

Abstract
Plasmid-encoded His-tagged glucose permease of Escherichia coli, the enzyme IIBCGlc (IIGlc), exists in two physical forms, a membrane-integrated oligomeric form and a soluble monomeric form, which separate from each other on a gel filtration column (peaks 1 and 2, respectively). Western blot analyses using anti-His tag monoclonal antibodies revealed that although IIGlc from the two fractions migrated similarly in sodium dodecyl sulfate gels, the two fractions migrated differently on native gels both before and after Triton X-100 treatment. Peak 1 IIGlc migrated much more slowly than peak 2 IIGlc. Both preparations exhibited both phosphoenolpyruvate-dependent sugar phosphorylation activity and sugar phosphate-dependent sugar transphosphorylation activity. The kinetics of the transphosphorylation reaction catalyzed by the two IIGlc fractions were different: peak 1 activity was subject to substrate inhibition, while peak 2 activity was not. Moreover, the pH optima for the phosphoenolpyruvate-dependent activities differed for the two fractions. The results provide direct evidence that the two forms of IIGlc differ with respect to their physical states and their catalytic activities. These general conclusions appear to be applicable to the His-tagged mannose permease of E. coli. Thus, both phosphoenolpyruvate-dependent phosphotransferase system enzymes exist in soluble and membrane-integrated forms that exhibit dissimilar physical and kinetic properties.
The bacterial phosphoenolpyruvate (PEP)-dependent phosphotransferase system (PTS) consists of many integral membrane permeases and sugar phosphotransferases, each specific for a different sugar, and a set of cytoplasmic energy-coupling proteins (19, 30, 31). The PTS permeases are called enzymes II, and the three best characterized such enzymes are the glucose permease (IIGlc) (4, 7, 20), the mannitol permease (IIMtl) (14, 18, 21-23, 26, 45), and the mannose permease (IIMan) (8, 9, 11-13, 27, 33, 34). These enzymes exist in the membrane as oligomers, most probably as homodimers (5, 12, 23, 28, 31, 36, 37, 39, 47). They catalyze two vectorial chemical reactions, sugar phosphorylation using PEP as the phosphoryl donor, dependent on Enzyme I, HPr, and IIA as well as IIBC or IIBCD (the PEP reaction), and transphosphorylation using a sugar phosphate (glucose-6-phosphate [glucose-6-P] for IIGlc and IIMan; mannitol-1-P for IIMtl) as the phosphoryl donor, dependent only on IIBC or IIBCD (the TP reaction) (16, 25, 35, 40-42). Thus, for example, the two reactions catalyzed by IIGlc are:
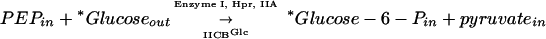 |
and
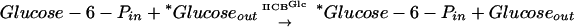 |
Enzyme I, HPr, and IIA are the energy-coupling proteins of the PTS (31). Although these reactions occur in a vectorial fashion in intact cells and bacterial membrane vesicles, they can also be demonstrated in vitro by using broken cell extracts or purified enzymes (28, 33, 38, 40-43).
In a recent communication (2) we presented evidence for two physically distinct forms of the enzymes II, one the membrane-integrated form, extensively characterized previously, and the other a “soluble” form not previously identified. When crude extracts of French-pressed Escherichia coli cells or osmotically shocked E. coli spheroplasts were centrifuged in an ultracentrifuge at high speed, a small fraction of the Enzyme II activity remained in the high-speed supernatant, and passage of the nonsedimented material through a gel filtration column gave two activity peaks, one in the void volume exhibiting high PEP-dependent and TP activities and a second included peak with high PEP-dependent activities and high (IIMan), moderate (IIGlc), or negligible (IIMtl) TP activities. The peak 1 enzyme probably consists of tiny membrane vesicles or membrane fragments with integrated enzymes II in a physical state similar to that of pelleted enzymes II.
Long-term exposure of cells to chloramphenicol resulted in selective loss of the soluble fraction with retention of much of the pelleted activity concomitant with extensive protein degradation, suggesting that both forms are present in intact cells. Western blot analyses showed that the soluble IIGlc exhibited a subunit size of about 45 kDa, and the soluble forms of all three native enzymes II eluted from the gel filtration column with apparent molecular masses of 40 to 50 kDa. We proposed that enzymes II of the PTS exist in two physically distinct forms in the intact E. coli cell, one integrated into the membrane and one either soluble or loosely associated with the membrane. We proposed that the membrane-integrated enzymes II are largely dimeric, whereas the soluble enzymes II that were retarded during passage through the gel filtration column are largely monomeric. It was considered likely that the membrane-integrated form catalyzes vectorial sugar phosphorylation, while the soluble form catalyzes the nonvectorial process.
In this communication we report further characterization of these two forms of the glucose-specific Enzyme II (IIGlc). Using a His-tagged IIGlc (his-IIGlc) (7, 44) and monoclonal antibody against the His tag, we show that His-IIGlc exists in the same two forms observed previously for the wild-type enzyme. On sodium dodecyl sulfate (SDS) gels, both preparations migrate as a single species, but on native gels the two preparations migrate differently, both before and after detergent treatment, consistent with oligomeric and monomeric forms. We further show that the two forms exhibit different kinetic properties. While the transphosphorylation reaction of the membrane-integrated form exhibits the characteristic substrate inhibition observed previously (41), the soluble form does not. Further, the two forms of the enzyme show different responses to pH. These observations lead to the suggestion that the catalytic properties of a PTS Enzyme II, including the characteristic of substrate inhibition, are strongly influenced by the enzyme oligomeric state in the membrane.
MATERIALS AND METHODS
Growth and assay conditions.
Unless otherwise indicated, strains (Table 1) were grown in 1 liter of Luria-Bertani (LB) broth in 2-liter conical flasks and incubated with shaking (275 rpm) at 37°C for 18 h. Strain ZSC112L(pJFGH11) expresses the ptsG gene constitutively under the control of its own promoter rather than the lac or ara promoter. This plasmid is a low-copy-number plasmid. Consequently, His-tagged IIGlc is not appreciably overproduced.
