

Abstract
Background and Purpose
The neutral amino acid transporter B0AT1 (SLC6A19) has recently been identified as a possible target to treat type 2 diabetes and related disorders. B0AT1 mediates the Na+‐dependent uptake of all neutral amino acids. For surface expression and catalytic activity, B0AT1 requires coexpression of collectrin (TMEM27). In this study, we established tools to identify and evaluate novel inhibitors of B0AT1.
Experimental Approach
A CHO‐based cell line was generated, stably expressing collectrin and B0AT1. Using this cell line, a high‐throughput screening assay was developed, which uses a fluorescent dye to detect depolarisation of the cell membrane during amino acid uptake via B0AT1. In parallel to these functional assays, we ran a computational compound screen using AutoDock4 and a homology model of B0AT1 based on the high‐resolution structure of the highly homologous Drosophila dopamine transporter.
Key Results
We characterized a series of novel inhibitors of the B0AT1 transporter. Benztropine was identified as a competitive inhibitor of the transporter showing an IC50 of 44 ± 9 μM. The compound was selective with regard to related transporters and blocked neutral amino acid uptake in inverted sections of mouse intestine.
Conclusion And Implications
The tools established in this study can be widely used to identify new transport inhibitors. Using these tools, we were able to identify compounds that can be used to study epithelial transport, to induce protein restriction, or be developed further through medicinal chemistry.
Abbreviations
- CHO‐BC
CHO cells stably transfected with B0AT1 and collectrin
- FGF21
fibroblast growth factor 21
- GIP
gastric inhibitory peptide
- GLP‐1
glucagon‐like peptide 1
- MW
molecular weight
- NMDG
N‐methyl‐d‐glucamine
Tables of Links
| TARGETS | |
|---|---|
| Transporters | |
| ASCT2 | PROT, SLC6A7 |
| B0AT1, SLC6A19 | LAT1 |
| DAT, SLC6A3 | LAT2 |
| LIGANDS |
|---|
| Benztropine |
| Nimesulide |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Protein restriction is a powerful signal to regulate cellular and organismic metabolism. It is known to induce longevity in many species and to improve metabolic health (Mirzaei et al., 2014; Solon‐Biet et al., 2014). While the concept is appealing, it has not yet resulted in pharmacological approaches. Instead of reducing nutritional protein intake, blocking the absorption of amino acids in the intestine may provide an approach to the pharmacological modulation of protein nutrition. In the intestine, amino acids, di‐ and tri‐peptides are the final products of protein digestion. Uptake of these nutrients is mediated by the peptide transporter PepT1 and a variety of amino acid transporters located in the apical membrane of enterocytes (Daniel, 2004; Broer, 2008). B0AT1 is the major apical neutral amino acid transporter in the intestine and kidney, and its expression appears to be limited to these tissues (Broer et al., 2004). It mediates the uptake of all neutral amino acids from the lumen of the intestine and the proximal tubule in the kidney. Pharmacological inhibition of B0AT1 thus not only reduces protein absorption but also increases loss of amino acids through the urine.
Genetic disruption of the Slc6a19 gene (encoding B0AT1) in mice induced greater glycaemic control and resistance to high‐fat diet induced obesity (Jiang et al., 2015). This is thought to be mediated through an amino acid starvation response in the liver, which in turn causes up‐regulation of the metabolic hormone FGF21. FGF21 increases metabolism of fat to generate ketone bodies and also induces browning of white adipose tissue (Kharitonenkov and Adams, 2014). Human clinical trials with FGF21 mimetics are consistent with this role (Reitman, 2013). Reduced absorption of amino acids from the lumen of the intestine has additional benefits, such as increased secretion of the incretins glucagon‐like peptide 1 (GLP‐1) and GIP (Jiang et al., 2015). Together, these results suggested that a pharmacological block of B0AT1 could replicate this metabolic phenotype, thus providing a new target to improve glycaemic control in type 2 diabetes and improve metabolic health in the obese. In contrast to the therapeutic use of FGF21 and GLP‐1 – which have to be administered subcutaneously as modified peptides to ensure extended biological half‐life – a low MW inhibitor of B0AT1 could be administered orally and could induce expression of FGF21 and GLP‐1. Humans with inactivating mutations in B0AT1 are known to have Hartnup disorder, a largely benign condition that in some cases is associated with a pellagra‐like skin rash (Broer, 2009). As a result, it appears reasonably safe to inhibit this transporter in adult humans. It is not known whether humans with Hartnup disorder have a metabolic phenotype similar to that of B0AT1‐deficient mice.
B0AT1 mediates Na+‐dependent transport of neutral amino acids with a preference for large neutral aliphatic amino acids (Bohmer et al., 2005). The transporter is electrogenic, that is, it carries one positive sodium ion per amino acid into the cytosol during transport (Bohmer et al., 2005). Accordingly, substrate‐dependent membrane depolarisation is observed upon expression in Xenopus laevis oocytes. Similar to many other transporters, B0AT1 requires ancillary proteins for trafficking to the cell surface and for catalytic function (Fairweather et al., 2015). In the kidney, this role is fulfilled by the type I membrane protein collectrin (TMEM27) (Danilczyk et al., 2006; Malakauskas et al., 2007). In the intestine where collectrin is missing, the exopeptidase angiotensin converting enzyme 2 (ACE2) carries out the same role (Kowalczuk et al., 2008). The peptidase domain of ACE2 is not required for trafficking and catalytic function of B0AT1 (Fairweather et al., 2015). Mice lacking ACE2 or collectrin each exhibit some, but not all, aspects of the global B0AT1 knockout phenotype (Malakauskas et al., 2009; Bernardi et al., 2015).
Little is known about the pharmacology of B0AT1. The approved drug and cyclooxygenase inhibitor nimesulide has been identified as an inhibitor of B0AT1 (Pochini et al., 2014), while loratadine was reported as an inhibitor of the related transporter B0AT2 (Cuboni et al., 2014). B0AT1 is a member of the solute carrier family 6 (SLC6A19), which comprises transporters for neurotransmitters, amino acids and related compounds (Broer and Gether, 2012). The pharmacology of the 5‐HT, dopamine and noradrenaline transporters in this family is well developed (Alexander et al., 2015). Notably, tricyclic antidepressants and related compounds bind to these transporters with high affinity.
While precise structures of B0AT1 are not available, high‐resolution structures are available for the sequence‐related bacterial amino acid transporter LeuT (Penmatsa and Gouaux, 2014); the dopamine transporter from Drosophila melanogaster (dDAT), which has pharmacological properties similar to human DAT (Penmatsa et al., 2013); and the human 5‐HT transporter (SERT; Coleman et al., 2016). Interestingly, a binding site for tricyclic antidepressants has been identified in LeuT. LeuT is mechanistically and functionally closely related to the amino acid transporter B0AT1. Both mediate the Na+‐cotransport of a large variety of neutral amino acids. Importantly, LeuT and dDAT have been crystallized in outside open conformation, which is the most relevant for drug binding. Although the overall sequence homology is limited, residues around the substrate binding site are highly conserved. As a result, reliable homology models of the binding site can be generated allowing computational screening for novel inhibitors. The binding site of LeuT or dDAT is characterized by a group of hydrophobic residues that enclose the side chain of the transported amino acid. The α‐carboxyl and α‐amino group of the substrate forms contacts with protein backbone carbonyl‐oxygens and side‐chain hydroxyl‐groups of neighbouring residues. Notably, the substrate α‐carboxyl‐group forms part of the binding site of one of the Na+ ions (Na1 site). The second Na+‐binding site (Na2 site) is not in direct contact with the substrate. It is not known whether it is used in B0AT1. The substrate binding site in LeuT/dDAT is accessible through a narrow vestibule. Access is constricted by an aspartate (D486 in B0AT1) and an arginine (R57 in B0AT1) residue that form an ion pair when the transporter is closed on the outside.
Here, we used a set of readily available tools to identify novel inhibitors of B0AT1, some of which bind to the transporter with 100‐fold greater affinity than the endogenous substrates. These tools can be applied to a variety of transporters.
Methods
Cell lines and media
Generation of CHO‐B0AT1‐collectrin cells
The CHO Flp‐In host cell line (Life Technologies) was used to generate a cell model stably expressing human B0AT1 and human collectrin (Life Technologies) via Flp recombinase‐mediated DNA recombination at a defined target site using Flp‐In™ vector constructs. CHO Flp‐In host cells were first transfected with the plasmid pcDNA5/FRT‐hSLC6A19 using LTX‐Lipofectamine (Life Technologies). The successfully transfected cells were selected by hygromycin B (0.33 mg·mL−1 Life Technologies) and the expression of human SLC6A19 was demonstrated by RT‐PCR. These cells were subsequently co‐transfected with the plasmid pcDNA3.1‐collectrin and selected by their resistance to hygromycin B and geneticin (G418 sulfate 0.275 mg·mL−1, Life Technologies). The co‐transfected CHO‐B0AT1‐collectrin cells (CHO‐BC) were subcloned by limited dilution and analysed for expression of human SLC6A19 and collectrin at the cell surface as well as for sodium‐dependent uptake of leucine and isoleucine.
