of mRNAs generally comprises those that must respond quickly in abundance to changing stimuli. In fact, almost half of the changes in physiologically rapid inducible gene expression occur at the level of mRNA stability (Cheadle et al., 2005). Many physiologically potent proteins are encoded by short-lived mRNAs with half-lives of minutes, in which mRNA destabilization is conferred by AREs in the 3′ untranslated region (3′UTR). A common ARE motif consists of the sequence AUUUA, typically repeated multiple times in the 3′UTR, often contiguously (Moore et al., 2014). The ARE is purely a cis-acing element that serves as a binding site for regulatory proteins known as AU-rich binding proteins (AUBPs) which bind the ARE with high affinity and control mRNA stability or translation. Several AUBPs have been well studied to date, and all act by recruiting mRNA decay, mRNA stabilizing or translation arrest proteins (Gratacos and Brewer, 2010). AUBPs also have different and overlapping target ARE-mRNAs (Garneau et al., 2007; Kim et al., 2009). ARE-mRNAs are thought to encode more than 5% of the protein expressed genome (Gruber et al., 2010).
AU-rich binding factor 1 (AUF1, HNRNPD) binds certain ARE-mRNAs to promote their rapid degradation through association with trans-acting RNA binding proteins (Moore et al., 2014). AUF1 primarily functions as an ARE-mRNA decay factor (Moore et al., 2014); von Roretz et al., 2011). AUF1 consists of four related protein isoforms named for their molecular weights (p37, p40, p42, p45), derived by differential exon splicing of a common pre-mRNA (Wagner et al., 1998). While not fully understood, the different isoforms target ARE-mRNAs for decay and interact in a variety of homo- and hetero-complexes (Moore et al., 2014). AUF1 isoforms have also been implicated in transcriptional activation of several genes based on their multimer formation and cellular distribution (Pont et al., 2012; Zucconi et al., 2010), as AUF1 isoforms can shuttle between the nucleus and cytoplasm at different efficiencies (He and Schneider, 2006; Sarkar et al., 2003a; Sarkar et al., 2003b).
We previously developed an auf1−/− mouse to better understand its physiological roles (Lu et al., 2006). We now report that auf1−/− mice undergo accelerated skeletal muscle wasting as adults. Skeletal muscle maintenance and regeneration in adults requires activation and differentiation of normally quiescent satellite cells, a skeletal muscle specific stem cell population (Lipton and Schultz, 1979). Satellite cells reside under the basal lamina, adjacent to myofibers, the bundles of muscle fibers that provide muscle mass and strength. Following muscle injury, satellite cells respond through rapid proliferation and differentiation, recapitulating the process of myogenesis (the formation of muscle tissue), including fusion to form myofibers (Brack, 2014; Gunther et al., 2013; Kudryashova et al., 2012; Lepper et al., 2009). Satellite cells must also self-renew and quiesce to prevent their depletion. Satellite cells therefore divide asymmetrically, enabling a small number of stem cells to return to quiescence, in part mediated through interaction with the satellite cell niche (Kuang et al., 2007). The satellite cell niche is loosely defined as the intact laminin-basement membrane structure that provides poorly characterized extrinsic factors crucial for their maintenance (Bernet et al., 2014; Carlson and Conboy, 2007; Collins et al., 2005; Kuang et al., 2007; Montarras et al., 2005; Zammit et al., 2004).
Skeletal muscle regeneration is impaired in age-related diseases such as sarcopenia, which is marked by reduced muscle regenerative capacity with aging, partly due to a reduction in satellite cells (Bernet et al., 2014; Carlson and Conboy, 2007; Shefer et al., 2006). Loss of the satellite cell population may result from a release of cell cycle inhibition (Sousa-Victor et al., 2014) and/or loss of the niche (Gopinath and Rando, 2008). These events are also common to certain adult late onset myopathies, including Limb Girdle Muscular Dystrophy (LGMD). LGMD is a heterogeneous family of diseases characterized clinically by adult onset rapid loss of skeletal muscle in the pelvic, scapular, shoulder, and limb regions of the body (Brack, 2014; Gunther et al., 2013; Kudryashova et al., 2012; Lepper et al., 2009).
We therefore sought to elucidate the role of AUF1 in skeletal muscle maintenance with age. Here we show that AUF1-mediated control of satellite cell ARE-mRNA stability is a key regulator of muscle stem cell fate and regenerative capacity in mice. Auf1−/− satellite cells show increased expression of key regenerative ARE-mRNAs, including mmp9, that regulate integrity of the muscle stem cell niche. By altering the niche, impaired auf1−/− satellite cell self-renewal leads to impaired muscle regeneration, satellite cell depletion and ultimately promotes late onset myopathy.
RESULTS
Auf1−/− mice increasingly lose muscle mass and strength with age
A severe loss in skeletal muscle mass was apparent in auf1−/− (auf1 Knockout, KO) skeletal muscle relative to wild type (WT) littermates by 6 months of age, as evident in anatomical images (Figure 1A). The progressive loss of skeletal muscle mass in auf1−/− mice was quantified by dual energy x-ray absorptiometry (DEXA), which measures body composition, including skeletal muscle mass (Harada, 2013; Marinangeli and Kassis, 2013). DEXA analysis showed a reduction in auf1−/− mouse skeletal muscle mass of 50% by 6 months of age, and 85% by 9 months (Figure 1B). Thus, there is a severe, progressive loss of skeletal muscle mass associated with age in auf1−/− mice. We therefore determined whether the loss of muscle mass corresponds to a loss of muscle strength as well, using two traditional approaches. The inverted cage flip quantifies the amount of time a mouse can hold on to the bottom of a cage. This is a measure of both limb and pectoral strength. The grip strength test quantifies fore limb strength in countering an opposite pull. The age-related reduction in skeletal muscle mass corresponded to a 92% loss in whole body skeletal muscle strength, measured by the inverted cage flip test (Figure 1C; Video S1, S2). The significant loss of limb muscle strength was independently validated by the grip strength test (Figure S1).
Figure 1. Auf1−/− mice show accelerated muscle wasting.

(A) Visual comparison of skeletal muscle mass between 4 month old WT and KO mice.
(B) DEXA measurement of lean muscle mass normalized to total mass in young (3–5 months old), mid-range (6–8 months old), and old (9–12 months old) WT (black) and KO (grey) mice. Five mice per genotype per age group were tested in triplicate. * P<0.05, ** P<0.005, unpaired t-test.
(C) Results of limb and pectoral strength test by inverted cage flip lasting up to 60 sec in young (3–5 months old), mid-range (6–8 months old), and old (9–12 months old) WT (black) and KO (grey) mice. Five mice per genotype per age group were tested in triplicate. *P<0.05 unpaired t-test.
(D) Immunofluorescence analysis of skeletal muscle laminin (AF488, green) and nuclei (DAPI, blue) in 4 and 8 month old mice. Representative centralized nuclei are denoted by arrowheads (<). TA muscles were frozen in OCT, 5 images from 3 sections were analyzed per mouse and 3 mice were studied per genotype (scale bar 50 μm).
(E) Quantification of centralized nuclei in 4 month old and 8 month old WT (black) and KO (patterned) mice. **P<0.005, by 2-way ANOVA.
