

Abstract
Objective
Sporadic inclusion body myositis (sIBM) pathogenesis is unknown; however, rimmed vacuoles (RVs) are a constant feature. We propose to identify proteins that accumulate within RVs.
Methods
RVs and intact myofibers were laser microdissected from skeletal muscle of 18 sIBM patients and analyzed by a sensitive mass spectrometry approach using label-free spectral count-based relative protein quantification. Whole exome sequencing was performed on 62 sIBM patients. Immunofluorescence was performed on patient and mouse skeletal muscle.
Results
213 proteins were enriched by >1.5X in RVs compared to controls and included proteins previously reported to accumulate in sIBM tissue or when mutated cause myopathies with RVs. Proteins associated with protein folding and autophagy were the largest group represented. One autophagic adaptor protein not previously identified in sIBM was FYCO1. Rare missense coding FYCO1 variants were present in 11.3% of sIBM patients compared with 2.6% of controls (p=0.003). FYCO1 co-localized at RVs with autophagic proteins such as MAP1LC3 and SQSTM1 in sIBM and other RV myopathies. One FYCO1 variant protein had reduced co-localization with MAP1LC3 when expressed in mouse muscle.
Interpretation
This study used an unbiased proteomic approach to identify RV proteins in sIBM that included a novel protein involved in sIBM pathogenesis. FYCO1 accumulates at RVs and rare missense variants in FYCO1 are overrepresented in sIBM patients. These FYCO1 variants may impair autophagic function leading to RV formation in sIBM patient muscle. FYCO1 functionally connects autophagic and endocytic pathways supporting the hypothesis that impaired endolysosmal degradation underlies the pathogenesis of sIBM.
Keywords: Sporadic inclusion body myositis, rimmed vacuoles, FYCO1
INTRODUCTION
Sporadic inclusion body myositis (sIBM) is the most common idiopathic inflammatory myopathy (IIM) in people over 50 years of age. It causes progressive muscle weakness, especially of finger flexion and knee extension. It is uncertain whether sIBM is a primarily inflammatory or a degenerative myopathy. In contrast to other IIM, conventional immunosuppressants and immunomodulatory regimens have not been found to alter disease progression.1,2 Typical myopathological features are inflammatory and degenerative changes, accompanied by rimmed vacuoles (RV) and protein aggregates.2-6 The current manuscript intentionally focuses on these “degenerative features” in order to gain insight into one aspect of sIBM pathogenesis.
RVs are characteristic for the disease and are a useful diagnostic pathological feature; however their genesis remains enigmatic.2,5 They are reported to contain both sarcoplasmic and myonuclear proteins. It has been suggested that the origin of a RV is due to myonuclear breakdown since both nuclear and nuclear envelope proteins are present in or adjacent to the RV.7 Others have identified autophagic, lysosomal and endosomal protein markers within and surrounding RVs.4,8,9 This has led to the hypothesis that impairment of autophagic degradation underlies sIBM pathogenesis. Finally, many proteins present in protein aggregate diseases such as Alzheimer’s Disease, amyotrophic lateral sclerosis and protein aggregate myopathies are consistently found accumulated in sIBM muscle suggesting that sIBM is a degenerative proteinopathy akin to neurodegenerative disorders. RVs are typical of sIBM pathology, but they are not specific and are also found in a number of inherited myopathies including myofibrillar myopathies, hereditary distal myopathies and rare inherited inclusion body myopathies due to mutations in GNE and VCP.10,11 In these disorders, the disease associated mutant protein accumulates within degenerating myofibers often within or adjacent to RVs.
Previously we established a combined laser microdissection and label-free proteomic approach to analyze cytoplasmic protein aggregates in myofibrillar myopathies.11-13 Using this technique, we identified distinct protein signatures in the muscle of myofibrillar myopathy patients. In the present study, we utilize the same proteomic approach to identify the protein composition of RVs in sIBM. We hypothesize that RV enriched proteins are pathogenic mediators of disease and will inform both genetic association and sIBM biomarker development.
MATERIALS AND METHODS
Patients and muscle biopsies
Studies were performed on muscle samples from 18 patients with sIBM (6 women, 12 men, mean age 66.7±10.7 years, range 49-93 years) according to local ethics committee regulations (reg. number 3882-10). The diagnosis of sIBM was based on the recently published 2011 European Neuromuscular Centre international workshop criteria.5 After surgical procedure, skeletal muscle biopsies were divided into 0.5 cm3 pieces, embedded into Tissue Freezing Medium® (Leica Microsystems, Wetzlar, Germany) and flash frozen in liquid nitrogen.
DNA was collected on 62 patients with a diagnosis of sIBM.5 40/62 patients had been previously reported and targeted sequencing for hereditary causes of muscle disease had been performed.14 An additional 12 patients were identified in the Washington University Neuromuscular Clinic and 10 patients from Johns Hopkins University. All participants gave written informed consent, and study procedures were approved by the Human Studies Committee at Washington University.
Proteomic analysis
Laser microdissection and sample processing
A combined laser microdissection and label free mass spectrometry approach was applied as described with modifications.13,15 Ten μm thick frozen skeletal muscle sections were placed on polyethylene terephthalate (PET) membranes (Leica Microsystems, Wetzlar, Germany) and stained with hematoxylin and eosin (H&E). These sections were used to collect (from each patient) a total area of 250,000 μm2 of RV and surrounding sarcoplasm (hereinafter referred as RV sample) and the same area of vacuole-free sarcoplasm in normally looking muscle fibers (control sample) by laser microdissection (LMD 6500, Leica Microsystems, Wetzlar, Germany) into tubes containing 40 μl of 98-100% formic acid. After incubation for 30 min and sonification (35 kHz) for 5 min (RK31, BANDELIN electronic, Berlin, Germany), samples were centrifuged at 12,000 g for 10 min at 4°C and frozen at -80°C. The in-solution digestion and sample processing was performed as described.13,15
Nano high performance liquid chromatography (HPLC) and mass spectrometry (MS)
The HPLC-MS analysis was performed on a nano-HPLC system UltiMate 3000 RSLCnano (Dionex, Idstein, Germany) interfaced to a quadrupole orbitrap mass spectrometer (Q Exactive; Thermo Fischer Scientific, Germany). HPLC-analysis was performed as described before.13 After nano-HPLC separation peptides were ionized in a nano electrospray ionization source (ESI) and analyzed in data dependent scan mode in the Q Exactive mass spectrometer. Full MS spectra were scanned between 350 and 1,400 m/z with a resolution of 70,000 at 200 m/z (AGC target 3e6, 80ms maximum injection time). The capillary temperature was set at 250°C and the spray voltage at 1600 V (+). Lock mass polydimethylcyclosiloxane (m/z 445.120) was used for internal recalibration. The m/z values initiating MS/MS were set on a dynamic exclusion list for 30s and the 10 most intensive ions (charge +2, +3, +4) with intensity higher than 1.7e3 were selected for fragmentation. Tandem MS fragments were generated by higher energy collision induced dissociation and the fragmentation was performed with 27% normalized collision energy. The first MS/MS mass was fixed at 130.0 m/z and isolation window 2.2 m/z. The fragments were injected into the orbitrap analyzer with 35,000 resolution at 2,000 m/z (AGC 1e6, maximum injection time 120 ms).
