

Abstract
Background
Fusarium graminearum is one of the main causal agents of the Fusarium Head Blight, a worldwide disease affecting cereal cultures, whose presence can lead to contaminated grains with chemically stable and harmful mycotoxins. Resistant cultivars and fungicides are frequently used to control this pathogen, and several observations suggest an adaptation of F. graminearum that raises concerns regarding the future of current plant disease management strategies. To understand the genetic basis as well as the extent of its adaptive potential, we investigated the landscape of genomic diversity among six French isolates of F. graminearum, at single-nucleotide resolution using whole-genome re-sequencing.
Results
A total of 242,756 high-confidence genetic variants were detected when compared to the reference genome, among which 96% are single nucleotides polymorphisms. One third of these variants were observed in all isolates. Seventy-seven percent of the total polymorphism is located in 32% of the total length of the genome, comprising telomeric/subtelomeric regions as well as discrete interstitial sections, delineating clear variant enriched genomic regions- 7.5 times in average. About 80% of all the F. graminearum protein-coding genes were found polymorphic. Biological functions are not equally affected: genes potentially involved in host adaptation are preferentially located within polymorphic islands and show greater diversification rate than genes fulfilling basal functions. We further identified 29 putative effector genes enriched with non-synonymous effect mutation.
Conclusions
Our results highlight a remarkable level of polymorphism in the genome of F. graminearum distributed in a specific pattern. Indeed, the landscape of genomic diversity follows a bi-partite organization of the genome according to polymorphism and biological functions. We measured, for the first time, the level of sequence diversity for the entire gene repertoire of F. graminearum and revealed that the majority are polymorphic. Those assumed to play a role in host-pathogen interaction are discussed, in the light of the subsequent consequences for host adaptation. The annotated genetic variants discovered for this major pathogen are valuable resources for further genetic and genomic studies.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3524-x) contains supplementary material, which is available to authorized users.
Keywords: Fungal pathogen, Fusarium head blight, Whole genome re-sequencing, Genome-wide polymorphism, Single nucleotides polymorphism, Host-Pathogen interaction, Evolution, Two-speed genome
Background
The ascomycete Fusarium graminearum (teleomorphe Gibberella zeae) is a hemibiotrophic pathogen commonly described as one of the main causal agent of the Fusarium Head blight (FHB), a devastating disease affecting small grains cereals worldwide [1]. In addition to the defect on annual yield, major concerns arise from contamination of grains by stable and harmful fungal metabolites so-called mycotoxins which are present in feed and food constitute a real threat for consumers and livestock [2]. Molecules belonging to the type B family of trichothecenes (TCTB) are probably the most concerning due to their frequent occurrence and demonstrated toxic effects [3]. The genes acting in TCTB production, named Tri genes, are clustered for the majority and expressed after plant penetration with an implication in pathogenicity [4, 5]. Despite the wide array of trichothecenes potentially produced by F. graminearum isolates, the spectrum of production observed in individual strains is more limited, defining chemotypes [6, 7]. To date, three chemotypes of TCTB-producing isolates have been described according to their ability to produce deoxynivalenol along with 15-acetyldeoxynivalenol (DON/15-ADON), deoxynivalenol and 3-acetyldeoxynivalenol (DON/3-ADON), and nivalenol and acetylated form (NIV). These chemotypes are associated with quantitative difference in pathogenicity; the strains producing DON instead of NIV are, for example, more aggressive against wheat [8]. In some cases, levels of TCTB have also been found to be correlated with the visual symptoms on the spike [9, 10]. Nevertheless, other factors were identified in F. graminearum with the detection of 50 quantitative trait nucleotides linked to aggressiveness variation [11].
Cultivars resistant against FHB and mycotoxin accumulation as well as fungicides are frequently used to control this pathogen [12]. However, there is now evidence that F. graminearum is adapting to such strategies, as demonstrated by the emergence of fungicide-resistant strains [13, 14] and the rapid shift towards more aggressive isolates in some part of the world [15]. Cultural management practices must therefore keep up with the “arm race”, which requires a detailed knowledge of the fungus adaptive potential with a special focus on the evolution of pathogenicity-related traits.
Grounds for F. graminearum adaptation are certainly provided for by intensive gene flow and large amounts of genetic diversity between and within field populations [16–24]. In F. graminearum specifically, these elements are further supported by particular biological features that favor the emergence of genetic diversity, namely a mixed reproduction system based on clonality, selfing and outcrossing [16, 24, 25] as well as both local and long range dispersal of the different spores produced [26–30]. Such combination is indeed particularly efficient to create new haplotypes of which the favorable ones will rapidly spread [31]. The molecular mechanisms underlying the emergence of more aggressive isolates of F. graminearum remains remain sparsely documented.
Deep sequencing technologies have been successfully used to investigate genome-wide polymorphism in various fungi, highlighting the importance of genome organization for pathogen evolution and eventually leading to the proposition of candidate genes implicated in phenotype variations [32–40]. In the case of F. graminearum, an annotated genome of reference is available, based on the sequencings of a North-American isolate [41–43]. The latest version consists of 38 Mb distributed in four scaffolds assigned to the four expected chromosomes and has been predicted to contain 14,160 nuclear protein coding genes [41]. The function of the majority of these genes remains unknown [41]. Nevertheless, specific efforts of manually curated genome-mining coupled to proteomics and transcriptomics studies revealed a large arsenal of potential effectors, including potential secreted proteins or secondary metabolites other than the currently known mycotoxins [41, 44–48]. Concerning genome-wide diversity, the first insights have been given after re-sequencing of a second North American isolate at 0.4X, identifying more than 10,000 SNPs located preferentially in chromosomes ends and inner chromosomal locations [42]. Although partial, this first re-sequencing gave a preliminary picture of the organization of the polymorphism in the genome [42]. However, several unanswered questions remained. What are the patterns of polymorphism in the regions of the reference genome not covered by reads produced after re-sequencing? Is this genomic organization respected across worldwide isolates? What is the state of the diversity affecting the functional part of the genome, including the genes for which a role for adaptation could be assumed? In order to answer those questions we proposed to re-sequence six strains of F. graminearum originally isolated from various locations in France. These strains all belong to the DON/15-ADON chemotypes, respecting the overrepresentation of this chemotype from French cultivated wheat [20].
The first objective of our analysis is therefore to quantify the whole genomic diversity of French isolates compared to the reference genome. The second objective is to evaluate the potential contribution of this diversity for phenotypic diversity by a systematic variant annotation and an estimation of the encoding-effects for variants located within genes; with a special attention on genes potentially implicated, or previously suggested to be implicated for host-pathogen interaction. By doing so, we were able to conduct a multi-scaled analysis, highlighting the organization of polymorphism in a genome-wide manner and giving access to candidate and individual gene information. Overall, these results strengthen the idea that genome organization plays a major role in the evolution of this pathogen while establishing a solid resource for further targeted genomic and genetic investigations.
Results
SNPs and InDels discovery
Our strategy of genome re-sequencing applied to six F. graminearum strains generated a total of 125 million of read pairs of 100 base pairs (bp) in length, corresponding to 37.0–44.7 million raw reads per genome (Additional file 1: Table S1). Quality trimming and filtering of reads resulted in 35.5–42.9 million paired-end reads per genome with an average read length of 91 bp. Between 88.4% and 94.8% of these reads were aligned correctly on the reference genome – a total genome coverage of 98.8% (considering all reads produced, 99% for mitochondrial genomes) and sequencing depths ranging from 79.5 X to 93.2 X depending on the considered isolate (Additional file 1: Table S1 and Figure S1). Only 13 protein coding genes of the 14,160 described in the reference nuclear genome were not covered by read in any of the isolate genomes presented herein (Additional file 2). The majority of these genes are located in genomic regions (1 kb upstream and 1 kb downstream) exhibiting deficiency in genome coverage (Additional file 2). Amplification of those targeted genes suggested that those genes are actually absent from the 6 genomes (data not shown). All of these 13 genes were discarded for downstream analysis.
The locations of genetic variations were investigated (Table 1). Variants were called on the basis of a variation compared to the sequence of the reference genome (RRES v4.0). Variant calling was fine-tuned to detect preferentially short size variants, i.e., Single Nucleotide Polymorphisms (SNPs) and short Insertions or Deletions (InDels), and obtained a final dataset of 242,756 highly-confident variants, all strains considered, consisting of 234,151 SNPs (96%) and 8,605 InDels (Table 1, Additional file 3). Regarding the insertion and deletion events, 52% and 50% of them, respectively, concerned single nucleotide positions. The largest insertion is 25 nucleotide-long and the largest deletion is 36 nucleotide-long, with mean lengths for both events being 2.8 bp and -2.7 bp respectively (Additional file 4: Figure S2).
Table 1.
Variant calling statistics, considering strain-specific reads and considering total reads produced
| Isolate | INRA-156 | INRA-159 | INRA-164 | INRA-171 | INRA-181 | INRA-195 | Totala |
|---|---|---|---|---|---|---|---|
| % of reference genome callableb | 97.7% | 97.7% | 97.6% | 97.6% | 97.5% | 97.7% | 98.3% |
| Number of SNPs | 144,679 | 146,849 | 144,802 | 143,283 | 145,071 | 145,840 | 234,151 |
| Number of InDels | 4,929 | 5,073 | 4,953 | 4,844 | 4,938 | 4,970 | 8,605 |
| Total number of variants | 149,608 | 151,922 | 149,755 | 148,127 | 150,009 | 150,810 | 242,756 |
| Mean depth of sequencing at variant position (X) | 72.3 | 83.5 | 78.0 | 79.5 | 79.6 | 80.3 | 78.9 |
| Mean variant density (variants per kb) | 3.9 | 4.0 | 3.9 | 3.9 | 3.9 | 4.0 | 6.4 |
| Number of exonic variants | 70,850 | 71,488 | 70,259 | 69,130 | 70,659 | 71,676 | 111,975 |
| Number of intronic variants | 10,779 | 10,821 | 10,509 | 10,320 | 10,682 | 10,786 | 17,095 |
| Number of non-genic variants | 67,979 | 69,613 | 68,987 | 68,677 | 68,668 | 68,348 | 113,686 |
| Number of variants with French genomes | 66,726 | 69,040 | 66,873 | 65,245 | 67,127 | 67,928 | 159,874 |
a: considering all reads produced by whole genome sequencing of the six isolates
b: exluding the end of the chromosome IV from 7,953,943 bp onwards, corresponding to repeated RNA encoding sequence (see Methods)
The number of variants per strain ranges from 143,283–146,849 for SNPs, and from 4,844–5,073 for InDels (Table 1). Among them, 82,882 variants (34.1%) are common between all six French isolates. For simplicity purposes, this particular subset of variants will be referred to as the “common block of diversity”. Beside this baseline of diversity, each isolate differs from the other five French isolates by 67,157 genetic variations in average (65,157–69,040; Table 1). Pairwise comparison of isolates shows that INRA-156, with an average of 69,165 variants with each other French isolates, has the most polymorphic genome whereas the genomes of INRA-164 and INRA-181 are the least different with 35,153 variants identified (Table 2). Among the complete set of variable loci identified in this analysis, 1,235 (0.5%) presented different alleles between French alleles, all different that the reference one (i.e. multi-allelic variants).
