

Abstract
Extracellular vesicles (EVs) are cell-derived vesicles present in body fluids that play an essential role in various cellular processes, such as intercellular communication, inflammation, cellular homeostasis, survival, transport, and regeneration. Their isolation and analysis from body fluids have a great clinical potential to provide information on a variety of disease states such as cancer, cardiovascular complication and inflammatory disorders. Despite increasing scientific and clinical interest in this field, at the time of writing there are still no standardized procedures available for the purification, detection, and characterization of EVs. Advances in microfluidics allow for chemical sampling with increasingly high spatial resolution and under precise manipulation down to single molecule level. In this review, our objective is to give a brief overview on the working principle and examples of the isolation and detection methods with the potential to be used for extracellular vesicles. This review will also highlight the integrated on-chip systems for isolation and characterization of EVs.
Keywords: Microfluidics, Microchip, Exosomes, Extracellular Vesicles
1- Introduction
Extracellular vesicles (EVs) are semispherical structures composed of a fluid core encapsulated by a lipid bilayer, mainly enriched with disaturated lipids such as sphingomyelin and gangliosides that induce a higher rigidity in the lipid bilayer compared to its cell membrane of origin (Laulagnier 2004). These vesicles typically contain cytosol from the secreting cells as well as cytoskeletal and heat shock proteins, both in high abundance (Yanez-Mo 2015). The EV surface is covered with integrins, glycoproteins and transmembrane proteins, which are involved in vesicle trafficking (Raposo 2013). Due to their lipid bilayer structure and their protein covered surface, EVs can be identified from their distinctive combination of various physical and chemical (surface) properties, such as size, shape, density, membrane stiffness, surface potential and surface protein (binding) interactions (Figure 1C).
Figure 1.

(A) Schematic representation of biogenesis and release of EVs from cells. Direct budding produces MVs through the plasma membrane. Exosomes are formed initially as intraluminal vesicles (ILVs) by growing into endosomes and multivesicular endosomes (MVEs). They are later released through the fusion of MVEs with the plasma membrane. Arrows show the transport direction of proteins and lipids between organelles, MVEs, and plasma membrane for exosome secretion. Adapted with permission from ref (EL Andaloussi 2013). Copyright 2013, the Rockefeller University Press. (B) Schematic representation indicates the detailed structure EVs. EV is composed of a lipid-based bilayer that contains different transmembrane proteins, essential for transport, and cell targeting. Other proteins that are involved in biogenesis from endosome or plasma membrane together with genetic materials, which can be used as molecular markers for the detection of exosomes. Adapted with permission from ref [web_evpedia]. (C) A transmission electron microscopy image of extracellular vesicles extracted from human urine. Adapted with permission from ref [web_vdpol].
Cells from different classes of organisms (e.g. eukaryotes and prokaryotic) have been shown to produce vesicles for subsequent release into their extracellular environment (Raposo 2013). Such extracellular vesicles have been successfully isolated from cell cultures (Balaj 2011) and different bodily fluids, such as blood plasma (Ashcroft 2012) and -serum, saliva (Dalton 1975), amniotic fluid, breast milk, and urine (Wiggins 1987, Keller 2011). There are two main types of EVs based on their biogenesis pathway and secretion from the cell. For example, exosomes are vesicles of endocytic origin that are formed by the inward ‘budding’ of the multivesicular bodies (MVB) and fusion with the cell membrane. On the contrary, microvesicles (MVs) are formed by outward ‘budding’ of the cell membrane and they thus have plasma membrane origins (Figure 1A) (EL Andaloussi 2013) These vesicles are not only different in their origin, but also their size is different. Exosomes are approximately 30 to 100 nm in diameter (Théry 2009), MVs are in the range of 100 to 1000 nm (Gyorgy 2011). In addition to these two classes, apoptotic bodies are a type of MVs but produced from cells undergoing apoptosis, and they are approximately 500 to 4000 nm (Hristov 2004).
The characterization of EVs generally relies on different molecular markers that can be classified in three main categories. In the first group, the lipid composition of EVs, which depends on the characteristics of cells of origin, induces specific characteristics to the vesicle surface (Subra 2007). In the second group, the protein composition of EVs can provide information about the activation of certain signaling pathways or existence of the pathology, as they can, for example, carry tumor antigens or inflammatory mediators (Jakobsen 2015, Montaner 2014). Thus far many types of proteins composed of adhesion molecules, apoptosis proteins, and metabolic enzymes have been identified in EVs (Barral 2009). The third group contains nucleic acids, including small RNA, mRNA, and various sizes of miRNA, found in purified EVs (Figure 1B). However, recent studies suggest the possibility of contamination of isolated EVs with RNA and DNA from dead cells during the purification step (Crescitelli 2013). Therefore, a better analysis of membrane-encapsulated extracellular genetic material will become feasible once the sensitivities of sample handling are increased.
Possible applications of EVs have been studied in different fields. The immunomodulatory potential of EVs, such as presenting antigens and interacting with T-cells has been studied and consequently exosome vaccination has shown considerable anti-tumor effect (Cho 2005). In the field of organ transplantation, they can be used to induce immunotolerance and as a result of this, organ rejection can be prevented to some extent (Agarwal 2014, Monguió-Tortajada 2014). In the area of regenerative medicine, EVs can be applied to restore tissue and organ damage. Their effect can be partially explained by paracrine effects similar to stem cell-based therapeutic approaches (DeJong 2014). There are few studies available in which they have exploit the potential of loading therapeutic cargo in to the EVs for possible targeted delivery to certain type of tissues/cells (Kooijmans 2013). Their diagnostic properties have been confirmed in several studies in which they show increased amount of EVs in blood circulation during cardiovascular disease (Azevedo 2007, Tushuizen 2011) and various types of cancers, including glioblastoma, ovarian cancer, melanoma, and renal cancer (Lima 2009, Grange 2011) (Figure 2). With overall increased amounts of secreted EVs in body fluids under diseased conditions, variations in EV subpopulations that carry cell-specific biomarkers such as a certain type of surface protein, lipid composition or RNAs have also been detected (Del Boccio 2012, Lasser 2015, Lazar 2015). The latter can potentially be used in determining the state of malignancy of tumor tissue (Revenfeld 2014). Therefore, applying advanced purification technologies for isolating and sorting of these vesicles from body fluids would enable more detailed analyses of high quality samples that contain a high concentration of related genetic materials relevant to the state of malignancy.
Figure 2.

Applications of extracellular vesicles (EVs) in normal and pathological conditions. Adapted with permission from ref (De Toro 2015). Copyright 2015, Frontiers.
2- EV isolation techniques, an essential preparatory step in EVs handling
In general, there are many techniques that have been so far applied for isolation of various EVs populations. These techniques are mainly focused on fractionation of vesicles based on their biophysical properties such as size, density, morphology, membrane rigidity (lipid composition) and surface chemistry. As explained in previous paragraph, EVs are composed of vesicles with different size ranging from 30 to 1000 nm. Their lipid and membrane protein chemistry are dependent on cell type that they have been produced from. Therefore size, membrane fluidity (morphology) and the type of surface proteins are determining factors in the behavior of EVs in liquid environments, and as a consequence they also determine the type of isolation method that needs to be adapted for each of the different EV populations.
2-1 Conventional EV isolation and sorting techniques
At the moment the applied protocols for purification of EVs typically rely on expensive laboratory instrumentation, large scale sample preparation and multiple steps of ultracentrifugation, which are quite time consuming (Taylor 2014). Recently, several alternative methods (see Table 1) have been introduced that utilize more efficient techniques to isolate and purify EVs. Examples include so-called antibody-coated magnetic beads (Oksvold 2015), novel precipitation technologies (e.g. ExoQuick™) (Taylor 2014), and novel filtration technologies (Cheruvanky 2007). These examples will be discussed in more detail later in this paragraph.
Table 1.
A brief comparison among different conventional EV isolation methods.
| Methods | Approximate in-hand time (min) | Advantages | Disadvantages | References |
|---|---|---|---|---|
| Ultracentrifugation | 140 | Isolation of all vesicular particles, maintaining EVs structure | Time-consuming Expensive equipment Labor-intensive | (Scott 2005, Momen-Heravi 2012) |
| Sucrose density gradient | 250 | Divide EVs into different populations | Not preferable for samples with diverse EV populations | (Taylor 2011, Kalra 2013) |
| Ultrafiltration | 130 | easier to handle compared to ultracentrifugation | Retain contaminating proteins | (Cheruvanky 2007, Merchant 2010) |
| Exoquick-TC™ | 45b | Simple, fast, highly scalable | Low yield of EVs protein | (Taylor 2011, Alvarez 2012) |
| Immunoaffinity | 240a | Isolation of certain subpopulation of EVs | Nonspecific binding, recovery efficiency is less than 100% | (Clayton 2001, Tauro 2012) |
The rinsing time (~2–8 hrs) is not included.
Add overnight incubation time with ExoQuick-TC at 4 °C.
Differential centrifugation that includes several centrifugation or ultracentrifugation steps is currently the most commonly used protocol for isolation/purification of EVs (Taylor 2014). It consist of low speed centrifugation for a short time period as first step, to separate bigger particles or debris, followed by higher speed with possibly extended time period and finalized by ultra-high speed at 100,000g (ultra-centrifugation) to precipitate EVs. This method suffers from low (5% to 25%) recovery efficiency of starting material (EVs) and low purity (Witwer 2013, Lane 2015). However, each protocol can be optimized based on characteristics of the particles in solution and properties of rotors such as rotor type, geometrical parameters, path length, rotor speed and duration, to isolate the target population of vesicles with high yield and purity (Livshits 2015).
As a variation of above mentioned isolation method it is possible to add an additional density gradient centrifugation step, using sucrose or iodixanol (Optiprep™). Recent studies have shown improved purity in isolating exosomes from viruses or protein aggregates (Cantin 2008). One important factor which needs to be taken care of with this method is that it is important to keep the solution iso-osmolar at all densities, in order to better preserve the size of the particle (Dettenhofer 1999).
Ultrafiltration membranes are another type of conventional isolation method that can be combined with a differential centrifugation method, in which the first and second spinning steps can be replaced by filtration steps. In such a method, the EVs of certain pre-determined size are trapped by a nano-porous membrane. The common pore sizes of the membranes are between 0.1 to 0.001 μm. This method is usually convenient for isolation of EVs between 40 and 100 nm in small volume samples (Cheruvanky 2007). The method’s recovery efficiency is dependent on adherence interaction between EVs surface proteins and the membrane applied for separation. Therefore, applying a certain type of coated (hydrophilized) membranes is recommended in order to enhance the filtration efficacy (Merchant 2010).
So far, several biotechnology companies have developed new products aiming to further improve the recovery and product purity of their isolation methods using differential centrifugation. One of them these newly developed products is the so-called ExoQuick™ (System Bioscience, Mountain View, CA, USA). This is polymer based precipitation technique in which biological fluids are mixed with polymer containing reagents, incubated and treated following a shortened centrifugation at low speed (Yamada 2012). There are certain drawbacks connected to this method however. One of the drawbacks is the low purity of the isolated EVs population and another is that the presence of polymer in the isolated samples can potentially interfere with further characterization (down-stream analysis) of the isolated EVs.
In some studies, immunological based separation of EVs was carried out for further purification and potentially to eliminate a subpopulation of vesicles (Clayton 2001, Koga 2005, Tauro 2013). For this purpose, magnetic beads are applied, their surface coated with antibodies against cell-specific antigens that are present on certain EV sub-populations. This type of immunoaffinity based isolation of EVs can also be used as a tool for further characterization of isolated (bead-attached) EVs. In this way, the isolated bead-EV complexes can be combined with characterization techniques such as flow cytometry, electron microscopy or western blot analyses (Thery 2006). However, it is important to take into account that the immunoaffinity based isolation method is mainly suitable for pre-concentrated and small volume bio-fluids and thus cannot be applied for large scale EVs purification purposes (Thery 2006).