CHO‐BC cells were cultured in Ham's F‐12 GlutaMAX™ medium (Life Technologies) supplemented with 10% (v/v) FBS (Life Technologies) as well as antibiotics hygromycin B (0.33 mg·mL−1) and G418 (0.275 mg·mL−1), while CHO Flp‐In host cells were maintained in the same medium supplemented with 10% (v/v) FBS and Zeocin (0.1 mg·mL−1, Life Technologies). 143B TK− cells (human osteosarcoma cell line) were maintained in DMEM/Ham's F‐12 medium (Sigma) supplemented with 10% (v/v) FBS and 2 mM glutamine, while MCF‐7 cells (human breast adenocarcinoma cell line) were maintained in the same medium supplemented with 10% (v/v) FBS, 2 mM glutamine and 1× non‐essential amino acids (Life Technologies). The cells were passaged at about 80–90% confluence and the medium was changed every 3–4 days. All cells were kept in a humidified incubator at 37°C and 5% CO2.
Surface biotinylation and Western blotting
CHO Flp‐in host and CHO‐BC cells maintained in 60 mm dishes were washed three times with ice‐cold PBS buffer (137 mM NaCl, 2.7 mM KCl, 5 mM Na2HPO4, 0.9 mM CaCl2, 0.5 mM MgCl2; pH 8.0) followed by biotinylation in ice‐cold PBS (pH 8.0) buffer supplemented with 0.5 mg·mL−1 Sulfo‐NHS‐lc‐Biotin (Thermo Scientific) at room temperature on a slow shaker for 30 min. After washing three times with ice‐cold PBS buffer supplemented with 100 mM glycine to quench unbound reagent, the cells were collected in 1 mL lysis buffer (150 mM NaCl, 20 mM Tris–HCl, pH 7.6, 1% Triton X‐100) and incubated in a reaction tube on ice for 1.5 h with occasional inversions. The cell lysate was then centrifuged at 16 000 g in a benchtop centrifuge for 10 min, and the supernatant was transferred to a new reaction tube. After protein quantification, 100 μL of streptavidin beads (high‐capacity streptavidin agarose resin, Thermo Scientific) were added to each sample and incubated at 4°C on a rotating shaker overnight. After washing five times with lysis buffer, the streptavidin beads were mixed with 4× sample buffer, incubated at 95°C for 5 min and subjected to SDS‐PAGE analysis. The separated proteins were transferred onto nitrocellulose membranes (GE Healthcare) and bands of interest were detected by specific primary antibodies as follows: rabbit anti‐mouse B0AT1 (1:3000) (Custom antibody, Pineda Antibody Service); sheep anti‐mouse collectrin (1:3000) (R&D Systems); rabbit anti‐human Na/K‐ATPase (1:7000) (Abcam). The blots were then incubated with HRP‐conjugated secondary antibodies (donkey anti‐rabbit or anti‐sheep IgG) at the same dilution as the primary antibody. For image acquisition, HRP reagents were added to initiate the chemiluminescent reaction (Millipore HRP substrates Luminata Forte).
Transport assay using FLIPR
Membrane depolarisation induced by amino acid transport was monitored using a FLIPR® kit (Molecular Devices, R8042 BLUE DYE). Before the assay, CHO‐BC cells were seeded out at a density of 60 000 cells per well and maintained in a black‐wall 96‐well plate (Corning) overnight. After washing three times with HBSS+G (composition: 136.6 mM NaCl, 5.4 mM KCl, 0.44 mM KH2PO4, 2.7 mM Na2HPO4, 1.26 mM CaCl2, 0.5 mM MgCl2, 0.4 mM MgSO4, 10 mM HEPES, pH 7.5, supplemented with 5 mM glucose), cells in each well were incubated for 30–60 min at room temperature in 100 μL of HBSS + G containing compounds as indicated, together with 100 μL of dye‐loading buffer provided with the kit. Subsequently, the fluorescence signal was detected every 10 s before and after the addition of amino acids or other substrates as indicated in the figures. Fluorescence was detected at room temperature using a TECAN INFINITE M1000 PRO Plate Reader, with an excitation wavelength of 530 nm and emission wavelength of 565 nm (bandwidth 10 nm).
Transport assay using radiolabelled amino acids
CHO‐BC cells with passage numbers from 3 to 10 were used in radio‐labelled uptake assays. Before the assay, the cells were seeded out in 35 mm dishes (Corning) and maintained for 48–72 h until reaching 80–90% confluence. To initiate transport, the culture medium was removed and the cells were washed three times with HBSS + G. Subsequently, cells were incubated with HBSS + G, containing radiolabelled substrates (150 μM L‐[U‐14C]leucine or 150 μM L‐[U‐14C]isoleucine) and inhibitors as indicated in the figures or table, in a 37°C water bath for 6 min. To terminate transport, cells were washed three times with ice‐cold HBSS. For measuring sodium‐independent uptake, NaCl in HBSS + G was replaced by N‐methyl‐d‐glucamine (NMDG)‐Cl and Na+‐salts were replaced by K+‐salts. Cells were then harvested by homogenising with 500 μL of 0.1 M HCl. An aliquot of 400 μL was used for scintillation counting and the remainder for protein quantification. To investigate inhibition of ASCT2, uptake of 100 μM [14C]glutamine was measured in 143B cells in the absence and presence of inhibitor (at 300 μM). To measure inhibition of LAT1 in MCF‐7 cells, uptake of 100 μM [14C]leucine was measured in HBSS + G in which NaCl was replaced by NMDG‐Cl and Na+‐salts were replaced by K+‐salts. Inhibitors were added to yield a final concentration of 300 μM.
Animals
All animal care and experimental procedures complied with the National Health and Medical Research Council's (NHMRC) Australian Code for the Care and Use of Animals for Scientific Purposes and the ACT Animal Welfare Act 1992, and were approved by the Animal Experimentation ethics committee of the Australian National University (A2014/20 for the use of X. laevis and A2013/39 for the use of mice). The number of animals used was adjusted to the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Male and female C57/Bl6 mice (Animal services, Australian National University) were used at an age of 4–6 months. Mice were held in individually ventilated cages (<5 animals per cage) under specified pathogen‐free conditions under a 12 h light/12 h dark cycle and checked for general health once daily. Mice were killed by cervical dislocation before removal of organs. X. laevis were housed in tanks holding less than 20 animals per tank. The tank water was percolated and filtered in a closed system. About 10% of the total volume was replaced every day. Frogs were anesthetized by submersion in water containing MS‐222 (1.5 g l−1) until loss of reflexes. The surgical procedure and preparation of oocytes has been described in detail before (Broer, 2010).
Transport assay using sections of inverted intestine
For uptake assays with inverted sections of mouse small intestine, C57/BL6J female and male mice (4–6 months old) were killed by cervical dislocation (Animal ethics protocol A2013/39, Australian National University). The small intestine was removed and the intestinal lumen was rinsed with ice‐cold 0.9% NaCl supplemented with Roche complete protease inhibitor. Subsequently, the small intestine was inverted on a metal rod to expose the mucosa. The inverted intestine was then cut into 1 cm pieces and fitted onto enzyme spatulas, which were immersed in HBSS + G supplemented with 1 mM glutamine. After washing twice in modified HBSS + G (containing 10% NaCl + 90% NMDG‐Cl for Na+‐dependent uptake and 100% NMDG‐Cl for Na+‐independent uptake, both buffers supplemented with 1 mM glutamine) to remove residual compounds, the segments were pre‐incubated in the same buffer in the presence of 300 μM of transport inhibitor and protease inhibitors (Roche Complete) for 15 min. At the end of the pre‐incubation, the segments were transferred for 30 s into uptake buffer at 37°C (HBSS + G supplemented with 150 μM [14C]leucine and 300 μM transport inhibitor). For Na+‐dependent uptake HBSS + G was used, for Na+‐independent uptake NaCl was replaced by NMDG‐Cl. Subsequently, the segments were rinsed three times in ice‐cold HBSS pH 7.5 and lysed in 400 μL 10% SDS. After 2–3 h, the lysed tissues were analysed by scintillation counting.
Transport assay using X. laevis oocytes
Preparation of oocytes and transport experiments were performed as described in detail before (Broer, 2003). The following cRNA constructs were injected: 10 ng hB0AT1 cRNA per oocyte, 2 ng hcollectrin cRNA per oocyte. Transport experiments were conducted on oocytes 4–5 days after cRNA injection. Inhibitors were pre‐incubated for 30 min at the indicated concentration in ND96 (composition: 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, titrated with NaOH to pH 7.4). Uptake of [14C]leucine was measured at a total concentration of 100 μM for 30 min. The activity of non‐injected oocytes was subtracted before data analysis.
Docking protocol
AutoDock Tools (Morris et al., 2009) incorporating the Gasteiger PEOE method (Gasteiger and Marsili, 1980) was used to assign partial charges to all atoms in the DAT‐based B0AT1 structure and to all test‐ligand structures. As AutoDock does not explicitly support the Na+ atom type, parameters were approximated to Ca2+. Charge for Na+ was manually set to +1. Using AutoGrid, the docking search space was set to the region in between and inclusive of the Na+/substrate binding site 1 and the Arg57/Asp486 gate at the top of the binding pocket. All docking runs were carried out using AutoDock 4.2 compiled on the AVOCA IBM Blue Gene/Q supercomputer cluster (Norgan et al., 2011) maintained by VLSCI.