Table 2.
Genome-wide comparison of variants between pairs of isolates
| Strains | PH-1 | INRA-156 | INRA-159 | INRA-164 | INRA-171 | INRA-181 | INRA-195 |
|---|---|---|---|---|---|---|---|
| PH-1 | - | 149,608 | 151,922 | 149,755 | 148,127 | 150,009 | 150,809 |
| INRA-156 | 61.6% | - | 70,856 | 66,763 | 65,709 | 63,758 | 78,739 |
| INRA-159 | 62.6% | 29.2% | - | 48,501 | 55,771 | 62,330 | 71,769 |
| INRA-164 | 61.7% | 27.5% | 20.0% | - | 48,453 | 35,153 | 68,191 |
| INRA-171 | 61.0% | 27.1% | 23.0% | 20.0% | - | 36,719 | 66,139 |
| INRA-181 | 61.8% | 26.3% | 25.7% | 14.5% | 15.1% | - | 65,433 |
| INRA-195 | 62.1% | 32.4% | 29.6% | 28.1% | 27.2% | 27.0% | - |
Upper diagonal considers number of variants by pair, lower diagonal considers the part of the overall diversity (242,756 variants) in percent explained by this pair
Genomic distribution of variants
Variant average genome-wide density reached 6.6 variants per kilobase (kb) considering the all genomes, ranging from 3.9 to 4.0 variants per kb per individual genome (Table 1). The distribution of the variants is not uniform between and within chromosome. At the inter-chromosomal level, Chromosome II, with 5.4–5.6 variants per kb per genome always exhibits the greatest variant density (Fig. 1). The number of variants detected in the mitochondrial genomes dropped considerably (less than 0.3 variant per kb) compared to nuclear genomes, all variants being localized outside of annotated genic sequences (Additional file 3: Table S3). At the intra-chromosomal level, the contribution of chromosome segments to the overall polymorphism is not linear (Fig. 2a). Telomeric/subtelomeric ends and discrete interspersed interstitial regions participate actively to the total polymorphism. Polymorphic islands are distinguished easily (Fig. 2a, delimited by dot lines and dark stars; accounted for when longer than 200 kb and showing at least a two-fold increase in variant density compared to the genome-wide median density). Such regions present in average a 7.5-fold increase of variant density compared to others (16.0 variants/kb vs. 2.1 variants/kb). The additive length of these regions represents 31.5% of total nuclear genome length while containing 76.7% of the total polymorphism (Additional file 5: Table S4). The presence of polymorphic islands at both chromosome ends are a common feature between chromosomes, whereas the number and size of interstitial polymorphic regions differ: for example, chromosome I exhibits two distinct variant-rich regions, chromosome II has a long continuous variant-rich region spreading over one third of total chromosomic size, chromosome IV displays a single ~1 Mb-long variant-rich region, and chromosome III has none (Fig. 2a, b). The predicted positions of centromeres [41] also appear to collocate with variant-rich regions (Fig. 2b), whereas too short in length to be accounted for polymorphic islands. Variant density is not uniform within polymorphic islands either (Fig. 2b). General variant density profiles are conserved between genomes (Fig. 2b); and between the common block of diversity and the diversity recorded between French isolates (Fig. 2b). This tendency does not exclude occasional differences observed between strains (examples delimited by black rectangles, Fig. 2b). For instance, the region ranging from 7.8 Mb to 8 Mb on chromosome II is rich in variants in the genomes of INRA-156, INRA-159 and INRA-164 but not in those of the other three strains.
Fig. 1.

Average variant density by strain for the four chromosomes and the mitochondrial genome. Variant density is represented in variants/kb. The density of variants belonging to the common block of diversity (observed in all French isolates) is in red; the density of variant belonging to the diversity observed between French isolates is in blue
Fig. 2.

Profiles of variant distribution by chromosome. Density profiles were computed for non-overlapping 100 kb-long sliding windows along the four chromosomes of F. graminearum. a Cumulative variant density profiles, all polymorphism considered. Star-containing intervals delineated by dotted lines indicate polymorphic islands. b Variant density profiles along the four chromosomes of F. graminearum for each strain. The density of variants belonging to the common block of diversity (observed in all French isolates) is in red; the density of variant belonging to the diversity observed with other French isolates is in blue. Black rectangles highlight selected differences between isolates. The arrows indicate the positions of centromeres
Functional annotation of variants
All strains considered, 129,070 variants are found within genic (introns and exons) sequences and 113,686 variants are found elsewhere in the genome (Table 1). Although significant due to the large number of genes, variant density observed within genic sequences does not appear to be greatly reduced compared to the variant density of other sequences (1.05-fold; p-value < 0.001). Intronic variants (total: 17,095; per genome: 10,320–10,821) are overrepresented by 5.3-fold (p-value < 0.001) whereas exonic variants (total: 111,975; per genome: 69,130–71,676) are slightly underrepresented by 0.9-fold (p-value < 0.001). Considering all protein-coding nuclear genes (n = 14,147 excluding not covered genes), 80% present at least one mutation in at least one isolate - 69% of genes in average when strains are considered individually (Fig. 3). Median number of variants per gene per genome is 1, whereas the distribution of variant number per gene is skewed due to extreme variant content exhibited by a small percent of genes (Fig. 3).
Fig. 3.
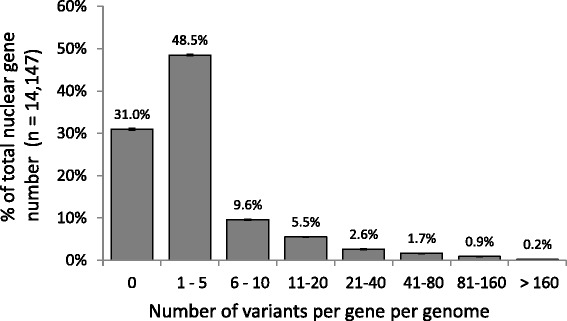
Distribution of average variant content per gene per genome. Values are expressed in percent of total nuclear protein encoding gene number (n = 14,147). Bars are mean values for the count of variant considered and error bars the standing deviations per genome
In order to identify biological functions possibly more affected than others by variants, we estimated the consequences of genic variants in all strains considered (including introns and exons; Fig. 4a and Additional file 3: Table S3). A little more than half of the variants (52.3%) are predicted to not change protein sequences because they are located in intergenic and intronic regions, outside of splicing sites. Another 28.3% have synonymous effects (i.e., a codon exchange leading to no change in amino acid), 0.7% of total variants have a predicted loss-of-function effect (LoF, in our case the introduction of a frameshift, a stop codon, the loss of the codon start or a critical mutation within the splicing-site), 18.7% have a non-synonymous effect (i.e., a codon exchange leading to a change in amino acid). Genes can also be organized according to their content in variants and their predicted effects (Fig. 4b and Additional file 6: Table S5). Four categories can be defined: the “non-functional” category consists of the 1,057 genes (7.5% of the protein-coding genes) that contain at least variants predicted to lead to a loss of function in at least one isolate; the “Modified Protein” and “Conserved Protein” categories includes 7,164 genes (50.6% of the protein-coding genes) with non-synonymous variant(s) and 3,085 genes (21.8% of the protein-coding genes) with synonymous variant(s) respectively; finally the “Highly Conserved Gene” category (Additional file 6: Table S5) includes genes with no variant identified in any of the isolates (n = 2,841, 20.1% of the protein-coding genes).
Fig. 4.
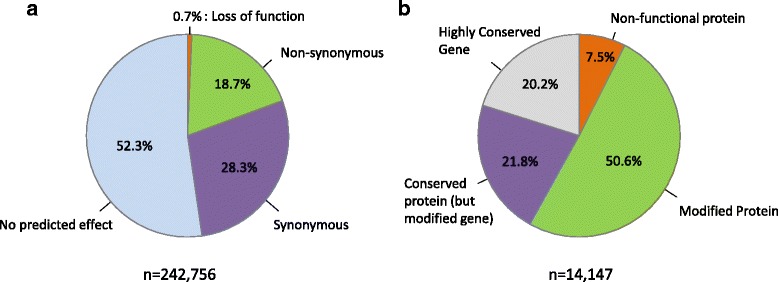
Variant effect prediction and subsequent gene classification. a Classification of variants according to their predicted effects (n = 242,756). Orange: variants leading to a loss of function (LoF) of the proteins; Green: variants with non-synonymous effects (including intronic and exonic variants); Purple: variants with no predicted effect; Blue: variants located outside of genic sequences. b Classification of genes according to the type of variant (predicted effect) they contain. Orange: genes containing at least variant(s) leading to a loss of function (LoF) of the proteins; Green: genes containing at least variants with non-synonymous effects (including intronic and exonic variants, and containing no LoF variant); Purple: genes containing only variants with no predicted effect; Blue: genes of which no variants have been detected
Biological functions that can be affected by genetic variants
We investigated the putative functions of the genes belonging to the different categories described above. A gene ontology (GO) term enrichment approach was used to discover top functions represented in gene lists belonging to each category. Results are summarized in Table 3. Category “Non-functional” is significantly enriched in genes implicated in chitin catabolism; category “Modified Protein” is enriched in genes involved in the regulation of transcription, in oxidation and reduction processes and in the regulation of primary metabolic process; category “Conserved Protein” is enriched in genes acting in signalization and communication, translation, protein transport and several process involved for example in carbohydrate metabolism; finally, the “Highly Conserved Gene” category is enriched in genes involved in more universal cellular process, such as cytoplasmic transport including Golgi vesicle transport, protein folding and macromolecule assemblies, translation, as well as several biosynthetic and catabolic processes (Table 3). GO term enrichment analyses are however prone to ontology mapping-related biases [49]. Forty five percent of the totality of nuclear protein-coding genes of F. graminearum lack GO term annotation [41]. Therefore, we developed a second approach that consist in using F. graminearum-specific gene lists compiled from transcriptomic experiments and genome-mining efforts and available from the literature: transcriptomic data from in planta experiment, genes coding for putative secreted proteins, genes belonging to predicted secondary metabolite clusters [41, 48, 50].
