

Abstract
The co-occurrence of epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) rearrangements constitutes a rare molecular subtype of non-small-cell lung cancer (NSCLC). Herein, we assessed the clinical outcomes and incidence of acquired resistance to tyrosine kinase inhibitors (TKIs) in this subtype. So we enrolled 118 advanced NSCLC treated with TKIs. EGFR mutations and ALK rearrangements were detected by DNA sequencing or Scorpion amplification refractory mutation system and fluorescence in situ hybridization respectively. Immunohistochemistry was used to evaluate the activation of associated proteins. We found that nine in ten patients with EGFR/ALK co-alterations had good response with first-line EGFR TKI, and the objective response rate (ORR) of EGFR TKIs was 80% (8/10) for EGFR/ALK co-altered and 65.5% (55/84) for EGFR-mutant (P = 0.57), with a median progression-free survival (PFS) of 11.2 and 13.2 months, (hazard ratio [HR]=0.95, 95% [CI], 0.49-1.84, P= 0.87). ORR of crizotinib was 40% (2/5) for EGFR/ALK co-altered and 73.9% (17/23) for ALK-rearranged (P= 0.29), with a median PFS of 1.9 and 6.9 months (hazard ratio [HR], 0.40; 95% [CI] 0.15-1.10, P = 0.08). The median overall survival (OS) was 21.3, 23.7, and 18.5 months in EGFR-mutant, ALK-rearranged, and EGFR/ALK co-altered (P= 0.06), and there existed a statistically significant difference in OS between ALK-rearranged and EGFR/ALK co-altered (P=0.03). Taken together, the first-line EGFR-TKI might be the reasonable care for advanced NSCLC harbouring EGFR/ALK co-alterations, whether or nor to use sequential crizotinib should be guided by the status of ALK rearrangement and the relative level of phospho-EGFR and phospho-ALK.
Keywords: non-small-cell lung cancer (NSCLC), epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), overall survival, cohort study
INTRODUCTION
Lung cancer is the leading cause of cancer-related death in the world [1]. However, recent discovery of oncogenic drivers has transformed the care of lung adenocarcinomas dramatically [2]. Epidermal growth factor receptor (EGFR), and anaplastic lymphoma kinase (ALK), are important oncogenic drivers in non-small-cell lung cancer (NSCLC) [2], and other oncogenic drivers in NSCLC, such as ROS1 and KRAS, have also been found [3, 4]. Advances in targeted therapeutic agents have already approved for the treatment of patients with EGFR mutation and ALK translocation [5]. Early studies suggested that EGFR mutations and ALK rearrangements are mutually exclusive [6]. However, isolated cases with concomitant EGFR and ALK alterations have been reported [7–20], and the co-occurrence of multiple oncogenic drivers was also published [21, 22]. These two oncogenic drivers might overlap, and little has been known on both molecular biology and the role of EGFR or ALK inhibitors in such EGFR/ALK co-altered NSCLC. The prevalence of EGFR/ALK co-alterations was different [17, 18, 20, 23, 24], and it increased when more sensitive methods of detection (next-generation sequencing, NGS) were applied [24]. The responses to EGFR and/or ALK inhibitors were varied [9, 11, 16–18, 20, 24]. One study showed that the frequency of EGFR/ALK co-alterations was 1.3% (13/977) in NSCLC [20]. Beyond evidence of co-occurrence, the efficacy of EGFR tyrosine kinase inhibitors (TKIs) and ALK inhibitors has been revealed to be predicted by the relative levels of phospho-EGFR and phospho-ALK [20]. However, little has been known about the overall survival (OS) and molecular mechanisms of resistance to EGFR or ALK inhibitors in EGFR/ALK co-altered NSCLC. To address these issues, we performed a retrospective investigation in a cohort of EGFR mutation, ALK rearrangement and EGFR/ALK co-altered patients. The design flow of this cohort study was seen in Figure 1.
Figure 1. The design flow of this cohort study.
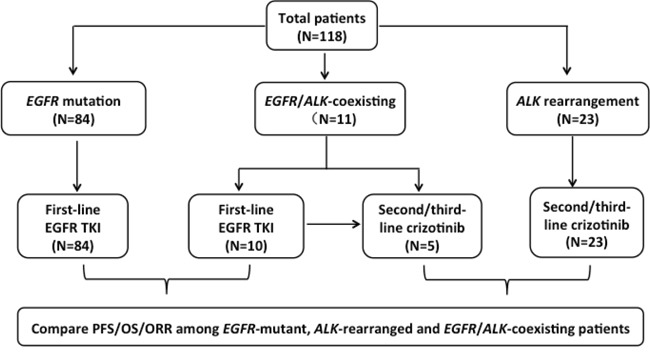
The three study populations and subsets analyzed are shown. TKIs, tyrosine kinase inhibitors. PFS, progression-free survival. OS, overall survival. ORR, objective response rate.EGFR, epidermal growth factor receptor.ALK, anaplastic lymphoma kinase.
RESULTS
Patient characteristics
In our previous study (2009-2011) of 11 EGFR/ALK co-altered patients treated with EGFR and/or ALK inhibitors, only objective response rate (ORR) and progression-free survival (PFS) were analyzed. And now, we aimed to investigate OS, the potential mechanisms of resistance to TKIs and the appropriate regimen for EGFR/ALK co-altered patients. As a comparison, totally, 84 advanced NSCLC patients with EGFR mutations treated with first-line EGFR TKIs, and 23 with only ALK rearrangements treated with second or further-line crizotinib, were retrospectively enrolled to investigate OS in the present study during the same period of time. From August 2009 to July 2011, 118 NSCLC patients treated with TKIs were enrolled, and the clinical characteristics of them were listed in Table 1. All patients were of Chinese ethnicity. The median ages were 63, 48 and 59 years for the EGFR-mutant, ALK-rearranged, and EGFR/ALK co-altered patients, respectively (P < 0.001).