Histologically, young auf1 knockout (KO) mice initially develop normal skeletal muscle, shown by laminin staining of muscle fibers at 4 months of age, indistinguishable from WT animals (Figure 1D). At this time, laminin completely outlines rounded skeletal muscle fibers with virtually all peripheral myonuclei (Figure 1E), a key measure of normal muscle development (Wicklund and Kissel, 2014). However, auf1 KO mice develop a drastic myopathy phenotype by 8 months of age, particularly showing signs of muscle wasting (Figure 1D). This is visually apparent in deficient and disrupted laminin staining indicative of a loss of muscle integrity, including skeletal muscle fibers that are incomplete with pronounced degradation. Notably, the 8 month old auf1 KO phenotype includes a >5-fold increase in centrally located nuclei within muscle fibers (Figure 1E), indicative of ongoing satellite cell regeneration efforts and a phenotypic hallmark of myopathic disease (Wicklund and Kissel, 2014).
AUF1 shows exclusive expression in activated satellite cells within the muscle
Studies have shown that AUF1 is expressed at extremely low or negligible levels in skeletal muscle fibers (Lu and Schneider, 2004) (Figure 2A, B). We therefore screened for AUF1 expression using immunofluorescence specifically in the quiescent and activated satellite cell population in vivo following injury, and in vitro on isolated skeletal muscle fibers. Quiescent satellite cells are identified by expression of PAX7 and Syndecan-4 (Sdc4), while activated satellite cells additionally gain expression of myogenic regulatory factors (MRFs), such as MyoD (Cornelison and Wold, 1997; Seale and Rudnicki, 2000). There was little or no detectable AUF1 expression in quiescent satellite cells prior to muscle injury. However, AUF1 was co-expressed in ~25% of the activated PAX7+ satellite cells 7 days post-injury (Figure 2A, Figure S2). In both the uninjured and the 7 days post-injury skeletal muscle, AUF1 expression was not observed in the skeletal muscle fibers (Figure 2A). AUF1 is therefore specifically expressed in a subset of activated satellite cells.
Figure 2. AUF1 is expressed in activated satellite cells.

(A) Immunofluorescence analysis for expression of laminin (AF488, green), PAX7 (AF 555, red), AUF1 (AF647, white) and nuclei (DAPI, blue) in uninjured (UI) or 7 day post-injury TA muscle in 4 month old WT mice. TA muscle was injured by BaCl2 injection. TAs were frozen in OCT, 5 images from 3 sections were analyzed per mouse (scale bar 50 μm). DAPI + 2nd is a background control, sections stained with DAPI and secondary antibody only.
(B) Immunofluorescence analysis for expression of AUF1 (AF488, green), MyoD (AF555, red), and nuclei (DAPI, blue) in myofibers isolated from WT skeletal muscle from 4 month old mice. Ten fibers were analyzed per mouse and three mice were studied (scale bar 50 μm).
To further validate restriction of AUF1 expression to activated satellite cells, skeletal muscle fibers were isolated, cultured, and screened for AUF1 co-expression with MyoD, an early time point MRF. The isolation of muscle fibers activates associated satellite cells that attempt to repair the sensed “wound” by differentiation. At 72 hours of culture, AUF1 was strongly co-expressed in >50% of the MyoD+ satellite cells (Figure 2B, Figure S2). Of note, AUF1 distribution was found to be nuclear and cytoplasmic, indicative of increased cytoplasmic ARE-mRNA decay function. AUF1 has been shown to shuttle between the nucleus and the cytoplasm; the cytoplasm being where it promotes ARE-mRNA decay. At steady-state AUF1 is primarily nuclear with export to the cytoplasm occurring as a result of specific mRNA association for decay (Moore et al., 2014; Sarkar et al., 2003a; Suzuki et al., 2005; Yoon et al., 2015; He and Schneider, 2006). Collectively, these data demonstrate that AUF1 is only expressed in activated satellite cells in skeletal muscle, and not in muscle fibers.
Independent confirmation of the AUF1 temporal expression profile was obtained using the murine myoblast C2C12 cell line. C2C12 cells can mimic the post-activated satellite cell state initiating at the progenitor myoblast level (Ho et al., 2015; Silva et al., 2015). The three largest AUF1 isoforms (p40, p42, p45) are expressed in the proliferating myoblast (Fig. S3) and significantly increased through the early stages of myoblast differentiation, similar to the differentiation of skeletal muscle satellite cells in the animal. The smallest isoform (p37) is minimally expressed in the fully differentiated myotubes (Figure S3).
AUF1 regulation of ARE-mRNA stability controls satellite cell fate following muscle injury
We next determined whether satellite cells and their regenerative capacity are functionally altered in the absence of AUF1-mediated regulation of ARE-mRNA stability. To activate quiescent satellite cells and initiate the program of muscle stem cell differentiation, the tibialis anterior (TA) muscle was injured by injection of BaCl2 in WT and auf1 KO mice at 4 months of age. BaCl2 causes rapid necrosis of the injected muscle. At this time point, as shown earlier, uninjured skeletal muscle from auf1 KO mice is morphologically normal (Figure 1E), and expression of AUF1 in the quiescent satellite cell population is almost undetectable (Figure 1D, Figure S4). Furthermore, there are no changes in the number of quiescent satellite cells present in the TA of WT and auf1 KO mice at 4 months of age (Figure 3C).
Figure 3. Auf1−/− satellite cells are unable to self-renew once activated.

(A) Immunofluorescence analysis for expression of laminin (AF-488, green), PAX7 expression (AF555, red), and nuclei (DAPI, blue) in 7 or 15 day post-injury skeletal muscle in 4 month old WT and KO mice. TA muscle was injured by BaCl2 injection. TA muscles were frozen in OCT, 5 images from 3 sections were analyzed per mouse (scale bar 50 μm). DAPI + 2nd is a control, sections stained with DAPI and secondary antibody only.
(B) Quantification of fiber size by Minimum Feret’s Diameter determined by ImageJ64 at 15 day post-injury in WT (black) and KO (grey) mice. Bars denoting significance represent the range in which the Minimum Feret’s Diameter is significantly different between WT and auf1 KO TA muscle. *P<0.05, **P<0.005, unpaired t-test.
(C) Quantification of PAX7 expression in WT and KO mice in uninjured (black), 7 day post-injury (patterned), and 15 day post-injury (grey) skeletal muscle. *P<0.05, **P<0.005, 2way ANOVA.
At 7 days post-injury the TA muscle from auf1 KO mice is unable to undergo repair compared to WT animals. There is sparse laminin expression with very few identifiable myofibers, indicative of loss of muscle fiber integrity and disruption of the satellite cell niche (Figure 3A). Evidence for loss of the satellite cell niche includes a >5-fold reduction in activated PAX7+ satellite cells in the auf1 KO TA muscle relative to WT (Figure 3A, C). At 15 days post-injury, auf1 KO TA muscle continues to show little regeneration in comparison to the near complete repair in WT mice. Laminin staining in the auf1 KO TA muscle indicates that significantly smaller and incomplete muscle fibers are formed (Figure 3A), quantified by determining the size (Minimum Feret’s Diameter) of muscle fibers (Figure 3B). There is also a significant depletion of the PAX7+ satellite cell population in the auf1 KO TA muscle at this time (Figure 3A), quantified by scoring the number of PAX7+ cells per mm2 (Figure 3C). AUF1 expression is therefore crucial for maintenance and re-establishment of both the satellite cell niche and the PAX7+ stem cell population.