Table 3.
Significant (p-value < 0.01) gene ontology enrichment of the categories built from their variant contents and downstream coding-effect
| Genes Lists | GO ID | GO Term | Genes in the GO list | Theoretical gene number | Observed gene number | Fold enrichment |
|---|---|---|---|---|---|---|
| "Non-functional" | GO:0006030 | chitin metabolic process | 21 | 1 | 5 | 4.9 |
| "Modified Protein" | GO:0006355 | regulation of transcription, DNA-dependent | 458 | 239 | 294 | 1.2 |
| GO:0055114 | oxidation-reduction process | 763 | 398 | 478 | 1.2 | |
| GO:0060255 | regulation of macromolecule metabolic process | 482 | 250 | 300 | 1.2 | |
| GO:0080090 | regulation of primary metabolic process | 497 | 258 | 307 | 1.2 | |
| “Conserved protein” | GO:0044262 | cellular carbohydrate metabolic process | 30 | 8 | 17 | 2.2 |
| GO:0007264 | small GTPase mediated signal transduction | 43 | 11 | 22 | 2.0 | |
| GO:0015031 | protein transport | 121 | 31 | 59 | 1.9 | |
| GO:0044723 | single-organism carbohydrate metabolic process | 82 | 21 | 36 | 1.7 | |
| GO:0072521 | purine-containing compound metabolic process | 90 | 23 | 38 | 1.6 | |
| GO:0006412 | translation | 181 | 47 | 76 | 1.6 | |
| GO:0007154 | cell communication | 140 | 36 | 56 | 1.5 | |
| GO:0044267 | cellular protein metabolic process | 509 | 132 | 179 | 1.4 | |
| "Highly conserved genes" | GO:0006888 | ER to Golgi vesicle-mediated transport | 8 | 1 | 6 | 4.5 |
| GO:0048193 | Golgi vesicle transport | 15 | 3 | 10 | 4.0 | |
| GO:0016482 | cytoplasmic transport | 26 | 4 | 12 | 2.8 | |
| GO:0034622 | cellular macromolecular complex assembly | 25 | 4 | 11 | 2.6 | |
| GO:0006457 | protein folding | 40 | 7 | 16 | 2.4 | |
| GO:0022607 | cellular component assembly | 35 | 6 | 14 | 2.4 | |
| GO:0044283 | small molecule biosynthetic process | 132 | 22 | 46 | 2.1 | |
| GO:1901136 | carbohydrate derivative catabolic process | 53 | 9 | 18 | 2.0 | |
| GO:0046394 | carboxylic acid biosynthetic process | 110 | 18 | 37 | 2.0 | |
| GO:0006996 | organelle organization | 66 | 11 | 22 | 2.0 | |
| GO:0006412 | translation | 181 | 30 | 59 | 1.9 | |
| GO:0008652 | cellular amino acid biosynthetic process | 86 | 14 | 27 | 1.9 | |
| GO:1901565 | organonitrogen compound catabolic process | 75 | 13 | 23 | 1.8 | |
| GO:0071840 | cellular component organization or biogenesis | 134 | 22 | 39 | 1.7 | |
| GO:1901566 | organonitrogen compound biosynthetic process | 190 | 32 | 54 | 1.7 | |
| GO:0044267 | cellular protein metabolic process | 509 | 85 | 128 | 1.5 | |
| GO:0044281 | small molecule metabolic process | 463 | 77 | 110 | 1.4 | |
| GO:1901564 | organonitrogen compound metabolic process | 400 | 67 | 95 | 1.4 | |
| GO:0044249 | cellular biosynthetic process | 788 | 132 | 174 | 1.3 | |
| GO:0019538 | protein metabolic process | 680 | 114 | 141 | 1.2 |
The first list derives from in planta transcriptomic experiments that identified genes showing unique host-specificity of expression (17% of total nuclear gene number, n = 2,353) by contrast with genes showing constitutive expression (36% of total nuclear gene number n = 5,029) suggested to correspond to basal and universal mechanism of host infection ([50], Additional file 6: Table S5). We observed a positive correlation between locations of polymorphisms and location of host-specific genes (Spearman rank order Rho = 0.55, Fig. 5 lane B). Host-specific genes are found overrepresented in the categories “Non-functional” and “Modified Protein” and underrepresented in the categories “Conserved Protein” and “Highly Conserved Gene” (Fig. 6a). This observation suggests than non-synonymous mutations tend to be accumulated into these genes. Indeed, loss-of-function and non-synonymous variants are particularly found within these genes with a 2.1-fold and 1.8-fold enrichment, respectively (Additional file 7). Conversely, the locations of genes expressed constitutively in all in planta conditions is negatively correlated to the locations of variants (Rho = - 0.60, Fig. 5 lane C). These genes are overrepresented in the categories “Highly Conserved Gene” and “Conserved Protein”, and underrepresented in the categories “Modified Protein” and “Non-functional” (Fig. 6b). Similarly, these genes contain less loss-of-function and other non-synonymous variants (5.6 times and 2.5 times respectively; Additional file 7).
Fig. 5.

Heatmap representation of variant and gene counts per 100 kb-long non-overlapping windows. Spearman rank order correlation coefficients were computed between variant and gene counts. The star * indicates that all correlations are significant at the threshold p = 0.01. A. Genetic variants (n = 242,756). B. Host-specific genes (n = 2,353) [50]. C. In planta-constitutive genes (n = 5,029) [50]. D. Secreted protein-encoding genes (n = 616) [41]. E. Secondary metabolite-encoding gene clusters (n = 67) [48]. The positions of the Tri cluster and the not-clustered Tri genes Tri1, Tri15 and Tri101 are indicated by arrows
Fig. 6.

Selected F. graminearum-specific gene content of each category of predicted variant effect. For each category, actual gene counts (colored bars) are compared to the theoretical counts expected under hypothesis of random distribution of variants (white). The star * means Chi-squared test was significant (p-value < 0.001). a Host-specific genes (n = 2,353) [50]. b In planta-constitutive genes (n = 5,029) [50]. c Secreted protein-encoding genes (n = 616) [41]. d Clustered secondary metabolite-encoding gene (n = 301)
The second list consists of genes with typical motifs suggesting that they code for secreted proteins that could therefore be potential effectors (n = 616; 126 have been shown to be expressed in a host-specific manner). The spatial distribution of these genes positively correlates with the genome-wide distribution of polymorphisms (Rho = 0.68, Fig. 5 lane D). These secreted protein-encoding genes are found overrepresented in the category “Modified Protein” and underrepresented in the category “Highly Conserved Gene” by 1.25 and 0.41-fold respectively (Fig. 6c). These genes are further enriched in non-synonymous mutations (other than loss-of-function) by 1.38 fold (Additional file 7).
Focus on secondary metabolites clusters and TCTB biosynthetic genes
Finally, we investigated genes predicted to be implicated in the biosynthesis of secondary metabolites and (mostly) organized in clusters on the genome (n = 301). The genomic distribution of these genes is significantly correlated with polymorphism (Rho = 0.38, Fig. 5 lane E). They are significantly overrepresented in the category “Modified Protein” and significantly underrepresented in the categories “Highly Conserved Gene” and “Conserved Protein” (Fig. 6d). These genes are indeed enriched in non-synonymous variants, but show in the other hand a reduction of LoF mutations (Additional file 7 and Additional file 8: Table S6). Still, 24 genes belonging to 20 different secondary metabolite clusters are affected by LoF variant(s) in at least one isolate (Additional file 8: Table S6). This is the case for example of the gene FGRRES_15980_M, coding a probable polyketide synthase involved in zearalenone biosynthesis, which contains a conserved loss of function variant in all French isolates (Additional file 8: Table S8). Remarkable secondary metabolites are the type B trichothecenes (TCTB), including the deoxynivalenol (DON), reported to be involved in pathogenicity [51]. We examined the polymorphisms affecting Tri genes (n = 15) involved in the biosynthesis of TCTB (12 of them are clustered on chromosome II as indicated on Fig. 5; Additional file 8: Table S7). An overall of 252 variants have been identified within the genic sequences and the intergenic sequences of Tri genes (located in the upstream and downstream sequences for the non-clustered Tri genes; Additional file 8: Table S8). Among these variants, 131 belong to the common block of diversity (observed in all six genomes analyzed herein). Only four of the rest of the variants are predicted to bring non-synonymous effects other than loss-of-function. All of them are located within the coding sequence of Tri15 and affect the strains INRA-159, INRA-164, INRA-171 and INRA-181 (Additional file 8: Table S7). Tri15 gene encodes a putative transcription factor and does not seem to be implicated in TCTB production [5].