Despite the overall similarities in how the various conventional EVs isolation strategies are adapted in the research labs, the exact details of the isolation/purification protocols for each of the (above-mentioned) methods can vary significantly between various studies. This large variation in the efficacy of the isolation/purification protocols is mainly due to the large differences in viscosity between each type of bio-fluid. Subsequently, these variations in the applied protocols and the lack of a standard methodology may lead to inconsistencies in the recovery of EVs, which severely complicates cross-study data comparison (Yuana 2011, Davies 2012).
In general, conventional isolation methods have two main drawbacks. One type of drawback is related to ‘infrastructure’, which includes high material costs, high energy consumption, large sample volumes for analysis and the need for expert operators. The other type of drawback is related to the sample quality, for example low EVs recovery efficiency, high risk of EVs deformation, and/or sample contamination with other impurities.
2-2 Microfluidic techniques for isolation and sorting of EVs
Non-invasive separation of EVs from their complex biological environment while preserving structure and composition of EVs intact is an essential step for any type of scientific research involving extracellular vesicles and processes related to them (e.g. drug delivery). The most optimal (and facile) conditions to investigate EVs are when they are suspended in (diluted) biological fluids. Microfluidic technology, which has previously shown unique advantages in genomics and proteomic analysis and quantitative biology, provides the ability to both process and analyze samples with precise dynamic control over analyte concentrations (Baker 2009). It gives the possibility to extend sample manipulations to small volumes, together with high throughput capability (He 2010).
The original idea of on-chip sample separation comes from capillary electrophoresis (CE), which has been applied for many types of molecular level separations (e.g. proteins). Micro-CE (μCE) takes advantage of system miniaturization. After its introduction by Manz and coworkers it has become a highly interesting area of research (Harrison 1993). Another boost for miniaturized separation approaches came from the revolution in genomics in the early 1990s and the sharply increased need for DNA separation devices (Tegenfeldt 2004). However, separation and isolation of larger biological “compartments”, such as vesicles or cells have since become a new area of focus and a large number of new on-chip separation methods have been developed in the past two decades.
2-2-1 Overview of techniques for on-chip isolation of EVs
In general, the working principle behind the miniaturized isolation techniques is based on variations in the intrinsic mechanical and physical properties of the various EV populations, including their size, shape, density, adhesive properties, and their deformability. A major motivation to shift toward applying microfluidics for separation purposes is to minimize the size, cost, and complexity of technologies by reducing the sample size, performing the reactions faster, and conducting multiple assays in small footprints simultaneously (Salieb-Beugelaar 2010, Jackson 2013).
Many different types of on-chip pre-concentration and isolation techniques have been developed, which are typically classified based on their different working principles (Table 2). In this review, we have chosen not to give a full ‘historic’ overview of the literature on (microfluidic) isolation techniques for EV containing samples, but instead we have emphasized on a selected number of recent publications that we believe have presented relevant new approaches among the various microfluidic EVs separation methods reported thus far. In most studies, isolation techniques are divided into two major categories: static or dynamic (Sueyoshi 2008, Lee 2011). Static pre-concentration techniques applicable for EVs combine the use of surface-binding techniques together with porous membrane (nanochannel/nanogap) techniques. The surface-binding techniques facilitate either the chemisorption (adsorption involving chemical bond formation) and/or physisorption (reversible adsorption) while the porous membrane is applied to focus the target analytes through filtering. Both of these techniques can be further categorized based on the exact details of their working mechanism (Lee 2011). Dynamic pre-concentration techniques do not involve the use of physical barrier/structures to separate analytes. Instead, they function based on the use of electro-kinetic or (occasionally) hydrodynamic equilibrium effects.
Table 2.
List of various methods used for separation of EVs.
| Methods | Mechanism | Separation marker | Advantage | Disadvantage | References |
|---|---|---|---|---|---|
| Passive | |||||
| Surface binding techniques | Specific binding to surface markers | Surface biomarkers and receptors | Highly specific | Requires cell-specific marker Dependent on antibody–ligand specificity | (Chen 2010) |
| Porous membrane technique (Sieving) | Pressure field gradient | Size | Short separation time | Saturation of filter EVs, low recovery | (Davies 2012) |
| Trapping on porous structures | Tunable hierarchical filtering structures | Size and shape | Intact EVs trapping with high purity | Saturation effect with high sample volume | (Wang 2013) |
| Dynamic | |||||
| Optical | Optical polarizability | Size, refractive index | Minimal sample preparation and labeling, non- invasive | Medium throughput | (MacDonald 2003) |
| Acoustic | Acoustic resonance (Ultrasonic standing waves) | Size, density | Label free, fast, high separation yields, non-invasive | Specific type of material to be used (expensive) | (Lee 2015, Evander 2015) |
| Dielectrophoretic (DEP) | Inhomogeneous electric field | Size (total membrane capacitance), polarizability (cell dielectric properties) | Label free, surface marker free, high throughput | Biological basis underexplored, DEP differences can be too subtle | (Zou 2008, Shim 2011) |
| Electrophoretic (EP) | Homogeneous electric field | Surface charge density, zeta potential | Label free | Complicated system, technical issues related to convection | (Akagi 2008) |
| Fluorescently labeled flow cytometry | Fluorescence | Fluorescent labels | High throughput | Influence on characteristics of EVs via labeling | (Pospichalova V 2015) |
| Magnetic | Magnetic field strength | Size, magnetic susceptibility (cell surface marker expression, magnetic label binding) | Can be highly specific | Low to medium throughput, time consuming, low recovery | (Berger 2001, Ingils 2004) |
The dynamic pre-concentration techniques can be divided into two major categories (Song 2006). The first type concentrates analytes by exploiting the differences in flow velocity across the boundary region in between the sample and the running buffer. The second category concentrates analytes by utilizing the so-called “focusing effect”, in which the net velocity of the particles becomes zero in a microchannel due to thermal or electrical forces. One example in this regard is the electric field gradient focusing (EFGF) technique, which uses electric field gradients for sample (charged species) separation and enrichment according to electrophoretic mobility (Kelly 2005). So far this technique has been established via applying different approaches, such as the introduction of cross-sectional areas to flow channels. This induces diffusion gradients across a membrane, either with buffer ions, or by inducing temperature gradients in buffer solutions by the application of electrode arrays (interdigitated) that change conductivity as a function of temperature. In a recent study Akbari and coworkers have used digital projection optics to concentrate and manipulate charged analytes in microfluidics independent of fluid flow. This is a good example of showing the synergy between optics and fluids. This technique enables dynamic and non-invasive optical control over the focused band location in fluid (Akbari 2012).
In a review by Bhagat and coworkers, different separation techniques were categorized into active and passive modes. Simply, active separation techniques depend on the interaction of intact vesicle or its labeled form with an external force field, such as magnetic or acoustic for operation (Plouffe 2012, Lee 2015). These types of techniques have since become extremely popular, due to their high sensitivity and efficiency. On contrary, passive techniques rely entirely on the characteristics inherent to the vesicle (e.g. electrical properties) or microfluidic channel geometry (e.g. hydrodynamic forces) for functionality (i.e. label free separation technique) (Figure 3). In this regard, inertial microfluidics is a good example, which is often used for particle separations in flow field as a consequence of the interaction between two inertial forces (Rafeie 2016). One is the shear gradient lift force (FS) and the other wall lift force (FL). Another example is deterministic lateral displacement technique where particles are separated based on their size and rigidity in a micropost array, which can be applied in devices with a pressure driven flow (parabolic flow) (Loutherback 2010). Common separation metrics, such as resolution, specificity, efficiency, and throughput rate have been specifically discussed for each method in previous work by other authors (Bhagat 2010).
Figure 3.

Heterogeneous EVs populations separated using microfluidic systems based on their intrinsic (passive) and extrinsic (dynamic) characteristics. (A) Dielectrophoresis (DEP)-based approach in which EVs are exposed to a non-uniform electric field and EVs are separated based on the difference between the DEP (FDEP) and (Fgrav) gravitational forces. (B) The hydrodynamic-based method that utilizes the competing hydrodynamic wall lift force (FL) and a shear gradient lift force (FS). (C) The immunoaffinity-based technique that depend on the interaction of cell surface molecules (red) with antibodies or other ligands (blue) functionalized on the channel surface. (D) Magnetic-based technique: an applied magnetic field is used to deflect and focus cells labeled with magnetic particles (green). Adapted with permission from ref (Jackson 2013), Copyright 2013, Elsevier.
2-2-2 Integrated techniques for on-chip isolation of EVs
There are also examples of combining two or more separation techniques in one device. In a recent study, Kim and coworkers showed that the pre-concentration and purification efficacy of nanoporous membranes on EVs could be improved by simultaneously applying an electric gradient over the membrane (i.e. electrophoresis driven filtration). This system is an example of the integration of two pre-concentration techniques (i.e. static and dynamic) into one (Davies 2012, Kim 2013). Another interesting example of an integrated technique for isolation of EVs is described in a study by Maruyama and coworkers (Maruyama 2010), where the combination of centrifugal force and a graduated mechanical gap allows for size dependent sorting and trapping of microparticles on-chip.
So far, the overall results on microfluidics based isolation of EVs have shown higher recovery and higher purity of the EVs compared to conventional isolation methods. Moreover, they have been proven to be compatible with concentrated blood samples and other type of body fluids. However, improving the throughput of on-chip isolation technologies while retaining their high particle sorting sensitivity is one of the ongoing challenges in this field.
3- Detection of EV-containing samples
Besides the efforts involved with the isolation of extracellular vesicles (EVs), distinguishing the chemical, biological or physical differences among EV subpopulations is also extremely challenging and requires the use of high quality processing and detection techniques (Santana 2014). Analysis (detection/characterization) of samples containing EVs in laboratory settings is currently common practice, though it requires relatively bulky and complex lab infrastructure. Therefore, there is a pressing need to develop miniaturized tools for analyzing EVs, that are portable, require small amounts of samples and can perform multiple assays (high content) in a high throughput manner. Ideally, this would mean full integration of EV sample analysis onto microfluidic or ‘on chip’ devices, as this would allow the use of EV analysis in (for example) future point of care medical devices, thereby significantly expanding their potential applications. Thus, effective detection/characterization of EVs on chip can be considered a key challenge to further improve the clinical utility of point care of devices.
Several recent proof-of-principle laboratory studies have already developed methods to detect and characterize EVs in a microfluidic setting (Chin 2007, Mark 2010, Matos Pires 2014). Thus far, various studies have also reported the adaptation of (advanced) detection methods for integration into microfluidic devices, aiming to implement on chip detection and characterization of EVs (Patel 1999, Mogensen 2004, Ring 2010). The microfluidic systems developed for detection/characterization EVs can be broadly categorized into six groups: (i) electrochemical, (ii) electrostatic potential, (iii) mechanical, (iv) electromechanical, (v) optical and (vi) non-optical based (Matos Pires 2014) (Table 3, vide infra, §4.8).
Table 3.