We chose the ZINC database (Version 12, (Irwin and Shoichet, 2005)) for virtual screening, which contains >35 million compounds. Due to computational restraints we selected a subset of 14 434 compounds based on the following criteria: (i) All available iterations of the National Cancer Institute diversity set, a catalogue of compounds designed to be representative of the chemical diversity of all known drug‐like molecules. (ii) All compounds known to bind to or significantly inhibit any member of the SLC6 family, and their structural analogues. (iii) The published B0AT1 inhibitor, nimesulide (Pochini et al., 2014), and its analogues. Conformations of each of the 14 434 docked compounds were ranked into clusters by AutoDock using a 2 Å root mean square deviation difference. The ratio of the mean docking score of the most abundant cluster of docking poses for each compound, to its molar mass, was used to determine the highest scoring 1000 compounds. To reduce compound numbers further, clusters of similar compounds were identified based on 2D Tanimoto coefficients (Rogers and Tanimoto, 1960), available through PubChem. The 40 highest scoring representatives of each cluster were requested from the Developmental Therapeutics Program of the National Cancer Institute, or when unavailable, compounds with an at least 85% similar structure (based on 2D Tanimoto coefficients), and better docking score were requested instead. In a second selection round, a set of 158 structural analogues of two compounds (NSC63912 and 186 059) showing the greatest inhibition in in vitro experiments were analysed by AutoDock as above resulting in 40 additional compound candidates.
Data and statistical analysis
Data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). A significant difference was assumed when random probability of likelihood was P < 0.05. Samples were randomised in 96 well plates, but not in other assays. Sample identity was known to the investigator (not blinded), because all results had fixed numerical values that are not open to interpretation. Normalized data, where the control did not have a variance, were analysed using non‐parametric tests. Data are shown as mean ± SD, transport activities indicated as Na+‐dependent net uptake, were calculated by subtracting the uptake activity in the absence of Na+ from the total uptake rate. The fluorescent signals measured in the FLIPR assay were normalized to baseline readings before the addition of substrates. Data are presented as percentage of baseline at each time point. The level of transporter overexpression declines systematically with increasing cell passage number. As a result, data were normalized against the base transport activity of the respective cell passage represented by three internal standards on each 96 well plate. Cells were used between passages 3–10. The number of experimental repeats (n) is indicated in the figure legends. Data analysis for each figure:
-
Fig. 1
ANOVA between Na+‐dependent uptake in parental cells (A) and Na+‐dependent uptake in CHO‐BC cells (B). F achieved P < 0.05 and there was no significant variance inhomogeneity.
-
Fig. 2
IC50 was calculated using the equation: .
-
Fig. 3
Computational data without variance.
-
Fig. 4
IC50 calculated as in Figure 2, Eadie‐Hofstee transformation of transport data in panel C and D.
-
Fig. 5
Panel A, non‐parametric test to normalized controls (not significant). Panel B,C: ANOVA of normalized data. Panel D: ANOVA of normalized data. F achieved P < 0.05 and there was no significant variance inhomogeneity.
Figure 2.
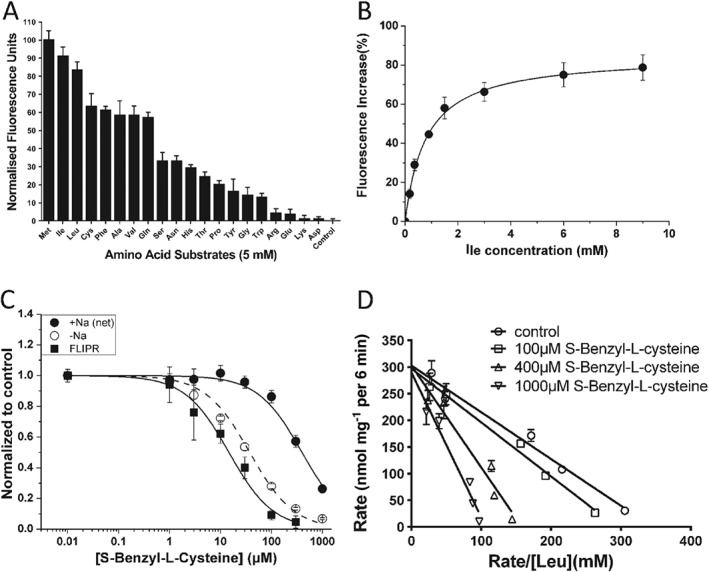
Characterization of substrate transport and substrate analogues. (A) The substrate specificity of B0AT1 expressed in CHO‐BC cells was determined using a variety of amino acids (5 mM) in the FLIPR assay. The transporter prefers large neutral amino acids and does not transport charged amino acids (n = 5). Data were normalized to the strongest signal elicited by methionine. (B) Isoleucine concentrations as indicated were used to determine its KM using the FLIPR assay (n = 5). (C) S‐Benzyl‐L‐cysteine inhibited [14C]leucine uptake (150 μM) via B0AT1 and the endogenous isoleucine transporter (n = 5). The data were compared to the action of S‐Benzyl‐L‐cysteine in the FLIPR assay. (D) The concentration dependence of [14C]leucine uptake was measured at increasing inhibitor concentrations to determine the mode of inhibition. S‐Benzyl‐L‐cysteine acts as a competitive inhibitor of leucine transport via B0AT1 (n = 5), altering the KM but not VMax. The letter ‘n’ refers to the number of independent experimental repeats for each experimental group.
Materials
Compounds were purchased from Sigma‐Aldrich, if available. Other compounds were requested from the Developmental Therapeutics Program of the National Cancer Institute. Purity and identity were confirmed by liquid chromatography and mass spectroscopy (Supporting Information Data S1 and S2). Other materials were sourced as listed in individual methods. Perkin Elmer (Waltham, MA) supplied the L[U‐14C]leucine (11.1 GBq mmol‐1), L[U‐14C]isoleucine (11.1 GBq mmol‐1)and L‐[U‐14C]glutamine (10Gbq mmol‐1).
Results
To identify novel inhibitors of B0AT1, we generated a cell line stably expressing B0AT1 and collectrin. As a host cell line, we selected CHO cells because endogenous uptake of isoleucine was entirely Na+‐independent (Figure 1A), that is, uptake activity was identical when measured in buffer containing Na+‐ions or in buffer where Na+‐ions were replaced by NMDG, a large non‐permeable cation. Uptake of [14C]isoleucine (unless mentioned otherwise all amino acids used in this study were L‐isomers) was suppressed by addition of an excess of unlabelled neutral amino acids (10 mM) such as phenylalanine and serine, but not by the cationic amino acid arginine. This is consistent with the presence of an amino acid transport system for large neutral amino acids (System L), which has been reported before (Oxender et al., 1977). Isoleucine is also one of the preferred substrates of the Na+‐dependent transporter B0AT1. To determine expression levels of B0AT1 in the recombinant cell line CHO‐B0AT1‐collectrin (CHO‐BC), we compared uptake of [14C]isoleucine in Hanks balanced salt solution (HBSS + G,) and in HBSS + G in which Na+ was replaced by NMDG+. To compare B0AT1 activity with the endogenous transport, we calculated the net Na+‐dependent uptake (activity in the presence of Na+ minus activity in absence of Na+, i.e., B0AT1 activity). This activity is undetectable in parental cells (Figure 1A), but was similar in capacity to the endogenous Na+‐independent transporter (System L) in CHO‐BC cells (Figure 1B). We used a 6 min time point for our transport studies, where net Na+‐dependent uptake is still proportional to time and most prominent compared to other activities (Figure 1C). The endogenous Na+‐independent System L activity is known to be electroneutral (Wagner et al., 2000), while the heterologously expressed B0AT1 is electrogenic. An assay based on a voltage‐sensitive dye (FLIPR Blue dye) would react to transport via B0AT1, but not to uptake via the endogenous transporter. Consistently, exposure of CHO‐BC cells to isoleucine generated a strong fluorescent signal, which was not observed in the parental cells (Figure 1D). Leucine generated a similar signal, but the background signal was slightly stronger (Figure 1E). The fluorescence signal was abolished when NaCl was replaced by NMDG‐Cl (Figure 1F), consistent with Na+‐dependence of amino acid uptake via B0AT1. Expression of B0AT1 and collectrin at the plasma membrane was confirmed by Western blotting after cell surface protein biotinylation (Figure 1G). Both proteins were absent in parental cells.
Figure 1.