Genes showing an excess of non-synonymous effect mutations
In order to identify genes accumulating non-synonymous effect mutations, we consider the total polymorphism detected in this analysis and extracted 797 genes that accumulated either or both non-synonymous (NS) and LoF mutations (NS + LoF > total number of mutation, minimum total number of mutation = 4; Additional file 6: Table S5). The large majority of them (64%) is located within polymorphic islands (Additional file 6: Table S5). Twenty-nine of them have been previously shown to be both expressed in planta and predicted to be secreted (Table 4). Fifteen have been shown to be expressed in a host-specific manner and only one has been shown to be expressed constitutively in all planta conditions tested (Table 4). Remarkably, all of them have no known function according to reference genome annotation [41], with the exception of FGRRES_04689 that code for a rhamnogalacturonase A, involved in cell wall polysaccharide degradation. Seven of them contain LoF variants (FGRRES_16333, FGRRES_03521, FGRRES_12210, FGRRES_04646_M, FGRRES_13876, FGRRES_07699, and FGRRES_09118). For FGRRES_04646_M, the mutation is present in every French isolates tested. This gene is unlikely to be an essential effector during infection of wheat as several strains of this sample have been shown to be highly aggressive (Table 5; Additional file 3: Table S3). In the other hand, the gene FGRRES_07699 is predicted to be non-functional in the highly aggressive strain INRA-156 only; the gene FGRRES_12210 is predicted to be non-functional in the less aggressive strain INRA-195 only. These genes represent interesting effectors that could have escaped from the host defense for the first case or implicated in aggressiveness reduction for the second case. The knowledge on the diversity of these genes might help further investigations.
Table 4.
Putative effectors showing an excess of non-synonymous effect mutations
| Ensembl gene ID | FGSG | Chrom | Gene start (bp) | Gene end (bp) | Gene description | InterPro ID | InterPro short description | Homology | Protein length | % cysteine | variants LoF | variants Non-Synonymous | Total variantsa | Ratio of non-synonymous effect mutations | Polymorphic Islands | In Planta Expression [50] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FGRRES_11675 | FGSG_11675 | I | 356,230 | 357,118 | Uncharacterized protein | - | - | - | 279 | 1.07 | 0 | 7 | 10 | 0.7 | Yes | Wheat |
| FGRRES_01778 | FGSG_01778 | I | 5,860,579 | 5,861,567 | Uncharacterized protein | - | - | - | 277 | 2.16 | 0 | 5 | 9 | 0.6 | Yes | Wheat |
| FGRRES_02228 | FGSG_02228 | I | 7,225,618 | 7,227,797 | Uncharacterized protein | IPR000120, IPR023631 | Amidase | glutamyl-trna amidotransferase subunit a [Fusarium langsethiae] | 664 | 0.75 | 0 | 10 | 19 | 0.5 | Yes | Wheat & Barley |
| FGRRES_02269 | FGSG_02269 | I | 7,357,559 | 7,358,332 | Uncharacterized protein | - | - | - | 237 | 2.1 | 0 | 4 | 7 | 0.6 | Yes | Wheat & Barley |
| FGRRES_13692 | FGSG_13692 | I | 9,626,040 | 9,628,066 | Uncharacterized protein | - | - | - | 499 | 1.6 | 0 | 4 | 7 | 0.6 | No | Wheat & Barley |
| FGRRES_07993 | FGSG_07993 | II | 110,904 | 113,251 | Uncharacterized protein | IPR001764, IPR002772, IPR017853, IPR026891, IPR026892 | Glycoside hydrolase/Fn3 like | exo- -beta-xylosidase bxlb [F. langsethiae] | 766 | 1.3 | 0 | 5 | 7 | 0.7 | Yes | Wheat & Barley |
| FGRRES_17022 | - | II | 1,652,257 | 1,656,378 | Uncharacterized protein | - | - | muc1-extracellular alpha- -glucan glucosidase [F. langsethiae] | 1340 | 0.3 | 0 | 5 | 6 | 0.8 | No | Wheat |
| FGRRES_16333 | - | II | 4,194,219 | 4,196,939 | Uncharacterized protein | - | - | - | 887 | 3.27 | 3 | 41 | 80 | 0.6 | Yes | Barley |
| FGRRES_03274 | FGSG_03274 | II | 4,695,334 | 4,698,042 | Uncharacterized protein | IPR029167 | Mug117 | - | 884 | 1.92 | 0 | 12 | 22 | 0.5 | Yes | Wheat & Barley |
| FGRRES_03521 | FGSG_03521 | II | 5,366,512 | 5,367,123 | Uncharacterized protein | IPR009327, IPR011051, IPR014710 | RmlC-like cupin domain | putative cupin family protein [Diaporthe ampelina] | 184 | 1.62 | 1 | 5 | 7 | 0.9 | Yes | Wheat |
| FGRRES_03612 | FGSG_03612 | II | 5,604,284 | 5,605,254 | Uncharacterized protein | IPR001087, IPR013830 | Lipase_GDSL, SGNH hydrolase-type esterase domain | gdsl lipase acylhydrolase [F. langsethiae] | 289 | 1.72 | 0 | 4 | 7 | 0.6 | Yes | Wheat & Barley |
| FGRRES_12405_M | FGSG_12405 | II | 5,622,275 | 5,622,943 | Uncharacterized protein | IPR003609 | Pan_apple | - | 222 | 4.04 | 0 | 8 | 9 | 0.9 | Yes | Wheat & Barley |
| FGRRES_03944 | FGSG_03944 | II | 6,465,510 | 6,466,808 | Uncharacterized protein | IPR011042 | Six-bladed beta-propeller, TolB-like | serum paraoxonase arylesterase [F. langsethiae] | 432 | 0.46 | 0 | 5 | 8 | 0.6 | Yes | Wheat & Barley |
| FGRRES_03972 | FGSG_03972 | II | 6,548,953 | 6,550,914 | Uncharacterized protein | IPR006094, IPR012951, IPR016166, IPR016169 | flavin adenine dinucleotide linked oxydase; Berberine & berberine-like; CO dehydrogenase flavoprotein-like | 6-hydroxy-d-nicotine oxidase [F. langsethiae] | 585 | 1.54 | 0 | 7 | 10 | 0.7 | Yes | Barley |
| FGRRES_04429 | FGSG_04429 | II | 7,989,077 | 7,992,064 | Uncharacterized protein | IPR003609 | Pan_apple | - | 995 | 5.22 | 0 | 4 | 7 | 0.6 | No | Wheat |
| FGRRES_12210 | FGSG_12210 | II | 8,620,515 | 8,622,358 | Uncharacterized protein | IPR003609 | Pan_apple | - | 596 | 2.18 | 1 | 10 | 18 | 0.6 | Yes | Wheat |
| FGRRES_04646_M | FGSG_04646 | II | 8,655,498 | 8,656,180 | Uncharacterized protein | - | - | - | 225 | 2.65 | 2 | 12 | 22 | 0.6 | Yes | Wheat |
| FGRRES_04689 | FGSG_04689 | II | 8,765,660 | 8,767,148 | Rhamnogalacturonase A | IPR000743, IPR011050, IPR012334 | Glycoside hydrolase, family 28; Pectin lyase | probable rhamnogalacturonase A precursor [Fusarium fujikuroi IMI 58289] | 446 | 2.46 | 0 | 7 | 13 | 0.5 | Yes | Wheat & Barley |
| FGRRES_05719 | FGSG_05719 | III | 3,177,333 | 3,180,794 | Uncharacterized protein | IPR029167 | Meiotically up-regulated gene 117 protein | - | 1153 | 1.73 | 0 | 4 | 6 | 0.7 | No | Wheat & Barley |
| FGRRES_05847 | FGSG_05847 | III | 3,549,004 | 3,550,475 | Uncharacterized protein | IPR001002, IPR002509, IPR011330, IPR018371 | Glycoside hydrolase/deacetylase, beta/alpha-barre; Chitin-binding, type 1 | bifunctional xylanase deacetylase [F. langsethiae] | 454 | 5.71 | 0 | 3 | 5 | 0.6 | No | Wheat & Barley & Maize |
| FGRRES_12835 | FGSG_12835 | III | 3,658,250 | 3,659,015 | Uncharacterized protein | - | - | - | 162 | 2.45 | 0 | 4 | 7 | 0.6 | No | Wheat |
| FGRRES_16623 | - | III | 4,751,122 | 4,755,561 | Uncharacterized protein | IPR013830 | SGNH hydrolase-type esterase domain | chitinase [Fusarium oxysporum Fo47] | 1461 | 1.92 | 0 | 6 | 11 | 0.5 | No | Wheat |
| FGRRES_13876 | FGSG_13876 | III | 6,370,523 | 6,371,374 | Uncharacterized protein | IPR013781 | glycosyl hydrolase catalytic domain | Glycoside hydrolase, superfamily [Cordyceps confragosa RCEF 1005] | 283 | 1.41 | 3 | 18 | 40 | 0.5 | Yes | Wheat |
| FGRRES_17469 | FGSG_13850/FGSG_13851 | III | 6,711,891 | 6,714,195 | Uncharacterized protein | - | - | related to DAN4-Cell wall mannoprotein [Fusarium proliferatum] | 719 | 1.53 | 0 | 7 | 12 | 0.6 | Yes | Barley |
| FGRRES_11379 | FGSG_11379 | III | 7,412,583 | 7,413,959 | Uncharacterized protein | - | - | - | 458 | 1.96 | 0 | 12 | 23 | 0.5 | Yes | Wheat |
| FGRRES_06610 | FGSG_06610 | IV | 582,670 | 585,011 | Uncharacterized protein | IPR018946, IPR029052, IPR032093 | Alkaline phosphatase D-related | alkaline phosphatase D precursor [Fusarium fujikuroi] | 631 | 0.79 | 0 | 5 | 9 | 0.6 | No | Wheat & Barley |
| FGRRES_07686_M | FGSG_07686 | IV | 4,196,885 | 4,197,838 | Uncharacterized protein | - | - | activator of stress protein 1 [F. langsethiae] | 317 | 2.2 | 0 | 8 | 14 | 0.6 | Yes | Wheat & Barley |
| FGRRES_07699 | FGSG_07699 | IV | 4,231,666 | 4,232,190 | Uncharacterized protein | - | - | - | 174 | 2.29 | 1 | 5 | 10 | 0.6 | Yes | Wheat & Barley |
| FGRRES_09118 | FGSG_09118 | IV | 7,533,260 | 7,536,888 | Uncharacterized protein | IPR001002, IPR001223, IPR011583, IPR013781, IPR017853, IPR018371, IPR029070 | Chitin binding; chitinase II | related to chitinase [Fusarium proliferatum] | 1159 | 3.62 | 3 | 10 | 21 | 0.6 | Yes | Wheat |
a total variant numbers include variant detected within the 100 base pairs located in upstream and downstream of the genic sequences
Table 5.