Database search and relative protein quantification
After ESI-MS/MS analysis mass spectrometric data were searched against a human protein database containing the entire UniProt/Swiss-Prot (release 2014/05/28, 20265 curated entries) using Mascot (version 2.5)16 and label-free relative protein quantification based on spectral counting was performed as described13 using PIA.17 To identify proteins that were overrepresented in RV within muscle fibers in sIBM, the ratios between the averaged proportions of proteins in RV and control samples were calculated and a two-tailed unpaired t-test (equal variances assumed) was performed for each protein. A protein was accepted as significantly overrepresented if the RV/control ratio was >1.5 and the p-value <0.05.
Validation of proteomic findings by immunofluorescence studies
Immunofluorescence studies were performed on muscle samples from five sIBM patients to validate the proteomic findings as described.6,13 Serial frozen skeletal muscle sections were incubated overnight at 4°C with primary antibodies against 21 proteins (Supplementary Table 1), followed by washing steps and incubation with isotope specific secondary antibodies conjugated with Alexa Fluor 488 (Dianova, Hamburg, Germany; dilution 1:1,000) or Texas Red (Dianova, Hamburg, Germany; dilution 1:400). Nuclei were visualized by 4′,6-diamidino-2-phenylindole (DAPI) staining (Roche Diagnostics, Indianapolis, IN, USA; dilution 1:10,000).
Whole exome sequencing (WES)
Indexed genomic DNA libraries were prepared from genomic DNA using TruSeq DNA Preparation Kit (Illumina, San Diego, CA, USA) and exome capture using TruSeq Exome Enrichment Kit (Illumina), according to the manufacturer’s protocol. Sequencing was performed with 100 bp paired-end reads on a HiSeq2000 (Illumina). Reads were aligned to the human reference genome with NovoAlign (Novocraft Technologies, Selangor, Malaysia) or Burrows-Wheeler Aligner.18 Variants were called with SAMtools19 and annotated with SeattleSeq. Coverage across genomic intervals was calculated using BEDTools.20 Genomic coordinates for regions targeted by the whole-exome capture kit were provided by Illumina. Whole exome sequences from 62 sIBM patients were filtered for variants that: 1) had a minor allele frequency of ≤0.001 in the ExAC Database; 2) generated a loss of function variant or a nonsynonymous change; and 3) fulfilled a strict sequence quality as defined by Genesis 2.0 software.
Mouse expression studies
GFP-FYCO-WT and mCherry-LC3 expressing plasmid constructs were obtained from Dr. Terje Johansen. The LIRmut (F1280A/I1283A), T1270A and P1302L point mutations were generated via site directed mutagenesis. For electroporation, mice were anesthetized using inhaled isoflurane. The skin overlying the TA muscle was shaved, and the animals were co-injected with 30 μg endotoxin-free expression plasmid (diluted in sterile phosphate-buffered saline (PBS) to a volume of 50 μl by using a 0.5 ml syringe fitted with a 29-gauge needle. Two-needle array electrodes (450121) (Harvard Apparatus, Holliston, MA, USA) were inserted into the muscle immediately after DNA delivery for electroporation. The distance between the electrodes was 5 mm, and the array was inserted longitudinally relative to the muscle fibers. In vivo electroporation parameters were the following: voltage, 75 V; pulse length, 50 ms; number of pulses, six pulses; pulse interval, 200 ms; desired field strength, 200 V/cm, given by a BTX ECM830 Electro Square Porator. Animals were allowed to recover for 7 days prior to muscle isolation which was frozen in liquid nitrogen cooled isopentane and processed into 10uM section. Slides were examined using a fluorescent microscope (80i upright; Nikon) and charge-coupled device camera (EZ monochrome; Roper Industries) with deconvolution software analysis (NIS Elements; Nikon). Image processing and analysis were performed with NIS Elements 4.0 software and Photoshop CS3 (Adobe). All images were performed on fixed tissue at room temperature using Prolong Gold mounting solution (Invitrogen). Objectives used for immunofluorescence were Apochromat 20× and 40×. For colocalization analysis, 40 random fibers from three experiments were selected. The Pearson’s colocalization coefficient was determined for each fiber using NIS Elements 4.0 software. All images were taken at the same gain and exposure intensity.
RESULTS
Proteomic analysis
In total 3873 different proteins were identified by mass spectrometric analysis including 213 proteins that showed a statistically significant over-representation in RV samples compared to controls (see Supplementary Table 1). The proteins were assigned to subgroups based on their main physiological cell function. The proportion of each subgroup related to the sum of spectral counts is shown in Fig. 1. Intermediate filaments were the most abundant overrepresented proteins, followed by proteins of the extracellular matrix and by proteins involved in cell stress response, protein quality control and protein degradation (Fig. 1). The latter group contains 29 different proteins including three proteins that were detected in >50% of RV samples but in none of the control samples: transaldolase (an enzyme of the pentose-phosphate pathway), p62 (SQSTM1), and FYVE and coiled-coil domain-containing protein 1 (FYCO1).
Figure 1. Functional classification of proteins identified as overrepresented in RV samples by proteomic analysis.
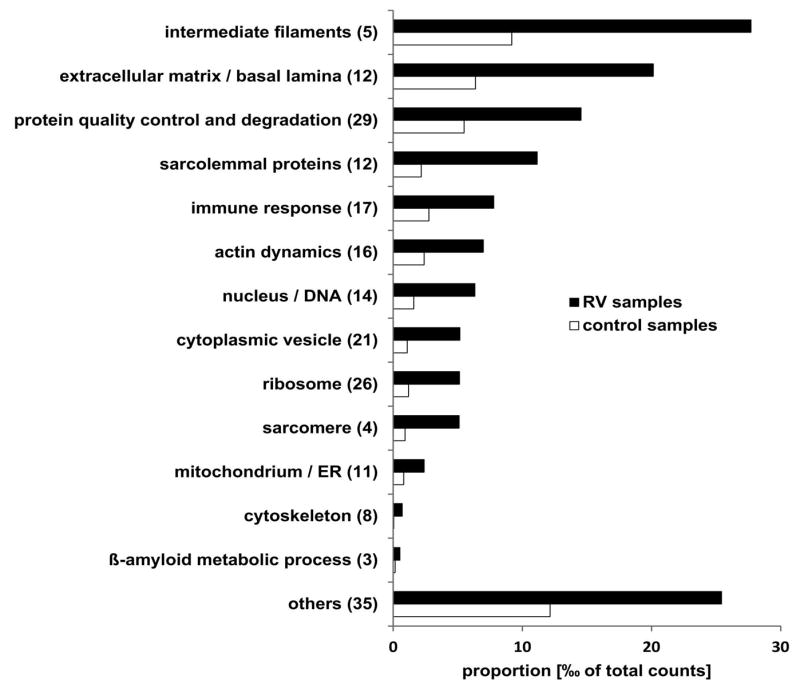
Bar chart showing the sum of the mean proportion of proteins assigned to a subgroup is given for RV and control samples in per mill of total spectral counts. The number of detected proteins is given in brackets behind the subgroup name. Black bars show the proportion in RV samples and grey bars the proportion in control samples. Details of proteins assigned to the different subgroups are provided in Supplementary Table 1. ER: endoplasmic reticulum.