TABLE 1.
Description of strains used in this study
| Strain | Description | Reference or source | Use in this study |
|---|---|---|---|
| S301 | An lpp-2 derivative of W3110, wild type with regard to phospholipid synthesis | 17 | Kinetics study of soluble versus membrane enzymes II |
| AD90 | A pssA::kan mutant strain derived from strain AD90/pDD72 by plasmid curing; the latter strain was derived from strain W3899 | W. Dowhan (6) | 4-h HSS was used as a source of soluble PTS enzymes (enzyme I, enzyme IIAGlc, and HPr) |
| ZSC112L(pJFGH11) (Ampr) | Carries plasmid pJFGH11 with IICBGlc with a C-terminal His tag; the chromosomal ptsG gene is inactivated | B. Erni (48) | Western blot study of EIIGlc using monoclonal antibody against His tag |
| ZSCΔmanXYZ (pTSP-H6M) (Cmr Ampr) | Carries plasmid pTSP-H6M (IICMan::H6IIDMan) with His tag at the N terminus of IIDMan; the chromosomal man operon is deleted | B. Erni (10) | Western blot study of EIIMan using monoclonal antibody against His tag |
When working with strain ZSC112L(pJFGH11) (Ampr), a loopful of a fresh culture from a plate with LB plus 100 μg of ampicillin/ml (LB-Amp) was used to inoculate 50 ml of LB-Amp broth in a 250-ml Erlenmeyer flask. Cells obtained were used to inoculate 1 liter of LB-Amp broth in a 2-liter flask. The flask was incubated at 37°C with shaking at 275 rpm for 18 h before harvesting. The extract, obtained by passage of the cell suspension through a French press at 10,000 lb/in2, was centrifuged at 12,000 × g for 10 min. The low-speed supernatant produced was recentrifuged at 200,000 × g for 2 h. The supernatant (2-h high-speed supernatant [HSS]), which exhibited low IIGlc activity comparable to that of an HSS from wild-type E. coli cells (reference 48 and unpublished observations), was loaded onto the gel filtration column for separation of the two forms of the enzyme (1).
When working with strain ZSCΔmanXYZ(pTSP-H6 M), conditions were as for strain ZSC112L(pJFGH11) except that in addition to ampicillin (100 μg/ml), chloramphenicol was added to a final concentration of 50 μg/ml and incubation was continued until the optical density at 600 nm reached 0.8; then, isopropyl-β-thiogalactopyranoside was added to a final concentration of 100 μM (10). Incubation was continued for 4 h. In all cases, cells were harvested, washed, and ruptured in a French press as described previously (1).
Gel filtration was conducted with a Biogel A-1 fine-grade column with an exclusion limit of <10,000 to 1,500,000 (Bio-Rad) as described previously (2). Both PEP and sugar phosphate-dependent sugar phosphorylation assays were conducted essentially as described previously (1, 2). When present, phosphatidylglycerol was included in the assay solution at a final concentration of 1 mg/ml.
Detergent treatment of IIGlc.
The sample of IIGlc (pellet fraction or peaks 1 and 2 from the gel filtration column) in either medium 63 (M63) adjusted to pH 8.9 or Tris-glycine, pH 8.9, containing 1 mM methyl-α-d-glucopyranoside was treated with Triton X-100 (2%) with stirring at 4°C for 1 h in order to solubilize IIGlc from the membrane according to the procedure of Waeber et al. (48). The sample was filtered through a 0.45-μm-pore-size Millipore filter. For gel filtration, the solubilized pellet was either applied directly and eluted under alkaline conditions (pH 8.9) in the presence of 0.1% Triton X-100 or it was first dialyzed three times against 2 liters of water for 36 h (total dialysis time) to reduce the detergent and salt concentrations and then loaded onto the column and eluted at neutral pH in the absence of additional detergent. For affinity purification of His-tagged proteins, the pH of the detergent-treated samples (e.g., peaks 1 and 2 after gel filtration of a 2-h HSS at neutral pH and in the absence of detergent) was adjusted to pH 8.0 before loading onto a nickel column (48).
Purification of His-tagged IIGlc on a Ni2+ column.
The nickel column was prepared with His-Bind resin from Novagen. A bed volume per column of 1.5 ml was produced from 3 ml of a resin suspension. Column preparation involved washing with sterile deionized water (three times with 1.5 ml, i.e., 4.5 ml), charging the column using charge buffer, 50 mM NiSO4 (five times with 1.5 ml [7.5 ml]), and equilibrating with buffer B (50 mM NaPi [pH 8], 300 mM NaCl, 1 mM methyl-α-d-glucopyranoside, 0.1% Triton X-100) (three times with 1.5 ml [4.5 ml]) (7, 48). For a 50-ml sample of the enzyme (peak 1 or peak 2 from the gel filtration column), the following protocol was used. The column was washed with buffer B, pH 8 (20 times with 1.5 ml [30 ml]), then washed with buffer B, pH 6 (10 times with 1.5 ml [15 ml]), and eluted with buffer B, pH 5 (10 times with 1.5 ml [15 ml]). The eluted sample was collected in fractions of about 1 ml and rapidly neutralized with 1 M NaOH before dialyzing against water. Fractions were tested for both activity and protein profile on SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Subsequently, the eluted sample was collected into two fractions, each of about 7.5 ml. These were concentrated using Centriprep columns (cutoff = 30,000 Da) by centrifugation at 1,500 × g for 15 min using a swinging angle rotor. The concentrated fractions were dialyzed against water (three times with 2 liters over a period of 36 h). Removal of detergent tended to cause the monomeric form of IIGlc produced from the solubilized pellet or solubilized peak 1 but not from peak 2 when treated similarly to oligomerize. Dithiothreitol was added to a final concentration of 5 mM after dialysis. Dialyzed fractions were assayed in the presence of phosphatidyl glycerol (1 mg/ml, final concentration) as described by Waeber et al. (48). The Centriprep column was equilibrated first with distilled water by centrifugation. For resin regeneration, the column was washed with strip buffer (0.5 M NaCl, 100 mM EDTA, 20 mM Tris-HCl; pH 7.9) (three times with 1.5 ml [4.5 ml]), and the resin was stored in strip buffer.