Phenotypic information and geographical origin for the six strains
| Strains | TCTB production in vitro | Aggressiveness on wheat | TCTB production in wheat | Location in France/Administrative division |
|---|---|---|---|---|
| INRA-156 | ++ | +++ | +++ | Center/Cher (18) |
| INRA-159 | ++++ | 0 | 0 | Center/Cher (18) |
| INRA-164 | ++++ | ++++ | ++++ | North/Seine Maritime (76) |
| INRA-171 | 0 | ++ | ++ | South-West/Gers (32) |
| INRA-181 | ++ | +++ | +++ | North/Eure (27) |
| INRA-195 | 0 | + | + | North-East/Meuse (55) |
Discussion
The presented work examines the level of variation that can be observed between the genomes of different F. graminearum isolates at the sequence level. In addition to describing genome-wide polymorphisms, this analysis proposes, for the first time, to quantify the downstream effects of the observed variants, particularly exonic as well as intronic variants that can lead to important consequences on the translation product [52]. Here, we applied a whole-genome reference-based DNA re-sequencing strategy rather than de novo assembly previously described as more sensitive to sequencing errors [53]. Using a re-sequencing method, the accuracy of variant calling greatly depends on the quality of the read alignments on the reference genome and the depth of read coverage per base. Filters must be applied to differentiate true variants from sequencing errors while keeping the false negative rate low. There is no « one size fit all » situation and settings must be adjusted according to the type of genetic variant investigated [53]. For F. graminearum, stringent filtration is further possible (and recommended) as this fungus is haploid and one allele is expected at the positon. A preliminary test-run indicated that SNPs and short InDels are mostly found in the genome of F. graminearum with very few variants of larger sizes (data not shown). Consequently, the filters applied to the analysis presented here were set as optimal for SNPs and short InDels detection for enhanced variant discovery.
F. graminearum genome-wide polymorphism is consistent with its pathogenic lifestyle
The comparison of six genomes of French isolates with the reference genome of the PH-1 strain of F. graminearum [41] produced a highly confident set of 242,756 distinct variants total. Each of the six genomes presented an average of 150,039 variations when compared to the PH-1 reference genome, and ~ 67,157 variations when compared to each other. This number is much higher than the 10,495 SNPs identified in the first published investigation of the genome-wide polymorphism between another North-American isolate and the PH-1 strain [42]. The much lower number of variants then observed is certainly linked to a very low fold-coverage, 0.4X, being insufficient for exhaustive and confident variant calling [42]. More recently, Walkowiak et al. [54] assembled two genomes of F. graminearum representative of the two chemotype-based populations observed in Canada, DON/3-ADON and DON/15-ADON. They reported 147,555 and 103,774 SNPs with the reference genome respectively, as well as 148,978 SNPs between the two Canadian isolates, approximating the level of polymorphism reported herein. Altogether, an estimate of ~150,000 SNPs seem to be a typical variant content expected to be observed between genomes of geographically distal isolates or belonging to different populations. Along the same line, the reduced level of polymorphism observed between French isolates may suggest that these strains are likely to belong to the same population.
On a broader scale, the genome-wide level of polymorphism observed between isolates of F. graminearum (~4 SNP/ Kb) is consistent with levels of polymorphism exhibited by other pathogenic fungi, as reported in the causal agent of poplar rust Melampsora larici-populina (~2 SNP/Kb; [37]), the causal agent of the wheat stripe rust Puccinia striiformis (~5 SNPs per Kb; [55]) or in the human pathogen Coccidioides immitis (~5 SNP/Kb); [56]). These levels are lower than the one revealed between isolates of Botrytis cinerea that could reach 10 variants per kb [38]. Authors suggested that such genome-wide diversity is linked to the ability of this pathogen to infect a remarkably broad range of hosts. In comparison, the human genome exhibits between 1.2 and 1.5 variants per kb [57]. The higher values observed in fungal pathogen genomes may be a consequence from their parasitic lifestyle that pushes their need to evolution up [58]. According to the criteria given by McDonald and Linde [23] to estimate the potential of evolution of fungal pathogen, F. graminearum can be considered as a high-adaptive potential pathogen. Paradoxically, the various analyses aiming at identifying parts of the genome that are under selection for diversification failed to identify genomic regions under strong selection [11, 59]. In the present analysis, we use total genome information and identify 797 candidate genes accumulating missense and nonsense mutations. The functions of these genes are unknown for the majority but their potential implication in pathogenicity and adaptation certainly calls for in-depth investigations.
The multi-scaled location of polymorphisms in the genome
The genome-wide average value of variant density is not a metric sufficient enough to fully comprehend the patterns of polymorphism in F. graminearum; indeed it does not describe the remarkable discrete variations that we observed at both inter- and intra- chromosomal dimensions. For example, chromosome II is more polymorphic than the other chromosomes. This higher speed of diversification of this individual chromosome has been previously reported in genome-wide comparisons of close species of the Fusarium genus [59]. The authors hypothesized that the chromosome II of F. graminearum could play a preponderant role for host infection and adaptation [59, 60]. Our analysis is in line with such a hypothesis. Distribution of polymorphisms is also highly uneven within each chromosome. The single-base resolution of our analysis enabled the accurate definition of polymorphism islands along chromosomes. Polymorphisms are preferentially located at the ends of chromosomes. Such phenomenon is common in fungal pathogens and more generally in eukaryotic genomes [58]. The interstitial polymorphic islands are, for their part, more original. These regions have been previously investigated and are described as telomeric-like and subtelomeric-like regions that originate from ancestral chromosomes fusion events [42, 60]. Remarkably, the chromosomal landscapes of polymorphism reported herein follow striking similitude with the lower-coverage analysis reported in 2007 [42]. Considering these patterns of polymorphism highly conserved between isolates, we may hypothesize that genome architecture plays a predominant role in shaping the polymorphism landscape, instead of evolution forces. Several mechanisms have been previously proposed as driving genome structure of fungal pathogens [61], among which the action of meiotic recombination may play an important role through the preferential shuffling of particular chromosome regions [62]. An inconsistency of recombination rate has been already reported along F. graminearum genome, and the increases of recombination activity seem to collocate with variant rich regions [42]. These same regions were further showed to be enriched in specific epigenetic mark [63], of which implication for meiotic recombination through chromatin remodeling has never been tested. Thus, the weight of individual contributions and inter-connections between the different proposed elements of regulation remains however unclear [61]. A comprehensive investigation of these different phenomena could shed light on the events driving the organization of F. graminearum genome and its evolution.
Finally, we observed that polymorphism rates are highly variable at gene level, with introns being more polymorphic than exons. Such situation could be the result of selective pressures since exonic variants can more directly affect protein function, as such, can be rapidly unselected [64]. Nevertheless, this does not mean that variants located in introns, and typically classified as synonymous mutations, have no contribution in protein polymorphism. Indeed, the demonstration has been done that mutations located in introns can have important effect, notably by altering the splicing process [52, 65]. Accordingly, the presented work takes all variants into consideration during the annotation process [66], identifying 1,647 variants with predicted loss of function effect and 45,196 variants with other predicted non-synonymous effects.
Further evidence of a two-speed genome organization in F. graminearum
Our data reveal a remarkable positive correlation between specific biological functions and polymorphism along the genome. For example, polymorphic islands are enriched in genes with roles in biotic and abiotic adaptation, these genes exhibiting a greater level of polymorphism than genes with basal and vital functions. This result is especially true for genes coding for secreted proteins or belonging to secondary metabolite biosynthesis clusters; both categories of genes that have been suggested to play preponderant roles during pathogenesis [41, 44]. As a whole, plant-specifically expressed genes, which translate host-specific mechanisms of infection [50], are overrepresented in these hotspots of diversity and are more diverse than other genes. In the line of our above hypothesis, we propose that this correlation arise from the preferential location of certain biological functions according to the organization of the polymorphism in the genome rather than a result of the historical and ongoing diversifying selection acting directly on these genes. Such genomic organization could argue in favor of contrasted abilities of evolution of F. graminearum gene repertoire - with genes implicated in basal process being placed in conserved compartments and genes with a bigger need of evolution being placed in highly diversifying chromosomal segments.
With the convergence of knowledge about fungal genomes, such “two-speed” organization seems to be a frequent feature in filamentous pathogens [58, 61, 67–70]. Nevertheless, in several pathogenic species, a faster speed of evolution has been attributed to gene sparse, repeat-rich compartment, as in Leptosphaeria maculans, or dedicated dispensable chromosomes, as observed in Zymoseptoria tritici [61, 67]. Compared to closely related species, as in Fusarium oxysporum, no dispensable chromosome has been ever observed in F. graminearum. Moreover, its genome is quite compacted and relatively poor in repeated sequences [41, 42]. This decreased number of repeated sequences compared to some other Fusarium species has been previously attributed to the action of the repeat induced mutation system (RIP) that introduces point mutations within repeated sequence and therefore protects the genome from the mobility of transposons [42]. Therefore, this system may have participated to the organization of the polymorphism in the genome of F. graminearum [42, 61]. Altogether, F. graminearum is an original and interesting model for the investigation of genome architecture in evolution as well as improve our understanding about the theory of two-speed genome evolution of fungal pathogens.
The added-value of genomic data to identify genes involved in pathogenicity
The level of polymorphism affecting gene sequence is remarkable. Indeed, up to 69% of the genes are polymorphic per genome compared to the reference sequence. Such polymorphism should be taken into account for genome editing approaches, often designed from the consensus sequence of the reference genome.