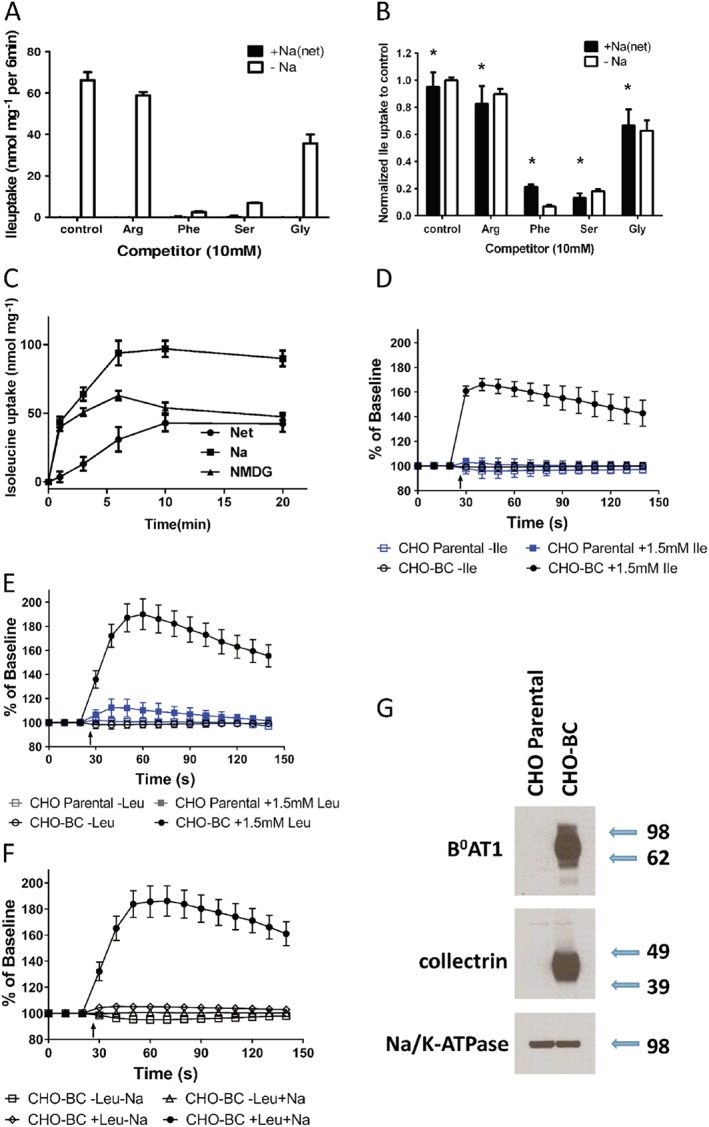
Cell line and assay for high throughput screening and inhibitor characterization. (A) Isoleucine transport in CHO parental cells was entirely Na+‐independent. Uptake of [14C]isoleucine (100 μM) was inhibited by 10 mM phenylalanine and serine, but not by arginine. Glycine was a partial inhibitor. Transport activity is shown for the Na+‐independent component of total uptake (−Na), and the net Na+‐dependent component of total uptake (+Na(net)) (n = 5). (B) CHO parental cells stably transfected with collectrin and B0AT1 (CHO‐BC) showed robust net Na+‐dependent isoleucine transport (+Na(net)), which was undetectable in the parental cells. Na+‐dependent uptake of [14C]isoleucine (100 μM) was inhibited by 10 mM phenylalanine and serine, but not by arginine. Glycine was a partial inhibitor, consistent with uptake being mediated by B0AT1 (n = 5). Please note the similar substrate specificity of the endogenous Na+‐independent transporter. (C) Uptake of [14C]isoleucine (100 μM) was measured in the presence and absence of Na+‐ions in CHO‐BC cells. Net Na+‐dependent uptake of isoleucine increased proportional to time over 10 min. Na+‐independent uptake was rapid initially, but equilibrated quickly (n = 5). (D) A membrane potential sensitive dye was used to detect Na+‐amino acid cotransport by B0AT1. In parental CHO cells, addition of 1.5 mM isoleucine (arrow) did not change the fluorescence (n = 5), while a large increase of fluorescence (depolarisation) was observed in CHO‐BC cells (n = 10). (E) Addition of 1.5 mM leucine generated a small signal in parental cells (n = 5) and a large signal CHO‐BC cells (n = 35). (F) Cell depolarisation was not only amino acid dependent, but also Na+‐dependent (n = 5). (G) Surface biotinylation was used to isolate membrane proteins followed by SDS‐PAGE and immunoblotting to detect collectrin and B0AT1. Both proteins were only detectable in CHO‐BC cells, Na,K‐ATPase served as a loading control (n = 5). Molecular weight in kDa is indicated next to the blot. *P < 0.05, significantly different from parental cells (A); n refers to the number of independent experimental repeats for each experimental group.
To confirm that the additional isoleucine uptake activity in CHO‐BC cells was indeed mediated by B0AT1, a basic characterization was performed. The substrate specificity was measured using the FLIPR assay, which measures depolarisation similar to electrophysiological recordings. Consistently, the order of substrate preference was virtually identical to that reported previously using the latter method (Figure 2A) (Bohmer et al., 2005; Camargo et al., 2005). The transporter showed a preference for methionine (normalized to 100) and branched‐chain amino acids over polar neutral amino acids. Anionic and cationic amino acids were not transported. The KM‐values for leucine and isoleucine were 0.5 ± 0.1 mM (data not shown) and 0.8 ± 0.1 mM (Figure 2B, Table 1) respectively. These values are also consistent with published data (Broer et al., 2004; Bohmer et al., 2005; Camargo et al., 2005).
Table 1.
Kinetic constants of substrates and inhibitors of B0AT1
| FLIPR assay | Radioactive uptake | |||||
|---|---|---|---|---|---|---|
| Compound | NSC | Purity (%) | KM | IC50 | KM | IC50 |
| Leucine | >98 | 500 ± 100 | 1050 ± 50 | |||
| Isoleucine | >98 | 700 ± 60 | 1400 ± 100 | |||
| S‐Benzyl‐L‐cysteine | 97 | 14 ± 2 | 398 ± 29 | |||
| O‐Benzyl‐L‐serine | >99 | 14 ± 5 | 810 ± 176 | |||
| S‐(4‐Tolyl)‐L‐cysteine | >95 | 23 ± 14 | 205 ± 46 | |||
| S‐Phenyl‐L‐cysteine | >97 | 49 ± 23 | ||||
| Nimesulide | >98 | 172 ± 26* | ||||
| Benztropine | 63 912 | >98 | 44 ± 9 | 71 ± 8 | ||
| 2‐Benzyl‐1‐(3‐phenylpropyl)piperidine | 22 789 | >98 | 90 ± 21 | 78 ± 16 | ||
| 1‐(1,2‐diphenylethyl)‐3‐methylpiperidine | 34 358 | >95 | 5 ± 1 | 17 ± 3* | ||
| 11‐dihydro‐5H‐dibenzo[a,d]cyclohepten‐5‐ylidene)methyl]‐1‐methyl‐pyrrolidine | 169 092 | >99 | 10 ± 9 | |||
| 1‐(4,4‐diphenylbut‐3‐enyl)piperidine | 39 706 | >99 | 13 ± 3 | 70 ± 21 | ||
| 4‐(dibenzo[1,2‐a:1′,2′‐e][7] annulen‐11‐ylidene)‐1‐methylpiperidine | 169 911 | >99 | 15 ± 3 | 67 ± 16* | ||
| 3‐[(2‐aminophenyl)sulfanyl]‐ 1‐[4‐(4‐chlorophenoxy) phenyl]pyrrolidine‐2,5‐dione | 201 503 | >99 | 17 ± 10 | |||
| N1,n1‐dimethyl‐4‐[[4‐(dimethylamino)phenyl](4‐nitrophenyl)methyl]aniline | 3323 | 94 | 33 ± 13 | |||
| 2‐chloro‐N‐(1,1,3,3,5‐ pentamethyl‐6‐(1‐pyrrolidinyl) ‐2,3‐dihydro‐1H‐inden‐4‐yl)benzamide | 321 496 | 92 | 37 ± 23 | 55 ± 22* | ||
Concentrations are given in μM. All compounds were preincubated with the cells for >30 min in the FLIPR assay. Some inhibitors required preincubation for 4 h to be effective (indicated by *). In the FLIPR assay, 1.5 mM leucine was used as substrate (n = 3); in the radioactive uptake assay, 150 μM isoleucine was used as substrate (n = 3). The chemical identification number (NSC) is shown for compound identification. Compound purity as provided by supplier or determined by liquid chromatography (see Supporting Information Data S1 and S2)
In order to identify novel inhibitors, we initially chose substrate analogues that were commercially available. These experiments revealed compounds such as S‐benzyl‐L‐cysteine, O‐benzyl‐L‐serine, S‐phenyl‐L‐cysteine and S‐(4‐tolyl)‐L‐cysteine as potential inhibitors of B0AT1 (Table 1). All compounds were analysed by FLIPR assay and selected candidates were further analysed by radiolabelled uptake assays. The radioactive uptake assay provides a suitable platform for an initial evaluation of inhibitor selectivity, as the endogenous Na+‐independent uptake has a substrate specificity similar to that of B0AT1 (Figure 1A,B). S‐benzyl‐L‐cysteine was the best performing substrate analogue inhibiting transport of leucine (1.5 mM) with an IC50 of 14 ± 2 μM as determined in the FLIPR assay (Table 1, Figure 2C). A much higher IC50 of 424 ± 38 μM was determined when inhibiting uptake of [14C]isoleucine (0.15 mM) (Figure 2C). The higher value is consistent with uptake experiments in oocytes, which yielded an IC50 of 254 ± 70 μM (inhibiting uptake of 0.1 mM leucine, n = 5). S‐benzyl‐L‐cysteine was an even better inhibitor of the endogenous Na+‐independent transporter (IC50 of 33 ± 3 μM, Figure 2C). As expected for a substrate analogue, kinetic analysis revealed a competitive mode of inhibition (Figure 2D), that is, the inhibitor increased the KM, but VMax remained constant. Moreover, S‐benzyl‐L‐cysteine was a substrate of the transporter as indicated by a strong depolarisation signal in the FLIPR assay (data not shown). These results suggested that simple substrate analogues can be used as inhibitors, but are unlikely to be specific.