Assessment of the detection capability of different methods for EVs detection, classified via underlying physical parameters
| Methods | Size detection range/detection limit | Size distribution* | Zeta potential* | Concentration* | Biochemical information* | Measurement time | References |
|---|---|---|---|---|---|---|---|
| Electrochemical | |||||||
| Western Blot/ELISA | NA | − | − | − | + | Hours | (Raposo 1996, Hoegger 2007, Logozzi 2009) |
| Impedance -based flow cytometry | ≥300nm | − | − | −/+ | − | Seconds | (Ito 2004, Zwicker 2009) |
| Electrostatic potential on-chip μCE system | + | + | + | + | − | Real time | (Kato 2013) |
| Mechanical | |||||||
| AFM | <1nm | + | − | −/+ | −/+ | Hours | (Siedlecki 1999, Yuana 2010, Sharma 2011) |
| Electromechanical | |||||||
| QNano | 70nm to 10um | + | − | + | − | (Momen-Heravi 2012) | |
| Optical | |||||||
| DLS | 1nm to 6um | + | − | − | − | Minutes | (Dragovic 2011, Gardiner 2013) |
| NTA | 50nm to 1 um | + | − | + | + | Minutes | (Harrison 2009, Gardiner 2013) |
| STED microscopy | 30nm | + | − | −/+ | + | Hours | (Willig 2006, Hein 2008) |
| nPLEX | NA | − | − | + | + | Minutes | (Im 2014) |
| Conventional flow cytometry | ≥300nm <300nm | − | − | + | + | Seconds | (Clayton 2001) |
| (binding to beads) | − | − | − | + | |||
| Fluorescent high resolution flow cytometry | ~100nm | − | − | + | + | Seconds | (Thery 2006, Nolte-'t Hoen 2012) |
| Raman spectroscopy Non-optical | 350nm | ? | − | −/+ | + | Hours | (Puppels 1990, Uzunbajakava 2003) |
| TEM | <1nm | + | − | − | + | Hours | (Yuana 2013) |
| SEM | ~1nm | + | − | − | + | Hours | (Sharma 2011) |
| μNMR system | 50–150nm | − | − | + | + | Minutes | (Shao 2012) |
+ indicates variable can be measured, − indicates it cannot.
4-1. Unimodal detection techniques
4-1-1 Electrokinetic detection
The electrokinetic potential of a colloidal dispersion, or as it is more frequently referred to: the ‘zeta potential’, is defined as the electric potential in the (colloidal) particle’s surface interfacial double layer, consisting of layers of adsorbed and screening ions respectively. This potential is dependent on the density of the particle’s surface charges and the composition of the (bulk) liquid. The zeta potential of a particle cannot be not measured directly, but is easily calculated by determining the electrophoretic mobility in liquid (Weiner 2013). The calculated zeta potential can be further applied to determine the surface charge density of the particles of interest, which in this work will be limited to extracellular vesicles (EVs) or similar structures. Recently, a patent for microcapillary electrophoresis of exosomes has been published. In this system exosomes were characterized based on zeta potential, which was calculated from the electrophoresis mobility of each exosome by using Smoluchowski equation (Ichiki 2014). In a study done by Kato and coworkers, an on-chip μCE system with an integrated laser dark-field microscope was utilized to evaluate the zeta potential of individual exosomes. The lipid type in exosome bilayer, surface antigen type and density were found to be determining factors for the exome’s zeta potential. Using this knowledge, the authors managed investigated the correlation between the measured zeta potential of the exosomes and their cell of origin (Kato 2013) (Figure 4).
Figure 4.

(A) Schematic representation of microcapillary electrophoresis (μCE system) on-chip integrated with a laser dark-field microscope. The system composed of different compartments such as electrodes, power supply, a 488-nm laser source, an electron multiplying charge-coupled device (EM-CCD) camera, and an inverted microscope (Nikon Ti-U). Adapted with permission from ref (Akagi 2014). Copyright 2014, IOP Science. (B, C) Schematic diagram of immuno-electrophoresis of EVs without and with antigen respectively on a μCE chip. Changes in zeta potential and electrophoretic mobility correlates with the immunoreactivity of individual EVs since the surface charge of the EV are modified upon antibody binding. The 2-methacryloyloxyethyl phosphorylcholine (MPC) polymer was applied to coat the inner surface of the flow channel to prevent electro-osmotic flow or non-specific adsorption of EVs. Adapted with permission from ref (Akagi 2015). Copyright 2015, PLOS ONE.
4-1-2 Mechanical detection
The application of mechanical systems with micron- and even nanometer-sized scales (cantilevers) as sensors has already been studied for several decades. Micro-cantilever technologies for bio-sensing applications have been developed along with research and development of so-called microelectromechanical systems (MEMS) and nanoelectromechanical systems (NEMS) (Waggoner 2007). These (bio)sensors have high sensitivity, high selectivity and label-free detection, and they can be applied to various analytes such as DNAs, marker proteins and pathogens. Cantilever-based devices can typically operate in one of two different modes upon contact of the analytes with the cantilever: (i) static mode or (ii) dynamic mode. Various physical techniques can be applied for sensing cantilever motion, such as use of piezoelectrics or laser-interferometer techniques. Cantilever technology has the potential to measure analyte binding and/or adsorption interaction, both in solution and in the air, while even the simultaneous measurement of multiple binding events through parallel channels has been reported (Ferrari 2005, Gruber 2011). One of the examples of such techniques can be found in the atomic force microscope (AFM), in which a mechanical cantilever is passed over a sample surface, during which the detection of minute variations in cantilever movement by the piezoelectric sensors is converted into a highly detailed profile of the measured sample surface. There are a number of studies that applied AFM technology for EV characterization (Sharma 2010, Yuana 2010). Hardij and coworkers have characterized EVs by comparing two different modes (air and liquid mode AFM analysis) and comparing the outcomes of the respective measurements (Hardij 2013) (Figure 5).
Figure 5.

(A) Schematic illustration of mica surface functionalization for AFM analyses in the air and liquid modes. (B) Air mode based cross-section image of the extracellular vesicles (EVs) derived from breast cancer cells using surface modified with anti-tissue factor (TF) antibodies. Image size: 1×1μm2. (C) Liquid mode based cross-section image of the same type EVs, using surface modified with TF. Image size: 3×3μm2. The colorimetric scale indicates the Z-dimension. Adapted with permission from ref (Hardij 2013). Copyright 2013, Co-Action Publishing.
4-1-3 Optical detection
Optical detection techniques are one of oldest techniques to be used for the detection of molecules, because the methodology is relatively simple and because the data acquired from a molecule’s interaction with the incident light is highly specific for that analyte. Optical detection techniques have been efficiently implemented on-chip, where they are used as a microfluidic biosensor formatted for online fluorescence (Ryu 2011), chemiluminescence (Guan 2015), surface plasmon resonance (SPR) based measurements (Luo 2008, Huang 2009) and surface enhanced Raman spectroscopy (SERS) based optical sensing (Qian 2008). Instead, other parts such as microscopes, spectrophotometers, charge-coupled devices (CCDs) and photomultiplier tubes (PMTs) have yet to be fully integrated to microfluidic devices, for now remaining off-chip because of difficulties in miniaturization (Schwarz 2001, Huang 2005, Myers 2008, Wolter 2008, Lee 2009, Huh 2009). If effectively integrated on chip in the future, optical detection methods, specifically SERS, could have the potential for online detection of all major clinically relevant properties of a single vesicles at high speed, with high sensitivity at micron- and nanometer dimensions (Huh 2009, Stremersch 2016).
When using optical detection methods in microfluidic devices it is necessary to take into account the behavior of light on interfaces of two or more (liquid) media with different refractive indexes. The behavior of light on interfaces between media is governed by two physical phenomena, namely: reflection, a change in direction of the wave front, and refraction, a change in direction of wave propagation when passing from one medium to another. In practice, more difference between the refractive index of microvesicles and its surrounding medium results in higher light scattering by a single particle and increased sensitivity. Light scattering-based measurements such as the frequently used ‘dynamic light scattering’ (DLS) technique, give information on the relative size of a particle compared to other particles in the suspension, thus over time eventually obtaining a relative size distribution of the particles in the suspension. The absolute size distribution can be calculated once the relative size distribution and microvesicle concentration in the suspension is known. Alternatively, the absolute size distribution of microvesicles can be obtained by more elaborate techniques, such as the so-called ‘nanoparticle tracking analysis’ (NTA) which also gives information about concentration (Carr 2013) (Figure 6). Though NTA has shown to be a promising technique for fundamental studies, it should be noted that the usage of high-resolution microscopes complicates the on-chip integration of the technique, thus making commercial application of NTA less likely in the near future.
Figure 6.
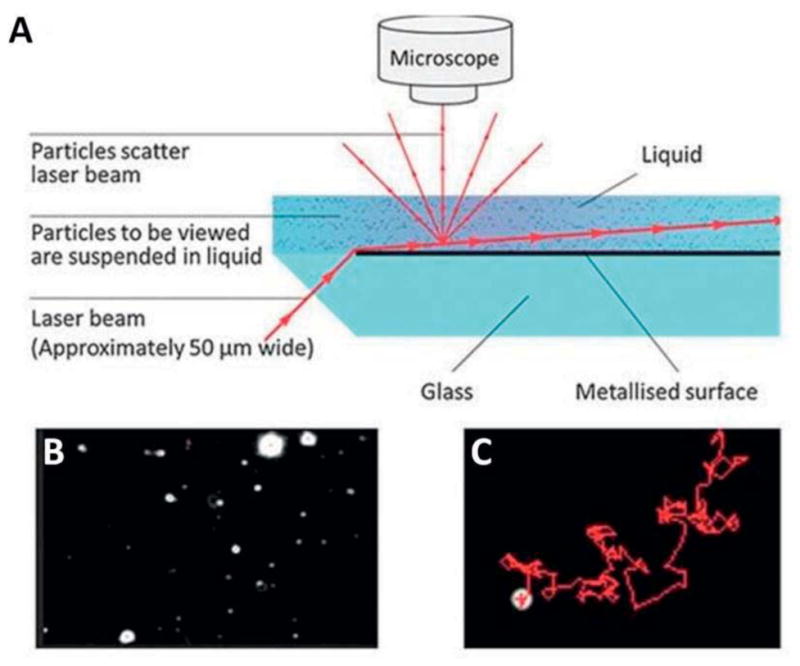
(A) Schematic representation of the Nanosight design. (B) A snapshot from a video taken by Nanosight. Each white spot is one nanoparticle dispersed in a liquid medium. (C) Brownian motion pattern of single nanoparticle traced by NTA software. Adapted with permission from ref (Carr 2013). Copyright 2013, Malvern Instruments Ltd.
4-1-4 Non-optical detection
Non-optical (detection) methods are typically categorized as such when the methods use electrons rather than photons to generate an image of the target analyte. The efficiency of the electron-based detection methods depends on the spatial stability of the electron beam, as well as on the structural stability of the sample under conditions of electron beam irradiation. Because the electron ‘wavelength’ is over three orders of magnitude shorter than the wavelength of visible light photons, the use of electrons results in a higher resolution, which therefore makes it possible to obtain more accurate information on the morphology of microvesicles or other analytes. In this context, despite its low throughput, transmission electron microscopy (TEM) can be considered as an example with high (<1 nm) imaging resolution, well suited to visualize nanoparticles, especially when those contain heavy elements (i.e. metals). Although electron microscopy is a powerful technique for characterization of microvesicles (MVs), fixation and dehydration steps during sample preparation might affect the morphology of the microvesicles as well as their size distribution. For example, possible aggregate formation during the drying process will result in a shifted size distribution and can thus cause biased counting of MVs on the grid (Binnig 1986, Pisitkun 2004, Yuana 2010). To solve this problem, Tai and coworkers fabricated a microchip nano-pipet on a hydrophilic SixNy thin film using semiconductor processing. The small geometry of the nano-pipet can puncture sample droplets and it can also suppress the capillary flow of the liquid during the vacuum drying process, thus resulting in a more homogenous nanoparticle distribution on the platform after drying. The dried sample is stable during ultra-high vacuum (UHV) treatment, which allows electron microscopy imaging or other sample (surface) analyses (Tai 2012).