Genetic Analysis
We reasoned that the proteins overrepresented in RVs may facilitate the identification of potential genetic risk alleles for sIBM. To test this, we identified rare missense or loss of function (LoF) variants in genes that encoded proteins that were present in ≥50% of RV samples (131 genes, see Supplementary Table 1) using WES of 62 patients with sIBM. This analysis identified 100 variants from 52 genes. 17 genes had variants in two or more sIBM patients (see Table 1). To see if the burden of variants within a single gene was increased in other diseased control populations, or if this enrichment was specific to sIBM, we determined the number of rare missense of LoF variants within these 17 genes in sporadic amyotrophic lateral sclerosis (ALS) patients from an existing WES dataset. Using this data we found that variants in only one gene were statistically enriched in sIBM patients with a p value of ≥0.01. Specifically, 7/62 (11.3%) of sIBM patients carried a rare missense or LoF FYCO1 variant as compared with 18/680 (2.6%) of ALS patients (p value=0.0029) (Table 1). Similarly, the burden of FYCO1 variants in sIBM was significantly enriched (p value=0.011) when compared with ethnically matched patient genomes within the 1000 genomes database with 17/503 carrying a rare missense or LoF variant in FYCO1. sIBM associated FYCO1 rare variants were present throughout the protein although two variants were adjacent to or within the LC3 interacting region (LIR) domain (Supplementary Table 2, Fig. 2).21
Table 1.
Rare missense variants in >2 of 62 sIBM patients compared with ALS patients
| Gene name | sIBM patients with rare variant | ALS patients with rare variant | Statistical Significance | ||||
|---|---|---|---|---|---|---|---|
| Number | Total | Percent | Number | Total | Percent | ||
| AHNAK | 7 | 62 | 11.3 | 77 | 680 | 11.3 | 1 |
| ALDH3A2 | 2 | 62 | 3.2 | 6 | 680 | 0.88 | 0.14 |
| ASAH1 | 2 | 62 | 3.2 | 4 | 680 | 0.59 | 0.08 |
| C3 | 2 | 62 | 3.2 | 8 | 680 | 1.2 | 0.2 |
| COL4A2 | 2 | 62 | 3.2 | 14 | 680 | 2.1 | 0.64 |
| DMD | 4 | 62 | 6.5 | 29 | 680 | 4.3 | 0.51 |
| DYSF | 4 | 62 | 6.5 | 28 | 680 | 4.1 | 0.51 |
| FYCO1 | 7 | 62 | 11.3 | 18 | 680 | 2.6 | 0.003 |
| GSN | 2 | 62 | 3.2 | 10 | 680 | 1.5 | 0.61 |
| HSPG2 | 10 | 62 | 16.1 | 56 | 680 | 8.2 | 0.06 |
| LAMA2 | 2 | 62 | 3.2 | 24 | 680 | 3.5 | 1 |
| LAMC1 | 2 | 62 | 3.2 | 16 | 680 | 2.4 | 0.45 |
| NRAP | 5 | 62 | 8.1 | 17 | 680 | 2.5 | 0.03 |
| SPTAN1 | 2 | 62 | 3.2 | 9 | 680 | 1.3 | 0.23 |
| SPTB | 4 | 62 | 6.5 | 33 | 680 | 4.9 | 0.8 |
| UNC45B | 3 | 62 | 4.8 | 9 | 680 | 1.3 | 0.07 |
| XIRP2 | 4 | 62 | 6.5 | 48 | 680 | 7.1 | 1 |
Figure 2. Scheme of FYCO1.
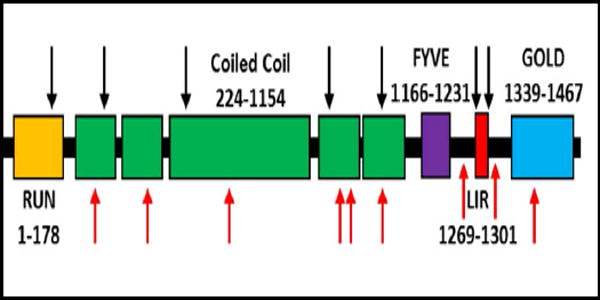
Black arrows denote sites of missense variants identified in patients with sIBM. Red arrows denote mutations previously identified in patients with congenital cataracts. Domains include RUN (GTPase interacting motif); Coiled Coil (dimerization motif); FYVE (phospholipid binding region); LIR (LC3 interacting region); and GOLD (golgi dynamics domain).
Validation of proteomic data by double immunofluorescence staining
Our proteomic and genetic data suggested that FYCO1 was an intriguing candidate protein to explore further. FYCO1 binds to LC3 and other vesicular cargo facilitating autophagic degradation.22 We found that RVs displayed immunoreactivity for FYCO1, the autophagic adaptor p62 and the autophagosome protein MAP1LC3 in sIBM patients, with a slightly stronger immunoreactivity for FYCO1 (Fig. 3, copy using magenta-green coloring for readers who are red-green color blind in Supplementary Fig. 1). Therefore, we used FYCO1 as a marker for RV in double immunofluorescence analyses to evaluate 18 additional proteins from different subgroups identified as enriched in RV samples by our proteomic analyses (Fig. 4 and 5, magenta-green copies in Supplementary Fig. 2 and 3). The immunoreactivity for these proteins was increased inside RVs or very closely around them, matching the area that has been collected by laser microdissection and thus confirming our mass spectrometric data.
Figure 3. Co-localization of FYCO1, p62 and LC3 in RVs of sIBM patients.
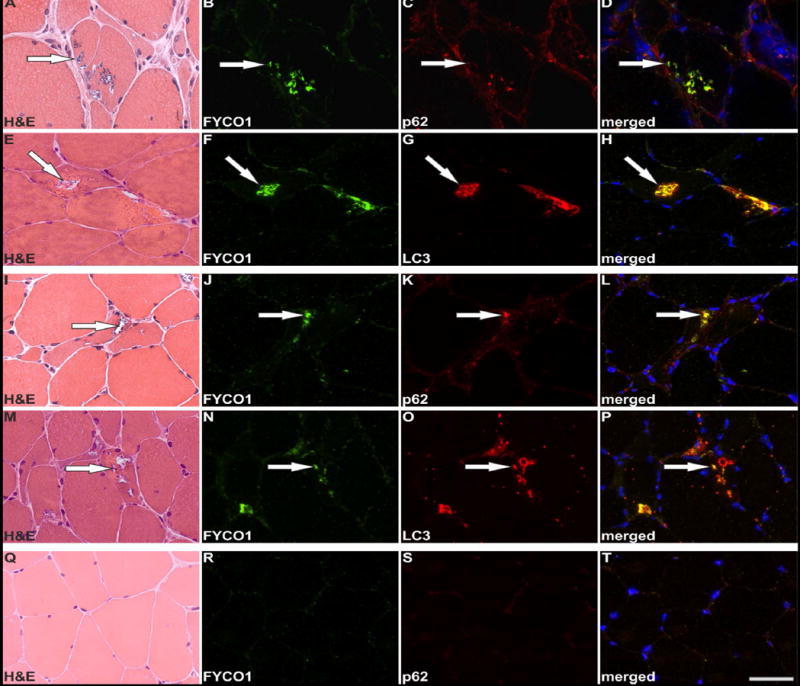
Serial skeletal muscle sections from two sIBM patients (patient 1: A-H, patient 2: I-P) and from a healthy control (Q-T) were stained with H&E and double-immunostained with primary antibodies directed against FYCO1 (green) and p62 or LC3 (red). Nuclei are stained with DAPI (blue). For each sIBM patient two different RV containing areas of the muscle samples are displayed. All RVs show a strong immunoreactivity for FYCO1, p62 and LC3. The co-localization of FYCO1 with p62 LC3 is indicated by yellow in the merged images. Scale bar = 50 μm.