Since the primary function of AUF1 is to target ARE-mRNAs for rapid decay, we sought to identify mRNAs with altered abundance in sorted satellite cells from auf1 KO mice compared to WT. We conducted genome-wide, satellite cell-specific RNA-Sequencing (RNA-seq) mRNA expression analysis. Satellite cells were isolated from auf1 WT and auf1−/− KO mouse whole hind limb skeletal muscle from 4–6 month old animals by fluorescence-activated cell sorting (FACS), gating on cells positive for satellite cell marker Sdc4 and negative for endothelial cell markers. Ninety-one mRNAs were altered in abundance in auf1 KO compared to WT satellite cells, with ~75% (~70 mRNAs) showing >2-fold increased or decreased abundance. Of these, 34/70, or almost half, were mRNAs containing 3′UTRs with putative AUF1/AUBP-binding AREs based on the ARE-motif AUUUA, typically with at least two contiguous AUUUA sequences required for AUF1 binding (Moore et al., 2014). Additionally, the majority of the ARE-mRNAs were increased in abundance, supporting the role of AUF1 in promoting ARE-mRNA decay in the stem cell population (Figure 4A, Table S1). Interestingly, 18 mRNAs were increased in abundance only in auf1 KO satellite cells and were not detectable in the WT, of which 8 contain 3′UTR multiple AREs, including established targets of AUF1 such as IL17 (Han et al., 2014). Other established ARE-mRNA targets of AUF1 increased in abundance in auf1 KO satellite cells compared to WT, and include IL10 (Sarkar et al., 2008), MMP9 (Liu et al., 2006), GBP1 and SAMSN1 (Sarkar et al., 2011), and IL1β (Pont et al., 2012).
Figure 4. MMP9 transcript levels are significantly higher in the auf1−/− satellite cells.

(A) Heat map of RNA-Seq analysis from sorted WT and KO satellite cells. Three mice per genotype were studied. Ninety-one genes were differentially expressed in KO satellite cells with the majority showing increased expression (red).
(B) IPA characterization of top cellular function and disease pathways for satellite cell ARE-mRNAs dysregulated in the absence of AUF1 expression. Numbers represent P-value x 10−5.
(C) mRNA levels of AUF1 and MMP9 from cultured C2C12 cells treated with vehicle (black) or siAUF1 (grey). Two siAUF1 targeting sequences were used. mRNA levels were normalized to GapDH. Each experiment was performed in triplicate. *P<0.05, **P<0.005, unpaired t-test.
(D) Relative MMP9 mRNA decay rate in cultured C2C12 cells treated with control (black) or siAUF1-1 (grey). Cells were collected post-actinomycin D treatment and RNA isolated per manufacturer instructions (TRIzol). Partial decay curve is shown. Inset: immunoblot of AUF1 levels post-silencing. *,P<0.001, unpaired t-test.
(E) RNA-Immunoprecipitation of IgG (black) or endogenous AUF1 (grey) in C2C12 cells analyzed for MMP9 and ITGB1 mRNA levels. Experiments were performed in triplicate. ***P<0.0005, paired t-test. ITGB1 mRNA levels were not statistically different.
In silco analysis was next performed to identify favorable AUF1-regulated ARE-mRNAs, focusing on mRNAs upregulated in the auf1 KO satellite cells consistent with the primary function of AUF1 in mediating ARE-mRNA decay. mRNAs with at least one canonical ARE motif (AUUUA) in the 3′-UTR were identified using ARESite (Table S1 identified by *). These mRNAs were further prioritized as AUF1-prefered targets based on established AUF1 preference for at least two ARE pentamers, often adjacent (Gratacos and Brewer, 2010)(Table S1 identified by **). The prioritized gene list was subjected to Ingenuity Pathway Analysis (IPA) to determine functional clusters. IPA assigns gene lists to experimentally authenticated biochemical and molecular networks.
IPA analysis revealed that upregulated mRNAs were enriched for functions including cell movement, cell-to-cell signaling, cell maintenance and cell growth (Figure 4B). These pathways provide crucial signaling for the proper activation, differentiation, and self-renewal of stem cells in adult tissue. Notably, the upregulated MMP9 transcript was identified in most of these cellular function pathways. We characterized the importance of the genes identified by IPA analysis by established function in skeletal muscle regeneration. Four ARE-mRNAs were identified (Table S2) with two (IL17, MMP9) having been previously shown to bind AUF1 in other cell types (Han et al., 2014).
MMP9 has a central importance in muscle regeneration and wound repair (Webster et al., 2015; Gu et al., 2005; Hindi et al., 2013; Murase and McKay, 2012) and was found in most of the relevant pathways analyses we conducted. MMP9 is a matrix metallopeptidase that degrades extracellular matrix (ECM) proteins, including skeletal muscle laminin, a component of the satellite cell niche (Gu et al., 2005; Hindi et al., 2013; Murase and McKay, 2012). While controlled remodeling of the ECM is required for skeletal muscle regeneration, excessive and/or continuous post-wounding MMP9 activity would be predicted to deregulate satellite cell function and impair stem cell regenerative capacity through chronic degradation of the surrounding matrix (Webster et al., 2015; Shiba et al., 2015). Accordingly, inhibition of MMP9 has been shown to improve skeletal muscle repair in certain models of muscular dystrophy (Hindi et al., 2013; Li et al., 2009; Shiba et al., 2015). Moreover, the extensive pathological effects of muscle wounding in auf1 KO mice are consistent with the predicted phenotype of increased MMP activity.
Importantly, the source of MMP9 expression during muscle wound repair and its pathological relevance when overexpressed have not been studied. We therefore first confirmed that changes in MMP9 and other mRNAs identified by genome-wide satellite cell RNA-seq analysis are in fact satellite cell autonomous. To do so, we conducted a genome-wide gene expression analysis of mRNAs in the WT and auf1 KO mouse skeletal muscle fibers taken from 4–6 month old animals (Figure S5). MMP9 mRNA was undetectable in both WT and KO skeletal muscle fibers, indicating that MMP9 expression is solely satellite cell-autonomous, and the source of MMP9 overexpression in auf1 KO mice.
Verification that AUF1 promotes MMP9 mRNA degradation was obtained in C2C12 myoblast cells, since it is not feasible to study mRNA decay rates in the animal satellite cell population. Silencing of AUF1 by two different siRNAs (~80%) increased MMP9 mRNA levels by ~4 fold (Figure 4C), consistent with that identified in the RNA-Seq data from satellite cells. MMP9 mRNA relative half-life, determined by addition of actinomycin D to block transcription, and qRT-PCR quantitation was increased from 1 h in vehicle treated controls to 4.5 h in C2C12 cells treated with siAUF1 (~80% silenced) (Figure 4D). To confirm that MMP9 mRNA destabilization is the result of AUF1 action on the ARE repeats in the 3′UTR, the longest contiguous ARE-rich region (~200 bps) was cloned into the 3′UTR of a luciferase reporter (pzeo-luc). The MMP9-3′-Luc reporter or its control (Empty-3′-Luc) were transfected into C2C12 cells with either non-silencing or siAUF1 treatment. The control mRNA had a relative stable half-life of 112 min, which was significantly reduced to 28 min by the insertion of the MMP9 3′-UTR ARE fragment (MMP9-3′-Luc) (Figure S6A). siAUF1 treatment, which partially silenced AUF1 by 50–70%, partially rescued MMP9-3′-Luc mRNA stability (81 min). These data confirm that AUF1 promotes the decay of the MMP9 mRNA through the AU-rich 3′-UTR (Figure S6A). Further validating these results, cells treated with siAUF1 showed significantly increased luciferase activity, validating the role of AUF1 in promoting MMP9 instability (Figure S6B).
Additionally, MMP9 mRNA was found strongly bound to immunoprecipitated AUF1 from WT C2C12 cells (Figure 4E, Figure S7A). A suspected AUF1 target mRNA in certain cell types, Integrinβ-1 (ITGB1), that was not altered in the satellite cell RNA-Sequencing data was used as a control. ITGB1 did not associate with AUF1 in the C2C12 cells, validating the interaction with MMP9 (Figure 4E). Moreover, silencing of AUF1 in C2C12 cells strongly increased secreted MMP9 protein levels (Figure S7B, C).