Table 1. Baseline demographic and clinicopathologic characteristics in the three subgroups.
| Characteristic | EGFR mu (%) | ALK re (%) | EGFR/ALK (+) (%) | Total (%) | P |
|---|---|---|---|---|---|
| Gender | 0.206 | ||||
| Male | 33(39.3%) | 13(56.5%) | 3(27.3%) | 49(41.5%) | |
| Female | 51(60.7%) | 10(43.5%) | 8(72.7%) | 69(58.5%) | |
| Age (median) | 63(38-85) | 48(26-75) | 59(40-71) | 60.5(26-85) | <0.001* |
| WHO PS | 0.061 | ||||
| 0-1 | 81(96.4%) | 19(82.6%) | 10(90.9%) | 110(93.2%) | |
| 2-3 | 3(3.6%) | 4(17.4%) | 1(9.1%) | 8(6.8%) | |
| Smoking history | 0.377 | ||||
| Non smoker | 69(82.1%) | 20(87.0%) | 11(100.0%) | 100(84.7%) | |
| Heavy smoker | 15(17.9%) | 3(13.0%) | 0(0.0%) | 18(15.3%) | |
| Histology | 1.000 | ||||
| AC | 82(97.6%) | 23(100%) | 11(100.0%) | 116(98.3%) | |
| SCC | 2(2.4%) | 0(0.0%) | 0(0.0%) | 2(1.7%) | |
| Clinical stage | 0.343 | ||||
| III | 2(2.4%) | 0(0.0%) | 1(9.1%) | 3(2.5%) | |
| IV | 82(97.6%) | 23(100.0%) | 10(90.9%) | 115(97.5%) | |
| Total | 84(71.2%) | 23(19.5%) | 11(9.3%) | 118(100.0%) |
Aberrations: mu, mutation; re, rearrangement; WHO PS, World Health Organization performance status; SCC, squamous cell carcinoma; AC, adenocarcinoma. EGFR, epidermal growth factor receptor; ALK, anaplastic lymphoma kinase; A heavy smoker was defined as a patient who had smoked >100 cigarettes in his or her lifetime or that had stopped smoking less than 15 years and had a total of ≥10 pack-year of smoking.
The age in three groups was analyzed with the use of Wilcoxon test
Beyond this difference, the three groups were well balanced with respect to demographic and baseline characteristics. The majority of patients (84.7%) were never-smokers, and the most common histological subtype was adenocarcinoma (98.3%).
Efficacy of TKI treatment
All patients were assessed for a TKI response; a detailed list of the treatments for each of the eleven patients with concomitant EGFR mutations and ALK rearrangements was given in Figure 1. The ORR of EGFR TKIs was 80% (8/10) for EGFR/ALK co-altered patients and 65.5% (55/84) for EGFR-mutant patients, respectively; this difference was not statistically significant (P= 0.57). Similarly, no statistical difference in the ORR to crizotinib between the EGFR/ALK co-altered (2/5; 40.0%) and ALK-positive (17/23; 73.9%) patients (P= 0.29).
The last follow-up time was July 13 2015, and the median follow-up time was 23.2 months. The median PFS was 13.2 months in EGFR-mutant patients and 11.2 months in EGFR/ALK co-altered patients treated with EGFR TKIs (hazard ratio [HR], 0.95; 95% confidence interval [CI], 0.49-1.84; P = 0.87) (Figure 2A). The median PFS was 6.9 months for ALK-rearranged patients and 1.9 months for EGFR/ALK co-altered patients treated with crizotinib (HR, 0.40; 95% CI, 0.15 - 1.10;P= 0.08) (Figure 2B). The median OS were 21.3, 23.7, and 18.5 months for EGFR-mutant, ALK-rearranged, and EGFR/ALK co-altered patients, respectively (P = 0.06). The OS of subgroup of EGFR-mutant and EGFR/ALK co-altered patients did not have statistically significant difference (HR, 0.71; 95% CI, 0.37-1.35;P=0.29) (Figure 2C), but the statistical difference between ALK-rearranged and EGFR/ALK co-altered was significant (HR, 0.43; 95% CI, 0.20 - 0.93; P=0.03) (Figure 2D). Among 11 EGFR/ALK co-altered patients, all received EGFR TKI, and ten received EGFR TKI as the first line, and nine of them had good response and prolonged survival, but the last one had progression disease and shorter PFS. One with second-line crizotinib had a good response; after progression, EGFR TKI was taken without good response and the tendency of PFS was shorter (Figure 2E). Five patients received both TKIs, four with first-line EGFR TKI, and three of them had clinical benefits and prolonged survival, but without benefits from the following TKI, the same as the one taking ALK TKI and subsequent EGFR TKI (Figure 2F).
Figure 2. Comparisons of survival among the three groups.