PAGE.
When conducting SDS-PAGE analyses, the standard method was used (1) except that the separating gel was 15% acrylamide. When conducting native gel analyses, a 6% acrylamide concentration was used and there was no stacking gel. The sample was loaded in Laemmli buffer that was free of SDS.
Western blot analyses.
Western blot analyses were conducted essentially as described previously (1). The first antibody was His tag monoclonal antibody purchased from Novagen (catalog no. 70796-3). It was applied at a dilution of 1:1,000. The secondary antibody was horseradish peroxidase (HRP)-labeled goat anti-mouse immunoglobulin G (catalog no. NEF822; NEN, Boston, Mass.) and was applied at a dilution of 1:5,000.
Hybridization protocol.
Separated protein bands on a gel were transferred to a membrane using the standard protocol described previously (1, 2) but with modifications as follows. The membrane was washed three times (each time for 5 min) in Tris-buffered saline (TBS; 10 mM Tris-HCl [pH 7.5], 7.5 mM NaCl) containing 1% Tween 20. The membrane was then blocked in TBS buffer containing 1% Tween 20 and 5% skim milk for 6 to 8 h at 4°C with gentle shaking. The membrane was then washed three times (5 min each) with TBS-1% Tween 20. It was then incubated with the first antibody and diluted in TBS-1% Tween 20-5% skim milk for 1 h, followed by washing three times (10 min each) with TBS-1% Tween 20. Finally, it was incubated with the secondary antibody diluted in TBS-1% Tween 20-5% skim milk for 1 h, followed by washing three times (10 min each) in TBS-1% Tween 20.
Chemiluminescent detection.
Detection of IIGlc and IIMan by using primary mouse anti-His tag antibody and secondary goat anti-mouse immunoglobulin G-HRP involved use of the chemiluminescent detection technique with Immune-Star HRP (catalog no. 170-5040; Bio-Rad) (24) as described by the manufacturer. The membrane was packed between two plastic sheets and developed with autoradiographic film (X-ray film). Exposure times ranged between 1 and 8 min.
Materials.
The materials used, their purities, and their sources were as described previously (1, 2) except when noted otherwise. Radioactive sugars were used at a specific activity of 5 μCi/μmol. Phosphatidylglycerol (PG) was obtained from Sigma Chemical Corporation. Acrylamide, bisacrylamide, Tris-glycine buffer, SDS, ammonium persulfate, and Laemmli buffer were from Bio-Rad.
RESULTS
Detection of soluble and membrane-integrated His-tagged Enzyme IIGlc by Western blotting.
The His-tagged Enzyme IIGlc in the 2-h HSS derived from strain ZSC112L(pJFGH11) was passed through the gel filtration column, yielding peaks 1 and 2 (Fig. 1) as reported previously for the wild-type enzyme from E. coli strain 301 (2). It was detected by the Western blotting technique using anti His tag monoclonal antibody. Both pooled fractions (peaks 1 and 2) were treated with detergent (2% Triton X-100) as described in Materials and Methods. Using the Coomassie blue stain, both gel filtration peaks 1 and 2 yielded multiple bands on SDS-PAGE (data not shown). The Western blotting results for peaks 1 and 2 following gel filtration and SDS-PAGE are shown in Fig. 2A. Both fractions yielded a single band corresponding to His-tagged IIGlc, and the migration rate was the same for IIGlc from both peaks. However, when IIGlc was analyzed using native gels, peaks 1 and 2 appeared in different positions, with peak 1 IIGlc running much more slowly than peak 2 IIGlc (Fig. 2B).
FIG. 1.

Profiles of PEP-dependent activity (○) and TP activity (□) as well as protein concentration (Δ) following gel filtration of a 2-h HSS from an extract of E. coli strain ZSC112L(pJFGH11). Elution was with M63, pH 7.0, containing 2 mM dithiothreitol. [14C]methyl-α-glucoside (20 μM for the PEP reaction; 100 μM for the TP reaction) plus either PEP (10 mM) and a 4-h HSS or glucose-6-P (10 mM) were used for the assay under standard conditions. The fractions pooled for enzyme analysis are shown by the horizontal black bars.
FIG. 2.

Western blot analysis of SDS-PAGE (A and C) and native PAGE (B and D) for His-tagged enzyme IIGlc. The proteins analyzed were either obtained directly from the gel filtration column (A and B) or after purification on a Ni2+ column (C and D). Prior to purification of the enzyme on the Ni2+ column, peak 1 IIGlc was solubilized with Triton X-100 as described in Materials and Methods, and peak 2 IIGlc was treated the same before being applied to the column.
The detergent-treated peaks 1 and 2 of His-tagged IIGlc were purified using a nickel (Ni2+) column. After passage of soluble IIGlc through the nickel column, only a few faint bands were observed in addition to the dominant band at about 50 kDa, expected for His-tagged IIGlc, when Coomassie blue stain was used (data not shown) (7, 48). Both purified fractions showed a single band with His tag-specific monoclonal antibody, regardless of whether analysis was performed with SDS-PAGE (Fig. 2C) or with native PAGE (Fig. 2D). Again, the detergent-treated nickel column-purified IIGlc from peak 1 ran more slowly than that from peak 2, and the positions were the same as before purification.
Based on these observations, we conclude that Enzyme II preparations from peaks 1 and 2 are in different conformational or oligomeric states. The results are consistent with our previous suggestion, based on other criteria (2), that the soluble forms of the enzymes II in peak 2 are monomeric while the membrane-integrated form(s) in peak 1 is oligomeric.
Activity measurements for His-tagged IIGlc following gel filtration and affinity purification.
Activity measurements for the His-tagged IIGlc from the gel filtration column showed two activity peaks essentially as reported previously for native IIGlc (2) (Fig. 1). Both peaks exhibited both PEP-dependent and TP sugar phosphorylation activities.