Figure 4. Validation of proteomic findings by immunolocalization studies I.
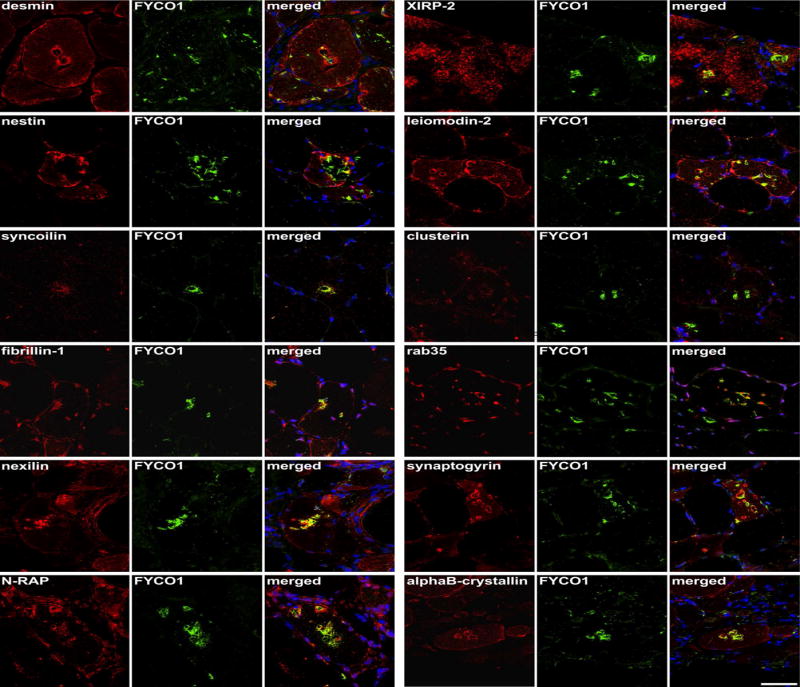
Serial sections from two sIBM patients were stained with H&E and double-immunostained with antibodies recognizing desmin, nestin, syncoilin, fibrillin-1, nexilin, N-RAP, XIRP-2, leiomodin-2, clusterin, rab35, synaptogyrin, and alphaB-crystallin. All proteins showed an accumulation in RV samples (red) and FYCO1 (green) as a positive control to localize RVs. Nuclei are stained with DAPI (blue). Increased immunoreactivity was observed with all proteins as indicated by yellow in the merged image. Scale bar = 50 μm.
Figure 5. Validation of proteomic findings by immunolocalization studies II.
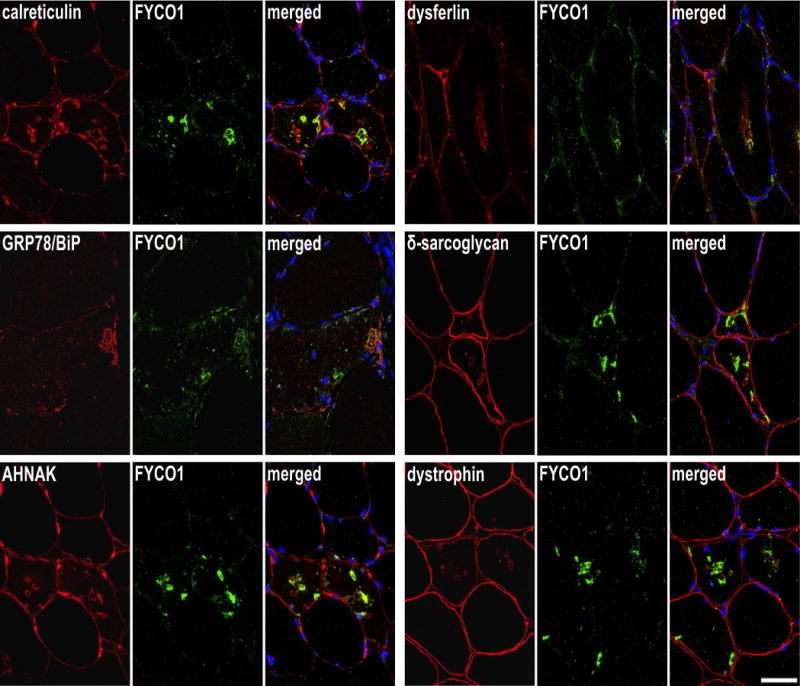
Serial sections from two sIBM patients were stained with H&E and double-immunostained with antibodies recognizing calreticulin, GRP78/BiP, AHNAK, dysferlin, δ-sarcoglycan and dystrophin All proteins showed an accumulation in RV samples (red) and FYCO1 (green) as a positive control to localize RVs. Nuclei are stained with DAPI (blue). Increased immunoreactivity was observed with all proteins as indicated by yellow in the merged image. Scale bar = 50 μm.
FYCO1 localization in other vacuolar myopathies and idiopathic inflammatory myopathies
To assess the specificity of FYCO1 for RV, we performed immunofluorescence staining on skeletal muscle sections from patients with hereditary inclusion body myopathy caused by GNE mutations, filaminopathy associated with a myofibrillar myopathy phenotype, glycogen storage disorder type II (Pompe’s disease) and normal control muscle tissue. The sarcolemma was marked with an antibody to spectrin (Fig. 6, magenta-green copy in Supplementary Fig. 4). FYCO1 was associated with RVs in hereditary inclusion body myopathy, glycogen storage disorder type II and filaminopathy. In filaminopathy, immunoreactivity for FYCO1 was increased in RV and in sarcoplasmic aggregates (Fig. 6). In dermatomyositis, some perifascicular muscle fibers showed an increased immunoreactivity for FYCO1 but “punched-out” areas of myofibrillar loss were not rimmed or markedly filled with FYCO1. In polymositis and in sIBM with a morphological phenotype of polymyositis, muscle fibers surrounded by inflammatory cells displayed a diffuse/punctate immunoreactivity for FYCO1. In addition, some fibers showed FYCO1 accumulations in subsarcolemmal areas similar to that found in RV areas in sIBM. These areas were basophilic in H&E staining (Fig.6). FYCO1 immunostaining and its co-localization to autophagic proteins (LC3 and p62) at RVs were similar in three sIBM patients carrying FYCO1 variants as compared with co-localization in sIBM patients not carrying a FYCO1 variant (data not shown).