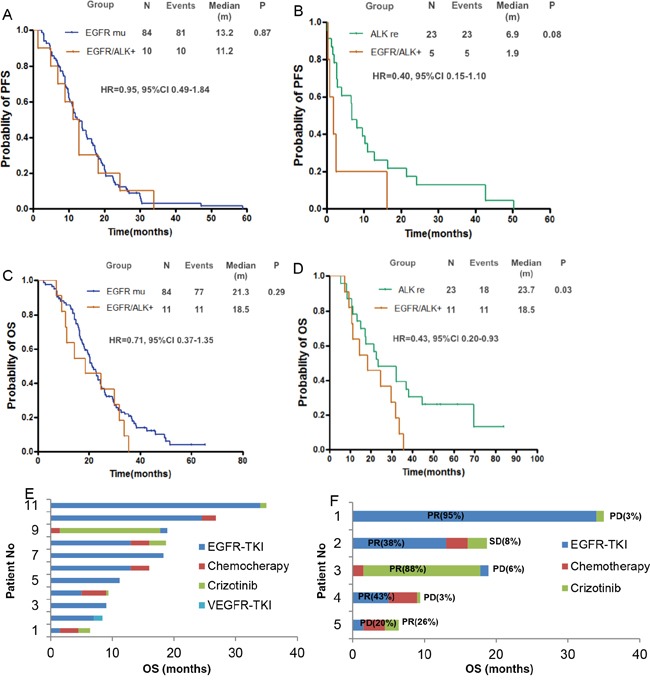
A. Kaplan-Meier curves for PFS of EGFR-mutant patients and EGFR/ALK-coexisting patients B. PFS curves of ALK-rearranged patients and EGFR/ALK-coexisting patients. C. Kaplan-Meier curves for OS of EGFR-mutant patients and EGFR/ALK-coexisting patients D. OS curves of ALK-rearranged patients and EGFR/ALK-coexisting patients E. The contributions of chemotherapy or TKIs to OS for 11 EGFR/ALK-coexisting patients F. PFS of two TKIs contributing to OS for the five patients who received both ALK and EGFR TKIs. VEGFR, vascular endothelial growth factor receptor. Chemo, chemotherapy. PR, partial response. SD, stable disease. PD, progressive disease. ALK, anaplastic lymphoma kinase. EGFR,epidermal growth factor receptor. mu, mutation. re, rearrangement. PFS, progression-free survival. OS, overall survival. TKI, tyrosine kinase inhibitor.
Potential mechanism of TKI resistance
The potential mechanisms of resistance to TKIs in the two EGFR/ALK co-altered patients were shown in Figure 3 and Figure 4. The first case received first-line erlotinib for nine months, achieving a partial response; however, after tumour progression, she was switched to intercalated erlotinib and chemotherapy for eight months, achieving stable disease. The patient then received crizotinib, and progressive disease occurred two months later, and re-biopsy was performed. Her genetic profiles were as follows: EGFR mutation with both an exon 19 deletion and exon 20 T790M mutation, negative ALK rearrangement (Figure 3). The level of phospho-EGFR decreased, while expression of phospho-ALK was increased, and the level of phospho-EGFR was higher than phospho-ALK after resistance to crizotinib (Figure 5A, 5B). EGFR exon 20 T790M mutation had been linked to resistance to erlotinib, and relative higher level of phospho-EGFR than phospho-ALK might be the result of resistance to crizotinib.
Figure 3. Changes of driver genes in EGFR/ALK-coexisting patient treated with crizotinib.
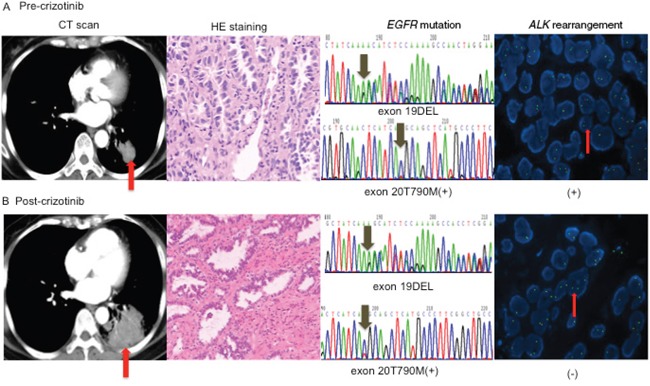
A. Pre-crizotinib histology was adenocarcinoma (original magnification×200), and genetic profiles showed EGFR exon 19 deletion and exon 20 T790M mutation detected by DNA sequencing, as well as positive ALK rearrangement detected by FISH B. After resistance to crizotinib, both the histology (original magnification×200) and EGFR mutation were the same as the baseline, but ALK rearrangement was negative. ALK, anaplastic lymphoma kinase. EGFR, epidermal growth factor. DEL, deletion.
Figure 4. Changes of driver genes in different lesions with EGFR-TKI.
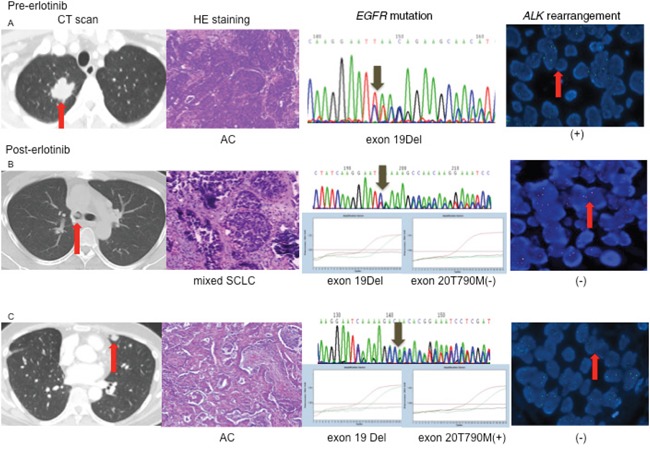
A. Before EGFR-TKI treatment, the pathology of lung lesion was adenocarcinoma, EGFR mutation detected by sequencing was exon 19 deletion, and ALK was positive B. After EGFR-TKI treatment, the pathology of the right main bronchus was mixed small-cell lung cancer with adenocarcinoma; EGFR mutation detected by ARMS was exon 19 deletion. ALK rearrangement was negative C. the pathology of the lesion in the left upper lung lobe was adenocarcinoma, EGFR mutation was exon 19 deletion and exon 20 T790M mutation. AC, adenocarcinoma. ALK, anaplastic lymphoma kinase. DEL, deletion.EGFR, epidermal growth factor receptor.SCLC, small-cell lung cancer.