The His-tagged peak 1 IIGlc was solubilized using Triton X-100 according to the method of Waeber et al. (48) (see Materials and Methods), and peak 2 IIGlc was similarly treated. These preparations were then applied to a Ni2+ affinity column as described above, yielding in each case fractions exhibiting both PEP-dependent and TP IIGlc activities.
The TP activities of the purified IIGlc in peaks 1 and 2 were assayed as a function of glucose-6-P concentration at a constant methyl-α-glucoside concentration (Fig. 3). It can be seen that the two enzyme preparations exhibited strikingly different behaviors. Thus, the peak 2 activity increased with glucose-6-P concentration up to 1,000 mM, while the peak 1 activity peaked at a glucose-6-P concentration of about 10 mM. These results show that these two enzyme preparations, which on the basis of native gel electrophoresis experiments appeared to be in different oligomeric states (Fig. 2), exhibit different kinetic properties.
FIG. 3.

TP activities of Ni2+ column-purified preparations of the IIGlc from gel filtration peaks 1 (Δ) and 2 (○). The enzyme preparation applied to the gel filtration column was a 2-h HSS from strain ZSC112L(pJFGH11). Activity was measured as a function of glucose-6-P concentration. The final methyl-α-glucoside concentration was 50 μM. The specific activities, when measured with a final [14C]methyl-α-glucoside concentration of 50 μM and a final glucose-6-P concentration of 5 mM with 1 μg of phosphatidylglycerol/μl present, were 16 and 0.7 pmol of sugar-P formed per mg of protein per min for the peak 1 and peak 2 enzyme preparations, respectively.
The radioactive product of the TP reaction obtained with the nickel column-purified IIGlc preparations was subjected to treatment with alkaline phosphatase (10 U in 200 μl; solution adjusted to pH 9.8). The solution was incubated at 37°C for 90 min, which resulted in conversion of greater than 95% of the anionic product into a neutral product. These results are consistent with the conclusion that the product of the TP reaction was methyl-α-glucoside-6-phosphate, as expected.
The Ni2+ column-purified His-tagged IIGlc from peak 2 was assayed for the TP reaction in the presence and absence of the ammonium salt of commercial l-α-phosphatidyl-d,l-glycerol from egg lecithin (catalog no. P5531; Sigma Chemical Corp.). For this purpose, 100 μM [14C]methyl-α-glucoside and 10 mM glucose-6-P were present as substrates under standard conditions (1). Addition of 20 μl of a 10-mg/ml PG solution in chloroform-methanol (98:2) to a final volume of 200 μl (final PG concentration of 1 mg/ml) stimulated the TP activity optimally. Stimulation was 3.5-fold. Control experiments showed that the organic solvent had no stimulatory effect. We conclude that the purified peak 2 enzyme is lipid deficient.
Characteristics of detergent-solubilized IIGlc.
Pelleted IIGlc (centrifuged at 200,000 × g for 2 h) from a crude extract of strain ZSC112L(pJFGH11) was treated with Triton X-100 (see Materials and Methods). The treated pellet and the original 2-h HSS were passed separately at alkaline pH and in the presence of 0.1% Triton X-100 through the gel filtration column, instead of at neutral pH in the absence of detergent, as performed in the experiment shown in Fig. 1. The elution profiles are shown in Fig. 4. The solubilized pellet fraction gave three TP activity peaks, while the HSS gave one broad peak. The elution profiles suggest that multiple molecular species of IIGlc are present in both preparations. The species from the Triton-solubilized pellet probably included at least two oligomeric forms as well as a monomeric species. The HSS fraction could be monomeric IIGlc associated with various amounts of lipid, detergent, and/or other proteins.
FIG. 4.

Elution profile of the 2% Triton-solubilized pellet fraction (Δ) and a 2-h HSS (□), eluted from the gel filtration column with M63, pH 8.9, containing 0.1% Triton X-100 and 2 mM dithiothreitol and monitored using the TP assay. The pellet fraction was solubilized at pH 8.9 with 2% Triton X-100, stirring at 4°C for 2 h, and then filtered through a 0.45-μm Millipore membrane filter. An aliquot of 2.5 ml of the solubilized pellet or 2.5 ml of the 2-h HSS was fractionated through the gel filtration column. The resultant fractions were dialyzed against water for 36 h (three changes; three 2-liter volumes; 12 h each) to reduce the detergent and salt concentrations and then assayed as follows: 100 μl from the dialyzed fractions was mixed with an aliquot of assay mix containing both TP substrates (100 μM [14C]methyl-α-glucoside and 10 mM glucose-6-P). Phosphatidylglycerol was incorporated at 200 μg per reaction mix (1 mg/ml) in the case of the solubilized pellet fractions only. Assay times were 2 and 1.5 h for the solubilized pellet fractions and the HSS fractions, respectively.
The TP kinetics of the different fractions shown in Fig. 4 as a function of glucose-6-P concentration are shown in Fig. 5. The crude solubilized pellet before gel filtration appeared to show a slight activity peak at about 20 mM glucose-6-P, but activity then increased further as the glucose-6-P concentration increased. Peak P1 from the gel filtration column (Fig. 4) showed maximal activity at about 10 mM glucose-6-P with inhibition at higher concentrations (Fig. 5). This behavior resembles that of membrane-integrated IIGlc (40, 41). By contrast, the low-molecular-weight enzyme (peak P2 in Fig. 4) showed no substrate inhibition at high glucose-6-P concentrations. These results reveal different kinetic behaviors for the different IIGlc preparations, comparable to that with the membrane-integrated and soluble forms of the enzyme obtained from a total crude extract.
FIG. 5.

TP activities of the detergent-solubilized pellet fraction (Δ), peak 1 of the solubilized pellet fraction eluted with M63, pH 8.9, containing 0.1% Triton X-100 (○), and peak 2 of the solubilized pellet fraction eluted with M63, pH 8.9, containing 0.1% Triton X-100 (×). All three fractions were dialyzed three times against 2 liters water for 12 h each before assay. The assay was performed as a function of glucose-6-P concentration with the [14C]methyl-α-glucoside concentration held constant at 50 μM. Phosphatidylglycerol dissolved in hexane-ethanol (9:1) was included in the assay mix at a final concentration of 1 mg/ml. The specific activities of the peak 1 and peak 2 enzyme preparations were 36 and 20 pmol of sugar-P formed per mg of protein per min, respectively, with [14C]methyl-α-glucoside at 50 μM and glucose-6-P at 5 mM.