MMP9 inhibition rescues depletion of auf1−/− satellite cells following injury
We determined in vivo in live animals whether loss of AUF1-targeted decay of the MMP9 ARE-mRNA is in large part responsible for the post-injury muscle regeneration defect, validating that this phenotype is caused by overexpression from auf1 KO satellite cells. We used live animal imaging to visualize MMP9 activity in auf1 WT and auf1−/− KO TA skeletal muscle at 24 h post-injury in 4 month-old mice. This corresponds to a time point at which satellite cells are activated in the absence of an immune infiltrate (Dumont et al., 2015). Mice were injected intraperitoneally (IP, abdominal cavity) with an optically silent collagen matrix analog designed for selective MMP9 cleavage starting 24 hours prior to injury. Once cleaved, the matrix releases a fluorophore localized to the site of MMP9 activity. MMP9 activity at the site of repeated needle IP injections is expected. Following BaCl2 TA muscle injury, MMP9 was strongly (>3-fold) more active in the injured TA skeletal muscle of auf1 KO mice compared to WT mice (Figure 5A). No MMP9 activity was evident in the uninjured right hind limb control in both the WT and auf1 KO mice (Figure 5A). Surgical excision of the injured TA muscle from WT and auf1 KO mice followed by bioluminescence imaging (Figure 5B, C) confirmed that there is an average 3-fold increase in continuous MMP9 activity in auf1 KO mice compared to WT mice. These data indicate that activated auf1 KO satellite cells secrete continuous and increased levels of MMP9 following muscle injury, which is likely exacerbated as the satellite cell population expands.
Figure 5. MMP9 is significantly more active in the auf1−/− skeletal muscle following injury.

(A) Bioluminescence (IVIS) images of representative 4 month old mice treated with MMPSense for 48 h to assess MMP9 activity 24 h following TA BaCl2 injury of left hind limb, compared to an uninjured control (right hind limb). Three mice per genotype were studied.
(B) IVIS images of representative WT (left) and KO (right) excised TA muscles treated with MMP-Sense for 48 h to assess MMP9 activity 24 h after injury.
(C) Quantification of MMP-Sense IVIS images in WT and KO injured TA muscles 24 h post-injury. *P<0.05, unpaired t-test.
We therefore sought to determine whether the increased expression and activity of MMP9 is responsible for the auf1 KO injury phenotype observed, particularly the severe loss of laminin and depletion of the satellite cell population. Chronically increased MMP9 activity may promote excessive ECM damage and subsequent disruption of the satellite cell niche, ultimately inhibiting satellite cell return to PAX7+ quiescence by interrupting crucial cell-niche crosstalk. To test this, a MMP9 small molecule inhibitor, SB-3CT (Jia et al., 2014; Sassoli et al., 2014), was administered through IP injection to auf1 KO mice in conjunction with BaCl2-mediated TA injury. SB-3CT blocks MMP9 activity through an irreversible covalent interaction (Jia et al., 2014; Sassoli et al., 2014). Mice were treated with 10 mg/kg SB-3CT daily starting 24 hours prior to injury in combination with MMP9-specific collagen matrix injections (Cai et al., 2015). Auf1 KO mice treated with SB-3CT showed near complete extinction of MMP9 activity at the site of IP injection and significantly reduced MMP9 activity in the injured TA muscle (Figure 6A). Bioluminescence analysis demonstrated a >5-fold reduction in MMP9 activity in SB-3CT treated mice post-injury (Figure 6B). The scale used to quantitate fluorescence is shown in Figure 5.
Figure 6. Inhibition of MMP9 activity in auf1−/− mice restores maintenance of the PAX7+ satellite cell population.

(A) IVIS images of 4 month old mice treated with MMP-Sense with (right, KO+SB-3CT) or without (left, KO) SB-3CT for 48 h to assess MMP9 activity 24 h after TA muscle BaCl2 injury (left hind limb) compared to an uninjured TA muscle (right hind limb). Three mice per treatment were studied.
(B) Quantification of MMP-Sense IVIS imaging in KO and KO+SB-3CT injured TA muscles 24 h post-injury. **P<0.005, unpaired t-test.
(C) Immunofluorescence for the expression of laminin (AF488, green), PAX7 (AF555, red), and nuclei (DAPI, blue) in 7 days post-injury skeletal muscle in 4 month old KO and KO+SB-3CT mice. TA muscle was injured through BaCl2 injection. TA muscles were frozen in OCT, 5 images from 3 sections were analyzed per mouse (scale bar 50 μm).
(D) Quantification of PAX7 expression in KO and KO+SB-3CT mice in 7 days post-injury skeletal muscle. *P<0.05, unpaired t-test.
The reduction in MMP9 activity by SB-3CT treatment in injured auf1 KO mice resulted in restoration of muscle wound repair. Laminin expression was strongly increased and near-normal muscle fibers were evident in injured, SB-3CT treated auf1 KO animals, consistent with repair of the satellite cell niche (Figure 6C). Furthermore, the PAX7+ satellite cell population underwent significant increased expansion 7 days post-injury only in MMP9 inhibited (SB-3CT treated) auf1 KO mice (Figure 6C). Specifically, a ~4-fold increase was found in the PAX7+ satellite cell population with SB-3CT treatment following injury (Figure 6D). These data demonstrate that the severe myopathic pathology of auf1 KO mice following skeletal muscle injury is due to loss of AUF1 targeted ARE-mRNA decay, resulting in increased and constitutive muscle tissue remodeling through elevated MMP9 activity and subsequent loss of stem cell maintenance. These findings further identify the source of late onset myopathy observed in aging auf1 KO mice - the accelerated depletion of the satellite cell population and increased degradation of laminin due to loss of AUF1-mediated regulation of ARE-mRNA decay. In both phenotypes, the source of increased MMP9 is the activated auf1 KO satellite cell, itself causing loss of self-renewal, making auf1−/− satellite cells act in a self-sabotaging manner.
DISCUSSION
The targeted decay of ARE-mRNAs by AUBPs has emerged as a major regulator of many complex physiological pathways and a source of disease when it goes awry (Moore et al., 2014). AUBPs have multiple poorly understood roles in orchestrating the process of myogenesis, whether during development or regeneration following wound repair. Studies indicate that the complex and temporally ordered process of muscle regeneration, including the regulation, differentiation and restoration of satellite cells in this process, involves a tightly regulated AUBP network (Dormoy-Raclet et al., 2013; Figueroa et al., 2003; Hausburg et al., 2015; Legnini et al., 2014; Panda et al., 2014; Singh et al., 2014). The individual AUBP molecular activities and coordination of their respective functions are very poorly understood, particularly in the context of stem cell mediated regeneration. Here we focused on the role of AUF1 in satellite cell mediated skeletal muscle repair, demonstrating that in the absence of functional AUF1, certain ARE-mRNAs in satellite cells are increased in abundance, disrupting satellite cell differentiation and self-renewal following wounding. The elevated expression of active MMP9, encoded by an AUF1 targeted ARE-mRNA, was found to uncontrollably degrade the surrounding skeletal muscle ECM, including laminin and the satellite cell niche, generating a myopathic phenotype similar to a variety of late onset human myopathic diseases.