Quantitative variations of mycotoxin production and aggressiveness have been previously observed within field populations of F. graminearum [71]. Although variable according to environment, heritability of these traits has been demonstrated and suggests the action of multiple genetic factors [11]. To associate genetic variation with phenotype changes remains however challenging. The polymorphisms of genes involved on TCTB production, as the biosynthetic Tri gene cluster, have been previously suggested to be responsible to aggressiveness variation in F. graminearum [8]. Likewise, a genome-wide association survey associated variants located in 27 different genes to variations in aggressiveness, all of them unlinked to mycotoxin production [11, 72]. The isolates used in the present survey presented contrasted level of aggressiveness in wheat, correlated to contrasted level of DON production (Table 5). These phenotypic characteristics were further shown to be stable, as repeatable and not dependent to the wheat variety inoculated (data not shown). Although these phenotypes may be related to genetic variation, none of the variants described previously were retrieved in French isolates; and may indicate that the genetic bases of aggressiveness and TCTB production may be more complex. As a preliminary investigation, we separated our sample into two discrete groups according to aggressiveness and DON production and consider the distribution of variants. Therefore, four hundred eighty four genes were conserved in highly toxinogenic and aggressive isolates, and accumulating non-synonymous mutation(s) in less toxinogenic and less aggressive isolates (data not shown). Genes coding for vesicle trafficking were found to be significantly impacted by mutation in the less toxinogenic and less aggressive isolates. This observation is consistent with the vesicle-mediated secretion of TCTB in F. graminearum [73], the regulation of secondary metabolism and cellular compartmentalization of biosynthesis pathways being tightly linked in fungi [74].
Conclusion
Whole-genome sequencing of six F. graminearum isolates revealed a remarkable number of polymorphisms, with an overall of 242,756 highly confident variants. Polymorphisms are preferentially found clustered in the genome and may play a role in the diversification of the gene repertoire implicated in host-pathogen interaction. We further hypothesize that fungal biological functions are organized in such a way that they take full advantage of the evolving dichotomy proposed by the intrinsic architecture of this pathogen’s chromosomes. The molecular control of intrinsic chromosome features remains however to be investigated. Our observations further emphasize the high-adaptive potential of this pathogen and defend the use of more integrative pest management. As a whole, this detailed description of the genetic and functional diversity of these genomes is a milestone on the path to dissect the genetic bases of important history-life traits of F. graminearum.
Methods
Fungal isolates
Six strains of Fusarium graminearum sensu stricto were isolated from wheat plants cultivated in several French regions between 2001 and 2002 ([75], Table 5). These strains exhibit various trends of pathogenicity and quantitative profiles of DON/15-ADON production that are representative of the genotypic and phenotypic diversity observed within a larger French collection of isolates ([75], Table 5). PH-1 strain was originally isolated from corn in Michigan (NRRL 31084). The strain has been shown to be highly fertile, produces trichothecenes and zearalenone, sporulates abundantly in pure culture and is highly pathogenic to wheat and barley [76, 77].
Extraction of genomic DNA and sequencing
Genomic DNA was extracted from ~50 mg of lyophilized mycelium previously grown for five days on potato dextrose agar (39 g/l, Difco). Mycelia were lysed in 600 μL of a buffer containing 100 mM Tris-HCl (pH 9.0), 10 mM EDTA, 1% sarkosyl, and proteinase K 200 μg/mL for 2 h at 65 °C. After centrifugation (10 min at 10,000 g), the supernatant was extracted successively with 1 volume of phenol, 1 volume of phenol: chloroform (50:50) and finally 1 volume of chloroform. Nucleic acids were precipitated with 0.1 volume of cold sodium acetate (pH 5.5, 3 M) and two volumes of isopropanol and agitated twice. Solution was then centrifuged 10 min at 10,000 g and supernatant were eluted. DNA precipitate was washed twice with 1 mL cold 70% ethanol for 5 min. After centrifugation, DNA was air dried 5 min using SpeedVac. DNA was dissolved in 100 μL nuclease-free water. Preparation of the libraries and sequencing was performed at the Montpellier GenomiX sequencing platform (France, http://www.mgx.cnrs.fr). Briefly, quantities of genomic DNA were measured using a Qubit® Fluorometer (Life Technologies) and DNA integrity was verified by electrophoresis on Bioanalyzer (Agilent). DNA libraries were prepared from one μg of DNA per strain using TruSeq DNA sample preparation kit (Illumina) following the manufacturer’s instructions for 6-plexed samples (library size 350 bp +/-50 bp). Sequencing was performed on one lane of Illumina HiSeq 2000 generating 100 bp-long paired-end reads. Post-run read quality was verified using FastQC ([78], v0.11.2).
SNP & InDel discovery and analysis
Reads were cleaned up using PRINSEQ v0.17.1 [79]. Briefly, duplicated reads were removed and the 9th first 5’ nucleotides were systematically trimmed due to skewed base composition introduced by sequencing preparation. Reads with an overall mean Phred-scaled value less than 20 were discarded. Remaining reads were further 3’ trimmed for quality (Phred scale threshold of 20); high quality paired-end reads with length greater than 20 nucleotides were aligned on the genome version RRes V4.0 ([41], [EMBL-EBI accessions HG970331, HG970332, HG970333, HG970334, and HG970335]) using BWA (v0.7.8) and BWA-MEM with standard parameters and a seed size of 15 nucleotides [80]. Invalid paired with aberrant insertion size or unpaired alignments were filtered out using Samtools v 0.1.19 [81]. Valid alignments were annotated using Picard tools (v1.88, http://broadinstitute.github.io/picard/) for further processing with the GATK suite (v2.4, [82]). Identification of genomic regions that could not be mapped or called for variants considering our sequencing set up was conducted using the CallableLoci module of GATK. Alignments on the last 1.38 Mb of the end of chromosome IV systematically showed poor quality scores and were removed from the analysis. This result arises from the large number of rRNA-encoding repetitive sequences units [41] which lengths approximate the length of the sequenced reads resulting in non-unique alignments that are excluded from the analysis. This specific region, however, does not contain known protein-coding genes.
The six BAM files were simultaneously used to discover SNPs and InDels using GATK and the Unified Genotyper walker in haploïd mode [82]. Depth of read coverage per 100 kb-long non-overlapping bins was calculated using the bamCoverage tool of the Bamtools suite (version 1.5.9.1.0). Positions of InDels were recalibrated using the RealignerTargetCreator and the IndelRealigner modules of GATK with standard parameters. A variant was called when observed on at least 80% of the reads aligned at the position with a read coverage more than 5 reads, and a confidence threshold greater than 250 (SelectVariants module of GATK, v2.4). Other parameters were standards. Variant densities for 100 kb-long non-overlapping windows and other statistics on the resulting Variant Call Format (VCF) files were obtained using Vcftools (v0.1.12a, [83]). Regions longer than 200 kb showing at least a two-fold increase in variant density compared to the genome-wide median density where considered as polymorphic islands.
Functional variant annotation
Genetic variants from specific genomic compartments (i.e., exons, introns, and intergenic regions) were annotated and their effect predicted using snpEff [66] and a custom database constructed from the reference genome (RRes v4.0) and the GTF file associated. Briefly, the software classifies variants found within genes (exons and introns) according to their downstream effect at the protein level (under the standard genetic code). A full list of mutation event classification is available (http://snpeff.sourceforge.net/SnpEff_manual.html). Using this classification, four categories of genes were distinguished: i) the category “Non-functional”, i.e., genes containing variant(s) predicted to lead to a loss of the function of the protein; ii) the category “Modified Protein”, i.e., genes containing only variant(s) predicted to bring one or few amino acid composition changes; iii) the category “Conserved Protein”, i.e., genes containing only variant(s) which do(es) not change amino composition of protein; iv) the category “Highly Conserved Gene”, i.e., genes with no variant detected. Then, we calculated the ratio between the number of mutations with non-synonymous effect (including non-synonymous exonic and intronic variants as well as loss-of-function variants) and the total number of variants within genic sequences (including 100 bp downstream and upstream). Genes were accepted to show an excess of mutation with non-synonymous effect if they contained at least 4 mutations and a ratio of non-synonymous effect mutation on total number of mutation greater than 0.5 (corresponding to a two-fold increase of the genome-wide median number). PROVEAN software [84] was finally used to estimate the functional importance of missense mutations found in genes coding for secreted proteins (as described in King et al., [41]), genes expressed in wheat, barley and/or maize [50], and genes predicted to belong to secondary metabolite pathways (retrieved from Sieber et al., [48]).
Statistical analyses
Annotation enrichment analysis was conducted using the Gene Ontology Enrichment tools proposed online by the EuPathDB project [43, 85] using Biological ontology and InterPro predictions. Enrichments were accepted for p-value lower or equal to 0.01. Chi-squared test was used to compare the observed distribution of the number of genes affected by mutations in the genome with the theoretical distribution of the number genes affected by mutations under the hypothesis of a random distribution of variants in the genome. Similarly, variant enrichment in the polymorphic islands or in genes was tested using the Chi-squared test. Over and underrepresentation were accepted for p-value < 0.001. Gene-enrichment in variant-rich region was determined by calculating the density of both variants and genes in non-overlapping 100 kb windows and testing each window for Spearman rank order correlation at the significance threshold of p-value < 0.001.
Acknowledgements
We would like to acknowledge the Genotoul bioinformatics platform Toulouse Midi-Pyrénées and Sigenae group for providing help along the bioinformatics analysis and for providing computing and storage resources (http://bioinfo.genotoul.fr/). We are grateful to Vessela Atanasova-Pénichon, Pauline Lassere-Züber and Richard Blanc for their help in strain characterizations.
Funding
This study was supported by a grant from the research department Plant Health and Environment of the French National Institute of Agricultural Research. BL received a PhD fellowship from the French Research Ministry.
Availability of data and materials
Strains are available from the International Center for Microbial Resources (CIRM, (http://www6.inra.fr/cirm)), with codes 1974, 1977, 1981, 1988, 1999 and 2013 for strains INRA-156, INRA-159, INRA-164, INRA-171, INRA-181 and INRA-195 respectively. The entire set of raw reads that was generated in this project has been deposited in fastq format at the GenBank Sequence Read Archive under the accession number SRP064374. The description and annotation of variant supporting the conclusions of this article is accessible in Additional file 3: Table S3.