Pelleted IIGlc from a crude extract of the His-tagged IICBGlc-producing strain, ZSC112L(pJFGH11) (Table 1), was solubilized with 2% Triton X-100 at pH 8.9, and 5 ml of the solubilized fraction was three times dialyzed against 2 liters of water to reduce the detergent and salt concentrations. An aliquot (2.5 ml) of this preparation was applied to the gel filtration column and eluted with M63, pH 7. Fractions from this column were assayed for TP activity with and without PG. When present, 4 μl of a 50-mg/ml PG solution in hexane-ethanol (9:1) (lyophilized PG was from Sigma Corp., catalog no. P8318) was added to the 200-μl assay mixture to give a final PG concentration of 1 mg/ml. As shown in Fig. 6, the addition of PG greatly increased the TP activity of the enzyme and shifted the activity peak towards higher-molecular-weight material. This stimulation by PG resembled that noted above for the affinity-purified soluble IIGlc. Control experiments showed that the solvent alone was without effect. Thus, both preparations are lipid deficient.
FIG. 6.

TP activities of gel filtration fractions of the detergent-solubilized and dialyzed pellet fraction eluted with M63, pH 7, and assayed in the presence (▪) or absence (□) of phosphatidylglycerol. Detergent-solubilized fractions of 5 ml were dialyzed three times against 2 liters of water for a total period of 36 h. Aliquots (2.5 ml) were loaded onto the gel filtration column and eluted with M63, pH 7. When present, phosphatidylglycerol was added to the assay mixture at a final concentration of 1 mg/ml. The substrates for assay were methyl-α-glucoside (100 μM) and glucose-6-P (10 mM).
Comparing Fig. 4 with Fig. 6 reveals that the gel filtration profile changed dramatically when the detergent-treated pellet fraction was eluted at alkaline pH in the presence of detergent (Fig. 4) versus when it was dialyzed against water and eluted from the gel filtration column at neutral pH in the absence of added detergent (Fig. 6). The multiple detergent-solubilized enzyme species evidently reassociate into higher-molecular-weight material when substantial amounts of the detergent are removed at neutral pH.
Kinetic properties of soluble versus membrane-integrated enzymes II.
As noted above and shown in Fig. 1, passage of the 2-h HSS protein fraction through the gel filtration column gave two activity peaks, an excluded peak (P1) and an included peak (P2). Both IIGlc and IIMan activities could be identified in both peaks. These enzyme preparations gave linear increases in IIGlc and IIMan activities when assayed as a function of protein concentration. Figure 7 shows the activities of IIGlc and IIMan as a function of the glucose-6-P concentration. Regardless of the radioactive substrate used (methyl-α-glucoside to assay IIGlc or 2-deoxyglucose to assay IIMan), the membrane-integrated enzymes (peak 1) exhibited substrate inhibition, while the soluble enzymes (peak 2) exhibited an essentially hyperbolic response to substrate concentration. Half-maximal activity was observed at about 10 mM glucose-6-P for both IIGlc and IIMan. It is therefore apparent that while the two enzyme preparations exhibit strikingly different kinetic characteristics, IIGlc and IIMan exhibit comparable behaviors. Peak 2 was assayed for sugar-phosphatase activity, but under the conditions of the assay, which included addition of fluoride to inhibit phosphatase activities, no such activity was detected (data not presented).
FIG. 7.

TP activities of peaks 1 and 2 obtained from gel filtration of a 2-h HSS of wild-type E. coli strain 301 a final fixed concentration of [14C]methyl-α-glucoside (50 μM) or [14C]2-deoxyglucose (25 μM) and as a function of glucose-6-P concentration. ○, peak 1 (methyl-α-glucoside); □, peak 1 (2-deoxyglucose); •, peak 2 (methyl-α-glucoside); ▪, peak 2 (2-deoxyglucose). The specific activities of the peak 1 and peak 2 IIGlc preparations were 0.9 and 0.09 pmol of sugar-P formed per mg of protein per min, respectively, with [14C]methyl-α-glucoside at 50 μM and glucose-6-P at 5 mM. The corresponding specific activities for IIMan assayed with [14C]2-deoxyglucose were 0.025 and 0.082 pmol/min/mg of protein for peaks 1 and 2, respectively, under the same assay conditions.
Distinct pH curves for the soluble and membrane-integrated enzymes II.
Figure 8 shows the pH profiles for the PEP-dependent sugar phosphorylation reactions for peaks 1 and 2 when methyl-α-glucoside (Fig. 8A) or 2-deoxyglucose (Fig. 8B) was the sugar substrate and PEP (2.5 mM) serving as the phosphoryl donor was used to assay IIGlc or IIMan, respectively. The results showed that the two peaks of both IIGlc and IIMan exhibit distinctive responses to pH.
FIG. 8.

PEP-dependent sugar phosphorylation activities of peaks 1 (white bars) and 2 (black bars) at different pH values from 4 to 10 with methyl-α-glucoside (A) or 2-deoxyglucose (B) as the sugar substrate. The assays were conducted as outlined in Materials and Methods, with PEP at 2.5 mM and the 14C-labeled sugar concentration at 10 μM. The enzyme preparations used were from strain 301, and a 4-h HSS of an extract from strain AD90 was used as a source of the soluble PTS energy-coupling enzymes. The experiments were conducted three times, and the standard deviation values are indicated by the vertical bars.
DISCUSSION
The PTS consists of integral membrane sugar-phosphorylating permeases (the enzymes II) and the cytoplasmic energy-coupling proteins (31). These PTS proteins are often fused together in various combinations and orders (32, 46). Fusion of PTS protein domains has important consequences with respect to catalytic activity and stability (44). The enzymes II are all believed to exist in the membrane in oligomeric states, and at least some of them are probably dimeric (31).