The finding that auf1−/− mice show accelerated skeletal muscle wasting with aging is likely a result of increased satellite cell-secreted MMP9 activity following accumulative minor wounds over time. Our findings demonstrate that continuous MMP9 activity damages the laminin and ECM structures, disrupting the quiescent satellite cell niche. This results in a relentless cycle of destructive degradation and repair established by an MMP9-driven muscle wounding response, which pre-maturely activates and depletes yet more satellite cells. Activated satellite cells then fuse to existing myofibers, as indicated by the increase in centrally located nuclei in 8 month old auf1−/− mice. Consequently, the loss of functional AUF1 specifically in satellite cells leads to a late onset myopathy, with no phenotype present at a young age. The chronic and increased expression of MMP9 in the absence of AUF1-mediated ARE-mRNA decay is therefore clearly a major driver of age-related and post-injury myopathy. Importantly, the disruption of the satellite cell niche by increased and unregulated MMP activity in auf1−/− mice leads to the partial depletion of the quiescent PAX7+ satellite cell population, culminating in the development of a late onset myopathy observed in aging and following muscle injury.
Although additional ARE-mRNAs other than MMP9 identified in the satellite cell RNA-seq analysis likely contribute to determination of satellite cell fate and the regulation of skeletal muscle integrity and regeneration, it is clear that AUF1 regulation of MMP9 ARE-mRNA decay defines a primary controlling step. In this regard, the ability to not only restore laminin expression, and therefore muscle regeneration, but also increase expansion of auf1−/− PAX7+ satellite cells by treatment with the MMP9 inhibitor SB-3CT underscores the important function of AUF1-mediated decay of a single ARE-mRNA (MMP9). This further validates the importance of AUF1-regulated ARE-mRNA decay in the activation and self-renewal of satellite cells, mediated through their interaction with the niche. Future studies will be directed to understanding the role of AUF1 in later stages of muscle regeneration, including expansion, differentiation and fusion of the satellite cell. Accordingly, MEF2C, a late stage MRF and AUF1 mRNA target (Panda et al., 2014), was not identified in our RNA-seq analysis, presumably due to the time point in regeneration at which auf1−/− satellite cells were selected and sorted for this study.
The chronic and increased expression of MMP9 in the absence of AUF1-mediated ARE-mRNA decay is therefore clearly a major driver of age-related and post-injury myopathy. Importantly, the disruption of the satellite cell niche by increased MMP9 activity in auf1−/− mice leads to the partial depletion of the quiescent PAX7+ satellite cell population, culminating in the development of a late onset myopathy observed in aging and following muscle injury.
This work addresses the importance of post-transcriptional control in the coordinated process of tissue regeneration. Studies could prove extremely beneficial to further understand the multiple roles of the different AUBPs in coordinating myogenesis and muscle regeneration. Clearly, AUF1 functions at different temporal points in the process of myogenesis, shown by work in C2C12 cells (Panda et al., 2014) and here. HuR, another AUBP that often opposes AUF1 action and stabilizes ARE-mRNAs (Figueroa et al., 2003), increases dramatically in satellite cells in the very early stages of activation (Legnini et al., 2014), at a time before the rise in AUF1 expression. HuR promotes the stability of certain MRFs such as myogenin and MyoD (Figueroa et al., 2003). HuR was also recently shown to stabilize the non-coding RNA linc-MD1, with high expression in the earliest stages of myogenesis (Legnini et al., 2014), and the mRNA hmgb1 following injury. HMGB1 promotes a motility program involved as an early activator of the skeletal muscle repair response (Dormoy-Raclet et al., 2013). Yet another AUBP, TTP, which is also an ARE-mRNA decay mediator, is highly expressed in only quiescent satellite cells, when AUF1 is not expressed. Furthermore, TTP shows immediate inactivation following injury when AUF1 expression increases dramatically (Hausburg et al., 2015). In the quiescent satellite cell, TTP mediates the rapid decay of the MyoD mRNA, preventing expansion of the satellite cell population. Previous studies have shown that AUF1 and TTP tend to show mutually exclusive expression or activity (Moore et al., 2014), consistent with these findings and our data that AUF1 is only expressed following satellite cell activation.
Reported data lead to the possibility that loss or mutation of AUF1 is related to the development of LGMD, a late onset human myopathy. Multiple family cohorts with LGMD type 1G have a mutation in the 4q21 locus which contains the auf1 gene, and one family was shown to have a mutation in HNRNPDL, a poorly described AUF1 homolog (Starling et al., 2004; Vieira et al., 2014). The age of onset for LGMD type 1G ranges from 30–47 years with no childhood history of myopathy (Starling et al., 2004; Vieira et al., 2014). As clinically described, LGMD disease shows a similar relative age of onset and histological representation as identified in the auf1−/− mouse.
In summary, our work identifies a myopathy of true satellite cell origin in an animal model and places the AUBP mRNA decay protein AUF1 as a key regulator of adult stem cell fate. These findings have important clinical implications. While healthy skeletal muscle can develop in the absence of functional AUF1, the satellite cell population is clearly altered and, once activated, is quickly depleted. Activated auf1−/− satellite cells secrete elevated levels of MMP9 that continuously breaks down the ECM and niche, causing premature satellite cell activation, satellite cell depletion, and subsequent development of myopathy with age. Consequently, a combination of MMP9 inhibition and potential AUF1-medated satellite cell therapy may have a role in regenerative medicine for chronic and acute adult myopathies.
EXPERIMENTAL PROCEDURES
Animal studies
All animal studies were approved by the NYU School of Medicine Institutional Animal Care and Use Committee (IACUC) and conducted in accordance with IACUC guidelines.
Dual energy x-ray absorptiometry (DEXA)
The Lunar Pixi DEXA was used to record lean tissue mass as per manufacturer recommendations. Male and female mice from 3–12 months of age were weighed for total body mass and scanned for lean body mass. Mice were divided into the following age groups: 3–5, 6–8, 9–12. A ratio of lean body mass to total body mass was used for analysis. Five mice per genotype per age group were analyzed in triplicate, then means and standard deviations calculated. Data were analyzed with an unpaired t-test. (Harada, 2013; Marinangeli and Kassis, 2013).
Cage flip
Male and female mice were placed on a grid for 30 sec to acclimate before being inverted for up to 60 sec at a height of 3 feet. The time until mice let go of the grid was recorded. Mice were divided into the following month age groups: 3–5, 6–8, 9–12. Five mice per genotype per age group were tested in triplicate, then means and standard deviations calculated. Data were analyzed with an unpaired t-test.
Strength grip
Male and female mice 6 months of age were tested as per manufacturer recommendations (Bioseb). In brief, mice were handled by their tails and allowed to grab a grid attached to a force monitor with their upper limbs. Mice were pulled off the grid by their tail and a measurement of upper limb strength was recorded. Five mice per genotype were tested in triplicate, then means and standard deviations calculated. Data were analyzed with an unpaired t-test (Ke et al., 2015; van Norren et al., 2015).
Immunofluorescence
Male and female mice 4–8 months of age (designated by experiment) had their TA muscles removed and frozen in OCT (Tissue-Tek). Samples were post-fixed in 4% paraformaldehyde and blocked in 3% BSA in TBS. Primary antibodies were incubated at 4°C overnight. Alexa Fluor donkey 488, 555, and 647 secondary antibodies were used at 1:500 and incubated for 1 hour at room temperature. Slides were sealed with Vectashield with DAPI. The following antibody dilutions were used: rat antibody to laminin (Sigma, L0663, 1:250), mouse antibody to PAX7 (in vivo: DSHB, 1:1000, PAX7 was deposited to the DSHB by Kawakami, Atsushi), goat antibody to hnRNPD (Santa Cruz Biotechnology, SC-22368, 1:250), rabbit antibody to MyoD (Santa Cruz Biotechnology, SC-760, 1:1000).
Microscopy, image processing and analysis
Images were acquired using a Zeiss LSM 700 confocal microscope, primarily with the 20X lens. Images were processed and scored using ImageJ64. If needed, color balance was adjusted linearly for the entire image and all images in experimental sets.