Figure 6. Localization of FYCO1 in hereditary myopathies with rimmed vacuoles and in idiopathic inflammatory myopathies.
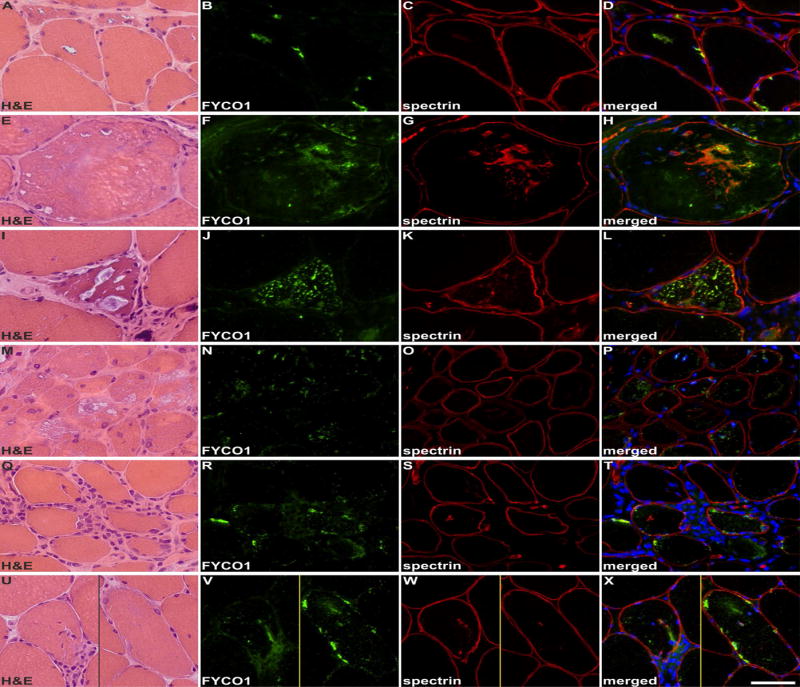
Shown are findings in patients with: GNE-related hereditary inclusion body myopathy (A-D), myofibrillar myopathy caused by FLNC mutation (E-H), glycogen storage disease type II (I-L), dermatomyositis (M-P), polymyositis (Q-T) and a morphological diagnosis of polymyositis but a typical sIBM clinical phenotype (U-X). Serial skeletal muscle sections were stained with H&E and double-immunostained with antibodies recognizing FYCO1 (green) and the constituent muscle protein spectrin (red). Nuclei were stained with DAPI (blue). RVs in hereditary inclusion body myopathy, myofibrillar myopathy and glycogen storage disease type II showed a strong immunoreactivity for FYCO1. In myofibrillar myopathy, FYCO1 was also located in cytoplasmic protein aggregates. In dermatomyositis, some perifascicular muscle fibers showed an increased immunoreactivity for FYCO1 but “punched-out” areas of myofibrillar loss were not rimmed or markedly filled with FYCO1. In polymositis and in sIBM with a morphological phenotype of polymyositis, muscle fibers surrounded by inflammatory cells displayed a diffuse/punctate immunoreactivity for FYCO1. In addition, some fibers showed FYCO1 accumulations in subsarcolemmal areas similar to that found in RV areas in sIBM. These areas were basophilic in H&E staining. Scale bar = 50 μm.
FYCO1 variant localization in mouse muscle
FYCO1 is reported to facilitate the transport of autophagosomes along microtubules via its association with LC3.22 Two identified missense variants, FYCO1-T1270A and FYCO1-P1302L, are adjacent to or within the LC3 interacting region domain (LIR) suggesting they may alter LC3 interaction (Supplementary Table 2, Fig. 2). We co-electroporated a plasmid expressing mCherry tagged LC3 with a plasmid expressing green fluorescent protein tagged FYCO1-WT in mouse tibialis anterior muscle. After seven days muscle was sectioned and visualized via fluorescent microscopy. GFP-FYCO1-WT was present as small puncta throughout the sarcoplasm that co-localized with mCherry-LC3 (Figure 7A-C). Expression of FYCO1 with two point mutations within the LIR domain that abolish LC3 interaction, GFP-FYCO1-LIRmut, demonstrated a similar pattern of GFP-FYCO1 puncta but diffuse mCherry-LC3 with reduced co-localization (Figure 7D-F). This pattern of reduced FYCO1/LC3 colocalization was also seen with the sIBM variant, GFP-FYCO1-P1302L (Figure 7G-I) but not with GFP-FYCO1-T1270A (Figure 7J-L). The degree of GFP-FYCO1/mCherry-LC3 co-localization was quantified from co-expressing fibers/condition from three independent experiments and was significantly reduced in GFP-FYCO1-P1302L expressing fibers compared to GFP-FYCO1-WT controls (Figure 7M).
Figure 7. Localization of GFP-FYCO1 and mCherry-LC3 in mouse tibialis anterior muscle.
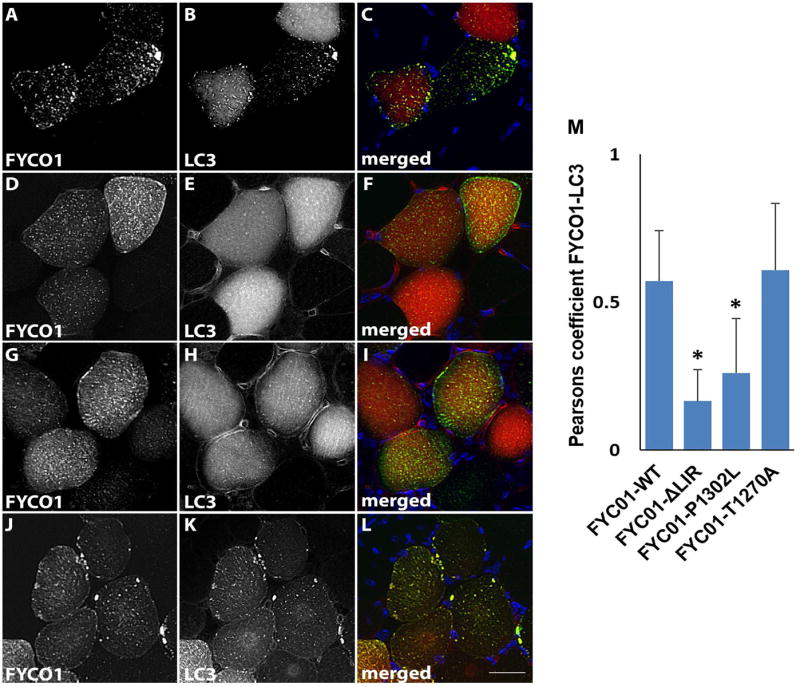
GFP-FYCO1-WT (A-C), GFP-FYCO1-LIRmut (D-F), GFP-FYCO1-P1302L (G-I) and GFP-FYCO1-T1270A (J-L) were co-expressed with mCherry-LC3 and visualized via fluorescence microscopy for FYCO1 (A, D, G, J in green on merged), LC3 (B, E, H, K in red on merged) and DAPI for nuclei (C, F, I, L in blue on merged). Scale bar = 25 μm. M) Quantitation of the Pearsons co-localization coefficient for FYCO1 and LC3 in 40 fibers from three independent experiments. Error bars are standard deviation and * denotes p value <0.001.