In a recent publication, we provided evidence for a novel form of the enzymes II (2). This novel form eluted from a gel filtration column as a monomeric protein and appeared to be either cytoplasmic or loosely associated with the membrane. The evidence presented suggested that both forms exist in intact wild-type E. coli cells and are not artifacts of enzyme preparation. No evidence for a precursor-product relationship during membrane biogenesis was obtained.
As reported in this communication, we have extended these studies by conducting comparative analyses of different forms of the His-tagged Enzyme II complexes with emphasis on the glucose-specific Enzyme II, IIGlc. The following conclusions resulted from these studies. First, by using the His-tagged IIGlc (7, 48) together with anti-His tag monoclonal antibody, we showed that the two forms of the protein migrated differently in native gels, both before and after detergent treatment, as expected assuming that the membrane-integrated form is oligomeric while the soluble form is monomeric. Both forms of IIGlc migrated at the same rate in denaturing SDS gels. These observations suggest that the gentle Triton X-100 extraction procedure described by Waeber at al (48) and Buhr et al. (7) does not cause extensive subunit dissociation.
Second, the two forms of the enzyme could be affinity purified on a Ni2+ column, yielding preparations that approached homogeneity. Both forms of the enzyme exhibited both PEP-dependent and sugar-P-dependent [14C]sugar phosphorylation under our standard assay conditions. However, on native gels, the detergent-solubilized, purified, membrane-integrated form still appeared to be oligomeric, while the purified soluble form seemed to be monomeric.
Third, the two forms of IIGlc exhibited different enzymatic characteristics. They displayed different pH curves for the PEP-dependent reaction and, most strikingly, only the membrane-integrated form exhibited the characteristic substrate inhibition when assayed using the TP reaction. This surprising result suggests that the characteristic of substrate inhibition, previously documented for all well-characterized Enzyme II complexes of the PTS, is dependent on enzyme oligomerization or integration into the membrane.
Fourth, in preliminary studies, results with IIMan suggested that the two forms of this nonhomologous enzyme complex exhibit differences similar to those studied in detail with IIGlc. Thus, the two forms of IIMan from strain 301 exhibited clear kinetic differences (Fig. 7 and 8 and our unpublished observations). It therefore seems likely that our observations with IIGlc will prove to be generally relevant to other Enzyme II complexes of the PTS.
Fifth, the soluble IIGlc exhibited low activity that could be stimulated by addition of phosphatidyl glycerol both before and after affinity purification. This result suggests that the soluble IIGlc is lipid deficient. It seems reasonable to propose that oligomerization is dependent on appropriate lipid associations (29).
Finally, several of the experimental results reported as well as our most recent unpublished results suggest that an equilibrium exists between the monomeric and oligomeric forms of IIGlc in vitro. The soluble monomer is favored by (i) detergent, (ii) low protein concentration, and (iii) high pH, while the membrane-integrated oligomer is favored by phospholipids, high protein concentration, and neutral pH. Since protein conformations and protein-protein interactions are often ligand dependent (3), it might be expected that a lipid-dependent oligomerization process would be dependent, or at least influenced by, protein conformational transitions. However, cause-and-effect relationships are often difficult to establish.
The studies reported here provide new insight into the potential of integral membrane enzymes to exist in alternative conformations. Different conformations of a single polypeptide chain may exhibit drastically different physicochemical and physiological characteristics (15). They may also provide a springboard for the appearance of divergent protein evolutionary pathways (15). To what extent the observations reported here will prove to be applicable to other types of integral membrane proteins has yet to be examined.
Acknowledgments
We thank Bernhard Erni for providing the E. coli strains and plasmids used to express His-tagged IIGlc and IIMan free of the native proteins and Mary Beth Hiller for her assistance in the preparation of the manuscript.
This work was supported by National Institutes of Health grant GM64368 from the National Institute of General Medical Sciences.
REFERENCES
- 1.Aboulwafa, M., and M. H. Saier, Jr. 2002. Dependency of sugar transport and phosphorylation by the phosphoenolpyruvate-dependent phosphotransferase system on membranous phosphatidyl glycerol in Escherichia coli: studies with a pgsA mutant lacking phosphatidyl glycerophosphate synthase. Res. Microbiol. 153:667-677. [DOI] [PubMed] [Google Scholar]
- 2.Aboulwafa, M., and M. H. Saier, Jr. 2003. Soluble sugar permeases of the phosphotransferase system in Escherichia coli: evidence for two physically distinct forms of the proteins in vivo. Mol. Microbiol. 48:131-141. [DOI] [PubMed] [Google Scholar]
- 3.Amar, P., P. Ballet, G. Barlovatz-Meimon, A. Benecke, G. Gernot, Y. Bouligand, P. Bourguine, F. Delaplace, J.-M. Delosme, M. Demarty, I. Fishov, J. Fourmentin-Guilbert, J. Fralick, J.-L. Giavitto, B. Gleyse, C. Godin, R. Incitti, F. Kepes, C. Lange, L. Le Sceller, C. Loutellier, O. Michel, F. Molina, C. Monnier, R. Natowicz, V. Norris, N. Orange, H. Pollard, D. Raine, C. Ripoll, J. Rouviere-Yaniv, M. Saier, Jr., P. Soler, P. Tambourin, M. Thellier, P. Tracqui, D. Ussery, J.-C. Vincent, J.-P. Vannier, P. Wiggins, and A. Zemirline. 2002. Hyperstructures, genome analysis and I-cells. Acta Biotheoretica 50:357-373. [DOI] [PubMed] [Google Scholar]