Authors’ contributions
BL carried out the bioinformatical and statistical analyses and wrote the manuscript. BL, MM and CS conducted the wet lab experiments. NP assisted to the data mining and manuscript writing. CB contributed to conceive and design the study. MFO was responsible for overall research management. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- 15-ADON
15-acetyldeoxynivalenol
- 3-ADON
3-acetyldeoxynivalenol
- DON
deoxynivalenol
- FHB
Fusarium head blight
- GO
Gene ontology
- InDel
Insertion or deletion
- LoF
loss of function
- NIV
nivalenol
- NS
non synonymous
- SNP
Single nucleotide polymorphism
- TCTB
Type B Trichothecenes
- VCF
variant call format
Additional files
Metrics and statistics of read sequencing and alignment on the reference genome (version RRES V4.0). Figure S1: Minimum and maximum read depth (mean number of reads at the position) calculated along sliding 100 kb bin windows of the four chromosomes of the RRES reference genome using the six bam files produced after re-sequencing of the French isolates. (XLSX 49 kb)
Genes described the reference genome (RRES v4.0) not covered by reads in any isolate genomes. (XLSX 16 kb)
Information and annotations for all genetic variants detected in this analysis. (XLSX 32569 kb)
{kind=link}
Distribution of the length of the variants identified in this analysis. Polymorphisms of length equal to zero are SNPs. (JPG 65 kb)
Details of the polymorphic islands delineated during the analysis. (XLSX 15 kb)
Gene annotation according to the polymorphism detected in the analysis. (XLSX 4054 kb)
{kind=link}
Variant enrichment of genes with candidate functions. Representation of the observed variant number (in color) compared to the theoretical number expected under hypothesis of random distribution of variants (white). A. Variants predicted to lead to a loss of function of the protein. B. Variants predicted to have non-synonymous effects on the protein. “*” means Chi-squared test was significant (p-value < 0.001). (JPG 57 kb)
Annotation of genes previously predicted to belong to secondary metabolite clusters. Table S7: Summary of known Tri genes implicated in trichothecene of type B (TCTB) production. Table S8: Annotation of Tri genes and surrounding sequences according to the polymorphism detected during this analysis. (XLSX 122 kb)
References
- 1.Trail F. For blighted waves of grain: Fusarium graminearum in the postgenomics era. Plant Physiol. 2009;149:103–10. doi: 10.1104/pp.108.129684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waskiewicz A, Golinski P. Mycotoxins in foods, feeds and their components. Krmiva. 2013;55:35–45. [Google Scholar]
- 3.Morgavi DP, Riley RT. Fusarium and their toxins: Mycology, occurrence, toxicity, control and economic impact. Anim Feed Sci Technol. 2007;137:199–200. doi: 10.1016/j.anifeedsci.2007.06.001. [DOI] [Google Scholar]
- 4.Brown NA, Bass C, Baldwin TK, Chen H, Massot F, Carion PW, et al. Characterisation of the Fusarium graminearum-wheat floral interaction. J Pathog. 2011;2011:626345. doi: 10.4061/2011/626345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kimura M, Tokai T, Takahashi-Ando N, Ohsato S, Fujimura M. Molecular and genetic studies of Fusarium trichothecene biosynthesis: pathways, genes, and evolution. Biosci Biotechnol Biochem. 2007;71:2105–23. doi: 10.1271/bbb.70183. [DOI] [PubMed] [Google Scholar]
- 6.Alexander NJ, McCormick SP, Waalwijk C, van der Lee T, Proctor RH. The genetic basis for 3-ADON and 15-ADON trichothecene chemotypes in Fusarium. Fungal Genet Biol. 2011;48:485–95. doi: 10.1016/j.fgb.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Lee T, Han YK, Kim KH, Yun SH, Lee YW. Tri13 and Tri7 determine deoxynivalenol- and nivalenol-producing chemotypes of Gibberella zeae. Appl Env Microbiol. 2002;68:2148–54. doi: 10.1128/AEM.68.5.2148-2154.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cumagun CJR, Bowden RL, Jurgenson JE, Leslie J, Miedaner T. Genetic mapping of pathogenicity and agressiveness of Gibberella zeae (Fusarium graminearum) toward wheat. Phytopathology. 2004;94:520–6. [DOI] [PubMed]
- 9.Jin F, Bai G, Zhang D, Dong Y, Ma L, Bockus W, et al. Fusarium-damaged kernels and deoxynivalenol in Fusarium-infected U.S. winter wheat. Phytopathology. 2014;104:472–8. doi: 10.1094/PHYTO-07-13-0187-R. [DOI] [PubMed] [Google Scholar]
- 10.Paul PA, Lipps PE, Madden LV. Relationship between visual estimates of Fusarium head blight intensity and deoxynivalenol accumulation in harvested wheat grain: a meta-analysis. Phytopathology. 2005;95:1225–36. doi: 10.1094/PHYTO-95-1225. [DOI] [PubMed] [Google Scholar]
- 11.Talas F, Kalih R, Miedaner T, McDonald BA. Genome-wide association study identifies novel candidate genes for aggressiveness, deoxynivalenol production and azole sensitivity in natural field populations of Fusarium graminearum. Mol Plant-Microbe Interact. 2016;10:1094–29. doi: 10.1094/MPMI-09-15-0218-R. [DOI] [PubMed] [Google Scholar]
- 12.Terzi V, Tumino G, Stanca AM, Morcia C. Reducing the incidence of cereal head infection and mycotoxins in small grain cereal species. J Cereal Sci. 2014;59:284–93. doi: 10.1016/j.jcs.2013.10.005. [DOI] [Google Scholar]
- 13.Chen Y, Zhou M-G. Characterization of Fusarium graminearum Isolates resistant to both carbendazim and a new fungicide JS399-19. Phytopathology. 2009;99:441–6. doi: 10.1094/PHYTO-99-4-0441. [DOI] [PubMed] [Google Scholar]
- 14.Talas F, McDonald BA. Significant variation in sensitivity to a DMI fungicide in field populations of Fusarium graminearum. Plant Pathol. 2015;64:664–70. doi: 10.1111/ppa.12280. [DOI] [Google Scholar]
- 15.Ward TJ, Clear RM, Rooney AP, O’Donnell K, Gaba D, Patrick S, et al. An adaptive evolutionary shift in Fusarium head blight pathogen populations is driving the rapid spread of more toxigenic Fusarium graminearum in North America. Fungal Genet Biol. 2008;45:473–84. doi: 10.1016/j.fgb.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Zeller KA, Bowden RL, Leslie JF. Population differentiation and recombination in wheat scab populations of Gibberella zeae from the United States. Mol Ecol. 2004;13:563–71. doi: 10.1046/j.1365-294X.2004.02098.x. [DOI] [PubMed] [Google Scholar]
- 17.Akinsanmi OA, Backhouse D, Simpfendorfer S, Chakraborty S. Genetic diversity of Australian Fusarium graminearum and F. pseudograminearum. Plant Pathol. 2006;55:494–504. doi: 10.1111/j.1365-3059.2006.01398.x. [DOI] [Google Scholar]
- 18.Gale LR, Ward TJ, Balmas V, Kistler HC. Population subdivision of Fusarium graminearum sensu stricto in the upper Midwestern United States. Phytopathology. 2007;97:1434–9. doi: 10.1094/PHYTO-97-11-1434. [DOI] [PubMed] [Google Scholar]
- 19.Leslie JF, Anderson LL, Bowden RL, Lee Y-W. Inter- and intra-specific genetic variation in Fusarium. Int J Food Microbiol. 2007;119:25–32. doi: 10.1016/j.ijfoodmicro.2007.07.059. [DOI] [PubMed] [Google Scholar]
- 20.Boutigny A-L, Ward T, Ballois N, Iancu G, Ioos R. Diversity of the Fusarium graminearum species complex on French cereals. Eur J Plant Pathol. 2014;138:133–48. doi: 10.1007/s10658-013-0312-6. [DOI] [Google Scholar]
- 21.Liang JM, Xayamongkhon H, Broz K, Dong Y, McCormick SP, Abramova S, et al. Temporal dynamics and population genetic structure of Fusarium graminearum in the upper Midwestern United States. Fungal Genet Biol. 2014;73:83–92. doi: 10.1016/j.fgb.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Van der Lee T, Zhang H, van Diepeningen A, Waalwijk C. Biogeography of Fusarium graminearum species complex and chemotypes: a review. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2015;32:453–60. doi: 10.1080/19440049.2014.984244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonald B, Linde C. The population genetics of plant pathogens and breeding strategies for durable resistance. Euphytica. 2002;124:163–80. doi: 10.1023/A:1015678432355. [DOI] [Google Scholar]
- 24.Talas F, McDonald B. Genome-wide analysis of Fusarium graminearum field populations reveals hotspots of recombination. BMC Genomics. 2015;16:996. doi: 10.1186/s12864-015-2166-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yun SH, Arie T, Kaneko I, Yoder OC, Turgeon BG. Molecular organization of mating type loci in heterothallic, homothallic, and asexual Gibberella/Fusarium species. Fungal Genet Biol. 2000;31:7–20. doi: 10.1006/fgbi.2000.1226. [DOI] [PubMed] [Google Scholar]
- 26.Keller M, Bergstrom G, Shields E. The aerobiology of Fusarium graminearum. Aerobiologia (Bologna) 2014;30:123–36. doi: 10.1007/s10453-013-9321-3. [DOI] [Google Scholar]
- 27.Maldonado-Ramirez SL, Schmale Iii DG, Shields EJ, Bergstrom GC. The relative abundance of viable spores of Gibberella zeae in the planetary boundary layer suggests the role of long-distance transport in regional epidemics of Fusarium head blight. Agric For Meteorol. 2005;132:20–7. doi: 10.1016/j.agrformet.2005.06.007. [DOI] [Google Scholar]
- 28.Prussin AJ, Li Q, Malla R, Ross SD, Schmale DG. Monitoring the long-distance transport of Fusarium graminearum from field-scale sources of inoculum. Plant Dis. 2013;98:504–11. doi: 10.1094/PDIS-06-13-0664-RE. [DOI] [PubMed] [Google Scholar]
- 29.Prussin AJ, Szanyi NA, Welling PI, Ross SD, Schmale DG. Estimating the Production and Release of Ascospores from a Field-Scale Source of Fusarium graminearum Inoculum. Plant Dis. 2013;98:497–503. doi: 10.1094/PDIS-04-13-0404-RE. [DOI] [PubMed] [Google Scholar]
- 30.Schmale Iii DG, Leslie JF, Zeller KA, Saleh AA, Shields EJ, Bergstrom GC. Genetic structure of atmospheric populations of Gibberella zeae. Phytopathology. 2006;96:1021–6. doi: 10.1094/PHYTO-96-1021. [DOI] [PubMed] [Google Scholar]