Figure 5. Dynamic changes of expression of protein in EGFR/ALK co-existing patients who received TKI.
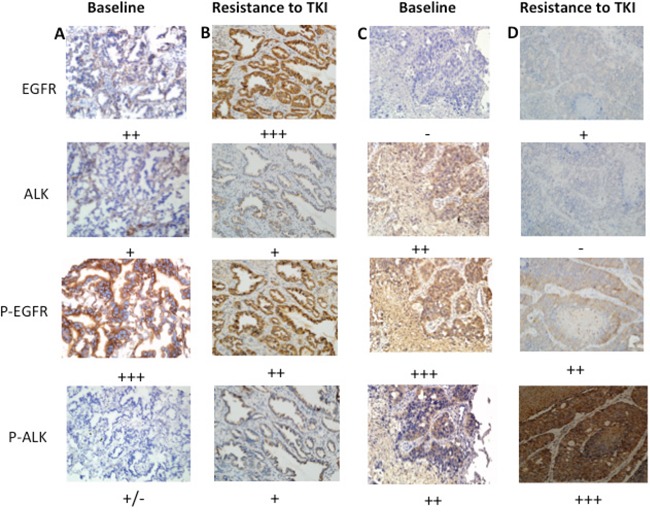
A. After resistance to erlotinib, P-EGFR was at higher level than P-ALK. B. After resistance to crizotinib, P-EGFR was higher than P-ALK. C. Pre-EGFR-TKI, P-EGFR was higher than P-ALK. D. After resistance to erlotinib, the level of P-ALK was higher than P-EGFR. P-, phospho-. ALK, anaplastic lymphoma kinase. EGFR, epidermal growth factor receptor.TKI, tyrosine kinase inhibitor.
The second patient received erlotinib for thirteen months, exhibiting gradual progression without any symptoms; therefore, he continued on erlotinib for an additional seven months until dramatic progression. CT scan showed new lesions in the left upper lung lobe and the right main bronchus, and wedge resection of the left upper lobe revealed a lung adenocarcinoma harbouring an EGFR exon 19 deletion and exon 20 T790M mutation. Two weeks later, he received bronchoscopy, and the result showed small-cell lung cancer (SCLC) mixed with an adenocarcinoma harbouring an EGFR exon 19 deletion. The two lesions were both negative for ALK rearrangements after treatment with erlotinib (Figure 4). Phospho-EGFR was decreased from its initial high level, but the level of phospho-ALK was higher than pre-treatment of erlotinib, and the level of phospho-ALK was higher than phospho-EGFR after resistance to erlotinib (Figure 5C, 5D).
Taken together, these data suggested the relative higher level of phospho-EGFR was the mechanism of resistance to crizotinib; the EGFR exon 20 T790M mutations, pathology transformation from adenocarcinoma to SCLC and the relative higher level of phospho-ALK as molecular mechanisms of resistance to EGFR TKIs.
DISCUSSION
Herein, we analysed OS in advanced NSCLC patients harbouring concomitant EGFR mutations and ALK rearrangements, as well as the potential mechanisms underlying acquired resistance to EGFR-TKI and/or crizotinib in two cases.
To EGFR/ALK co-altered patients with sequential both EGFR TKI and ALK TKI, EGFR TKI had good response and prolonged survival for most of them, but primary resistance occurred to the subsequent ALK TKI. It seemed that first-line EGFR TKI was a reasonable regimen for most of the subgroup, but single ALK TKI was excluded from the following eligible treatment. They exhibited a tendency towards relatively shorter OS than those with either an EGFR mutation or ALK rearrangement, and there existed statistically significant difference between the subgroup of ALK rearrangement and EGFR/ALK co-alterations. This difference should be related to the different first/second-line therapeutic regimens administered to these subgroups. Crizotinib is a standard of care for ALK-rearranged patients, and it can prolong survival, but as to EGFR/ALK co-altered patients in our study, crizotinb was only used as second- or further-line therapy and might not favor efficacy and survival. On the other hand, efficacy of crizotinib might be related to the abundance of ALK rearrangement determined by the sensitivity of test method. As to the first case with double TKIs, after resistance to EGFR TKI and chemotherapy, the level of phospho-EGFR was higher than phospho-ALK, although ALK rearrangement was positive, primary resistance to crizotinib occurred, and at that time the level of phospho-EGFR was still higher than phospho-ALK. The second case only receiving EGFR TKI had negative ALK rearrangement after resistance to EGFR TKI. The lack of efficacy of crizotinib in the subgroup of EGFR/ALK co-altered patients could be correlated with the lower activation of ALK than EGFR signaling pathway or relatively lower abundance of ALK rearrangement after resistance to EGFR-TKI and/or chemotherapy. A re-biopsy for the detection of EGFR mutation and ALK rearrangement would be necessary to select the appropriate therapeutic regimen for these patients. The second case only receiving EGFR TKI had negative ALK rearrangement after resistance to EGFR TKI, and the loss of ALK rearrangement might correlate with EGFR TKI. Previous research showed that one driver gene may be sufficient to drive resistance to TKIs in dual EGFR/ALK co-altered NSCLC [11]. Combination of both TKIs may be the potential choice after progression with first-line EGFR-TKI.