- 4.Beutler, R., M. Kaufmann, F. Ruggiero, and B. Erni. 2000. The glucose transporter of the Escherichia coli phosphotransferase system: linker insertion mutants and split variants. Biochemistry 39:3745-3750. [DOI] [PubMed] [Google Scholar]
- 5.Boer, H., R. H. ten Hoeve-Duurkens, and G. T. Robillard. 1996. Relation between the oligomerization state and the transport and phosphorylation function of the Escherichia coli mannitol transport protein: interaction between mannitol-specific Enzyme II monomers studied by complementation of inactive site-directed mutants. Biochemistry 35:12901-12908. [DOI] [PubMed] [Google Scholar]
- 6.Bogdanov, M., and W. Dowhan. 1995. Phosphatidylethanolamine is required for in vivo function of the membrane-associated lactose permease of Escherichia coli. J. Biol. Chem. 270:732-739. [DOI] [PubMed] [Google Scholar]
- 7.Buhr, A., K. Flükiger, and B. Erni. 1994. The glucose transporter of Escherichia coli. Overexpression, purification, and characterization of functional domains. J. Biol. Chem. 269:23437-23443. [PubMed] [Google Scholar]
- 8.Erni, B., B. Zanolari, P. Graff, and H. P. Kocher. 1989. Mannose permease of Escherichia coli. Domain structure and function of the phosphorylating subunit. J. Biol. Chem. 264:18733-18741. [PubMed] [Google Scholar]
- 9.Erni, B., B. Zanolari, and H. P. Kocher. 1987. The mannose permease of Escherichia coli consists of three different proteins: amino acid sequence and function in sugar transport, sugar phosphorylation, and penetration of phage λ DNA. J. Biol. Chem. 262:5238-5247. [PubMed] [Google Scholar]
- 10.Esquinas-Rychen, M., and B. Erni. 2001. Facilitation of bacteriophage lambda DNA injection by inner membrane proteins of the bacterial phosphoenolpuruvate:carbohydrate phosphotransferase system (PTS). J. Mol. Microbiol. Biotechnol. 3:361-370. [PubMed] [Google Scholar]
- 11.Garcia-Alles, L. F., A. Zahn, and B. Erni. 2002. Sugar recognition by the glucose and mannose permeases of Escherichia coli. Steady-state kinetics and inhibition studies. Biochemistry 41:10077-10086. [DOI] [PubMed] [Google Scholar]
- 12.Gutknecht, R., K. Flükiger, R. Lanz, and B. Erni. 1999. Mechanism of phosphoryl transfer in the dimeric IIABMan subunit of the Escherichia coli mannose transporter. J. Biol. Chem. 274:6091-6096. [DOI] [PubMed] [Google Scholar]
- 13.Huber, F., and B. Erni. 1996. Membrane topology of the mannose transporter of Escherichia coli K12. Eur. J. Biochem. 239:810-817. [DOI] [PubMed] [Google Scholar]
- 14.Jacobson, G. R., C. A. Lee, J. E. Leonard, and M. H. Saier, Jr. 1983. The mannitol-specific Enzyme II of the bacterial phosphotransferase system. I. Properties of the purified permease. J. Biol. Chem. 258:10748-10756. [PubMed] [Google Scholar]
- 15.James, L. C., and D. S. Tawfik. 2003. Conformational diversity and protein evolution—a 60-year-old hypothesis revisited. Trends Biochem. Sci. 28:361-368. [DOI] [PubMed] [Google Scholar]
- 16.Khandekar, S. S., and G. R. Jacobson. 1989. Evidence for two distinct conformations of the Escherichia coli mannitol permease that are important for its transport and phosphorylation functions. J. Cell. Biochem. 39:207-216. [DOI] [PubMed] [Google Scholar]
- 17.Kikuchi, S., I. Shibuya, and K. Matsumoto. 2000. Viability of an Escherichia coli pgsA null mutant lacking detectable phosphatidylglycerol and cardiolipin. J. Bacteriol. 182:371-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koning, R. I., W. Keegstra, G. T. Oostergetel, G. Schuurman-Wolters, G. T. Robillard, and A. Brisson. 1999. The 5 Å projection structure of the transmembrane domain of the mannitol transporter Enzyme II. J. Mol. Biol. 287:845-851. [DOI] [PubMed] [Google Scholar]
- 19.Kundig, W., and S. Roseman. 1971. Sugar transport. II. Characterization of constitutive membrane-bound enzymes II of the Escherichia coli phosphotransferase system. J. Biol. Chem. 246:1407-1418. [PubMed] [Google Scholar]
- 20.Lanz, R., and B. Erni. 1998. The glucose transporter of the Escherichia coli phosphotransferase system. Mutant analysis of the invariant arginines, histidines, and domain linker. J. Biol. Chem. 273:12239-12243. [DOI] [PubMed] [Google Scholar]
- 21.Lee, C. A., and M. H. Saier, Jr. 1983. Mannitol-specific Enzyme II of the bacterial phosphotransferase system III. The nucleotide sequence of the permease gene. J. Biol. Chem. 258:10761-10767. [PubMed] [Google Scholar]
- 22.Leonard, J. E., and M. H. Saier, Jr. 1981. Genetic dissection of catalytic activities of the Salmonella typhimurium mannitol enzyme II. J. Bacteriol. 145:1106-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leonard, J. E., and M. H. Saier, Jr. 1983. Mannitol-specific Enzyme II of the bacterial phosphotransferase system II. Reconstitution of vectorial transphosphorylation in phospholipid vesicles. J. Biol. Chem. 258:10757-10760. [PubMed] [Google Scholar]
- 24.Liu, Y., M. P. Patricelli, and B. F. Cravatt. 1999. Activity-based protein profiling: the serine hydrolases. Proc. Natl. Acad. Sci. USA 96:14694-14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lolkema, J. S., and G. T. Robillard. 1992. The Enzymes II of the phosphoenolpyruvate-dependent carbohydrate transport systems, p. 135-167. In J. J. H. H. M. De Pont (ed.), Molecular aspects of transport proteins. Elsevier, Amsterdam, The Netherlands.