4-2. Multimodal detection techniques
4-2-1 Electrochemical detection
Electrochemical detection is the study of the chemical response of a system to electrical stimulation. The electrochemical measurements are based on two major phenomena: (i) the promotion of chemical reactions by applying electrical current to the adjacent electrodes, (ii) electrode responses that are triggered by specific electro-chemical (i.e. redox) reactions occurring in the system. The resulting variations in the electrical signal, induced by the interaction of the analyte with the electrodes, can be easily measured and quantified (Srivastava 2013). So far, there have been many studies reporting on-chip cellular analysis methods that are based on advances in electrochemical detection (Johnson 2013), commonly used for the on-chip detection of inorganic and organic ions in solution (Matos Pires 2014).
In a study by Hoegger and coworkers (Hoegger 2007), the authors took advantage of on-chip ELISA assay to design a 8-channel immunochip cartridge that allow eight assays to run in parallel in the space of a few minutes, combined with a continuous amperometric flow monitoring and detection (Figure 3A). This is an example of the integration of immunoassay with impedance based flow sensors (immunosensors) that is capable of running several different assays in parallel, thus holding big promises for future point-of-care device applications.
Electrochemical biosensors are categorized into subgroups based on the method of measuring the electrical properties, such as voltage, current, and capacity. One example is impedance-based detection and quantification on a chip. Impedance is defined as the complex current-voltage ratio in an alternating current (AC) circuit, or in other words the phase dependent electrical resistance. This technique is based on electrochemical responses at the interface of the sensing electrode that are induced by applying a sigmoidal voltage at a particular frequency, and measured from the resulting current. The captured EVs on the probe can be detected by variations of the impedance at the interface. However, the detection limit of ≥ 300 nm will weaken its use to specific systems due to the introduced bias toward the detection of larger vesicles, such as microvesicles versus exosomes (Lvovich 2010). A similar approach has been recently applied using gold electrode arrays with an aptamer-functionalized surface for capturing intact exosomes. The changes in electrochemical signal were proportional to the concentration of exosomes captured (Zhou 2016) (Figure 7B). Another application of electrochemical sensing is amperometric detection at biomolecular level via electrode surface modification with antibodies or mRNA probes (Wei 2009). This approach can be further expanded with analyses of captured EVs extractions (e.g. proteins or mRNAs) in a microfluidic platform.
Figure 7.

Electrochemical detection of EVs. (A) The design of an 8-channel microfluidic device showing the microchannel cross-section with inlets, outlets, and the working electrodes. Adapted with permission from ref (Hoegger 2007). Copyright 2007, Springer. (B) Schematic of an aptamer-based exosome detection that includes a gold electrode array patterned and a flow chamber made in PDMS. The changes in electrochemical signal were proportional to the concentration of exosomes captured. Adapted with permission from ref (Zhou 2016). Copyright 2016, Elsevier.
4-2-2 Scanning ion occlusion spectroscopy (SIOS)
Scanning ion occlusion spectroscopy (SIOS) is an electromechanical detection technique, which is also referred to as resistive pulse sensing. It is an integrated system that monitors the ion current flow through a tunable nanopore membrane. This technique is used by researchers to measure the size, charge, and concentration of small biomolecules and/or biological particles (e.g. viruses and bacteria) that are suspended in ionic solution (Ito 2003). Sample particles are passed through the nano-sized pores in the membrane by applying (osmotic) pressure and/or electrical potential. Passage of each particle through the membrane pores causes a (temporary) local blockade, resulting in a partial drop in total current flow over the membrane, dependent on the size and number of the blocked nanopores. The fluctuations in the total current flow are continuously recorded and processed. The magnitude of blockade can be correlated to the volume of each particle, as the dimensions of the membrane pores are known. Duration of the blockade, which is proportional to the initial particle velocity, can be indirectly used to calculate the particles’ surface charge. The frequency of blockades is a measure of the amount of particles per membrane surface area, and can thus be used to measure the initial concentration of particles in solution (Ali 2011, Sowerby 2007). A good example of SIOS-based detection systems is reported by Roberts and his coworkers where IZON’s qNanotechnology was applied to detect nanoparticles. The work shows the high performance of this instrument, with its added gating ability to measure the size and count individual nanoparticles in real time over a wide range (bimodal and trimodal) of particle distributions (Roberts 2010) (Figure 8).
Figure 8.

Application of scanning ion occlusion spectroscopy for the detection of EVs. (A) qNano (a bench-top) instrument. Adapted with permission from ref [web_izon]. (B) A fluid cell, consisting of adjustable jaws, which are holding at the center, a tunable membrane composed of nanopores. (C) Schematic cross-section of pore, which depicts the principles of selective transport for a bimodal particle suspension. Adapted with permission from ref (Roberts 2010). Copyright 2010, John Wiley & Sons, Inc.
4-2-3 Electro-optical detection
As discussed previously, electrically simulated nanopore techniques have recently become quite popular for single molecule detection applications. Passage of a molecule or particle through the opening correlates with the production of a specific signal blockade signature (Clarke 2009, Rosen 2014). In addition it has been shown that microscope based imaging of particles and visualization of their movements is possible by using fluorescent labeling (Soni 2010, Kurz 2013). The potential integration of the two above mentioned particle-specific detection methods opens avenues toward the development of a new detection method, suitable for more complex systems that contain several different types of particles dispersed in a fluid. To this end, Liu and coworkers (Liu 2014) developed a new ‘electro-optofluidic’ platform which was sensitive enough to detect subpopulations in heterogeneous biological suspensions by using computational cross77correlation of optical and electrical signals. The dual-mode single-particle detection method was shown to be applicable to different types of biological suspensions. Moreover, it was shown that various different methods of optical detection could be successfully applied in this integrated dual-mode detection platform (Figure 9).
Figure 9.

Application of nanopore gated electrical sensor combined with optical detection in a microfluidic platform. (A) Schematic view on particle movement through the nanopore gate. Liquid flow direction shown with blue arrow. Red area demonstrates the optical excitation volume. (B) Movement of biological particle through electrically gated nanopore. (C) Two characteristic signals, electrical (black) and optical fluorescence (red), for separate detection events related to a single particle. The electrical current dip and the fluorescence spike appear separated due to time need for a particle to travel (flow) from the nanopore towards the optical excitation spot (Δt). (D) Computational cross-correlation of detection events (particles), adjusted for delay time (Δt). Adapted with permission from ref (Liu 2014). Copyright 2014, ACS.
4- Integrating different detection and characterization components on-chip
The ultimate goal of microfluidics is a challenging one indeed: to combine sample (pre-) treatment, -processing and -detection steps in a workable way onto silicon chip barely the size of a bankcard. For this reason the various components must be designed in such a manner that each of the components is compatible with every other. To achieve such mutual compatibility between the components is extremely challenging. Each of different components on a chip, from microvesicle filtering to extraction, and molecular level characterization, requires its own specific microenvironment to perform best. It is important to take into account that vast majority of different individual components on-chip are mutually incompatible (Ismagilov 2003, Chin 2012). The other challenge is large-scale integration (LSI) on microfluidics, which contains interconnected microtube networks, as well as hundreds of nano-sized mechanical valves and multiple individually accessible reaction- or mixing chambers. In this regard, the design of integrated microfluidic systems resembles electronic integrated circuits, though fabricated in large-scale (Thorsen 2002). The studies of Quake and coworkers used this analogy between microfluidic analytical systems and miniaturized integrated electronic circuits to present a potential solution for large-scale analysis of multiple assays on microfluidic platforms (Thorsen 2002). An in-depth discussion about their findings is unfortunately beyond of scope of this paper. However, we encourage readers attentive to this topic to study the referenced paper for a more detailed overview.
Microfluidic devices capable of single step extracellular vesicle (EV) capture directly from serum, facilitate more rapid sample analyses with less contamination. To this end, Chen and coworkers developed a device to capture EVs based on immuno-affinity interactions (Chen 2010) (Figure 10A, i). The device is reported to offer capability for direct on-chip lysis of EVs for RNA extraction. However, it currently suffers from low microvesicles capture yield, requiring additional engineering efforts in order to increase the surface area available for interaction as well as the dimensions of the microfluidic channels. In another study, Ashcroft and coworkers developed a microfluidic system based on similar approach, enabling the device to capture and isolate EVs that present the specific CD41 antigen, followed by direct analysis of their size distribution using AFM (Ashcroft 2012) (Figure 10A, ii–iii). The authors managed to increase AFM sensitivity by increasing the concentration of grafted EVs on the active surface (Ashcroft 2012). In general this type of systems allow less invasive, less contaminated isolation of EVs directly from blood samples, in contrast to the magnetic-bead based capture and isolation of EVs that is currently a common lab practice.
Figure 10.

(A) The first microfluidic device for the separation of microvesicles: (i) after flowing the serum samples through the microchannels at optimized flow rates, microvesicles attached to the surface, and later fixed for SEM or lysed for RNA extraction. Adapted with permission from ref (Chen 2010). Copyright 2010, Royal Society of Chemistry; (ii) purified blood plasma sample run over antibody coated mica surface on PDMS chip; (iii) followed by removal of mica surface and imaging with AFM analysis. Adapted with permission from ref (Ashcroft 2012). Copyright 2012, Springer Link. (B) Miniaturized nuclear magnetic resonance (μNMR) system, consisting microchannels for sample manipulations, integrated with a portable magnet for magnetic field generation, micro coil array, and miniaturized electronics. Adapted with permission from ref (Lee 2008). Copyright 2008, Nature Publishing Group. (C) A microfluidic prototype comprised of multiple microchannels for the immunomagnetic capture of circulating exosomes, on-chip lysis, and characterization. Ports 1–4 show the inlet junctions for (in order): beads for exosome capture, washing/lysis buffer, beads for protein capture, ELISA reagents. Adapted with permission from ref (He 2014). Copyright 2014, Royal Society of Chemistry.
Another highly sensitive and rapid integrated on-chip system has been described by Lee and coworkers, which makes use of micro-nuclear magnetic resonance (μNMR) in order to create and on-chip magnetic sensing platform. Over the past few years, research on miniaturized NMR detectors, has had huge impact on development of this type systems and improved detection sensitivity. The system uses magnetic nanoparticles (MNPs) that have been surface grafted with specific antibodies to capture EVs of interest in fluid flow, followed by their quantitative analysis using μNMR (Figure 10B). Using this device, the authors managed to distinguish between tumor-derived EVs and EVs derived from normal cells. Differentiating between EVs from tumor and normal cells is particularly valuable, especially when it can be done on-chip, making this device a promising concept for use in point of care settings for early diagnoses of cancer in patients (Lee 2008). In another type of study by He and coworkers, the authors followed a similar approach on chip, namely immuno-magnetic isolation of tumor exosomes, followed by chemical lysis and intra-vesicular protein analysis, thereby applying enzymatic chemo-fluorescence detection (miniaturized ELISA) (He 2014) (Figure 10C). It is important to note that, in these types of systems, surface binding to the immuno-magnetic beads playing main role in outcome of experiment. Therefore, flushing and mixing steps are essential, and this directly correlates with the engineering and design of microchannels and flow rate in those channels.
Flow field-flow fractionation (F-FFF) is a method that uses various types of external fields perpendicular to laminar flow with a parabolic profile for macromolecule separation. In a study done by Korgel and coworkers, they managed to successfully combine flow field-flow F-FFF with multi-angel light scattering (MALS) for simultaneous size and concentration measurement of the fractionated vesicles (Korgel 1998). In their study, the authors showed the capacity of this method by effectively separating a mix of 27 nm and 37 nm diameter microvesicles. This separation was followed by MALS (or DLS) measurements to determine the average vesicle size and to confirm the monodisperse size distribution of the vesicles. In a similar study by Kang and coworkers, the authors highlighted the importance of fractionation before detection, since the relative abundance is different for each fraction of exosomes, this can affect the protein pattern, which has analytically important implications from less abundant exosome population. For this reason, they have first used size fractionation via applying F-FFF and further the morphological characterization of exosome subpopulations was accomplished using TEM and last step followed by in-solution digestion of each fraction and subsequent shotgun proteomic analysis by nanoflow LC-ESI-MS-MS. This study is a complete example of integrated technique on microchip and its wide application (Kang 2008).