We have previously reported that LeuT can serve as a highly suitable template for the generation of a homology model of B0AT1 (O'Mara et al., 2006; Fairweather et al., 2015). The homology model is likely to be reliable in the regions around the binding site in the core of the transporter, which show high sequence similarity, while loops are less conserved (Broer and Gether, 2012). Because of the higher sequence similarity between dDAT and B0AT1 (31% identical residues, 49% similar), we decided to use a homology model based on the Drosophila protein for computational docking studies to identify novel inhibitors of B0AT1 with better selectivity (Penmatsa et al., 2013). A set of ~14 000 ligands (selected as outlined in Methods) was docked to the dDAT‐based homology model of B0AT1 using AutoDock4 (Morris et al., 2009). The trend in docking scores for these compounds is summarized in Figure 3A, which shows a histogram of the number of ligands within 1 kcal·mol−1 bins. As AutoDock reports free energy values, a more negative score represents stronger binding. Over 99% of compounds had binding energies ranging from −3 to −12 kcal·mol−1, with the bulk of all compounds lying between −7 and −8 kcal·mol−1. We highlight compounds NSC63912 and NSC22789, which are analysed in more detail below, with the transporter substrate leucine shown for comparison.
Figure 3.
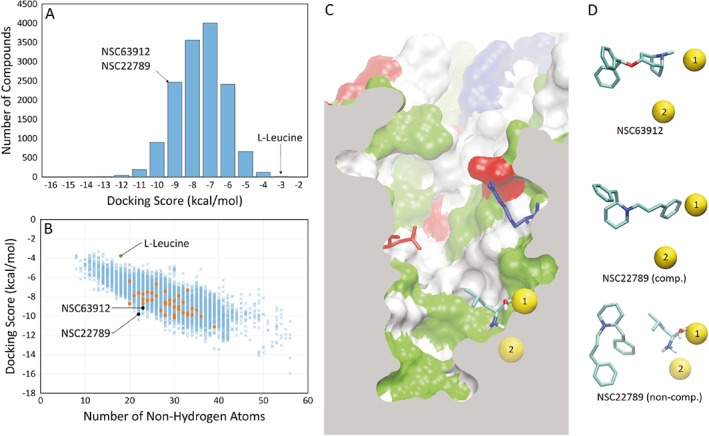
Trends in molecular docking scores and poses of specific B0AT1 inhibitors (A). Histogram of the docking score of all compounds screened, sorted into 1 kcal·mol−1 bins. (B) Docking score of all compounds screened compared to their molecular weight as approximated by the number of non‐hydrogen atoms present in the compound. The first batch of 40 compounds was chosen for functional testing (orange points). Substrate L‐leucine and two identified inhibitors (NSC63912 and NSC22789) are highlighted. (C) Cutaway of B0AT1 homology model showing a side profile of the substrate binding site. Highlighted are sodium ions 1 (accessible to substrate) and 2 (inaccessible to substrate) in their binding sites (yellow spheres), and the Arg57 and Asp486 residues that form the extracellular gate to the binding site. The protein surface is represented, where white represents non‐polar amino acids, green represents polar amino acids, red represents acidic amino acids and blue represents basic amino acids. (D) The most favourable docked pose of NSC63912 and NSC22789, respectively, alongside the sodium ions for reference. A second major pose for NSC22789 highlights a non‐competitive binding location, showing leucine at the substrate binding site for reference.
The docking score tended to increase with the molecular weight of the ligand as approximated by the number of non‐hydrogen atoms (Figure 3B). In Figure 3B, we highlight in orange the first batch of 40 compounds selected for functional testing. These represent compounds with the highest docking score that were readily available from the Developmental Therapeutics Program – National Cancer Institute. Following in vitro, tests the two best performing inhibitors were selected to identify structurally similar compounds, which were also analysed in vitro as shown in detail below. NSC63912 and NSC22789 represent a good compromise between molecule size and binding score, and are commercially available.
Figure 3C shows the B0AT1 homology model, detailing the binding site, along with the docked pose of L‐leucine close to the Na+‐binding site 1 (yellow). Na+‐binding site 2 (yellow) was not accessible by the substrate. Highlighted in blue and red are the gate‐forming residues Arg57 and Asp486 respectively. Figure 3D shows docking poses of NSC63912 and NSC22789 relative to the two Na+‐binding sites. NSC63912 is shown docked to the substrate binding site, a pose that AutoDock predicts in 88% of all poses. NSC22789 is also shown docked to the same site, but this is predicted to be only 50% of all poses. A majority of the other poses (48%) follow an alternative binding location, shown alongside leucine, in the substrate binding site in the lower panel of Figure 3D.
The most promising compounds are listed in Table 1. Pharmacological studies have consistently shown that most drugs targeting monoamine transporters act as competitive inhibitors (Broer and Gether, 2012). However, the presence of a second substrate binding site has been proposed (Zhao et al., 2011) and was subsequently confirmed experimentally (Coleman et al., 2016). For further characterization, we selected benztropine (NSC63912) and 2‐benzyl‐1‐(3‐phenylpropyl)piperidine (NSC22789), which showed IC50‐values of 44 ± 9 μM and 90 ± 21 μM respectively (FLIPR assay). In both cases, radiolabelled flux assays revealed similar IC50 values of 71 ± 8 and 78 ± 16 μM (Figure 4A,B). More importantly, both compounds showed good selectivity against the endogenous Na+‐independent transporter (Figure 4A,B, open circles). We used our assay to compare these novel inhibitors to nimesulide. Surprisingly, nimesulide only inhibited B0AT1 after preincubation for 4 h with an IC50 of 178 ± 40 μM (Table 1). Thus, our novel compounds inhibit B0AT1 with higher potency and instantaneously. Loratadine, which has previously been reported as an inhibitor of the related transporter B0AT2, did not inhibit transport via B0AT1 (data not shown). Further characterization revealed that NSC63912 acted as a competitive inhibitor, while NSC22789 was non‐competitive (Figure 4C,D). Notably, NSC63912 (Figure 4E) showed a strong preference for the single docking pose depicted in Figure 3D. NSC22789 (Figure 4F), by contrast, docked to two different areas of the binding pocket (Figure 3D), only one of which overlapped with that of leucine. This may explain the non‐competitive type of inhibition observed by this compound. Although X. laevis oocytes are widely used to characterize transport proteins, hydrophobic compounds often show much higher IC50‐values for heterologous targets in oocytes compared to heterologous expression in cells (Kiehn et al., 1996). We made similar observations with inhibitors NSC22789 and NSC63912, which caused partial inhibition of B0AT1 at 300 μM where inhibition is complete in CHO‐BC cells and in addition required preincubation with the inhibitor for at least 30 min (Figure 5A).
Figure 4.
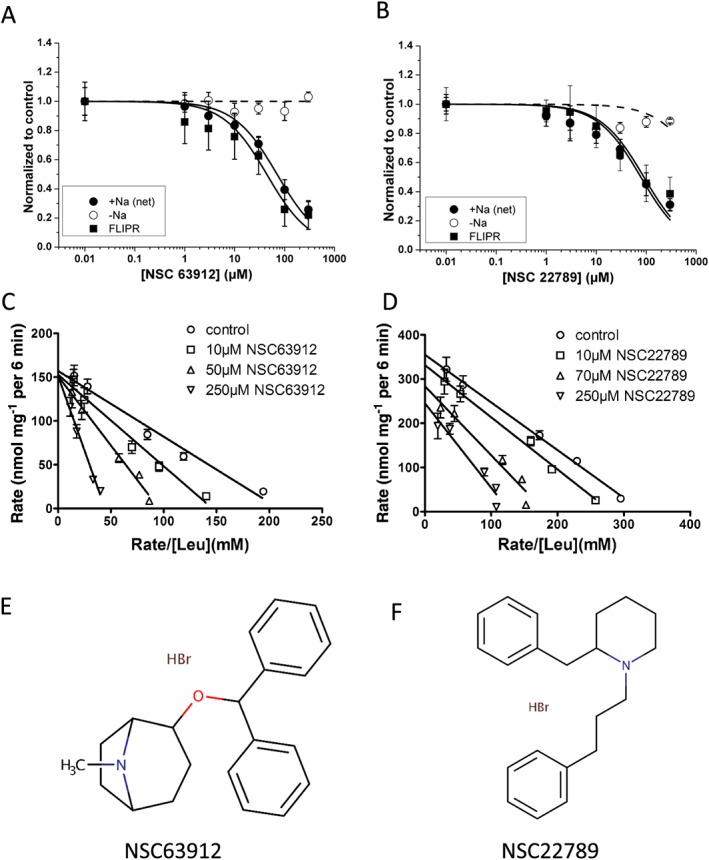
Characterization of specific B0AT1 inhibitors. (A, B) Radioactive flux experiments showed that NSC63912 (A) and NSC22789 (B) inhibited Na+‐dependent leucine transport in CHO‐BC cells, but did not block the endogenous Na+‐independent transporter (n = 5). Inhibition of leucine transport in the FLIPR assay is shown for comparison (closed squares). (C) NSC63912 acts as a competitive inhibitor by affecting the KM, but not the Vmax of the transporter as shown in an Eadie‐Hofstee plot (n = 5). (D) NSC22789, by contrast, selectively reduced the Vmax without affecting the KM and therefore qualifies as a non‐competitive inhibitor (n = 5). (E,F) Chemical structures of NSC63912 and NSC22789. The letter ‘n’ refers to the number of independent experimental repeats for each experimental group.