The result of the study demonstrated that first-line EGFR TKI might be the reasonable care for advanced NSCLC patients harboring concomitant EGFR mutations and ALK rearrangements, but the result of the other study was conflicting, suggesting that ALK inhibitors appeared to be effective for patients with EGFR/ALK co-alterations [24]. In our study, we detected EGFR mutation with DNA direct-sequencing, and in the other study, a more sensitive test method, called as the next-generation sequencing, was applied [24]. The abundance and the intratumoral heterogeneity of EGFR mutation and ALK rearrangement probably correlated with the efficacy of EGFR TKI and/or ALK TKI in EGFR/ALK co-altered patients [25]. The diagnosis of driver gene status might be affected by the sensitivity of detection methods, and therapy selection may make tumors become more heterogeneous for intratumoral genetic heterogeneity, which might be the major reason for resistance to TKIs [26]. As to EGFR/ALK co-altered patients, both oncogenic drivers status should be evaluated for the appropriate management of patients using more sensitive assays. The abundance of EGFR mutation and ALK rearrangement and the levels of phosphorylation of downstream proteins are detected, and treatment strategies might be optimized for patients with concomitant EGFR and ALK alterations in the future.
Despite the favourable efficacy of TKIs in patients with EGFR mutations or ALK rearrangements, resistance to these drugs is inevitable. Both EGFR-mutant and ALK-rearranged lung cancers utilise common mechanisms of resistance, including the development of secondary mutations in the tyrosine kinase domain, driver gene amplification, phenotypic alterations, and reactivation of the primary signalling pathways through alternative signalling molecules [27–29]. Of these many different mechanisms of resistance, approximately half of all lung carcinomas with acquired resistance to EGFR TKIs develop a T790M point mutation in exon 20 of EGFR [30, 31]. The T790M mutation is thought to reactivate EGFR signaling by increasing the receptor's affinity for ATP over TKIs [32] This mutation is also thought to amplify the expression of components of the hepatocyte growth factor receptor and mesenchymal epithelial transition (MET) pathways [27, 33–36] In addition, PIK3CA and BRAF mutations, as well as tumour morphological changes such as transformation from NSCLC to SCLC small cell lung cancer and epithelial to mesenchymal transitions have been observed, though the mechanisms by which they develop and lead to resistance are incompletely understood [27, 34, 37]. The mechanisms of acquired resistance to crizotinib in NSCLC include secondary mutations within the ALK kinase domain, amplification of the ALK gene, and bypass of important signalling mechanisms via an increase in EGFR phosphorylation or KIT amplification, and so on [23, 28, 38]. The frequency of ALK negative tumor was observed in patients with resistance to crizotinib was 18.2% [23], given that the emergence of an ALK negative tumor was associated with other separate oncogenic drivers, or the percentage of positive cells in this case was not zero, consistent with background noise in the break apart FISH assay as previously described [7]. Given the limited tumor sample of the first case, we were unable to detect the presence of other oncogenic drivers. Though the molecular mechanisms underlying acquired resistance to EGFR TKI and crizotinib in EGFR/ALK co-altered patients were similar to those in typical EGFR mutation and ALK rearrangement patients, such as EGFR exon 20 T790M mutation and pathologic transformation from adenocarcinoma to SCLC small-cell lung cancer have been linked to resistance to erlotinib. As well as the relative higher level of phospho-ALK than phospho-EGFR is related to resistance to EGFR TKI. In addition to phospho-EGFR is higher than phospho-ALK is also related to resistance to crizotinib. The mechanisms of acquired resistance in patients with more than one positive driver genes were inevitably more complex. As to EGFR/ALK co-alterations, status of EGFR mutations and ALK rearrangements should be known to us, the downstream proteins of the two pathways should also be detected, and we could have the eligible regimen for the special group through detection of dynamic changes of the downstream proteins of the two pathways.
The level of phosphorylation of downstream protein is related to the activation of the signaling pathway, and detection of the relative phosphorylation levels of EGFR and ALK might help to guide the selection of TKIs in clinical practice [20]. The dynamic change of the level of the phosphorylation, for example, increased or decreased, can be detected by IHC, and the variation of staining intensity might predict the efficacy of TKIs [20, 38]. As to the tumor with 2 oncogenic drivers, which agent becomes more effective might depend on the levels of the relevant gene alterations. Which signaling pathway is the dominant one should be investigated initially, and re-biopsy is very important to analyze the potential mechanisms of resistance to TKIs.
To the best of our knowledge, this is the first study examining OS and the molecular mechanism of resistance to EGFR or ALK TKI in EGFR/ALK co-altered patients. However, this study had a few limitations. First, this was a retrospective, single-institution study, which could not accurately reflect the population with EGFR/ALK co-alterations at large. Second, the numbers of phospho-EGFR and phospho-ALK tests were small. Finally, the lack of sufficient biopsy samples limited our investigation of the mechanisms underlying TKI resistance.
In summary, first-line EGFR TKI might be the reasonable care for advanced NSCLC patients harbouring concomitant EGFR mutations and ALK rearrangements. As to this special gene profile patients, the molecular mechanisms underlying acquired resistance to EGFR TKI or crizotinib were similar to those of EGFR mutation and ALK rearrangement patients, but whether or nor to use sequential or in combination with crizotinib should be guided by the status of ALK rearrangement and the relative level of phospho-EGFR and phospho-ALK. Additional investigations are needed to overcome resistance to EGFR or ALK TKI and to improve OS in patients harbouring EGFR/ALK co-alterations.