In a recent study by Reategui and coworkers, a novel on-chip platform with a temperature sensitive interface for tumor-specific recovery of exosomes through immuno-affinity immobilization was introduced. In their work, an interface, consisting of an ultrathin gelatin membrane (135 nm) and functionalized with nanoparticles was introduced in order to maximize the EVs and membrane surface interaction. Moreover, the temperature sensitive characteristic of the membrane coating surface was used to enable selective recovery of EVs from microfluidic system upon heating several minutes at 37°C. Further biochemical analysis on separated EVs was performed to detect tumor specific microRNAs (Réategui 2015). With their methodology, they were able to detect and isolate 21% of tumor-derived spiked exosomes in healthy human serum at processing flow rates of 1 ml/h.
In a more innovative study by Im et al, the authors introduced a label free detection technique, (nPLEX) sensor on-chip, which is a basic sensing unit, consisting of a periodic lattice pattern of nanosized holes in a gold film coated with polyethylene glycol and specific antibodies for molecular profiling of exosomes. The gold thin film enables label free surface plasmon resonance (SPR)-based detection of exosomes that have been immuno-captured in the periodic nanoholes. This system is a good example of a microarray-type sensor for large scale parallel detection and analysis of captured exosomes with a high sensitivity (Im 2014) (Figure 11A).
Figure 11.

(A) An SEM micrograph of nanoholes in the nPLEX sensor: (i) the structure was embedded in a 200 nm thick gold film, placed on a glass substrate. The inset is a zoomed image; (ii) Enhancement of the electromagnetic field strength, limited to the nanohole surface. An increased in detection sensitivity is determined which is due to the overlap of electromagnetic filed with the size of captured exosomes at nanohole surface; (iii) a schematic representation of polyethylene glycol (PEG) coated gold surface for conjugation with exosome specific antibodies on top; (iv) A prototype nano-plasmonic sensor integrated with miniaturized imaging system. Adapted with permission from ref (Im 2014). Copyright 2014, Nature Publishing Group. (B) Antibody coated magnetic beads applied for cancer exosome capture and enrichment: (i) exosomes capturing and lysis. The extracted RNA in the flow channels are captured (adsorbed) via a passing through a filter composed of packed glass beads. Next the RNA is collected (eluted) and RT-PCR is applied (on-chip) for quantitation; (ii) SEM images of antibody-functionalized magnetic microbeads after exosome capture. Microbeads (left, 3 μm) functionalized with affinity ligands; (iii) A prototype of iMER system. The microfluidic chamber was engineered to integrate all the processing components for multiplexed detection. Scale bar, 1 cm. Adapted with permission from ref (Shao 2015). Copyright 2015, Nature Publishing Group.
As mentioned previously, fluctuations in the expression level of certain genes that have been secreted into exosomes can correlate to a particular feature of a disease or an indication of disease response to certain type of treatment. For this reason Shao and coworkers, inspired by earlier work of Lee et al. (Lee 2008), developed the new idea of on-chip exosome capture, mRNA extraction and qPCR analyses (Shao 2015). In their novel microfluidic platform, a similar approach of applying magnetic nanoparticles (MNPs) was used to capture and quantify specific tumor derived exosomes. Additionally, they integrated new steps of mRNA extraction from exosomes together with on-chip real time qPCR analyses (Figure 11B). For this novel microfluidic platform, they used the term immuno-magnetic exosome RNA (iMER) analyzer. Using this technology, the authors managed to monitor dynamic sequential changes in key exosomal mRNA markers of cancer and correlate those to treatment responses, thus increasing their value in clinical applications
5- High throughput capability and standardization of microfluidics
Microfluidics have a great value in improving the performance of point of care clinical testing as it can perform various regular laboratory operations using a small fraction of the original sample and time. Its unique potential for large scale bioassays (high throughput) has been proven with various applications for (Guo 2012) single-molecule, and single-cell sensitivity (Novak 2011, Witters 2013), functional integration (Chen 2010, Shuga 2013) and automation (Jin 2015). To consider high throughput capability on-chip, the design of a system for manipulation of fluids flow plays a main role. Regarding the fluid manipulation on-chip, so far two main options have been discussed in the literature, namely continuous flow- and droplet flow-based systems. Continuous flow is a common technique, which is easy to implement and suitable for uncomplicated biochemical applications. The laminar flow behavior and control over the flow characteristics allow for the generation of detailed concentration gradients, due to presence of net forces that cause spatial separation of analytes from their original flow path. There are several limitations however, such as the relatively high volumes of sample/analyte consumption, poor scalability due to the closed-channel configuration, slow mixing rate due to laminar flow, and flow-based interdependency of system. (Tuantranont 2013).
Droplet-based microfluidics (also referred as digital microfluidics) has recently been developed to overcome the above-mentioned shortcomings of continuous flow devices. These microfluidic devices create, in a highly controlled manner, ‘droplets’ in a biphasic system (e.g. oil in water, gas in liquid, etc.) that can be individually manipulated, thus effectively generating a multitude of tiny droplet-sized micro-reactors suitable for rapid parallel on-chip analyses (Nisisako 2007, Bardin 2011). Droplet-based devices typically use bi-phasic liquids, in which each individual assay can be compartmentalized in an aqueous micro-droplet (volumes ranging from 1 pL to 10 nL) that is emulsified by immiscible oil. An important advantage of this technique is related to the isolation of the droplets, both in terms of physical separation and chemical stability, which decreases the risk of cross contamination via unintended mixing of droplets and reagents.
Another advantage is that the device allows for the manipulation of droplets at high-throughput, without the need for additional valves and pumps, by using intelligent microchannel design. Additionally, it is possible to incubate droplets with certain reagents off-chip and then reintroduce them into the flow system for further processing and analysis. This is also referred to as the ‘discrete droplet’ based approach. In earlier studies, the individual parameters that are required for the development of a digital microfluidic platform have been analyzed in detail. These parameters include (among others) the following: droplet sorting (Ahn 2006); droplet mixing (Sarrazina 2007); droplet merging (Ahn 2006, Niu 2008); in droplet cell encapsulation (Clausell-Tormos 2008, Köster 2008); introduction of different reagents to droplets (Boedicker 2008) and on-chip incubation (Song 2003). However, each of these droplet manipulation steps have thus far only been demonstrated separately, and they have not yet been combined into a functioning integrated system on-chip. In addition, thus far there have yet to be studies that attempt to combine multiple analytical detection methods in sequence on-chip, using labeled (fluorescence or colorimetric) droplet technology to create a more universal ‘on-chip’ device, and to build up ‘droplet’ libraries to facilitate high-throughput screening runs. Although the throughput and rate of droplet production and manipulation is already very high (Shim 2013), the unprecedented capabilities of microfluidics for parallelization (Lim 2013) should be considered in future ‘droplet’ type microfluidic designs.
The parallelization at droplet production combines the fine control on size, frequency, and dispersion with ultra-high-throughput. To reduce the number of inlets and outlets for parallel production, microfluidic systems based on multiple layer microfabrication have been proposed (Lim 2015). In a recent study, Lim et al demonstrated “pipette-and-play” like technology, which combines a microfluidic chip and a pressure chamber, offering the possibility of simultaneously manipulating ten different disperse phases on a single-layer device. Their microfluidic chip is composed of one inlet for the continuous phase and ten parallel flow focusing droplet makers, each comprising a loading well for the disperse phases and for the outlet. The dispersed phases are loaded directly into the microfluidic chip mini-wells by simple pipetting. The pressure chamber enables controlled driving of the loaded samples without the need of complex connections (Lim 2015) (Figure 12A). This allows the ultra-high-throughput production (up to 110000 droplets/s) of highly monodisperse emulsions with user-defined chemical composition. In these types of multichannel network, resistance, and pressure distribution over channels play main role in performance of system. In another study by Brouzes et al, the authors showed a similar droplet -based approach for high-throughput single cell screening, by applying cell viability assay on digital microfluidic format. In this way, the authors produced optically indexed droplets, which carry a single cell at a very high throughput level (Brouzes 2009) (Figure 12B). The system they have developed has the capacity to be adjusted for different types of on-chip operations, such as screening of EVs. Additionally, many different droplet bar-coding techniques (high content) can be used, depending on the screening technique. Finally, there is the possibility for combinatorial screening, for example by combining two or more libraries, which makes the system an ideal platform for “-omics” (e.g., genomics, proteomics, lipidomics, etc.) studies (Brouzes 2009).
Figure 12.

(A) Ultra-high-throughput and small droplets production system, which allows the production (up to 110,000 droplets/s) of highly monodisperse droplets (channel width: 100 μm). Adapted with permission from ref (Lim 2015). Copyright 2015, AIP Publishing. (B) Droplet-based microfluidic platform composed of 5 integrated modules for high-throughput cell viability assay: (i) a set of 2 nozzles with Y junction which enables mixing of the streams via alternating one droplet type with another type (interdigitation); (ii) a fusion module that delivered an (alternating current) AC field permitted, which enables electrically-controlled merging of pairs of droplets (100 μm deep); (iii) a mixing module that is used for enhancing droplets mixing in a shorter time frame (100 μm deep); (iv) a delay line module that is utilized for incubation (15 min) of droplets to stain the cells on-chip (260 μm deep); (v) a detection module that is used for trapping droplets vertically and laterally to excite them with a laser for the fluorescent signals collection (100μm deep). Adapted with permission from ref (Brouzes 2009). Copyright 2009, PubMed Central.
When viewing the current status of research related to extracellular vesicles (EVs), it is noticeable that scientific literature contains a wide variety of many technical protocols, some more suited than others based on the particular research aim. The dilemma is which protocol to select, which to best adapt.
In general, EVs research has been broadly divided into four categories, each of which requires a different level of quality control and different emphasis on operating procedures. The four categories of research are: (I) ‘Discovery research’, which isolates and characterizes EV based on size, charge, content, lipid composition and other characteristics. Attention must be paid to less aggressive and more sensitive methods of assay development, in order to enhance experimental reproducibility and to minimize the detection of “off-target” EVs, such as background EVs originating from e.g. platelet- and neutrophil cells. (II) ‘Diagnostic research’, which involves the quantitative and preferentially qualitative search for biomarkers that are related to a specific type of disease. Therefore, internal geometric and spatial characteristics of flow tubes are one of the main parameters to guarantee sub population separation with minimum EVs consolidation. (III) ‘Preparative research’, which involves studies toward purification and preparation of clinically grade EVs for their use as smart nanocarrier system, or as a vaccine for patients. Using EVs in patients might involve a role of antigen delivery to provoke certain immune responses against tumor tissue (Lamparski 2002). (IV) ‘Mechanistic research’, which deals with the basic understanding on functional role of secreted EVs in normal or diseased condition. Therefore, it requires very high standards of sample handling, processing and selection steps, aims to preserve the function of the EVs and to selectively purify EVs from cell or micro-organism of origin (Witwer 2013, Momen-Heravi 2012).
6- Conclusion remarks and future perspective
Microvesicles and exosomes are small particles that exist in body fluids and contain complex RNA and protein cargos. The potential of using exosomes for diagnostic applications and understanding of intracellular communications have resulted in a growing interest among the scientific community. A wide range of technologies applied for extraction, detection, and characterization of extracellular vesicles (EVs) are discussed in this review. It is clear that the concept of integrated device miniaturization is still an ongoing research avenue that requires the development of novel materials and new detection methods to improve both temporal and spatial resolution, combined with increasing control of fluid dynamics at the nanoscale. Also, there are numerous challenges to be addressed in the development of integrated platforms that are capable of multistep operations in fully automated manners.