Figure 5.
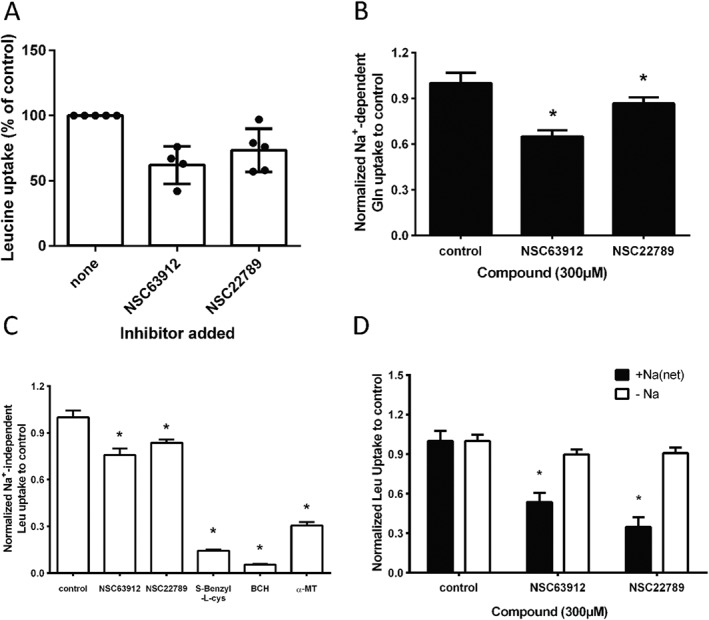
Specificity of B0AT1 inhibitors. (A) NSC63912 and NSC22789 at a concentration of 300 μM, were used to test for inhibition of leucine transport in X. laevis oocytes expressing B0AT1 and collectrin. The same inhibitors (300 μM) were tested in 143B osteosarcoma cells (B), where Na+‐dependent glutamine transport is largely mediated by ASCT2 (n = 5), and in MCF‐7 cells (C) where Na+‐independent transport of leucine is largely mediated by LAT1 (n = 5). Moderate inhibition was observed in both cell lines. Substrate analogues such as S‐benzyl‐L‐cysteine and BCH, by contrast, fully blocked leucine transport in MCF‐7 cells. (D) Both compounds also inhibited Na+‐dependent leucine transport in inverted sections of mouse intestine an in situ preparation with significant B0AT1 activity (n = 5). * P < 0.05, significantly different from control; n refers to the number of independent experimental repeats for each experimental group.
While the endogenous system L activity served as an initial test for inhibitor selectivity, we used two additional cell lines to more thoroughly investigate inhibitor selectivity. To measure inhibition of ASCT2, 143B osteosarcoma cells were used. To this end, glutamine uptake was measured in the presence and absence of Na+, and compared to the same transport activities in the presence of inhibitor. In Figure 5B the inhibition of net Na+‐dependent glutamine transport is shown, which is largely mediated by ASCT2 (Broer et al., 2016). Leucine transport in MCF‐7 cells, by contrast, is largely mediated by system L (molecular isoforms LAT1 or LAT2), which can be inhibited by the amino acid analogue 2‐aminobicyclo[2.2.1]heptane‐2‐carboxylic acid (BCH, Figure 5C) and α‐methyl‐tyrosine. The latter inhibitor affects LAT2 less than LAT1 (Khunweeraphong et al., 2012), demonstrating that leucine uptake is indeed largely mediated by LAT1. Testing the two selected compounds revealed that compound NSC63912 partially inhibited ASCT2 and LAT1 activity, while NSC22789 showed better selectivity in both cell lines with little inhibition of either ASCT2 or LAT1 (Figure 5B/C). Substrate analogues, such as S‐benzyl‐L‐cysteine again showed lack of selectivity. To confirm that our cell‐based in vitro test systems were indeed reliable indicators of B0AT1 function, we tested NSC63912 and NSC22789 in transport assays using inverted sections of mouse intestine. We have previously demonstrated B0AT1 activity in this preparation (Broer et al., 2011). As shown in Figure 5D both compounds significantly inhibited Na+‐dependent leucine uptake in the intestine, which is mediated by B0AT1.
Discussion
In agreement with earlier findings (Danilczyk et al., 2006; Malakauskas et al., 2007; Kowalczuk et al., 2008; Camargo et al., 2009), stable expression of B0AT1 requires co‐expression of collectrin or ACE2. The catalytic domain of ACE2 is irrelevant for B0AT1 expression (Fairweather et al., 2015), we therefore chose collectrin due to its smaller size. CHO cells do not express detectable amounts of ACE2 (Iwata et al., 2009) to replace collectrin. Heteromeric design of transporters is a common occurrence (Ballatori et al., 2013; Fotiadis et al., 2013; Halestrap, 2013), which needs to be taken into consideration when designing cell lines for HTS. Incidentally, the substrate specificity of B0AT1 is very similar to that of the endogenous transporter, which is most likely the hamster isoform of LAT2. As a result, it is very difficult to suppress the endogenous transporter using compounds like BCH, which also inhibits B0AT1 (Broer et al., 2004). However, more recently developed inhibitors of LAT1 may prove to be more selective (Khunweeraphong et al., 2012). In this study, however, the endogenous transporter provided a useful first screen for the selectivity of any compound. The endogenous activity could be differentiated from transport via B0AT1 as it is not dependent on the presence of Na+.
Membrane potential sensitive dyes have been used to screen for ion channel inhibitors (Whiteaker et al., 2001; Wolff et al., 2003; Joesch et al., 2008) and have more recently also been used to screen for transporter inhibitors (Benjamin et al., 2005; Ruggiero et al., 2012). Here, we established the FLIPR system as a suitable assay to screen large compound libraries and to characterize amino acid transport inhibitors. Alternatively, FRET sensors can be used to detect amino acid transport (Whitfield et al., 2015; Vanoaica et al., 2016). However, these monitor amino acid accumulation through any transport system and therefore require secondary screening to ensure targeting is correct. For some transporters, fluorescent substrates are available that can be used for screening purposes (Landowski et al., 2003). As expected, the FLIPR assay was insensitive to any electroneutral move of amino acids across the membrane and hence selective for the heterologously expressed B0AT1. It is important to choose the appropriate substrate as shown by the better signal produced by isoleucine as compared to leucine. Leucine can, to some extent, be transported by electrogenic transporters such as system A, while isoleucine is more selective due to its earlier carbon chain branch (Shotwell et al., 1981). We also noted that the FLIPR assay usually yielded lower IC50‐values than radioactive uptake measurements. Due to technical reasons the inhibitor has to be incubated with the dye for 30 min on the cells. This can cause non‐specific effects such as depolarisation due to the activation or inhibition of ion channels or quenching of fluorescence. It can also lead to lower IC50‐values when significant diffusion barriers are observed in the transporter vestibule or if the inhibitor needs to cross the lipid bilayer before binding. It is therefore important to complement the FLIPR assay with a robust assay that is less prone to false positives, such as radiolabelled uptake assays.
Due to significant progress in computational power and high‐resolution protein structures virtual screening has become a widely used tool to identify novel inhibitors (Moitessier et al., 2008). Computational screening has a number of shortcomings, such as exclusion of protein flexibility, incomplete accounting of water molecules in binding and incomplete accounting of specialized interactions (Moitessier et al., 2008). However, many programs successfully select strong binders from a series of decoys (Warren et al., 2006). As a result, computational screening is particularly powerful when used in combination with medium and high through‐put in vitro assays to identify false positives. Loratadine, for instance, inhibits B0AT2 (Cuboni et al., 2014), but was inactive as an inhibitor of B0AT1. However, it docks as well to B0AT1 as expected. It should be noted that benztropine, identified here as an inhibitor of B0AT1, has also been reported as an inhibitor of the proline transporter PROT (SLC6A7) and the dopamine transporter (SLC6A3) (Yu et al., 2009). However, these transporters are unlikely to be coexpressed with B0AT1 in biological preparations, due to highly tissue‐specific promoters (Tumer et al., 2013).