MATERIALS AND METHODS
Patients and treatment
Patients were eligible for inclusion in the study if they were 18 years old or older, had histologically or cytologically confirmed clinical stage IIIB or IV NSCLC with histologic feathers of adenocarcinoma or squamous cell carcinoma. All patients provided written informed consent; separate consent was provided for the assessment of EGFR and ALK biomarkers. An independent ethics committee at the institution approved the study protocol. The study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonization Guidelines for Good Clinical Practice and applicable requirements. We recruited 118 patients from the Guangdong Lung Cancer Institute (GLCI) between August 2009 and July 2011. Among these patients, we identified 11 patients with concomitant EGFR mutations and ALK rearrangements treated with EGFR TKIs, crizotinib, or both, These EGFR/ALK co-altered patients were the same as those having evaluable clinical data as previously published [20]. Two additional groups, consisting of 23 ALK-positive patients receiving second or later-line crizotinib therapy as part of the A8081005 (NCT0093245) and A8081007 (NCT0093289) trials [39, 40], A total of 4 cases with ALK rearrangement also enrolled in our previous study [20]. and 84 EGFR-mutant patients enrolled in the CTONG 0901 trial receiving the first-line EGFR TKIs gefitinib and erlotinib [41] were used as controls, once progressive disease occurred, the further therapy after progression of the disease was at the physician's discretion.
EGFR mutation analysis
Genetic profiles were identified in the Department of Pathology of GGH and the GLCI. All tumour samples were routinely assessed by sectioning, haematoxylin-eosin staining, and visualisation under a microscope to ensure a tumour content of at least 50%. A mutation analysis of the EGFR tyrosine kinase domain was performed by DNA directed sequencing and/or the amplification refractory mutation system (ARMS) as described previously [42, 43] and according to the manufacturer's protocol, patients were considered to be positive for the EGFR mutation if 1 of 29 EGFR mutations was detected with the above methods.
RT-PCR and RACE-PCR sequencing for AK fusion analysis
Total RNA was extracted from tissue samples using the RNeasy Kit (Qiagen). Reverse-transcriptase PCR (RT-PCR) and 5′ rapid amplification c-DNA ends (RACE) coupled PCR plus sequencing was detected as described previously [8]. The products of PCR were then sequenced with a 3730XL Genetic Analyser (Applied Biosystems). Target sequences of interest were aligned with the ALK reference sequence (NM_004304.3) to determine whether the fusion with another gene appeared.
Fluorescence in situ hybridization (FISH) assays for ALK rearrangements
Tumour histology was classified using World Health Organisation criteria. Interphase molecular cytogenetic studies were performed using a commercially available ALK probe (Vysis LSI ALK Dual Colour, Break Apart Rearrangement Probe; Abbott Molecular Inc.) using 4-μm thick paraffin-embedded tissue sections. Samples were deemed to FISH-positive, if > 15% of the scored tumour cells had split ALK 5′ and 3′ probe signals or isolated 3′ signals [44].
Immunohistochemistry (IHC) for EGFR, ALK and downstream molecules
Protein expression was detected by IHC in serial sections of formalin-fixed paraffin-embedded tumour samples according to the manufacturer's instructions for each antibody. The samples were stained with antibodies against total EGFR, phospho-EGFR (Y1068), total ALK, and phospho-ALK (Y1604) (Cell Signalling Technology), followed by incubation in rabbit monoclonal anti-human ALK antibody (#3633 WP1-01; clone D5F3) at a dilution of 1:100. The staining intensity was scored from 0 to 3+; tumours with no expression (0) were described as negative for ALK protein expression, while tumours scored as 1+, 2+, or 3+ were deemed positive [45, 46], and the staining intensity was scored according to the following criteria: 3+, intense, granular cytoplasmic staining; 2+, moderate, smooth cytoplasmic staining; 1+, faint cytoplasmic staining in 10% of tumor cells; and 0, no staining [45], and high phosphorylation of proteins means IHC ++ or +++; low phosphorylation of proteins means IHC + or +/− NA, not available. -/NA, IHC negative because of not available testing [20, 38].
Assessments and statistical analysis
Evaluation of efficacy was assessed every 6 to 8 weeks according to Response Evaluation Criteria in Solid Tumours (RECIST1.1) [47]. PFS was measured from the initial treatment with an EGFR TKI or crizotinib to radiographic or clinical progression, as determined by means of the RECIST1.1′s criteria, or death from any cause. OS was calculated from the date of first-line therapy to death from any cause, or the date of last follow-up. ORR was analysed by the χ2-test, and PFS and OS were analysed with the use of the Kaplan-Meier curves, and hazard ratios for progression or death were calculated using the Cox proportional-hazards model. Subgroup analyses were performed to compare PFS and OS between treatments in groups defined according to the WHO performance status, age, disease stage at screening, pathology and smoking status. Detection the status of biomarkers and treatment with covariates were used to identify predictive factors by assessing whether there was a significant difference in the biomarkers and treatment effect for PFS and OS (hazard ratio for progression or death) between subgroups. The χ2 test, Fisher's exact test, and Wilcoxon test were used to compare categorical variables. All statistical tests were two-sided, with P value < 0.05 considered statistically significant.
Acknowledgments
We thank the participants and the staff of GLCI for their contribution to this study.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicts of interest.
GRANT SUPPORT
This study was supported by grants from the Project of the National Natural Science Funding of China (Grant No. 81472207), the Guangdong Provincial Key Laboratory of Lung Cancer Translational Medicine (Grant No. 2012A061400006), the Special Fund for Research in the Public Interest from the National Health and Family Planning Commission of PRC (Grant No. 201402031), and the Research Fund from the Guangzhou Science and Technology Bureau (Grant No. 2011Y2-00014).