DISCUSSION
In this study, we applied for the first time a highly sensitive proteomic approach to analyze the composition of a characteristic histopathological feature in sIBM. We used laser microdissection to collect RV and intraindividual control samples from muscle sections of sIBM patients. Mass spectrometric analysis and relative protein quantification allowed us to identify 213 proteins accumulated in RV samples compared to controls. Forty of these overrepresented proteins have already been described in sIBM (see Supplementary Table 1). Thus, the results of these previous studies validate our method and findings. Many of the proteins overlap with those identified in other protein aggregate myopathies11-13 making most of the identified proteins not specific for sIBM but rather highlights converging pathogenic mechanisms with other muscle diseases. We also identified 173 proteins which have not been described in sIBM previously, which provides a basis for future studies to further investigate disease mechanisms. Our approach of label-free proteomic analysis combined with next generation sequencing has enabled us to identify a possible new genetic risk factor for sIBM.
We categorized the overrepresented RV proteins by their main cellular function and performed extensive immunofluorescence studies to further evaluate selected members of the different protein subgroups. These analyses confirmed and validated our proteomic data. We found that intermediate filaments were the most abundant overrepresented components in RV samples and immunolocalization analysis proved that distinct intermediate filaments accumulate in RV areas. Very few studies have investigated intermediate filaments in sIBM.23-26 Olivé et. al. showed an accumulation of desmin in muscle fibers from patients with sIBM.23 One proteomic study revealed an increase of vimentin in whole muscle biopsies24 and two studies described nestin and vimentin in regenerating muscle fibers.25,26 The surprising abundance of intermediate filaments in RVs is new but their exact role in RV genesis needs further investigation. Intermediate filaments are engaged in the formation and organization of aggregated and misfolded proteins.27 Intracellular protein aggregates are transported along microtubules to form larger aggresomes finally removed by autophagic degradation.27,28 The intermediate filament protein vimentin forms a cage-like structure around aggresomes and is also responsible for lysosome organization and transport.28,29 The abundance of intermediate filaments around RVs may suggest that they support autophagic degradation in muscle fibers of sIBM patients. It is notable that mutations in the intermediate filament desmin causes a myopathy with rimmed vacuoles.10
The largest group of proteins identified as overrepresented in RV areas is those associated with protein folding and degradation. Protein homeostasis or “proteostasis” likely plays an important role in sIBM pathogenesis and is thus a tractable therapeutic target.30 Our study reveals that several new chaperone components may be relevant and refine therapeutic strategies. Small heat shock proteins and other molecular chaperones are of particular interest since they facilitate proper protein folding and degradation of misfolded and aggregated proteins. αB-crystallin, 78 kDa glucose-regulated protein and calreticulin have already been described in sIBM.31-34 A new finding is the enrichment of components of the TCP-1 ring complex (TRiC, also called CCT for chaperonin containing TCP1). Interestingly, an over-representation of these proteins was not detected in other protein aggregate myopathies (11-13 and unpublished data) and it may therefore be a specific feature of sIBM that needs further investigations. TRiC is a central chaperonin complex that interacts with approximately 5-10% of cytosolic proteins35 and seems to be important for the prevention of protein aggregation and toxicity.35-37 Moreover, TRiC is a regulator of the heat shock transcription factor 1 (HSF1) in muscle fibers.38,39 It directly interacts with HSF1 and represses HSF1 activity.38 It would be interesting to know if TRiC affects the efficacy of arimoclomol – a pharmacological agent that prolongs the activation of HSF1. Arimoclomol is currently in clinical trials for the treatment of sIBM.30
Of the 213 proteins found enriched at RVs, disease mutations in twenty-two of these proteins lead to muscle related phenotypes including nine proteins associated with myopathies containing prominent RV pathology. This may not be surprising since several studies have demonstrated the utility of proteomics for the identification of accumulated proteins that are ultimately found to be the genetic cause of the disease.40,41 These previous reports led us to evaluate the burden of rare coding variants in RV accumulated proteins in sIBM patients. Current genetic studies in sIBM are limited. Some studies have performed targeted genetic mutation analysis in small cohorts of sIBM patients14 whereas other studies have focused on modifier genes within populations of sIBM patients such as HLA subtypes as means to correlate MHC gene alleles with sIBM severity and prognosis.42 However, no studies have identified a clear risk allele for sIBM. One challenge to sIBM genetics relates to the rarity of the disease and its late onset which usually precludes obtaining parental DNA. Therefore obtaining patient samples with the statistical power necessary to perform genome wide associations is difficult. To circumvent this, the current study uses proteomic candidates to explore potential genetic risk factors. Indeed, rare variants in FYCO1 were found in 11.3% of sIBM patients as compared with 2.6% of disease controls and 3.4% of population controls. We suggest that rare variants in FYCO1 are associated with risk of developing sIBM. This would be similar to other recently identified genetic risk variants such as TREM2 and TBK1 in sporadic amyotrophic lateral sclerosis where there is an overrepresentation of rare missense and LoF variants in cases as compared to matched controls.43,44 It is important to note several limitations of our genetic study. In particular, the small sample size of 62 sIBM patients. Future studies will be necessary to explore whether FYCO1 remains a risk factor for sIBM in larger cohorts. It will also be interesting to see whether FYCO1 variants explain the pathogenesis or modify the phenotype of other myopathies with RVs.
FYCO1 belongs to an emerging group of autophagic adaptor proteins.45 These adaptors facilitate autophagic cargo loading, autophagosome to lysosome maturation or in the case of FYCO1, autophagosome/endosome trafficking.45 FYCO1 binds to LC3 and Rab7 on the surface of autophagosomes and endosomes, respectively, links them to microtubules via kinesin, and enables transport of autophagosomes along microtubules to acidic lysosomes.22,46 Depletion of FYCO1 or point mutations within its LC3 interacting region (LIR) domain lead to the accumulation of autophagosomes and autophagic cargo that have failed to mature to autolysosomes and be degraded.21 We identified two rare FYCO1 variants (T1270A and P1302L) that resided in or adjacent to the LIR domain. In mouse skeletal muscle, FYCO1-WT co-localizes with LC3 puncta. In contrast, deletion of the LIR domain or expression of the FYCO1-P1302L variant failed to recruit and localize to LC3 puncta suggesting that this sIBM associated FYCO1 missense variants may disrupt its function. Missense variants in FYCO1 have been previously identified in rare patients with congenital cataracts.47 Interestingly, expression of these variants did not disrupt FYCO1’s association with LC3 suggesting that loss of LC3 interaction is not the only mechanism by which FYCO1 mutations can disrupt its function. Indeed, the role of FYCO1 in normal skeletal muscle is currently unknown. The fact that FYCO1 localizes throughout the myofiber in control mouse muscle and in myopathies lacking RVs such as dermatomyositis and polymyositis supports a role for FYCO1 in processes unrelated to its pathologic accumulation at RVs.