We regularly use X. laevis oocytes as a system to characterize transporter activity and function. To our surprise, the inhibitors described here were much less effective when B0AT1/collectrin was expressed in oocytes or required considerable preincubation. The differences appear to be more prominent when hydrophobic compounds are considered, while substrate specificity and substrate affinity of natural amino acids was identical to previous studies in oocytes (Broer et al., 2004; Bohmer et al., 2005). Notably, substrate specificity of the neutral amino acid transporter LAT1 is different in cell lines as compared to oocytes (Khunweeraphong et al., 2012). Two possible explanations come to mind. First, the vitellin layer surrounding oocytes could form a barrier for hydrophobic compounds, which has also been observed for HERG channel inhibitors (Kiehn et al., 1996). Second, some compounds may inhibit the transporter from the inside and are captured by the abundant egg‐yolk inside the oocyte. An example of the latter is the monocarboxylate transport inhibitor AR‐C155858 (Ovens et al., 2010).
A binding site for tricyclic antidepressants has already been found in the prokaryotic LeuT (Singh et al., 2007). In agreement with an evolutionary conserved binding site for antidepressants, we found a significant number of compounds that resembled tricyclic antidepressants (e.g. NSC22789 and NSC63912). The vast majority of compounds identified in the virtual screen contain one or more aromatic rings attached to a polar functional group, such as alcohol groups, nitro groups or sulfones. Several compounds are also entirely nonpolar, with no atoms capable of involvement in hydrogen bonding. Compounds with polar or charged functional groups tended to orient these functional groups towards the most polar surfaces in the protein, that is, the Na+ binding site 1 or the aspartate and arginine residues that restrict the vestibule before the substrate binding site. Hydrophobic parts of the ligands tended to confine themselves to the hydrophobic pocket (white surface in Figure 3) located between the Na+/substrate binding site and the aspartate and arginine residues in the vestibule. In the LeuT structure, the antidepressant binding site was located outside aspartate and arginine residues in the vestibule. In our docking simulations, none of the compounds tested bound at this site, most likely because we used the outside‐open structure. The vestibule in the outside‐open conformation is wider allowing less contacts. This second binding site may only become available after binding of leucine (Rudnick, 2007). In general, binding of tricyclic antidepressants to human monoamine transporters appears to be competitive, consistent with the positioning of these compounds in the structures of a bacterial‐human hybrid transporter (Wang et al., 2013), the Drosophila (Penmatsa et al., 2013) and human dopamine transporter (Coleman et al., 2016). However, the second binding site has also been observed in the human dopamine transporter structure, when the first binding site is occupied (Coleman et al., 2016).
Previous docking studies using homology models have largely identified substrate homologues as potential inhibitors (Geier et al., 2013; Colas et al., 2015). Here, we demonstrate that computational screening using openly accessible docking programs in combination with suitable transport assays can identify novel inhibitors that show good affinity and selectivity for B0AT1. The compounds identified here form the base of a more comprehensive structure–activity approach to identify high‐affinity compounds for this transporter. In combination with molecular dynamics studies, this should lead to significantly improved compounds that also have improved selectivity. Pharmacological inhibition of B0AT1 should replicate the metabolic phenotype of B0AT1 deficient mice, which includes greater glycaemic control, low levels of triglycerides and cholesterol and resistance to diet induced obesity (Jiang et al., 2015). If these results can be replicated in humans, inhibitors of B0AT1 have the potential to be used as drugs to treat metabolic disorders.
Author contributions
Q. C. and N. S. contributed equally to this manuscript, carried out the majority of experiments and docking studies, analysed data, prepared data for publication and drafted parts of the manuscript. A. B., S. F., Y. J. and D. S. supported the study with specific expertise. B. C. and S. B. designed the study and planned experiments together with all other authors. S. B. compiled and wrote the manuscript, which was reviewed and edited by all authors.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Data S1 Certificate of analysis provided by Sigma‐Aldrich, the commercial supplier of benztropine used in this study (NSC63912) showing purity as determined by HPLC.
Data S2 Identity and purity of compounds used in this study provided by DTP‐NCI, as determined by electrospray ionisation mass spectrometry and liquid chromatography in full scan mode at 214 nm absorption wavelength, using 500 nm as a reference. See table 1 for purity values and chemical names (NSC number shown).
Acknowledgements
The work reported here was supported by a sponsored research contract with Sanofi (Germany). Computational studies were supported by computer time on the Peak Computing Facility at the Victorian Life Sciences Computation Initiative (VLSCI) via a grant through the National Merit Allocation Scheme of the National Computational Infrastructure.
Cheng, Q. , Shah, N. , Bröer, A. , Fairweather, S. , Jiang, Y. , Schmoll, D. , Corry, B. , and Bröer, S. (2017) Identification of novel inhibitors of the amino acid transporter B0AT1 (SLC6A19), a potential target to induce protein restriction and to treat type 2 diabetes. British Journal of Pharmacology, 174: 468–482. doi: 10.1111/bph.13711.
References
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015). The concise guide to PHARMACOLOGY 2015/16:Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatori N, Christian WV, Wheeler SG, Hammond CL (2013). The heteromeric organic solute transporter, OSTalpha‐OSTbeta/SLC51: a transporter for steroid‐derived molecules. Mol Aspects Med 34: 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin ER, Skelton J, Hanway D, Olanrewaju S, Pruthi F, Ilyin VI et al. (2005). Validation of a fluorescent imaging plate reader membrane potential assay for high‐throughput screening of glycine transporter modulators. J Biomol Screen 10: 365–373. [DOI] [PubMed] [Google Scholar]
- Bernardi S, Tikellis C, Candido R, Tsorotes D, Pickering RJ, Bossi F et al. (2015). ACE2 deficiency shifts energy metabolism towards glucose utilization. Metabolism 64: 406–415. [DOI] [PubMed] [Google Scholar]
- Bohmer C, Broer A, Munzinger M, Kowalczuk S, Rasko JE, Lang F et al. (2005). Characterization of mouse amino acid transporter B0AT1 (slc6a19). Biochem J 389: 745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broer A, Juelich T, Vanslambrouck JM, Tietze N, Solomon PS, Holst J et al. (2011). Impaired nutrient signaling and body weight control in a Na + neutral amino acid cotransporter (Slc6a19)‐deficient mouse. J Biol Chem 286: 26638–26651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broer A, Klingel K, Kowalczuk S, Rasko JE, Cavanaugh J, Broer S (2004). Molecular cloning of mouse amino acid transport system B0, a neutral amino acid transporter related to Hartnup disorder. J Biol Chem 279: 24467–24476. [DOI] [PubMed] [Google Scholar]
- Broer A, Rahimi F, Broer S (2016). Deletion of amino acid transporter ASCT2 (SLC1A5) reveals an essential role for transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to sustain glutaminolysis in cancer cells. J Biol Chem 291: 13194–13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broer S (2003). Xenopus laevis Oocytes. Methods Mol Biol 227: 245–258. [DOI] [PubMed] [Google Scholar]
- Broer S (2008). Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev 88: 249–286. [DOI] [PubMed] [Google Scholar]
- Broer S (2009). The role of the neutral amino acid transporter B0AT1 (SLC6A19) in Hartnup disorder and protein nutrition. IUBMB Life 61: 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broer S (2010). Xenopus laevis oocytes. Methods Mol Biol 637: 295–310. [DOI] [PubMed] [Google Scholar]
- Broer S, Gether U (2012). The solute carrier 6 family of transporters. Br J Pharmacol 167: 256–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo SM, Makrides V, Virkki LV, Forster IC, Verrey F (2005). Steady‐state kinetic characterization of the mouse B(0)AT1 sodium‐dependent neutral amino acid transporter. Pflugers Arch 451: 338–348. [DOI] [PubMed] [Google Scholar]
- Camargo SM, Singer D, Makrides V, Huggel K, Pos KM, Wagner CA et al. (2009). Tissue‐specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology 136: 872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colas C, Grewer C, Otte NJ, Gameiro A, Albers T, Singh K et al. (2015). Ligand discovery for the alanine‐serine‐cysteine transporter (ASCT2, SLC1A5) from homology modeling and virtual screening. PLoS Comput Biol 11: e1004477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JA, Green EM, Gouaux E (2016). X‐ray structures and mechanism of the human serotonin transporter. Nature 532: 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuboni S, Devigny C, Hoogeland B, Strasser A, Pomplun S, Hauger B et al. (2014). Loratadine and analogues: discovery and preliminary structure–activity relationship of inhibitors of the amino acid transporter B(0)AT2. J Med Chem 57: 9473–9479. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel H (2004). Molecular and integrative physiology of intestinal peptide transport. Annu Rev Physiol 66: 361–384. [DOI] [PubMed] [Google Scholar]