REFERENCES
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II, Varella-Garcia M, Franklin WA, Aronson SL, Su PF, Shyr Y, Camidge DR, Sequist LV, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311:1998–2006. doi: 10.1001/jama.2014.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, Massion PP, Siwak-Tapp C, Gonzalez A, Fang R, Mark EJ, Batten JM, Chen H, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863–870. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santos E, Martin-Zanca D, Reddy EP, Pierotti MA, Della Porta G, Barbacid M. Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science. 1984;223:661–664. doi: 10.1126/science.6695174. [DOI] [PubMed] [Google Scholar]
- 5.Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, Jenkins RB, Kwiatkowski DJ, Saldivar JS, Squire J, Thunnissen E, Ladanyi M. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. Arch Pathol Lab Med. 2013;137:828–860. doi: 10.5858/arpa.2012-0720-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gainor JF, Varghese AM, Ou SH, Kabraji S, Awad MM, Katayama R, Pawlak A, Mino-Kenudson M, Yeap BY, Riely GJ, Iafrate AJ, Arcila ME, Ladanyi M, Engelman JA, Dias-Santagata D, Shaw AT. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res. 2013;19:4273–4281. doi: 10.1158/1078-0432.CCR-13-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camidge DR, Kono SA, Flacco A, Tan AC, Doebele RC, Zhou Q, Crino L, Franklin WA, Varella-Garcia M. Optimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin Cancer Res. 2010;16:5581–5590. doi: 10.1158/1078-0432.CCR-10-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, Zhang S, Yang X, Yang J, Zhou Q, Yin L, An S, Lin J, Chen S, Xie Z, Zhu M, Zhang X, Wu YL. Fusion of EML4 and ALK is associated with development of lung adenocarcinomas lacking EGFR and KRAS mutations and is correlated with ALK expression. Mol Cancer. 2010;9:188. doi: 10.1186/1476-4598-9-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JK, Kim TM, Koh Y, Lee SH, Kim DW, Jeon YK, Chung DH, Yang SC, Kim YT, Kim YW, Heo DS, Bang YJ. Differential sensitivities to tyrosine kinase inhibitors in NSCLC harboring EGFR mutation and ALK translocation. Lung Cancer. 2012;77:460–463. doi: 10.1016/j.lungcan.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 10.Boland JM, Jang JS, Li J, Lee AM, Wampfler JA, Erickson-Johnson MR, Soares I, Yang P, Jen J, Oliveira AM, Yi ES. MET and EGFR mutations identified in ALK-rearranged pulmonary adenocarcinoma: molecular analysis of 25 ALK-positive cases. J Thorac Oncol. 2013;8:574–581. doi: 10.1097/JTO.0b013e318287c395. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, Lathan C, Marcoux JP, Du J, Okuda K, Capelletti M, Shimamura T, Ercan D, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71:6051–6060. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baldi L, Mengoli MC, Bisagni A, Banzi MC, Boni C, Rossi G. Concomitant EGFR mutation and ALK rearrangement in lung adenocarcinoma is more frequent than expected: report of a case and review of the literature with demonstration of genes alteration into the same tumor cells. Lung Cancer. 2014;86:291–295. doi: 10.1016/j.lungcan.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 13.Kuo YW, Wu SG, Ho CC, Shih JY. Good response to gefitinib in lung adenocarcinoma harboring coexisting EML4-ALK fusion gene and EGFR mutation. J Thorac Oncol. 2010;5:2039–2040. doi: 10.1097/JTO.0b013e3181f43274. [DOI] [PubMed] [Google Scholar]
- 14.Popat S, Vieira de Araujo A, Min T, Swansbury J, Dainton M, Wotherspoon A, Lim E, Nicholson AG, O'Brien ME. Lung adenocarcinoma with concurrent exon 19 EGFR mutation and ALK rearrangement responding to erlotinib. J Thorac Oncol. 2011;6:1962–1963. doi: 10.1097/JTO.0b013e31822eec5e. [DOI] [PubMed] [Google Scholar]
- 15.Chiari R, Duranti S, Ludovini V, Bellezza G, Pireddu A, Minotti V, Bennati C, Crino L. Long-term response to gefitinib and crizotinib in lung adenocarcinoma harboring both epidermal growth factor receptor mutation and EML4-ALK fusion gene. J Clin Oncol. 2014;32:e30–32. doi: 10.1200/JCO.2012.47.7141. [DOI] [PubMed] [Google Scholar]
- 16.Santelmo C, Ravaioli A, Barzotti E, Papi M, Poggi B, Drudi F, Mangianti M, Salvi M, Crino L. Coexistence of EGFR mutation and ALK translocation in NSCLC: literature review and case report of response to gefitinib. Lung Cancer. 2013;81:294–296. doi: 10.1016/j.lungcan.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Miyanaga A, Shimizu K, Noro R, Seike M, Kitamura K, Kosaihira S, Minegishi Y, Shukuya T, Yoshimura A, Kawamoto M, Tsuchiya S, Hagiwara K, Soda M, et al. Activity of EGFR-tyrosine kinase and ALK inhibitors for EML4-ALK-rearranged non-small-cell lung cancer harbored coexisting EGFR mutation. BMC Cancer. 2013;13:262. doi: 10.1186/1471-2407-13-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Zhang J, Hu Q, Li X, Zhou C. A case of lung adenocarcinoma harboring exon 19 EGFR deletion and EML4-ALK fusion gene. Lung Cancer. 2013;81:308–310. doi: 10.1016/j.lungcan.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Zhao N, Zheng SY, Yang JJ, Zhang XC, Xie Z, Xie B, Su J, Chen ZH, Chen SL, Zhang N, Lou NN, Dong S, Wu YL. Lung adenocarcinoma harboring concomitant EGFR mutation and EML4-ALK fusion that benefits from three kinds of tyrosine kinase inhibitors: a case report and literature review. Clin Lung Cancer. 2015;16:e5–9. doi: 10.1016/j.cllc.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Yang JJ, Zhang XC, Su J, Xu CR, Zhou Q, Tian HX, Xie Z, Chen HJ, Huang YS, Jiang BY, Wang Z, Wang BC, Yang XN, et al. Lung cancers with concomitant EGFR mutations and ALK rearrangements: diverse responses to EGFR-TKI and crizotinib in relation to diverse receptors phosphorylation. Clin Cancer Res. 2014;20:1383–1392. doi: 10.1158/1078-0432.CCR-13-0699. [DOI] [PubMed] [Google Scholar]