BaCl2 hindlimb injury
Male and female mice 4 months of age were injected with 50 μl of filtered 1.2% BaCl2 in saline directly to the left TA muscle. The right TA muscle remained uninjured as a control. Mice were monitored and sacrificed by protocol at 7 and 15 days post-injection. Injured and uninjured TA muscles were surgically removed and frozen in OCT (Tissue-Tek) post-sacrifice. Three mice per genotype per time point were studied (Bernet et al., 2014; Cornelison et al., 2001).
Myofiber preparation
Myofibers were harvested from WT and KO mice at 4 months of age. In brief, muscles beneath the TA muscle, including the extensor digitorum longus (EDL) and soleus, were dissected from the hindlimb and digested in 1.5 U/ml Collagenase Type I (Worthington) for 1.5 h in a 37°C water bath. Myofibers were cultured for 72 h in F12 media (Corning) supplemented with 15% horse serum (Gibco), 1% penicillin streptomycin (Life Technologies), and 2.5 μg/μl FGF (Sigma) at 37 C in a 5% CO2 tissue culture incubator (Tanaka et al., 2009).
C2C12 cell culture
C2C12 cells were maintained in DMEM (Corning), 20% FBS (Gibco), and 1% penicillin streptomycin (Life Technologies). To differentiate cells, media was switched to DMEM (Corning), 2% Horse Serum (Gibco), and 1% penicillin streptomycin (Life Technologies) (Panda et al., 2014).
Fluorescence activated cell sorting (FACS)
Whole hindlimb skeletal muscle from male and female mice ages 4–6 months of age was digested in Collagenase Type I (Worthington) and sorted for the Sdc4+cd45−cd31−Sca1− population using a Beckman Coulter MoFlo XDP. Lineage markers were excited by a Coherent Sapphire 488nm laser and collected via 530/40 band-pass filter. Sdc4 (conjugated to Q-Dot800 using SiteClick antibody conjugation kit) was also excited by Coherent Sapphire 488 nm laser and collected via 740 long-pass filter. DAPI was used to select for live/intact cells and was excited by a JDSU Xcyte 355 nm laser and collected via 450/65 band-pass filter. Events were run through a 70 μm cyto-nozzle at approximately 10,000 events per second.
RNA-sequencing and analysis
RNA was extracted from FACs sorted satellite cells using TRIzol (Thermo Fisher Scientific) and purified using the RNeasy Mini Kit (Quiagen) as per manufacture instructions. Whole hindlimb skeletal muscle of male and female mice ages 4–6 months old was processed. Three mice were analyzed per genotype. RNA-sequencing was completed through the NYU School of Medicine Genome Technology Core using the Illumina Hi-Seq 2500 Single Read and analyzed through the NYU-CTSI Bioinformatics Core.
Affymetrix genome-wide mRNA analysis
RNA was extracted from whole hindlimb skeletal muscle with TRIzol (Thermo Fisher Scientific) and purified using the RNeasy Mini Kit (Quiagen) as per manufacture instructions. Whole hindlimb skeletal muscle of male and female mice aging 4–6 months old was processed. Five mice were analyzed per genotype. Affymetrix mouse genome 430 2.0 array chips were used. Affymetrix chips were processed by the NYU School of Medicine Genome Technology Core and analyzed through the NYU School of Medicine Bioinformatics Core.
In vitro AUF1 silencing
AUF1 was transiently silenced in vitro using 50 μM Ambion siRNA (s62815, s201078) and Life Technology Lipofectamine 2000 for 48 h. For extended studies, cells were treated with siRNA every 72 h as needed.
MMP9 ELISA
Tissue culture media was collected and tested using Molecular Probes EnzChek Gelatinase/Collagenase Assay Kit as per manufacture instructions. Experiments were analyzed in triplicate, then means and standard deviation calculated. Data were analyzed by unpaired t-test.
In vivo MMP9 activity
WT and KO male and female mice 4 months of age were given an IP injection with PerkinElmer MMPSense 750 solution 24 h prior to injury and the time of BaCl2 injection, 24 h prior to imaging. Animals were imaged using IVIS L-III through the NYU School of Medicine Small Animal Imaging Core. Three mice per genotype were analyzed, then means and standard deviations calculated. Data were analyzed with an unpaired t-test.
SB-3CT treatment
KO male and female mice 4 months of age were given an IP injection with 25 mg/kg SB-3CT (Sigma-Aldrich) every 24 h, starting 24 h prior to BaCl2 injury with MMPSense injection. Three mice per treatment were analyzed, then means and standard deviations calculated. Data were analyzed with an unpaired t-test.
Statistical analysis
Unpaired t-test or two-way ANOVA were used when applicable to determine significance. Data were analyzed using Prism 6.0f. Significant values are considered P<0.05 as noted by one asterisk (*), P<0.005 as noted by two asterisks (**), or P<0.0005 as noted by three asterisks (***).
Supplementary Material
Highlights.
AUF1 regulates muscle stem cell function by targeted degradation of specific mRNAs
Auf1−/− mice undergo skeletal muscle wasting and impaired regeneration following injury
AUF1 control of mRNA decay is a mechanism for regulating tissue regeneration
Mutations in AUF1 are implicated in human muscle wasting diseases
Acknowledgments
We thank Dr. Bruce Cronstein (NYU) for use of the DEXA. Technical assistance was provided by the NYU-CTSI Bioinformatics Core, Histopathology Core, Microscopy Core, Rodent Behavior Core, Small Animal Imaging Core, supported in part by grant UL1 TR00038 from NCATS. This work was supported by NIH grants GM085693 and R24OD018339 to R.J.S., NIH T32 13-A0-S1-090476 to D.M.C, and NIH grant AR049446 to B.B.O, who is supported as a Senior Scholar in Aging Research by the Ellison Medical Foundation.
Footnotes
ACCESSION NUMBERS
The gene expression data accession number GSE83555 at GEO contains all RNA-seq data that are presented in this study.