The first step toward developing such a high content and high throughput “sample-to-result” platform is to acquire a firm knowledge in sample pretreatment based on research aim. Nowadays, biotechnology companies are actively involved in commercializing customized EV diagnostic tools that mostly take advantage of immunoaffinity based isolation and purification of EVs from whole blood. Additional research into non-standard methods and specific design of flow channel with certain geometries can potentially open up interesting new possibilities for microfluidic based sample pre-treatment and isolation.
Besides the need for more effective EV sample pretreatment, a second step toward developing an efficient high-throughput microfluidic platform involves detection and characterization of EVs. These are essential parts of the capture-to-diagnosis path. Therefore, many of the current research efforts are focused toward the development and improvement of on-chip detection and sample analysis. Demonstrating the full potential of on-chip biological component analyses, as discussed in this review, will require establishing active collaboration between engineers, physicists, chemists, and clinicians.
Highlights.
A brief overview on the working principle and examples of the isolation and detection methods with the potential to be used for extracellular vesicles are provided.
Integrated on-chip systems for isolation and characterization of extracellular vesicles (EVs) are highlighted.
Challenges and future perspectives of microfluidic-based systems for characterization of EVs are summarized.
Acknowledgments
Dr. Daniel Stellwagen is kindly thanked for the interesting discussions and his valuable suggestions on this work. Dr. Edwin van der Pol is thanked for providing us high resolution TEM image of extracellular vesicles extracted from human urine. Dr. Mohsen Akbari acknowledges Natural Sciences and Engineering Research Council of Canada (NSERC) for funding.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [web_evpedia] (http://evpedia.info)
- [web_izon] (http://www.izon.com/products/qnano/)
- [web_vdpol] (http://www.edwinvanderpol.com)
- Agarwal AFG, Letizia M, Tung SL, Boardman D, Lechler R, Lombardi G, Smyth LA. Front Immunol. 2014;5:555. doi: 10.3389/fimmu.2014.00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, Kerbage C, Hunt TP, Westervelt RM, Link DR, Weitz DA. Appl Phys Lett. 2006;88:264105. [Google Scholar]
- Akagi T, Ichiki T. Anal Bioanal Chem. 2008;391:2433–2441. doi: 10.1007/s00216-008-2203-9. [DOI] [PubMed] [Google Scholar]
- Akagi T, Kato K, Hanamura N, Kobayashi M, Ichiki T. Jpn J Appl Phys. 2014;53(6S):06J. L01 1–4. [Google Scholar]
- Akagi T, Kato K, Kobayashi M, Kosaka N, Ochiya T, Ichiki T. PLoS One. 2015;10(1371):1–13. doi: 10.1371/journal.pone.0123603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbari M, Bahrami M, Sinton D. Microfluidics and Nanofluidics. 2012;12(1):221–228. [Google Scholar]
- Ali N. Thesis. Victoria University of Wellington; 2011. Detection of inorganic and metal nanoparticles using qNano – IZON science’s nanoparticle analysis system. [Google Scholar]
- Alvarez ML, Khosroheidari M, Kanchi Ravi R, DiStefano JK. Kidney Int. 2012;82:1024–1032. doi: 10.1038/ki.2012.256. [DOI] [PubMed] [Google Scholar]
- Ashcroft BA, de Sonneville J, Yuana Y, Osanto S, Bertina R, Kuil ME, Oosterkamp TH. Biomed Microdevices. 2012;4(14):641–649. doi: 10.1007/s10544-012-9642-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo LC, Pedro MA, Laurindo FR. Recent Pat Cardiovasc Drug Discov. 2007;2:41–51. doi: 10.2174/157489007779606121. [DOI] [PubMed] [Google Scholar]
- Baker CA, Doung CT, Grimley A, Roper MG. Bioanalysis. 2009;5(1):967–975. doi: 10.4155/bio.09.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, Skog J. Nat Commun. 2011;180(1–2):1–19. doi: 10.1038/ncomms1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardin D, Martz TD, Sheeran PS, Shih R, Dayton PA, Lee AP. Lab Chip. 2011;11(23):3990–3998. doi: 10.1039/c1lc20615j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral AM, Von Herrath MG. Current Immunology Reviews. 2009;1:1–6. [Google Scholar]
- Berger M, Castelino J, Huang R, Shah M, Austin RH. Electrophoresis. 2001;18(22):3883–3892. doi: 10.1002/1522-2683(200110)22:18<3883::AID-ELPS3883>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Bhagat AA, Bow H, Hou HW, Tan SJ, Han J, Lim CT. Med Biol Eng Comput. 2010;48(10):999–1014. doi: 10.1007/s11517-010-0611-4. [DOI] [PubMed] [Google Scholar]
- Binnig G, Quate CF, Gerber C. Phys Rev Let. 1986;56(9):930–933. doi: 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- Boedicker JQ, Li L, Kline TR, Ismagilov RF. Lab Chip. 2008;8(8):1265–1272. doi: 10.1039/b804911d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouzes E, Medkova M, Savenelli N, Marran D, Twardowski M, Hutchison JB, Rothberg JM, Link DR, Perrimon N, Samuels ML. Proc Natl Acad Sci USA. 2009;106(25):14195–14200. doi: 10.1073/pnas.0903542106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin R, Diou J, Bélanger D, Tremblay AM, Gilbert C. J Immunol Methods. 2008;338(30):21–30. doi: 10.1016/j.jim.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Carr B, Wright M. In House Publishing. 2013. Nanoparticle Tracking Analysis (NTA) - The first 1000 reports and applications & usage of NTA - Chapter 1, NanoSight Ltd. [Google Scholar]
- Chen C, Skog J, Hsu CH, Lessard RT. Lab Chip. 2010 Feb 21;10:505–511. doi: 10.1039/b916199f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheruvanky A, Zhou H, Pisitkun T, Kopp JB, Knepper MA, Yuen PS, Star RA. Am J Physiol Renal Physiol. 2007;292(5):1657–1661. doi: 10.1152/ajprenal.00434.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CD, Linder V, Sia SK. Lab Chip. 2012 Jan 27;12:2118–2134. doi: 10.1039/c2lc21204h. [DOI] [PubMed] [Google Scholar]
- Cho JA, Yeo D, Son HY, Kim HW, Jung DS, Ko JK, Koh JS, Kim YN, Kim CW. Int J Cancer. 2005 Apr 20;114:613–622. doi: 10.1002/ijc.20757. [DOI] [PubMed] [Google Scholar]
- Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H. Nature Nanotechnol. 2009;4(22):265–270. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- Clausell-Tormos J, Lieber D, Griffiths AD, Merten CA, et al. Chem Biol. 2008;5(15):427–437. doi: 10.1016/j.chembiol.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Clayton A, Court J, Navabi H, Adams M, Mason MD, Hobot JA, Newman GR, Jasani B. J Immunol Methods. 2001;247(1–2):163–174. doi: 10.1016/s0022-1759(00)00321-5. [DOI] [PubMed] [Google Scholar]
- Crescitelli R, Lässer C, Szabó TG, Kittel A, Eldh M, Dianzani I, Buzas EI, Lotvall J. J Extracell Vesicles. 2013;2(12):1–10. doi: 10.3402/jev.v2i0.20677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton AJ. J Natl Cancer Inst. 1975 Jan 31;54:1137–1148. doi: 10.1093/jnci/54.5.1137. [DOI] [PubMed] [Google Scholar]
- Davies RT, Kim J, Jang SC, Choi EJ, Gho YS, Park J. Lab Chip. 2012 Dec 21;12:5202–5210. doi: 10.1039/c2lc41006k. [DOI] [PubMed] [Google Scholar]
- De Jong OC, Van Balkom BW, Schiffelers RM, Bouten CV, Verhaar MC. Front Immunol. 2014 May 4;5(608):1–12. doi: 10.3389/fimmu.2014.00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Toro J, Herschlik L, Waldner C, Mongini C. Front Immunol. 2015;6:203. doi: 10.3389/fimmu.2015.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Boccio P, Raimondo F, Pieragostino D, Morosi L, Cozzi G, Sacchetta P, Magni F, Pitto M, Urbani A. Electrophoresis. 2012 Feb 4;33:689–696. doi: 10.1002/elps.201100375. [DOI] [PubMed] [Google Scholar]
- Dettenhofer M, Yu XF. J Virology. 1999 Feb 2;73:1460–1467. doi: 10.1128/jvi.73.2.1460-1467.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas D, Taylor SS. Methods. 2014 Oct 1;87:3–10. [Google Scholar]
- Dragovic RA, Gardiner C, Harrison P, Sargent IL, et al. Nanomedicine. 2011 Dec 6;7:780–788. doi: 10.1016/j.nano.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EL Andaloussi S, Mäger I, Breakefield XO, Wood MJ. Nat Rev Drug Discov. 2013 May 12;5:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- Evander M, Gidlof O, Olde B, Erlinge D, Laurell T. Lab Chip. 2015;15:2588–2596. doi: 10.1039/c5lc00290g. [DOI] [PubMed] [Google Scholar]
- Ferrari M. Nat Rev Cancer. 2005 Mar 3;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- Gardiner C, Ferreira YJ, Dragovic RA, et al. J Extracell Vesicles. 2013 Feb 15;2:1–11. doi: 10.3402/jev.v2i0.19671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, Tetta C, Bussolati B, Camussi G. Cancer Research. 2011 Aug 1;71:5346–5356. doi: 10.1158/0008-5472.CAN-11-0241. [DOI] [PubMed] [Google Scholar]
- Gruber K, Horlacher T, Castelli R, Mader A, Seeberger PH, Hermann BA. ACS Nano. 2011 Mar 9;5(5):3670–3678. doi: 10.1021/nn103626q. [DOI] [PubMed] [Google Scholar]
- Guan W, Zhang C, Liu F, Liu M. Biosens Bioelectron. 2015 Oct 15;72:114–120. doi: 10.1016/j.bios.2015.04.064. [DOI] [PubMed] [Google Scholar]
- Guo MT, Rotem A, Heymna JA, Weitz DA. Lab Chip. 2012 Jan 11;12:2146–2155. doi: 10.1039/c2lc21147e. [DOI] [PubMed] [Google Scholar]
- György B, Szabó TG, Falus A, Buzás EI, et al. Cell Mol Life Sci. 2011 Aug 16;68:2667–2688. doi: 10.1007/s00018-011-0689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardij J, Cecchet F, Berquand A, Gheldof D, Chatelain C, Mullier F, Chatelain B, Dogné J-M. J Extracell Vesicles. 2013 Aug 27;2:1–9. doi: 10.3402/jev.v2i0.21045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DJ, Fluri K, Seiler K, Fan Z, Effenhauser CS, Manz A. Science. 1993 Aug 13;261(5123):895–897. doi: 10.1126/science.261.5123.895. [DOI] [PubMed] [Google Scholar]