- 21.Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap) Ann Oncol. 2013;24:2371–2376. doi: 10.1093/annonc/mdt205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kris MG, Johnson BE, Kwiatkowski DJ, Iafrate AJ, Wistuba II, Aronson SL, Engelman JA, Shyr Y, Khuri FR, Rudin CM, Garon EB, Pao W, Schiller JH, et al. Identification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: the NCI's Lung Cancer Mutation Consortium (LCMC) J Clin Oncol. 2011;29 (suppl; CRA7506) [Google Scholar]
- 23.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, Varella-Garcia M, Camidge DR. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–1482. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Won JK, Keam B, Koh J, Cho HJ, Jeon YK, Kim TM, Lee SH, Lee DS, Kim DW, Chung DH. Concomitant ALK translocation and EGFR mutation in lung cancer: a comparison of direct sequencing and sensitive assays and the impact on responsiveness to tyrosine kinase inhibitor. Ann Oncol. 2015;26:348–354. doi: 10.1093/annonc/mdu530. [DOI] [PubMed] [Google Scholar]
- 25.Cai W, Lin D, Wu C, Li X, Zhao C, Zheng L, Chuai S, Fei K, Zhou C, Hirsch FR. Intratumoral Heterogeneity of ALK-Rearranged and ALK/EGFR Coaltered Lung Adenocarcinoma. J Clin Oncol. 2015;33:3701–3709. doi: 10.1200/JCO.2014.58.8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bourzac K. Biology: Three known unknowns. Nature. 2014;509:S69–71. doi: 10.1038/509S69a. [DOI] [PubMed] [Google Scholar]
- 27.Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, Riely GJ. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lovly CM, Pao W. Escaping ALK inhibition: mechanisms of and strategies to overcome resistance. Sci Transl Med. 2012;4:120ps122. doi: 10.1126/scitranslmed.3003728. [DOI] [PubMed] [Google Scholar]
- 29.Ke EE, Zhou Q, Wu YL. Emerging paradigms in targeted treatments for Asian patients with NSCLC. Expert Opin Pharmacother. 2015;16:1167–1176. doi: 10.1517/14656566.2015.1040391. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 31.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 36.Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, Del Grammastro M, Sciarrotta MG, Buttitta F, Incarbone M, Toschi L, Finocchiaro G, Destro A, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009;27:1667–1674. doi: 10.1200/JCO.2008.19.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, Pan Y, Wang L, de Stanchina E, Shien K, Aoe K, Toyooka S, Kiura K, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109:E2127–2133. doi: 10.1073/pnas.1203530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, Drew L, Saeh JC, Crosby K, Sequist LV, Iafrate AJ, Engelman JA. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4:120ra117. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O'Byrne KJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 40.Ou SH, Janne PA, Bartlett CH, Tang Y, Kim DW, Otterson GA, Crino L, Selaru P, Cohen DP, Clark JW, Riely GJ. Clinical benefit of continuing ALK inhibition with crizotinib beyond initial disease progression in patients with advanced ALK-positive NSCLC. Ann Oncol. 2014;25:415–422. doi: 10.1093/annonc/mdt572. [DOI] [PubMed] [Google Scholar]
- 41.Yang JJ, Zhou Q, Yan HH, Zhang XC, Chen HJ, Tu HY, Wang Z, Xu CR, Su J, Huang YS, Wang BC, Jiang BY, Bai XY, et al. A randomized controlled trial of erlotinib versus gefitinib in advanced non-small-cell lung cancer harboring EGFR exon 19 or 21 mutations (CTONG0901) WCLC2015. (suppl; abstr MINI16.13) [Google Scholar]
- 42.Jiang SX, Yamashita K, Yamamoto M, Piao CJ, Umezawa A, Saegusa M, Yoshida T, Katagiri M, Masuda N, Hayakawa K, Okayasu I. EGFR genetic heterogeneity of nonsmall cell lung cancers contributing to acquired gefitinib resistance. Int J Cancer. 2008;123:2480–2486. doi: 10.1002/ijc.23868. [DOI] [PubMed] [Google Scholar]
- 43.Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, Smith JC, Markham AF. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS) Nucleic Acids Res. 1989;17:2503–2516. doi: 10.1093/nar/17.7.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodig SJ, Mino-Kenudson M, Dacic S, Yeap BY, Shaw A, Barletta JA, Stubbs H, Law K, Lindeman N, Mark E, Janne PA, Lynch T, Johnson BE, Iafrate AJ, Chirieac LR. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res. 2009;15:5216–5223. doi: 10.1158/1078-0432.CCR-09-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yi ES, Boland JM, Maleszewski JJ, Roden AC, Oliveira AM, Aubry MC, Erickson-Johnson MR, Caron BL, Li Y, Tang H, Stoddard S, Wampfler J, Kulig K, Yang P. Correlation of IHC and FISH for ALK gene rearrangement in non-small cell lung carcinoma: IHC score algorithm for FISH. J Thorac Oncol. 2011;6:459–465. doi: 10.1097/JTO.0b013e318209edb9. [DOI] [PubMed] [Google Scholar]
- 46.Mino-Kenudson M, Chirieac LR, Law K, Hornick JL, Lindeman N, Mark EJ, Cohen DW, Johnson BE, Janne PA, Iafrate AJ, Rodig SJ. A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clin Cancer Res. 2010;16:1561–1571. doi: 10.1158/1078-0432.CCR-09-2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1. 1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]