Supplemental information includes 7 figures, 2 tables, Supplemental Figure Legends, Experimental Procedures and References, and can be found with this article online at:
AUTHOR CONTRIBUTIONS
D.M.C., R.J.S., B.B.O. and A.B.C. designed the studies, D.M.C. carried out the majority of the studies; A.B.C. and T.L.A. participated in carrying out hindlimb injury studies and satellite cell FACs sorting; L.C.L. assisted in all mouse maintenance, phenotype analysis, and injury with SB-3CT rescue experiments; J.W. conducted bioinformatics analysis from RNA-Seq and Affymetrix platforms; R.J.S. directed the project; D.C.M and R.J.S. wrote the manuscript; B.B.O. and A.B.C. participated in manuscript revision.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, Olwin BB. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nature medicine. 2014;20:265–271. doi: 10.1038/nm.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AS. Pax7 is back. Skeletal muscle. 2014;4:24. doi: 10.1186/s13395-014-0024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Mu Z, Jiang Z, Wang Y, Yang GY, Zhang Z. Hypoxia-controlled matrix metalloproteinase-9 hyperexpression promotes behavioral recovery after ischemia. Neurosci Bull. 2015;31:550–560. doi: 10.1007/s12264-015-1533-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson ME, Conboy IM. Loss of stem cell regenerative capacity within aged niches. Aging Cell. 2007;6:371–382. doi: 10.1111/j.1474-9726.2007.00286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle C, Fan J, Cho-Chung YS, Werner T, Ray J, Do L, Gorospe M, Becker KG. Control of gene expression during T cell activation: alternate regulation of mRNA transcription and mRNA stability. BMC Genomics. 2005;6:75. doi: 10.1186/1471-2164-6-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA, Morgan JE. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. 2005;122:289–301. doi: 10.1016/j.cell.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Cornelison DD, Filla MS, Stanley HM, Rapraeger AC, Olwin BB. Syndecan-3 and syndecan-4 specifically mark skeletal muscle satellite cells and are implicated in satellite cell maintenance and muscle regeneration. Dev Biol. 2001;239:79–94. doi: 10.1006/dbio.2001.0416. [DOI] [PubMed] [Google Scholar]
- Cornelison DD, Wold BJ. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev Biol. 1997;191:270–283. doi: 10.1006/dbio.1997.8721. [DOI] [PubMed] [Google Scholar]
- Dormoy-Raclet V, Cammas A, Celona B, Lian XJ, van der Giessen K, Zivojnovic M, Brunelli S, Riuzzi F, Sorci G, Wilhelm BT, et al. HuR and miR-1192 regulate myogenesis by modulating the translation of HMGB1 mRNA. Nat Commun. 2013;4:2388. doi: 10.1038/ncomms3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont NA, Wang YX, Rudnicki MA. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development. 2015;142:1572–1581. doi: 10.1242/dev.114223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa A, Cuadrado A, Fan J, Atasoy U, Muscat GE, Munoz-Canoves P, Gorospe M, Munoz A. Role of HuR in skeletal myogenesis through coordinate regulation of muscle differentiation genes. Mol Cell Biol. 2003;23:4991–5004. doi: 10.1128/MCB.23.14.4991-5004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- Gopinath SD, Rando TA. Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell. 2008;7:590–598. doi: 10.1111/j.1474-9726.2008.00399.x. [DOI] [PubMed] [Google Scholar]
- Gratacos FM, Brewer G. The role of AUF1 in regulated mRNA decay. Wiley interdisciplinary reviews. RNA. 2010;1:457–473. doi: 10.1002/wrna.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AR, Fallmann J, Kratochvill F, Kovarik P, Hofacker IL. AREsite: a database for the comprehensive investigation of AU-rich elements. Nucleic Acids Res. 2010;39:D66–69. doi: 10.1093/nar/gkq990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, Lipton SA. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401–6408. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther S, Kim J, Kostin S, Lepper C, Fan CM, Braun T. Myf5-positive satellite cells contribute to Pax7-dependent long-term maintenance of adult muscle stem cells. Cell stem cell. 2013;13:590–601. doi: 10.1016/j.stem.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Yang Q, Lin L, Xu C, Zheng C, Chen X, Han Y, Li M, Cao W, Cao K, et al. Interleukin-17 enhances immunosuppression by mesenchymal stem cells. Cell Death Differ. 2014;21:1758–1768. doi: 10.1038/cdd.2014.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A. Evaluation of muscle mass using Dual energy X-ray absorptiometry. Clinical calcium. 2013;23:361–364. [PubMed] [Google Scholar]
- Hausburg MA, Doles JD, Clement SL, Cadwallader AB, Hall MN, Blackshear PJ, Lykke-Andersen J, Olwin BB. Post-transcriptional regulation of satellite cell quiescence by TTP-mediated mRNA decay. Elife. 2015;4:e03390. doi: 10.7554/eLife.03390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Schneider R. 14-3-3sigma is a p37 AUF1-binding protein that facilitates AUF1 transport and AU-rich mRNA decay. The EMBO journal. 2006;25:3823–3831. doi: 10.1038/sj.emboj.7601264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindi SM, Shin J, Ogura Y, Li H, Kumar A. Matrix metalloproteinase-9 inhibition improves proliferation and engraftment of myogenic cells in dystrophic muscle of mdx mice. PLoS One. 2013;8:e72121. doi: 10.1371/journal.pone.0072121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TC, Chiang YP, Chuang CK, Chen SL, Hsieh JW, Lan YW, Tsao YP. PEDF-derived peptide promotes skeletal muscle regeneration through its mitogenic effect on muscle progenitor cells. Am J Physiol Cell Physiol. 2015 doi: 10.1152/ajpcell.00344.2014. ajpcell 00344 02014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia F, Yin YH, Gao GY, Wang Y, Cen L, Jiang JY. MMP-9 inhibitor SB-3CT attenuates behavioral impairments and hippocampal loss after traumatic brain injury in rat. J Neurotrauma. 2014;31:1225–1234. doi: 10.1089/neu.2013.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke YD, van Hummel A, Stevens CH, Gladbach A, Ippati S, Bi M, Lee WS, Kruger S, van der Hoven J, Volkerling A, et al. Short-term suppression of A315T mutant human TDP-43 expression improves functional deficits in a novel inducible transgenic mouse model of FTLD-TDP and ALS. Acta Neuropathol. 2015 doi: 10.1007/s00401-015-1486-0. [DOI] [PubMed] [Google Scholar]
- Kim MY, Hur J, Jeong S. Emerging roles of RNA and RNA-binding protein network in cancer cells. BMB reports. 2009;42:125–130. doi: 10.5483/bmbrep.2009.42.3.125. [DOI] [PubMed] [Google Scholar]
- Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudryashova E, Kramerova I, Spencer MJ. Satellite cell senescence underlies myopathy in a mouse model of limb-girdle muscular dystrophy 2H. The Journal of clinical investigation. 2012;122:1764–1776. doi: 10.1172/JCI59581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legnini I, Morlando M, Mangiavacchi A, Fatica A, Bozzoni I. A feedforward regulatory loop between HuR and the long noncoding RNA linc-MD1 controls early phases of myogenesis. Molecular cell. 2014;53:506–514. doi: 10.1016/j.molcel.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepper C, Conway SJ, Fan CM. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature. 2009;460:627–631. doi: 10.1038/nature08209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Mittal A, Makonchuk DY, Bhatnagar S, Kumar A. Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum Mol Genet. 2009;18:2584–2598. doi: 10.1093/hmg/ddp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton BH, Schultz E. Developmental fate of skeletal muscle satellite cells. Science. 1979;205:1292–1294. doi: 10.1126/science.472747. [DOI] [PubMed] [Google Scholar]
- Liu W, Rosenberg GA, Liu KJ. AUF-1 mediates inhibition by nitric oxide of lipopolysaccharide-induced matrix metalloproteinase-9 expression in cultured astrocytes. J Neurosci Res. 2006;84:360–369. doi: 10.1002/jnr.20895. [DOI] [PubMed] [Google Scholar]