- 26.Lolkema, J. S., R. H. ten Hoeve-Duurkens, and G. T. Robillard. 1993. Steady state kinetics of mannitol phosphorylation catalyzed by Enzyme IImtl of the Escherichia coli phosphoenolpyruvate-dependent phosphotransferase system. J. Biol. Chem. 268:17844-17849. [PubMed] [Google Scholar]
- 27.Mao, Q., T. Schunk, K. Flükiger, and B. Erni. 1995. Functional reconstitution of the purified mannose phosphotransferase system of Escherichia coli into phospholipid vesicles. J. Biol. Chem. 270:5258-5265. [DOI] [PubMed] [Google Scholar]
- 28.Meins, M., B. Zanolari, J. P. Rosenbusch, and B. Erni. 1988. Glucose permease of Escherichia coli. Purification of the IIGlc subunit and functional characterization of its oligomeric forms. J. Biol. Chem. 263:12986-12993. [PubMed] [Google Scholar]
- 29.Opekarova, M., and W. Tanner. 2003. Specific lipid requirements of membrane proteins—a putative bottleneck in heterologous expression. Biochim. Biophys. Acta 1610:11-22. [DOI] [PubMed] [Google Scholar]
- 30.Postma, P. W., J. W. Lengeler, and G. R. Jacobson. 1993. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57:543-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Postma, P. W., J. W. Lengeler, and G. R. Jacobson. 1996. Phosphoenolpyruvate:carbohydrate phosphotransferase systems, p. 1149-1174. In F. C. Neidhardt et al. (ed.), Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, D.C.
- 32.Reizer, J., A. Reizer, M. J. Lagrou, K. R. Folger, C. K. Stover, and M. H. Saier, Jr. 1999. Novel phosphotransferase systems revealed by bacterial genome analysis: the complete repertoire of pts genes in Pseudomonas aeruginosa. J. Mol. Microbiol. Biotechnol. 1:289-293. [PubMed] [Google Scholar]
- 33.Rephaeli, A. W., and M. H. Saier, Jr. 1978. Kinetic analyses of the sugar phosphate:sugar transphosphorylation reaction catalyzed by the glucose Enzyme II complex of the bacterial phosphotransferase system. J. Biol. Chem. 253:7595-7597. [PubMed] [Google Scholar]
- 34.Rhiel, E., K. Flükiger, C. Wehrli, and B. Erni. 1994. The mannose transporter of Escherichia coli K12: oligomeric structure and function of two conserved cysteines. Biol. Chem. Hoppe-Seyler 375:551-559. [DOI] [PubMed] [Google Scholar]
- 35.Robillard, G. T., and J. Broos. 1999. Structure/function studies on the bacterial carbohydrate transporters, Enzymes II, of the phosphoenolpyruvate-dependent phosphotransferase system. Biochim. Biophys. Acta 1422:73-104. [DOI] [PubMed] [Google Scholar]
- 36.Roossien, F. F., and G. T. Robillard. 1984. Mannitol-specific carrier protein from the Escherichia coli phosphoenolpyruvate-dependent phosphotransferase system can be extracted as a dimer from the membrane. Biochemistry 23:5682-5685. [DOI] [PubMed] [Google Scholar]
- 37.Roossien, F. F., W. van Es-Spiekman, and G. T. Robillard. 1986. Dimeric Enzyme IImtl of the E. coli phosphoenolpyruvate-dependent phosphotransferase system. Cross-linking studies with bifunctional sulfhydryl reagents. FEBS Lett. 196:284-290. [DOI] [PubMed] [Google Scholar]
- 38.Saier, M. H., Jr. 1977. Bacterial phosphoenolpyruvate:sugar phosphotransferase systems: structural, functional, and evolutionary interrelationships. Bacteriol. Rev. 41:856-871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saier, M. H., Jr. 1980. Catalytic activities associated with the Enzymes II of the bacterial phosphotransferase system. J. Supramol. Struct. 14:281-294. [DOI] [PubMed] [Google Scholar]
- 40.Saier, M. H., Jr., D. F. Cox, and E. G. Moczydlowski. 1977. Sugar phosphate:sugar transphosphorylation coupled to exchange group translocation catalyzed by the Enzyme II complexes of the phosphoenolpyruvate:sugar phosphotransferase system in membrane vesicles of Escherichia coli. J. Biol. Chem. 252:8908-8916. [PubMed] [Google Scholar]
- 41.Saier, M. H., Jr., B. U. Feucht, and W. K. Mora. 1977. Sugar phosphate:sugar transphosphorylation and exchange group translocation catalyzed by the Enzyme II complexes of the bacterial phosphoenolpyruvate:sugar phosphotransferase system. J. Biol. Chem. 252:8899-8907. [PubMed] [Google Scholar]
- 42.Saier, M. H., Jr., M. J. Newman, and A. W. Rephaeli. 1977. Properties of a phosphoenolpyruvate:mannitol phosphotransferase system in Spirochaeta aurantia. J. Biol. Chem. 252:8890-8898. [PubMed] [Google Scholar]
- 43.Saier, M. H., Jr., and M. R. Schmidt. 1981. Vectorial and nonvectorial transphosphorylation catalyzed by enzymes II of the bacterial phosphotransferase system. J. Bacteriol. 145:391-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siebold, C., K. Flükiger, R. Beutler, and B. Erni. 2001. Carbohydrate transporters of the bacterial phosphoenolpyruvate:sugar phosphotransferase system (PTS). FEBS Lett. 504:104-111. [DOI] [PubMed] [Google Scholar]
- 45.Sugiyama, J. E., S. Mahmoodian, and G. R. Jacobson. 1991. Membrane topology analysis of Escherichia coli mannitol permease by using a nested-deletion method to create mtlA-phoA fusions. Proc. Natl. Acad. Sci. USA 88:9603-9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tchieu, J. H., V. Norris, J. S. Edwards, and M. H. Saier, Jr. 2001. The complete phosphotransferase system in Escherichia coli. J. Mol. Microbiol. Biotechnol. 3:329-346. [PubMed] [Google Scholar]
- 47.van Montfort, B. A., G. K. Schuurman-Wolters, R. H. Duurkens, R. Mensen, B. Poolman, and G. T. Robillard. 2001. Cysteine cross-linking defines part of the dimer and B/C domain interface of the Escherichia coli mannitol permease. J. Biol. Chem. 276:12756-12763. [DOI] [PubMed] [Google Scholar]
- 48.Waeber, U., A. Buhr, T. Schunk, and B. Erni. 1993. The glucose transporter of E. coli: purification and characterization by Ni+ chelate affinity chromatography of the IIBCGlc subunit. FEBS Lett. 324:109-112. [DOI] [PubMed] [Google Scholar]