- Harrison P, Dragovic R, Albanyan A, Lawrie AS, Murphy M, Sargent I. J Thromb Haemost. 2009;7(Suppl) OC-TU-056. [Google Scholar]
- He M, Crow J, Roth M, Zeng Y, Godwin AK. Lab Chip. 2014 Oct 7;14(19):3773–3780. doi: 10.1039/c4lc00662c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Herr AE. Nat Protoc. 2010 Oct 28;5(11):1844–1856. doi: 10.1038/nprot.2010.142. [DOI] [PubMed] [Google Scholar]
- Hein B, Willing KI, Hell SW. Proc Natl Acad Sci USA. 2008 May 6;105(38):14271–14276. doi: 10.1073/pnas.0807705105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoegger D, Morier P, Vollet C, Heini D, Reymond F, Rossier JS. Anal Bioanal Chem. 2007 Jan 1;387:267–275. doi: 10.1007/s00216-006-0948-6. [DOI] [PubMed] [Google Scholar]
- Hristov M, Erl W, Linder S, Weber PC. Blood. 2004 Nov 1;104:2761–2766. doi: 10.1182/blood-2003-10-3614. [DOI] [PubMed] [Google Scholar]
- Huang C, Bonroy K, Reekmans G, Laureyn W, Verhaegen K, De Vlaminck I, Lagae L, Borghs G. Biomed Microdevices. 2009 Aug 11;4:893–901. doi: 10.1007/s10544-009-9306-8. [DOI] [PubMed] [Google Scholar]
- Huang X, Ren J. Electrophoresis. 2005 Oct 26;19:3595–3601. doi: 10.1002/elps.200500076. [DOI] [PubMed] [Google Scholar]
- Huh YS, Chung AJ, Erickson D. Microfluidics and Nanofluidics. 2009 Jan 8;6:285–297. [Google Scholar]
- Ichiki T, Akagi T, Shiono H. WO2014030590A1 Patent. 2014
- Im H, Shao H, Park YI, Peterson VM, Castro CM, Weissleder R, Lee H. Nature Biotechnology. 2014 May 5;32:490–495. doi: 10.1038/nbt.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingils DW, Riehn R, Austin RH, Sturm JC. Appl Phys Lett. 2004;85(21):5093–5095. [Google Scholar]
- Ismagilov RF. Angew Chem Int Ed. 2003;42(35):4130–4132. doi: 10.1002/anie.200301660. [DOI] [PubMed] [Google Scholar]
- Ito T, Sun L, Crooks RM. Analytical Chemistry. 2003 Apr 19;75(10):2399–2406. doi: 10.1021/ac034072v. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Lu H. Curr Opin Chem Eng. 2013 Nov 10;2(4):398–404. doi: 10.1016/j.coche.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen KR, Paulsen BS, Bæk R, Varming K, Sorensen BS, Jorgensen MM. J Extracell Vesicles. 2015;4(26659):1–10. doi: 10.3402/jev.v4.26659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SH, Jeon HH, et al. Lab on chip. 2015 Jul 23;15:3677–3686. doi: 10.1039/c5lc00651a. [DOI] [PubMed] [Google Scholar]
- Johnson AS, Selimovic A, Martin RS. Anal Bioanal Chem. 2013 Apr 10;405:3013–3020. doi: 10.1007/s00216-012-6682-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra H, Adda CG, Liem M, Ang CS, Mechler A, Simpson RJ, Hulett MD, Mathivanan S. Proteomics. 2013 Nov 22;13:3354–3364. doi: 10.1002/pmic.201300282. [DOI] [PubMed] [Google Scholar]
- Kang D, Oh S, Ahn SM, Lee BH, Moon MH. J Proteome Res. 2008 Aug 8;7:3475–3480. doi: 10.1021/pr800225z. [DOI] [PubMed] [Google Scholar]
- Kato K, Kobayashi M, Hanamura N, Akagi T, Kosaka N, Ochiya T, Ichiki T. Jpn J Appl Phys. 2013;52:06GK10. [Google Scholar]
- Keller S, Ridinger J, Rupp AK, Janssen JW, Altevogt P. J Transl Med. 2011 Jun 8–9;86:5876–5879. doi: 10.1186/1479-5876-9-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RT, Woolley AT. J Sep Sci. 2005 Sep 14;28:1985–1993. doi: 10.1002/jssc.200500228. [DOI] [PubMed] [Google Scholar]
- Kim M, Kim T. Analyst. 2013 Oct 21;138:6007–6015. doi: 10.1039/c3an00965c. [DOI] [PubMed] [Google Scholar]
- Koga K, Matsumoto K, Akiyoshi T, Kubo M, Yamanaka N, Tasaki A, Nakashima H, Nakamura M, Kuroki S, Tanaka M, Katano M. Anticancer Res. 2005 Nov-Dec;6A:3703–3707. [PubMed] [Google Scholar]
- Kooijmans SA, Stremersch S, Braeckmans K, de Smet SC, Hendrix A, Wood MJ, Schifflers RM, Raemdonck K, Vader P. J Control Release. 2013 Nov 28;172(1):229–238. doi: 10.1016/j.jconrel.2013.08.014. [DOI] [PubMed] [Google Scholar]
- Korgel BA, Van Zanten JH, Monbouquette HG. Biophys J. 1998;74(6):3264–3272. doi: 10.1016/S0006-3495(98)78033-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster S, Angilè FE, Griffiths AD, Weisz DA, et al. Lab Chip. 2008 Jul 8;7:1110–1115. doi: 10.1039/b802941e. [DOI] [PubMed] [Google Scholar]
- Kurz V, Nelson EM, Shim J, Timp G. ACS Nano. 2013 Apr 22;7(5):4057–4069. doi: 10.1021/nn400182s. [DOI] [PubMed] [Google Scholar]
- Lamparski HG, Metha-Damani A, Yao JY, Patel S, Hsu DH, Ruegg C, Le Pecq JB. J Immunol Methods. 2002 Dec 15;270(2):211–226. doi: 10.1016/s0022-1759(02)00330-7. [DOI] [PubMed] [Google Scholar]
- Lane RE, Korbie D, Anderson W, Vaidyanathan R, Trau M. Sci Rep. 2015;5:7639. doi: 10.1038/srep07639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasser C. Expert Opin Biol Ther. 2015 Jan 1;15:103–117. doi: 10.1517/14712598.2015.977250. [DOI] [PubMed] [Google Scholar]
- Laulagnier K, Motta C, Hamdi S. Biochemical Journal. 2004 May 15;380:161–171. doi: 10.1042/BJ20031594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar I, Clement E, Ducoux-Petit M. Pigment Cell Melanoma Res. 2015 Jul 28;4:464–475. doi: 10.1111/pcmr.12380. [DOI] [PubMed] [Google Scholar]
- Lee H, Sun E, Ham D, Weissleder R. Nature Med. 2008 Aug 14;8:869–874. doi: 10.1038/nm.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LM, Cui X, Yang C. Biomed Microdevices. 2009 Oct 11;5:951–958. doi: 10.1007/s10544-009-9312-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Shao H, Weissleder R, Lee H. ACS Nano. 2015 Mar 24;9(3):2321–2327. doi: 10.1021/nn506538f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Caen O, Vrignon J, Konrad M, Taly V, Baret JC. Biomicrofluidics. 2015;9(3):034101. doi: 10.1063/1.4919415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Gruner P, Konrad M, Baret JC. Lab Chip. 2013 Jan 30;13(8):1472–1475. doi: 10.1039/c3lc41329b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima LG, Chammas R, Monteiro RQ, Moreira ME, Barcinski MA. Cancer lett. 2009 Oct 8;283(2):168–175. doi: 10.1016/j.canlet.2009.03.041. [DOI] [PubMed] [Google Scholar]
- Lin CC, Hsu JL, Lee GB. Microfluidics and Nanofluidics. 2011 Jul 2;10(3):481–511. [Google Scholar]
- Liu S, Zhao Y, Parks JW, Deamer DW, Hawkins AR, Schmidt H. Nano Lett. 2014 Aug 13;14(8):4816–4820. doi: 10.1021/nl502400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livshits MA, Khomyakova E, Evtushenko EG, Lazarev VN, Kulemin NA, Semina SE, Generozov EV, Govorun VM. Sci Rep. 2015;5:17319. doi: 10.1038/srep17319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logozzi M, De Milito A, Rivoltini L, Fais S, et al. PLoS One. 2009;4(4):e5219. doi: 10.1371/journal.pone.0005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loutherback K, Chou KS, Newman J, Puchalla J, Austin RH, Sturm JC. J Microfluidics and Nanofluidics. 2010;9(6):1143–1149. [Google Scholar]
- Luo Y, Yu F, Zare RN. Lab Chip. 2008;8(5):694–700. doi: 10.1039/b800606g. [DOI] [PubMed] [Google Scholar]
- Lvovich V, Srikanthan S, Silverstein RL. Biosens Bioelectron. 2010 Oct 15;26(2):444–451. doi: 10.1016/j.bios.2010.07.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald MP, Spalding GC, Dholakia K. Nature. 2003;426(6965):421–424. doi: 10.1038/nature02144. [DOI] [PubMed] [Google Scholar]
- Mark D, Haeberle S, Roth G, von Stetten F, Zengerle R. Chem Soc Rev. 2010;39(3):1153–1182. doi: 10.1039/b820557b. [DOI] [PubMed] [Google Scholar]
- Maruyama H, Sakuma S, Yamanishi Y, Arai F. J Robotics and Mechatronics. 2010;22(3):280–285. [Google Scholar]
- Matos Pires NM, Dong T, Hanke U, Hoivik N. Sensors. 2014;14(8):15458–15479. doi: 10.3390/s140815458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant ML, Powell DW, Wilkey DW, Cummins TD, Deegens JK, Rood IM, McAfee KJ, Fleischer C, Klein E, Klein JB. Proteomics Clin Appl. 2010;4(1):84–96. doi: 10.1002/prca.200800093. [DOI] [PubMed] [Google Scholar]
- Mogensen KB, Henning K, Kutter JP. Electrophoresis. 2004;25(21–22):3498–3512. doi: 10.1002/elps.200406108. [DOI] [PubMed] [Google Scholar]
- Momen-Heravi F, Balaj L, Alian S, Trachtenberg AJ, Hochberg FH, Skog J, Kuo WP. Front Physiol. 2012;28(3):162. doi: 10.3389/fphys.2012.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momen-Heravi F, Balaj L, Alian S, Mantel PY, et al. Biol Chem. 2013 Oct 10;394:1253–1262. doi: 10.1515/hsz-2013-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monguió-Tortajada M, Lauzurica-Valdemos R, Borràs FE. Front Immunol. 2014;5:416. doi: 10.3389/fimmu.2014.00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaner S, Galiano A, Trelis M, Martin-Jaular L, DelPortillo HA, Bernal D, Marcilla A. Front Immunol. 2014;5:433. doi: 10.3389/fimmu.2014.00433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers FB, Lee LP. Lab Chip. 2008;8(12):2015–2031. doi: 10.1039/b812343h. [DOI] [PubMed] [Google Scholar]
- Nisisako T, Torii T. Advanced Materials. 2007;19(11):1489–1493. [Google Scholar]
- Niu X, Gulati S, Edel JB, deMello AJ. Lab Chip. 2008;8:1837–1841. doi: 10.1039/b813325e. [DOI] [PubMed] [Google Scholar]
- Nolte-'t Hoen EN, van der Vlist EJ, Wauben MH, et al. Nanomedicine. 2012;8(5):712–720. doi: 10.1016/j.nano.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak R, Zeng Y, Shuga J, Venugopalan G, Fletscher DA, Smith MT, Mathies RA. Angew Chem Int Ed. 2011;50(2):390–395. doi: 10.1002/anie.201006089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksvold MP, Neurauter A, Pedersen KW. Methods Mol Biol. 2015;1218:465–481. doi: 10.1007/978-1-4939-1538-5_27. [DOI] [PubMed] [Google Scholar]