Our study identified an overrepresentation of rare missense coding variants in FYCO1 in sIBM patients and suggests that a failure in autophagosome/endosome trafficking may underlie sIBM pathogenesis. The role of impaired vesicular trafficking along microtubules in vacuolar myopathies is further supported by patients and animals treated with colchicine, a microtubule destabilizing agent. Mice chronically treated with colchicine develop an autophagic vacuolar myopathy with the accumulation of LC3, p62 and late endosomal markers such as Lamp2.48 This also occurs in patients receiving toxic doses of colchicine.48 Similarly, RVs in sIBM seem to agglomerate, which hint at ineffective or inadequate transport mechanisms along the cytoskeleton. It is intriguing that dominantly inherited mutations in two other proteins (VCP and p62/SQSTM1) also responsible for autophagosome maturation/degradation lead to RV myopathies.49-52 Notably, VCP and p62 were both identified as RV enriched proteins in the current study. Moreover, rare missense pathogenic variants in both VCP and SQSTM1 have been found in patients with sIBM.14,53
The pathogenesis of sIBM is uncertain and likely due multiple contributing factors. Specifically, a combination of environmental, genetic and aged risk factors needs to be present for disease manifestation. Although our study has identified a potential risk allele in a gene associated with autophagic degradation, this does not preclude the possibility that a primary immune process instigates sIBM pathogenesis. Indeed, a FYCO1 missense variant is not sufficient for disease pathogenesis since all of our sIBM patients lacked a family history of sIBM or weakness. However, it is intriguing that several genes associated with autophagosome-lysosome processing are emerging as risk alleles in sIBM.14,53 Future therapies aimed at improving autophagic function may be beneficial in sIBM patients, especially when used in conjunction with other therapies addressing immune dysfunction.
Supplementary Material
Acknowledgments
This research was supported by the Ruhr-University Bochum (FoRUM F755-12 to A.G, K.K., K.M.), the German Research Foundation (DFG Research Unit 1228 to R.A.K., A.S.), NIH AG031867 (C.C.W.), NIH AG042095 (C.C.W.), the Myositis Association (C.C.W.), the National Institute for Health Research University College London Hospitals Biomedical Research Centre (JLH), the Muscular Dystrophy Campaign (SB), Myositis UK (SB, JLH), the German Federal Ministry of Education and Research (BMBF grant de.NBI - German Network for Bioinformatics Infrastructure, FKZ 031 A 534A, to J.U.) and related to PURE, projects of North Rhine-Westphalia (M.E.). The authors thank the patients for participation in this study and Prof. Dr. Dieter O. Fürst and Dr. Peter F. M. van der Ven, Bonn, Germany, for the generous gift of N-RAP and XIRP-2 antibodies.
Footnotes
AUTHOR CONTRIBUTIONS
A.K.G., S.B., A.M., A.S., M.T., J.H., M.V., C.C.W., K.M., R.A.K. contributed significantly to the conception and design of the study. A.K.G., S.B., K.K., A.M., J.U., M.E., A.S., S.G., J.M.R., M.B.H., T.E.L., M.V., C.C.W., K.M., R.A.K. contributed significantly to data acquisition and analysis. A.K.G., S.B., K.K., A.M., M.V., C.C.W., R.A.K. contributed significantly to drafting the manuscripts or figures.
POTENTIAL CONFLICTS OF INTEREST
Nothing to report
References
- 1.Machado PM, Dimachkie MM, Barohn RJ. Sporadic inclusion body myositis: new insights and potential therapy. Curr Opin Neurol. 2014;27:591–598. doi: 10.1097/WCO.0000000000000129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dimachkie MM. Idiopathic inflammatory myopathies. J Neuroimmunol. 2011;231:32–42. doi: 10.1016/j.jneuroim.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 3.Dalakas MC. Mechanisms of disease: signaling pathways and immunobiology of inflammatory myopathies. Nat Clin Pract Rheumatol. 2006;2:219–227. doi: 10.1038/ncprheum0140. [DOI] [PubMed] [Google Scholar]
- 4.Brady S, Squier W, Sewry C, et al. A retrospective cohort study identifying the principal pathological features useful in the diagnosis of inclusion body myositis. BMJ open. 2014;4:e004552. doi: 10.1136/bmjopen-2013-004552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rose MR. 188th ENMC International Workshop: Inclusion Body Myositis 2-4 December 2011 Naarden, The Netherlands. Neuromuscul Disord. 2013;23:1044–1055. doi: 10.1016/j.nmd.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Güttsches A, Jacobsen F, Theiss C, et al. Human beta-defensin-3 correlates with muscle fibre degeneration in idiopathic inflammatory myopathies. Innate Immun. 2014;20:49–60. doi: 10.1177/1753425913481820. [DOI] [PubMed] [Google Scholar]
- 7.Pinkus JL, Amato AA, Taylor JP, Greenberg SA. Abnormal distribution of heterogeneous nuclear ribonucleoproteins in sporadic inclusion body myositis. Neuromuscul Disord. 2014;24:611–616. doi: 10.1016/j.nmd.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 8.Weihl CC, Pestronk A. Sporadic inclusion body myositis: possible pathogenesis inferred from biomarkers. Curr Opin Neurol. 2010;23:482–488. doi: 10.1097/WCO.0b013e32833d3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Askanas V, Engel WK. Molecular pathology and pathogenesis of inclusion-body myositis. Microsc Res Tech. 2005;67:114–120. doi: 10.1002/jemt.20186. [DOI] [PubMed] [Google Scholar]
- 10.Weihl CC, Iyadurai S, Baloh RH, et al. Autophagic vacuolar pathology in desminopathies. Neuromuscul Disord. 2015;25:199–206. doi: 10.1016/j.nmd.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kley RA, Maerkens A, Leber Y, et al. A combined laser microdissection and mass spectrometry approach reveals new disease relevant proteins accumulating in aggregates of filaminopathy patients. Mol Cell Proteomics. 2012:215–227. doi: 10.1074/mcp.M112.023176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maerkens A, Kley RA, Olivé M, et al. Differential proteomic analysis of abnormal intramyoplasmic aggregates in desminopathy. J Proteomics. 2013;90:14–27. doi: 10.1016/j.jprot.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maerkens A, Olivé M, Schreiner A, et al. New insights into the protein aggregation pathology in myotilinopathy by combined proteomic and immunolocalization analyses. Acta Neuropathol Commun. 2016;4:8. doi: 10.1186/s40478-016-0280-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weihl CC, Baloh RH, Lee Y, et al. Targeted sequencing and identification of genetic variants in sporadic inclusion body myositis. Neuromuscul Disord. 2015;25:289–296. doi: 10.1016/j.nmd.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kley RA, van der Ven PF, Olivé M, et al. Impairment of protein degradation in myofibrillar myopathy caused by FLNC/filamin C mutations. Autophagy. 2013;9:422–423. doi: 10.4161/auto.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 17.Uszkoreit J, Maerkens A, Perez-Riverol Y, et al. PIA: An Intuitive Protein Inference Engine with a Web-Based User Interface. J Proteome Res. 2015;14:2988–2997. doi: 10.1021/acs.jproteome.5b00121. [DOI] [PubMed] [Google Scholar]