- Danilczyk U, Sarao R, Remy C, Benabbas C, Stange G, Richter A et al. (2006). Essential role for collectrin in renal amino acid transport. Nature 444: 1088–1091. [DOI] [PubMed] [Google Scholar]
- Fairweather SJ, Broer A, Subramanian N, Tumer E, Cheng Q, Schmoll D et al. (2015). Molecular Basis for the interaction of the mammalian amino acid transporters B0AT1 and B0AT3 with their ancillary protein collectrin. J Biol Chem 290: 24308–24325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotiadis D, Kanai Y, Palacin M (2013). The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med 34: 139–158. [DOI] [PubMed] [Google Scholar]
- Gasteiger J, Marsili M (1980). Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 36: 3219–3228. [Google Scholar]
- Geier EG, Schlessinger A, Fan H, Gable JE, Irwin JJ, Sali A et al. (2013). Structure‐based ligand discovery for the large‐neutral amino acid transporter 1, LAT‐1. Proc Natl Acad Sci U S A 110: 5480–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP (2013). The SLC16 gene family – structure, role and regulation in health and disease. Mol Aspects Med 34: 337–349. [DOI] [PubMed] [Google Scholar]
- Irwin JJ, Shoichet BK (2005). ZINC‐‐a free database of commercially available compounds for virtual screening. J Chem Inf Model 45: 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata M, Silva Enciso JE, Greenberg BH (2009). Selective and specific regulation of ectodomain shedding of angiotensin‐converting enzyme 2 by tumor necrosis factor alpha‐converting enzyme. Am J Physiol Cell Physiol 297: C1318–C1329. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Rose AJ, Sijmonsma TP, Bröer A, Pfenninger A, Herzig S et al. (2015). Mice lacking neutral amino acid transporter B0AT1 (Slc6a19) have elevated levels of FGF21 and GLP‐1 and improved glycaemic control. Mol Metabol 4: 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joesch C, Guevarra E, Parel SP, Bergner A, Zbinden P, Konrad D et al. (2008). Use of FLIPR membrane potential dyes for validation of high‐throughput screening with the FLIPR and microARCS technologies: identification of ion channel modulators acting on the GABA(A) receptor. J Biomol Screen 13: 218–228. [DOI] [PubMed] [Google Scholar]
- Kharitonenkov A, Adams AC (2014). Inventing new medicines: The FGF21 story. Mol Metab 3: 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khunweeraphong N, Nagamori S, Wiriyasermkul P, Nishinaka Y, Wongthai P, Ohgaki R et al. (2012). Establishment of stable cell lines with high expression of heterodimers of human 4F2hc and human amino acid transporter LAT1 or LAT2 and delineation of their differential interaction with alpha‐alkyl moieties. J Pharmacol Sci 119: 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehn J, Lacerda AE, Wible B, Brown AM (1996). Molecular physiology and pharmacology of HERG. Single‐channel currents and block by dofetilide. Circulation 94: 2572–2579. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczuk S, Broer A, Tietze N, Vanslambrouck JM, Rasko JE, Broer S (2008). A protein complex in the brush‐border membrane explains a Hartnup disorder allele. FASEB J 22: 2880–2887. [DOI] [PubMed] [Google Scholar]
- Landowski CP, Han HK, Lee KD, Amidon GL (2003). A fluorescent hPept1 transporter substrate for uptake screening. Pharm Res 20: 1738–1745. [DOI] [PubMed] [Google Scholar]
- Malakauskas SM, Kourany WM, Zhang XY, Lu D, Stevens RD, Koves TR et al. (2009). Increased insulin sensitivity in mice lacking collectrin, a downstream target of HNF‐1alpha. Mol Endocrinol 23: 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malakauskas SM, Quan H, Fields TA, McCall SJ, Yu MJ, Kourany WM et al. (2007). Aminoaciduria and altered renal expression of luminal amino acid transporters in mice lacking novel gene collectrin. Am J Physiol Renal Physiol 292: F533–F544. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaei H, Suarez JA, Longo VD (2014). Protein and amino acid restriction, aging and disease: from yeast to humans. Trends Endocrinol Metab 25: 558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil CR (2008). Towards the development of universal, fast and highly accurate docking/scoring methods: a long way to go. Br J Pharmacol 153 (Suppl 1): S7–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS et al. (2009). AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30: 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgan AP, Coffman PK, Kocher JP, Katzmann DJ, Sosa CP (2011). Multilevel parallelization of AutoDock 4.2. J Cheminform 3: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Mara M, Oakley A, Broer S (2006). Mechanism and putative structure of B(0)‐like neutral amino acid transporters. J Membr Biol 213: 111–118. [DOI] [PubMed] [Google Scholar]
- Ovens MJ, Davies AJ, Wilson MC, Murray CM, Halestrap AP (2010). AR‐C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7‐10. Biochem J 425: 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxender DL, Lee M, Moore PA, Cecchini G (1977). Neutral amino acid transport systems of tissue culture cells. J Biol Chem 252: 2675–2679. [PubMed] [Google Scholar]
- Penmatsa A, Gouaux E (2014). How LeuT shapes our understanding of the mechanisms of sodium‐coupled neurotransmitter transporters. J Physiol 592: 863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A, Wang KH, Gouaux E (2013). X‐ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503: 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochini L, Seidita A, Sensi C, Scalise M, Eberini I, Indiveri C (2014). Nimesulide binding site in the B0AT1 (SLC6A19) amino acid transporter. Mechanism of inhibition revealed by proteoliposome transport assay and molecular modelling. Biochem Pharmacol 89: 422–430. [DOI] [PubMed] [Google Scholar]
- Reitman ML (2013). FGF21 mimetic shows therapeutic promise. Cell Metab 18: 307–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers DJ, Tanimoto TT (1960). A computer program for classifying plants. Science 132: 1115–1118. [DOI] [PubMed] [Google Scholar]
- Rudnick G (2007). What is an antidepressant binding site doing in a bacterial transporter? ACS Chem Biol 2: 606–609. [DOI] [PubMed] [Google Scholar]
- Ruggiero AM, Wright J, Ferguson SM, Lewis M, Emerson KS, Iwamoto H et al. (2012). Nonoisotopic assay for the presynaptic choline transporter reveals capacity for allosteric modulation of choline uptake. ACS Chem Nerosci 3: 767–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shotwell MA, Jayme DW, Kilberg MS, Oxender DL (1981). Neutral amino acid transport systems in Chinese hamster ovary cells. J Biol Chem 256: 5422–5427. [PubMed] [Google Scholar]
- Singh SK, Yamashita A, Gouaux E (2007). Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature 448: 952–956. [DOI] [PubMed] [Google Scholar]
- Solon‐Biet SM, McMahon AC, Ballard JW, Ruohonen K, Wu LE, Cogger VC et al. (2014). The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum‐fed mice. Cell Metab 19: 418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumer E, Broer A, Balkrishna S, Julich T, Broer S (2013). Enterocyte‐specific regulation of the apical nutrient transporter SLC6A19 (B(0)AT1) by transcriptional and epigenetic networks. J Biol Chem 288: 33813–33823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoaica L, Behera A, Camargo SM, Forster IC, Verrey F (2016). Real‐time functional characterization of cationic amino acid transporters using a new FRET sensor. Pflugers Arch 468: 563–572. [DOI] [PubMed] [Google Scholar]
- Wagner CA, Broer A, Albers A, Gamper N, Lang F, Broer S (2000). The heterodimeric amino acid transporter 4F2hc/LAT1 is associated in Xenopus oocytes with a non‐selective cation channel that is regulated by the serine/threonine kinase sgk‐1. J Physiol (Lond) 526: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Goehring A, Wang KH, Penmatsa A, Ressler R, Gouaux E (2013). Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature 503: 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH et al. (2006). A critical assessment of docking programs and scoring functions. J Med Chem 49: 5912–5931. [DOI] [PubMed] [Google Scholar]
- Whiteaker KL, Gopalakrishnan SM, Groebe D, Shieh CC, Warrior U, Burns DJ et al. (2001). Validation of FLIPR membrane potential dye for high throughput screening of potassium channel modulators. J Biomol Screen 6: 305–312. [DOI] [PubMed] [Google Scholar]
- Whitfield JH, Zhang WH, Herde MK, Clifton BE, Radziejewski J, Janovjak H et al. (2015). Construction of a robust and sensitive arginine biosensor through ancestral protein reconstruction. Protein Sci 24: 1412–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff C, Fuks B, Chatelain P (2003). Comparative study of membrane potential‐sensitive fluorescent probes and their use in ion channel screening assays. J Biomol Screen 8: 533–543. [DOI] [PubMed] [Google Scholar]
- Yu XC, Zhang W, Oldham A, Buxton E, Patel S, Nghi N et al. (2009). Discovery and characterization of potent small molecule inhibitors of the high affinity proline transporter. Neurosci Lett 451: 212–216. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Terry DS, Shi L, Quick M, Weinstein H, Blanchard SC et al. (2011). Substrate‐modulated gating dynamics in a Na + −coupled neurotransmitter transporter homologue. Nature 474: 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Certificate of analysis provided by Sigma‐Aldrich, the commercial supplier of benztropine used in this study (NSC63912) showing purity as determined by HPLC.
Data S2 Identity and purity of compounds used in this study provided by DTP‐NCI, as determined by electrospray ionisation mass spectrometry and liquid chromatography in full scan mode at 214 nm absorption wavelength, using 500 nm as a reference. See table 1 for purity values and chemical names (NSC number shown).