- Patel N, Sanders GHW, Shakesheff KM, Cannizzaro SM, Davies MC, Langer R, Roberts CJ, Tendler SJB, Williams PM. Langmuir. 1999;15(21):7252–7257. [Google Scholar]
- Pisitkun T, Shen RF, Knepper MA. Proc Natl Acad Sci USA. 2004 Sep 7;101(36):13368–13373. doi: 10.1073/pnas.0403453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe BD, Mahalanabis M, Lewis LH, Klapperich CM, Murthy SK. Anal Chem. 2012;84(3):1336–1344. doi: 10.1021/ac2022844. [DOI] [PubMed] [Google Scholar]
- Pospichalova V, Svoboda J, Dave Z. J Extracell Vesicles. 2015 Mar 31;4:25530, 1–10. doi: 10.3402/jev.v4.25530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppels GJ, de Mul FF. Nature. 1990 Sep 20;347:6290, 301–303. doi: 10.1038/347301a0. [DOI] [PubMed] [Google Scholar]
- Qian XM, Nie SM. Chem Soc Rev. 2008 Mar 26;37(5):912–920. doi: 10.1039/b708839f. [DOI] [PubMed] [Google Scholar]
- Rafeie M, Zhang J, Asadnia M, Li W, Ebrahimi Warkiani M. Lab Chip. 2016;16:2791–2802. doi: 10.1039/c6lc00713a. [DOI] [PubMed] [Google Scholar]
- Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CF, Geuze HJ. J Exp Med. 1996 Mar 1;183(3):1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Stoorvogel W. J Cell Biol. 2013 Feb 18;200(4):373–838. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Réategui E, Lai CP, Van der Vos K, Zeilani M, Breakefield XO, Stott SL. Specific isolation of tumour-derived extracellular vesicles using microfluidic technologies. ISEV Abstract 2015 [Google Scholar]
- Revenfeld AL, Bæk R, Nielsen MH, Stensballe A, Varming K, Jørgensen M. Clinical Therapeutics. 2014 Jun 1;36:830–846. doi: 10.1016/j.clinthera.2014.05.008. [DOI] [PubMed] [Google Scholar]
- Ring EA, de Jonge N. Microsc Microanal. 2010;16(5):622–629. doi: 10.1017/S1431927610093669. [DOI] [PubMed] [Google Scholar]
- Roberts GS, Kozak D, Anderson W, Broom MF, Vogel R, Trau M. Small. 2010;6(23):2653–2658. doi: 10.1002/smll.201001129. [DOI] [PubMed] [Google Scholar]
- Rosen CB, Larrea DR, Bayley H. Nat Biotechnol. 2014;32(2):179–181. doi: 10.1038/nbt.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu G, Huang J, deMello AJ, Bradley DC, et al. Lab Chip. 2011 Mar 24;9:1664–1670. doi: 10.1039/c0lc00586j. [DOI] [PubMed] [Google Scholar]
- Salieb-Beugelaar GB, Simone G, Arora A, Philippi A, Manz A. Anal Chem. 2010 Jun 15;82(12):4848–4864. doi: 10.1021/ac1009707. [DOI] [PubMed] [Google Scholar]
- Santana SM, Antonyak MA, Cerione RA, Kirby BJ. Biomed Microdevices. 2014;16(6):869–877. doi: 10.1007/s10544-014-9891-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin F, Prat L, Di Micelia N, Cristobal G, Link DR, Weitz DA. Chemical Engineering Science. 2007 Oct 26;62:1042–1048. [Google Scholar]
- Schwarz MA, Hauser PC. Lab Chip. 2001 Jun 13;1:1–6. doi: 10.1039/b103795c. [DOI] [PubMed] [Google Scholar]
- Scott D, Harding SE, Rowe A, editors. Analytical Ultracentrifugation: Technique and Methods. The Royal Society of Chemistry; 2005. pp. 273–276. [Google Scholar]
- Shao H, Chung J, Weissleder R, Lee H, et al. Nature Med. 2012 Nov 11;18(12):1835–1840. doi: 10.1038/nm.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Chung J, Lee K. Nature Commun. 2015;11(6):6999. doi: 10.1038/ncomms7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Gillespie BM, Palanisamy V, Gimzewski JK. Langmuir. 2011 Dec 6;27(23):14394–14400. doi: 10.1021/la2038763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Rasool HI, Palanisamy V, Mathisen C, Schmidt M, Wong DT, Gimzewski JK. ACS Nano. 2010;7(7):5955–5964. doi: 10.1021/nn901824n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim S, Gascoyne P, Noshari J, Hale KS. Integr Biol (Camb) 2011;3(8):850–862. doi: 10.1039/c1ib00032b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim JU, Ranasinghe RT, et al. ACS Nano. 2013;7(7):5955–5964. doi: 10.1021/nn401661d. [DOI] [PubMed] [Google Scholar]
- Shuga J, Zeng Y, Mathies RA, Smith MT, et al. Nucl Acids Res. 2013 Jul 19;41(16):e159. doi: 10.1093/nar/gkt613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siedlecki CA, Wang IW, Higashi JM, Kottke-Marchant K, Marchant RE. Biomaterials. 1999;20(16):521–529. doi: 10.1016/s0142-9612(99)00065-4. [DOI] [PubMed] [Google Scholar]
- Song H, Tice JD, Ismagilov RF. Angew Chem Int Ed. 2003;42(7):768–772. doi: 10.1002/anie.200390203. [DOI] [PubMed] [Google Scholar]
- Song S, Singh AK. Anal Bioanal Chem. 2006;384(1):41–43. doi: 10.1007/s00216-005-0206-3. [DOI] [PubMed] [Google Scholar]
- Soni GV, Singer A. Rev Sci Instrum. 2010;81(1):014301. doi: 10.1063/1.3277116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowerby SJ, Broom MF, Petersen GB. Sensors and Actuators B: Chemical. 2007;123(1):325–330. [Google Scholar]
- Srivastava SA, Ali A, Solanki PR, Chavhan PM, Pandey MK, Mulchandani A, Srivastava AM, Malhotra BD. RSC Adv. 2013;3:228–235. [Google Scholar]
- Stremersch S, Marro M, Pinchasik BE, Baatsen P, Hendrix A, De Smedt SC, Loza-Alvarez P, Skirtach AG, Raemdonck K, Braeckmans K. Small. 2016;12(24):3292–3301. doi: 10.1002/smll.201600393. [DOI] [PubMed] [Google Scholar]
- Subra C, Laulagnier K, Perret B, Record M. Journal of Biochimie. 2007;89(2):205–212. doi: 10.1016/j.biochi.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Sueyoshi K, Kitagawa F, Otsuka K. J Sep Sci. 2008;31(14):2650–2666. doi: 10.1002/jssc.200800272. [DOI] [PubMed] [Google Scholar]
- Tai LA, Kang YT, Yew T-R, Yang C-S, et al. Anal Chem. 2012 Aug 7;84:6312–6316. doi: 10.1021/ac301523n. [DOI] [PubMed] [Google Scholar]
- Tauro BJ, Greening DW, Mathias RA, Mathivanan S, Scott AM, Simpson RJ. Methods. 2012;56(2):293–304. doi: 10.1016/j.ymeth.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Tauro BJ, Greening DW, Mathias RA, Mathivanan S, Ji H, Simpson RJ. Mol Cell Proteomics. 2013;12(3):587–598. doi: 10.1074/mcp.M112.021303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DD, Zacharias W, Gercel-Taylor C. Methods Mol Biol. 2011;728:235–246. doi: 10.1007/978-1-61779-068-3_15. [DOI] [PubMed] [Google Scholar]
- Tegenfeldt JO, Prinz C, Cao H, Huang RL, Austin RH, Chou SY, Cox EC, Sturm JC. Anal Bioanal Chem. 2004;378(7):1678–1692. doi: 10.1007/s00216-004-2526-0. [DOI] [PubMed] [Google Scholar]
- Théry C, Amigorena S, Raposo G, Clayton A. Curr Protoc Cell Biol Chapter. 2006;3(Unit 3.22):1–29. doi: 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
- Théry C, Ostrowski M, Segura E. Nature Rev Immunology. 2009 Aug 9;8:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- Thorsen T, Maerkl SJ, Quake SR. Science. 2002;298(5593):580–584. doi: 10.1126/science.1076996. [DOI] [PubMed] [Google Scholar]
- Tuantranont A, editor. Application of Nanomaterials in Sensors and Diagnostics. Springer-Verlag; Berlin Heidelberg: 2013. [Google Scholar]
- Tushuizen ME, Diamant M, Sturk A, Nieuwland R. Arterioscler Thromb Vasc Biol. 2011;31:4–9. doi: 10.1161/ATVBAHA.109.200998. [DOI] [PubMed] [Google Scholar]
- Uzunbajakava N, Lenferink A, Kraan Y, Volokhina E, Vrensen G, Greve J, Otto C. Biophys J. 2003;84(6):3968–3968. doi: 10.1016/S0006-3495(03)75124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner PS, Craighead HG. Lab Chip. 2007 Jul 25;7(10):1238–1255. doi: 10.1039/b707401h. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wu HJ, Fine D, Schmulen J, Hu Y, Godin B, Zhang JXJ, Liu X. Lab Chip. 2013 Aug 7;13(15):2879–2882. doi: 10.1039/c3lc41343h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Patel P, Liao W, et al. Clin Cancer Res. 2009 Jul 1;15(13):4446–4452. doi: 10.1158/1078-0432.CCR-09-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner BB. Measuring Size & Surface Charge of Exosomes, Microvesicles & Liposomes, Application Note. Brookhaven Instruments Holtsville; NY: 2013. [Google Scholar]
- Wiggins R, Glatfelter A, Kshirsagar B, Beals T. Lab Invest. 1987;56(3):264–272. [PubMed] [Google Scholar]
- Willig KI, Rizzoli SO, Westphal V, Jahn R, Hell SW. Nature. 2006 Apr 13;440:935–939. doi: 10.1038/nature04592. [DOI] [PubMed] [Google Scholar]
- Witters D, Knez K, Ceyssens F, Puers R, Lammertyn J. Lab Chip. 2013 Jun 7;13(11):2047–2054. doi: 10.1039/c3lc50119a. [DOI] [PubMed] [Google Scholar]
- Witwer KW, Buzás EI, Wauben MA, Hochberg F, et al. J Extracell Vesicles. 2013 May 27;2:20360. [Google Scholar]
- Wolter A, Niessner R, Seidel M. Anal Chem. 2008 Aug 1;80(15):5854–5863. doi: 10.1021/ac800318b. [DOI] [PubMed] [Google Scholar]
- Yamada T, Inoshima Y, Matsuda T, Ishiguro N. J Vet Med Sci. 2012;74(11):1523–1525. doi: 10.1292/jvms.12-0032. [DOI] [PubMed] [Google Scholar]
- Yanez-Mo M, Siljander PR-M, Wauben MHM, De Wever O, et al. J Extracell Vesicles. 2015;4:27066. doi: 10.3402/jev.v4.27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuana Y, Bertina RM, Osanto S. Thromb Haemost. 2011 Dec 21;105(3):396–408. doi: 10.1160/TH10-09-0595. [DOI] [PubMed] [Google Scholar]
- Yuana Y, Koning RI, Kuil ME, Rensen PC, Koster AJ, Bertina RM, Osanto S. J Extracell Vesicles. 2013 Dec 31;2:21494. doi: 10.3402/jev.v2i0.21494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuana Y, Oosterkamp TH, Bahatyrova S, et al. J Thromb Haemost. 2010;8(2):315–323. doi: 10.1111/j.1538-7836.2009.03654.x. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Rahimian A, Son K, Shin DS, Patel T, Revzin A. Methods. 2016 Mar 15;97:88–93. doi: 10.1016/j.ymeth.2015.10.012. [DOI] [PubMed] [Google Scholar]
- Zou Z, Lee S, Ahn CH. IEEE Sens. 2008 May 23;8(5):527–535. [Google Scholar]
- Zwicker JI, Liebman HA, Neuberg D. Clin Cancer Res. 2009 Nov 15;15(22):6830–6840. doi: 10.1158/1078-0432.CCR-09-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]