- 18.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–42. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olsvik HL, Lamark T, Takagi K, et al. FYCO1 Contains a C-terminally Extended, LC3A/B-preferring LC3-interacting Region (LIR) Motif Required for Efficient Maturation of Autophagosomes during Basal Autophagy. J Biol Chem. 2015;290:29361–29374. doi: 10.1074/jbc.M115.686915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Ma Z, Jiao X, et al. Mutations in FYCO1 cause autosomal-recessive congenital cataracts. Am J Hum Genet. 2011;88:827–838. doi: 10.1016/j.ajhg.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olivé M, Goldfarb L, Dagvadorj A, et al. Expression of the intermediate filament protein synemin in myofibrillar myopathies and other muscle diseases. Acta Neuropathol. 2003;106:1–7. doi: 10.1007/s00401-003-0695-0. [DOI] [PubMed] [Google Scholar]
- 24.Parker KC, Kong SW, Walsh RJ, et al. Fast-twitch sarcomeric and glycolytic enzyme protein loss in inclusion body myositis. Muscle Nerve. 2009;39:739–753. doi: 10.1002/mus.21230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wanschitz JV, Dubourg O, Lacene E, et al. Expression of myogenic regulatory factors and myo-endothelial remodeling in sporadic inclusion body myositis. Neuromuscul Disord. 2013;23:75–83. doi: 10.1016/j.nmd.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Güttsches A, Balakrishnan-Renuka A, Kley RA, et al. ATOH8: a novel marker in human muscle fiber regeneration. Histochem Cell Biol. 2015;143:443–452. doi: 10.1007/s00418-014-1299-6. [DOI] [PubMed] [Google Scholar]
- 27.Pérez-Sala D, Oeste CL, Martinez AE, et al. Vimentin filament organization and stress sensing depend on its single cysteine residue and zinc binding. Nat Commun. 2015;6:7287. doi: 10.1038/ncomms8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watabe M, Nakaki T. Protein kinase CK2 regulates the formation and clearance of aggresomes in response to stress. J Cell Sci. 2011;124:1519–1532. doi: 10.1242/jcs.081778. [DOI] [PubMed] [Google Scholar]
- 29.Wong ESP, Tan JMM, Soong W, et al. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum Mol Genet. 2008;17:2570–2582. doi: 10.1093/hmg/ddn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmed M, Machado PM, Miller A, et al. Targeting protein homeostasis in sporadic inclusion body myositis. Sci Transl Med. 2016;8:331ra41. doi: 10.1126/scitranslmed.aad4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nogalska A, Engel WK, McFerrin J, et al. Homocysteine-induced endoplasmic reticulum protein (Herp) is up-regulated in sporadic inclusion-body myositis and in endoplasmic reticulum stress-induced cultured human muscle fibers. J Neurochem. 2006;96:1491–1499. doi: 10.1111/j.1471-4159.2006.03668.x. [DOI] [PubMed] [Google Scholar]
- 32.Vattemi G, Engel WK, McFerrin J, Askanas V. Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am J Pathol. 2004;164:1–7. doi: 10.1016/S0002-9440(10)63089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li K, Pu C, Huang X, et al. Proteomic study of sporadic inclusion body myositis. Proteome Sci. 2014;12:45. doi: 10.1186/s12953-014-0045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muth IE, Barthel K, Bahr M, et al. Proinflammatory cell stress in sporadic inclusion body myositis muscle: overexpression of alphaB-crystallin is associated with amyloid precursor protein and accumulation of beta-amyloid. J Neurol Neurosurg Psychiatry. 2009;80:1344–1349. doi: 10.1136/jnnp.2009.174276. [DOI] [PubMed] [Google Scholar]
- 35.Yam AY, Xia Y, Lin HJ, et al. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol. 2008;15:1255–1262. doi: 10.1038/nsmb.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitamura A, Kubota H, Pack C, et al. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nature Cell Biol. 2006;8:1163–1170. doi: 10.1038/ncb1478. [DOI] [PubMed] [Google Scholar]
- 37.Tam S, Geller R, Spiess C, Frydman J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nature Cell Biol. 2006;8:1155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neef DW, Jaeger AM, Gomez-Pastor R, et al. A direct regulatory interaction between chaperonin TRiC and stress-responsive transcription factor HSF1. Cell Rep. 2014;9:955–966. doi: 10.1016/j.celrep.2014.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guisbert E, Czyz DM, Richter K, et al. Identification of a tissue-selective heat shock response regulatory network. PLoS Genet. 2013;9:e1003466. doi: 10.1371/journal.pgen.1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greenberg SA, Salajegheh M, Judge DP, et al. Etiology of limb girdle muscular dystrophy 1D/1E determined by laser capture microdissection proteomics. Ann Neurol. 2012;71:141–145. doi: 10.1002/ana.22649. [DOI] [PubMed] [Google Scholar]
- 41.Schessl J, Zou Y, McGrath MJ, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest. 2008;118:904–912. doi: 10.1172/JCI34450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mastaglia FL, Needham M, Scott A, et al. Sporadic inclusion body myositis: HLA-DRB1 allele interactions influence disease risk and clinical phenotype. Neuromuscul Disord. 2009;19:763–765. doi: 10.1016/j.nmd.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 43.Cirulli ET, Lasseigne BN, Petrovski S, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, et al. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 2014;71:449–453. doi: 10.1001/jamaneurol.2013.6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wild P, McEwan DG, Dikic I. The LC3 interactome at a glance. J Cell Sci. 2014;127:3–9. doi: 10.1242/jcs.140426. [DOI] [PubMed] [Google Scholar]
- 46.Pankiv S, Alemu EA, Brech A, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188:253–269. doi: 10.1083/jcb.200907015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pankiv S, Johansen T. FYCO1: linking autophagosomes to microtubule plus end-directing molecular motors. Autophagy. 2010;6:550–552. doi: 10.4161/auto.6.4.11670. [DOI] [PubMed] [Google Scholar]
- 48.Ching JK, Ju JS, Pittman SK, Margeta M, Weihl CC. Increased autophagy accelerates colchicine-induced muscle toxicity. Autophagy. 2013;9:2115–2125. doi: 10.4161/auto.26150. [DOI] [PubMed] [Google Scholar]
- 49.Ju JS, Fuentealba RA, Miller SE, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009;187:875–888. doi: 10.1083/jcb.200908115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 51.Bucelli RC, Arhzaouy K, Pestronk A, et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology. 2015;85:665–674. doi: 10.1212/WNL.0000000000001864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 53.Gang Q, Bettencourt C, Machado PM, et al. Rare variants in SQSTM1 and VCP genes and risk of sporadic inclusion body myositis. Neurobiol Aging. 2016 doi: 10.1016/j.neurobiolaging.2016.07.024. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.