- Lu JY, Sadri N, Schneider RJ. Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes & development. 2006;20:3174–3184. doi: 10.1101/gad.1467606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JY, Schneider RJ. Tissue distribution of AU-rich mRNA-binding proteins involved in regulation of mRNA decay. The Journal of biological chemistry. 2004;279:12974–12979. doi: 10.1074/jbc.M310433200. [DOI] [PubMed] [Google Scholar]
- Marinangeli CP, Kassis AN. Use of dual X-ray absorptiometry to measure body mass during short- to medium-term trials of nutrition and exercise interventions. Nutrition reviews. 2013;71:332–342. doi: 10.1111/nure.12025. [DOI] [PubMed] [Google Scholar]
- Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, Partridge T, Buckingham M. Direct isolation of satellite cells for skeletal muscle regeneration. Science. 2005;309:2064–2067. doi: 10.1126/science.1114758. [DOI] [PubMed] [Google Scholar]
- Moore AE, Chenette DM, Larkin LC, Schneider RJ. Physiological networks and disease functions of RNA-binding protein AUF1. Wiley interdisciplinary reviews. RNA. 2014;5:549–564. doi: 10.1002/wrna.1230. [DOI] [PubMed] [Google Scholar]
- Murase S, McKay RD. Matrix metalloproteinase-9 regulates survival of neurons in newborn hippocampus. The Journal of biological chemistry. 2012;287:12184–12194. doi: 10.1074/jbc.M111.297671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda AC, Abdelmohsen K, Yoon JH, Martindale JL, Yang X, Curtis J, Mercken EM, Chenette DM, Zhang Y, Schneider RJ, et al. RNA-binding protein AUF1 promotes myogenesis by regulating MEF2C expression levels. Mol Cell Biol. 2014;34:3106–3119. doi: 10.1128/MCB.00423-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pont AR, Sadri N, Hsiao SJ, Smith S, Schneider RJ. mRNA decay factor AUF1 maintains normal aging, telomere maintenance, and suppression of senescence by activation of telomerase transcription. Molecular cell. 2012;47:5–15. doi: 10.1016/j.molcel.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar B, Lu JY, Schneider RJ. Nuclear import and export functions in the different isoforms of the AUF1/heterogeneous nuclear ribonucleoprotein protein family. The Journal of biological chemistry. 2003a;278:20700–20707. doi: 10.1074/jbc.M301176200. [DOI] [PubMed] [Google Scholar]
- Sarkar B, Xi Q, He C, Schneider RJ. Selective degradation of AU-rich mRNAs promoted by the p37 AUF1 protein isoform. Mol Cell Biol. 2003b;23:6685–6693. doi: 10.1128/MCB.23.18.6685-6693.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Han J, Sinsimer KS, Liao B, Foster RL, Brewer G, Pestka S. RNA-binding protein AUF1 regulates lipopolysaccharide-induced IL10 expression by activating IkappaB kinase complex in monocytes. Mol Cell Biol. 2011;31:602–615. doi: 10.1128/MCB.00835-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Sinsimer KS, Foster RL, Brewer G, Pestka S. AUF1 isoform-specific regulation of anti-inflammatory IL10 expression in monocytes. J Interferon Cytokine Res. 2008;28:679–691. doi: 10.1089/jir.2008.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassoli C, Nosi D, Tani A, Chellini F, Mazzanti B, Quercioli F, Zecchi-Orlandini S, Formigli L. Defining the role of mesenchymal stromal cells on the regulation of matrix metalloproteinases in skeletal muscle cells. Exp Cell Res. 2014;323:297–313. doi: 10.1016/j.yexcr.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Seale P, Rudnicki MA. A new look at the origin, function, and “stem-cell” status of muscle satellite cells. Dev Biol. 2000;218:115–124. doi: 10.1006/dbio.1999.9565. [DOI] [PubMed] [Google Scholar]
- Shefer G, Van de Mark DP, Richardson JB, Yablonka-Reuveni Z. Satellite-cell pool size does matter: defining the myogenic potency of aging skeletal muscle. Dev Biol. 2006;294:50–66. doi: 10.1016/j.ydbio.2006.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba N, Miyazaki D, Yoshizawa T, Fukushima K, Shiba Y, Inaba Y, Imamura M, Takeda S, Koike K, Nakamura A. Differential roles of MMP-9 in early and late stages of dystrophic muscles in a mouse model of Duchenne muscular dystrophy. Biochim Biophys Acta. 2015;1852:2170–2182. doi: 10.1016/j.bbadis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Silva KA, Dong J, Dong Y, Dong Y, Schor N, Tweardy DJ, Zhang L, Mitch WE. Inhibition of stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. The Journal of biological chemistry. 2015;290:11177–11187. doi: 10.1074/jbc.M115.641514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Xia Z, Bland CS, Kalsotra A, Scavuzzo MA, Curk T, Ule J, Li W, Cooper TA. Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Molecular cell. 2014;55:592–603. doi: 10.1016/j.molcel.2014.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–321. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- Starling A, Kok F, Passos-Bueno MR, Vainzof M, Zatz M. A new form of autosomal dominant limb-girdle muscular dystrophy (LGMD1G) with progressive fingers and toes flexion limitation maps to chromosome 4p21. European journal of human genetics: EJHG. 2004;12:1033–1040. doi: 10.1038/sj.ejhg.5201289. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Iijima M, Nishimura A, Tomozoe Y, Kamei D, Yamada M. Two separate regions essential for nuclear import of the hnRNP D nucleocytoplasmic shuttling sequence. FEBS J. 2005;272:3975–3987. doi: 10.1111/j.1742-4658.2005.04820.x. [DOI] [PubMed] [Google Scholar]
- Tanaka KK, Hall JK, Troy AA, Cornelison DD, Majka SM, Olwin BB. Syndecan-4-expressing muscle progenitor cells in the SP engraft as satellite cells during muscle regeneration. Cell stem cell. 2009;4:217–225. doi: 10.1016/j.stem.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Norren K, Rusli F, van Dijk M, Lute C, Nagel J, Dijk FJ, Dwarkasing J, Boekschoten MV, Luiking Y, Witkamp RF, et al. Behavioural changes are a major contributing factor in the reduction of sarcopenia in caloric-restricted ageing mice. J Cachexia Sarcopenia Muscle. 2015;6:253–268. doi: 10.1002/jcsm.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira NM, Naslavsky MS, Licinio L, Kok F, Schlesinger D, Vainzof M, Sanchez N, Kitajima JP, Gal L, Cavacana N, et al. A defect in the RNA-processing protein HNRPDL causes limb-girdle muscular dystrophy 1G (LGMD1G) Hum Mol Genet. 2014;23:4103–4110. doi: 10.1093/hmg/ddu127. [DOI] [PubMed] [Google Scholar]
- von Roretz C, Di Marco S, Mazroui R, Gallouzi IE. Turnover of AU-rich-containing mRNAs during stress: a matter of survival. Wiley interdisciplinary reviews. RNA. 2011;2:336–347. doi: 10.1002/wrna.55. [DOI] [PubMed] [Google Scholar]
- Wagner BJ, DeMaria CT, Sun Y, Wilson GM, Brewer G. Structure and genomic organization of the human AUF1 gene: alternative pre-mRNA splicing generates four protein isoforms. Genomics. 1998;48:195–202. doi: 10.1006/geno.1997.5142. [DOI] [PubMed] [Google Scholar]
- Webster MT, Manor U, Lippincott-Schwartz J, Fan CM. Intravital Imaging Reveals Ghost Fibers as Architectural Units Guiding Myogenic Progenitors during Regeneration. Cell stem cell. 2015 doi: 10.1016/j.stem.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicklund MP, Kissel JT. The limb-girdle muscular dystrophies. Neurol Clin. 2014;32:729–749. ix. doi: 10.1016/j.ncl.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Yoon JH, Jo MH, White EJ, De S, Hafner M, Zucconi BE, Abdelmohsen K, Martindale JL, Yang X, Wood WH, 3rd, et al. AUF1 promotes let-7b loading on Argonaute 2. Genes & development. 2015;29:1599–1604. doi: 10.1101/gad.263749.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zammit PS, Golding JP, Nagata Y, Hudon V, Partridge TA, Beauchamp JR. Muscle satellite cells adopt divergent fates: a mechanism for self-renewal? J Cell Biol. 2004;166:347–357. doi: 10.1083/jcb.200312007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucconi BE, Ballin JD, Brewer BY, Ross CR, Huang J, Toth EA, Wilson GM. Alternatively expressed domains of AU-rich element RNA-binding protein 1 (AUF1) regulate RNA-binding affinity, RNA-induced protein oligomerization, and the local conformation of bound RNA ligands. The Journal of biological chemistry. 2010;285:39127–39139. doi: 10.1074/jbc.M110.180182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.