- 31.Stukenbrock EH, McDonald BA. The origins of plant pathogens in agro-ecosystems. Annu Rev Phytopathol. 2008;46:75–100. doi: 10.1146/annurev.phyto.010708.154114. [DOI] [PubMed] [Google Scholar]
- 32.Pombert J-F, Xu J, Smith DR, Heiman D, Young S, Cuomo CA, et al. Complete genome sequences from three genetically distinct strains reveal high intraspecies genetic diversity in the microsporidian Encephalitozoon cuniculi. Eukaryot Cell. 2013;12:503–11. doi: 10.1128/EC.00312-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ojeda DI, Dhillon B, Tsui CKM, Hamelin RC. Single-nucleotide polymorphism discovery in Leptographium longiclavatum, a mountain pine beetle-associated symbiotic fungus, using whole-genome resequencing. Mol Ecol Resour. 2014;14:401–10. doi: 10.1111/1755-0998.12191. [DOI] [PubMed] [Google Scholar]
- 34.Zander M, Patel DA, Van de Wouw A, Lai KT, Lorenc MT, Campbell E, et al. Identifying genetic diversity of avirulence genes in Leptosphaeria maculans using whole genome sequencing. Funct Integr Genomics. 2013;13:294–308. doi: 10.1007/s10142-013-0324-5. [DOI] [PubMed] [Google Scholar]
- 35.Abdolrasouli A, Rhodes J, Beale MA, Hagen F, Rogers TR, Chowdhary A, et al. Genomic context of azole resistance mutations in Aspergillus fumigatus determined using whole-genome sequencing. MBio. 2015;6:e00536–15. doi: 10.1128/mBio.00536-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hane JK, Anderson JP, Williams AH, Sperschneider J, Singh KB. Genome sequencing and comparative genomics of the broad host-range pathogen Rhizoctonia solani AG8. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Persoons A, Morin E, Delaruelle C, Payen T, Halkett F, Frey P, et al. Patterns of genomic variation in the poplar rust fungus Melampsora larici-populina identify pathogenesis-related factors. Front Plant Sci. 2014;5. [DOI] [PMC free article] [PubMed]
- 38.Atwell S, Corwin JA, Soltis NE, Subedy A, Denby KJ, Kliebenstein DJ. Whole genome resequencing of Botrytis cinerea isolates identifies high levels of standing diversity. Front Microbiol. 2015;6:996. doi: 10.3389/fmicb.2015.00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDonald MC, Williams AH, Milgate A, Pattemore JA, Solomon PS, Hane JK. Next-generation re-sequencing as a tool for rapid bioinformatic screening of presence and absence of genes and accessory chromosomes across isolates of Zymoseptoria tritici. Fungal Genet Biol. 2015;79:71–5. doi: 10.1016/j.fgb.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 40.Fedorova ND, Khaldi N, Joardar VS, Maiti R, Amedeo P, Anderson MJ, et al. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 2008;4:e1000046. doi: 10.1371/journal.pgen.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.King R, Urban M, Hammond-Kosack M, Hassani-Pak K, Hammond-Kosack K. The completed genome sequence of the pathogenic ascomycete fungus Fusarium graminearum. BMC Genomics. 2015;16:544. doi: 10.1186/s12864-015-1756-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuomo CA, Guldener U, Xu JR, Trail F, Turgeon BG, Di Pietro A, et al. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science. 2007;317:1400–2. doi: 10.1126/science.1143708. [DOI] [PubMed] [Google Scholar]
- 43.Wong P, Walter M, Lee W, Mannhaupt G, Münsterkötter M, Mewes H-W, et al. FGDB: revisiting the genome annotation of the plant pathogen Fusarium graminearum. Nucleic Acids Res. 2011;39:D637–9. doi: 10.1093/nar/gkq1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown NA, Antoniw J, Hammond-Kosack KE. The predicted secretome of the plant pathogenic fungus Fusarium graminearum: a refined comparative analysis. PLoS One. 2012;7 doi: 10.1371/journal.pone.0033731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rampitsch C, Day J, Subramaniam R, Walkowiak S. Comparative secretome analysis of Fusarium graminearum and two of its non-pathogenic mutants upon deoxynivalenol induction in vitro. Proteomics. 2013;13:1913–21. doi: 10.1002/pmic.201200446. [DOI] [PubMed] [Google Scholar]
- 46.Lowe RGT, McCorkelle O, Bleackley M, Collins C, Faou P, Mathivanan S, et al. Extracellular peptidases of the cereal pathogen Fusarium graminearum. Front Plant Sci. 2015;6. [DOI] [PMC free article] [PubMed]
- 47.Phalip V, Delalande F, Carapito C, Goubet F, Hatsch D, Leize-Wagner E, et al. Diversity of the exoproteome of Fusarium graminearum grown on plant cell wall. Curr Genet. 2005;48:366–79. doi: 10.1007/s00294-005-0040-3. [DOI] [PubMed] [Google Scholar]
- 48.Sieber CMK, Lee W, Wong P, Münsterkötter M, Mewes H-W, Schmeitzl C, et al. The Fusarium graminearum genome reveals more secondary metabolite gene clusters and hints of horizontal gene transfer. PLoS One. 2014;9 doi: 10.1371/journal.pone.0110311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Timmons JA, Szkop KJ, Gallagher IJ, Keller P, Vollaard N, Gustafsson T, et al. Multiple sources of bias confound functional enrichment analysis of global -omics data. Genome Biol. 2015;16:46–59. doi: 10.1186/s13059-015-0761-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harris LJ, Balcerzak M, Johnston A, Schneiderman D, Ouellet T. Host-preferential Fusarium graminearum gene expression during infection of wheat, barley, and maize. Fungal Biol. 2016;120:111–23. doi: 10.1016/j.funbio.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 51.Bai GH, Desjardins AE, Plattner RD. Deoxynivalenol-nonproducing Fusarium graminearum causes initial infection, but does not cause disease spread in wheat spikes. Mycopathologia. 2002;153:91–8. doi: 10.1023/A:1014419323550. [DOI] [PubMed] [Google Scholar]
- 52.Pagani F, Baralle FE. Opinion: Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet. 2004;5:389–96. doi: 10.1038/nrg1327. [DOI] [PubMed] [Google Scholar]
- 53.Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15:121–32. doi: 10.1038/nrg3642. [DOI] [PubMed] [Google Scholar]
- 54.Walkowiak S, Bonner CT, Wang L, Blackwell B, Rowland O, Subramaniam R. Intraspecies Interaction of Fusarium graminearum Contributes to Reduced Toxin Production and Virulence. Mol Plant Microbe Interact. 2015;28:1256–67. doi: 10.1094/MPMI-06-15-0120-R. [DOI] [PubMed] [Google Scholar]
- 55.Cantu D, Segovia V, MacLean D, Bayles R, Chen X, Kamoun S, et al. Genome analyses of the wheat yellow (stripe) rust pathogen Puccinia striiformis f. sp. tritici reveal polymorphic and haustorial expressed secreted proteins as candidate effectors. BMC Genomics. 2013;14:270. doi: 10.1186/1471-2164-14-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neafsey DE, Barker BM, Sharpton TJ, Stajich JE, Park DJ, Whiston E, et al. Population genomic sequencing of Coccidioides fungi reveals recent hybridization and transposon control. Genome Res. 2010;20:938–46. doi: 10.1101/gr.103911.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gladieux P, Ropars J, Badouin H, Branca A, Aguileta G, de Vienne DM, et al. Fungal evolutionary genomics provides insight into the mechanisms of adaptive divergence in eukaryotes. Mol Ecol. 2014;23:753–73. doi: 10.1111/mec.12631. [DOI] [PubMed] [Google Scholar]
- 59.Sperschneider J, Gardiner DM, Thatcher LF, Lyons R, Singh KB, Manners JM, et al. Genome-wide analysis in three Fusarium pathogens identifies rapidly evolving chromosomes and genes associated with pathogenicity. Genome Biol Evol. 2015;7:1613–27. doi: 10.1093/gbe/evv092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma L-J, van der Does HC, Borkovich KA, Coleman JJ, Daboussi M-J, Di Pietro A, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464:367–73. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raffaele S, Kamoun S. Genome evolution in filamentous plant pathogens: why bigger can be better. Nat Rev Microbiol. 2012;10:417. doi: 10.1038/nrmicro2790. [DOI] [PubMed] [Google Scholar]
- 62.Croll D, Lendenmann MH, Stewart E, McDonald BA. The impact of recombination hotspots on genome evolution of a fungal plant pathogen. Genetics. 2015;201:1213–28. doi: 10.1534/genetics.115.180968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Connolly LR, Smith KM, Freitag M. The Fusarium graminearum histone H3 K27 methyltransferase KMT6 regulates development and expression of secondary metabolite gene clusters. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McVicker G, Gordon D, Davis C, Green P. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Supek F, Miñana B, Valcárcel J, Gabaldón T, Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014;156:1324–35. doi: 10.1016/j.cell.2014.01.051. [DOI] [PubMed] [Google Scholar]
- 66.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dong S, Raffaele S, Kamoun S. The two-speed genomes of filamentous pathogens: waltz with plants. Curr Opin Genet Dev. 2015;35:57–65. doi: 10.1016/j.gde.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 68.Raffaele S, Farrer RA, Cano LM, Studholme DJ, MacLean D, Thines M, et al. Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science. 2010;330:1540–3. doi: 10.1126/science.1193070. [DOI] [PubMed] [Google Scholar]
- 69.Croll D, McDonald BA. The accessory genome as a cradle for adaptive evolution in pathogens. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Galazka JM, Freitag M. Variability of chromosome structure in pathogenic fungi—of ‘ends and odds’. Curr Opin Microbiol. 2014;20:19–26. doi: 10.1016/j.mib.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Talas F, Kalih R, Miedaner T. Within-field variation of Fusarium graminearum isolates for aggressiveness and deoxynivalenol production in wheat head blight. Phytopathology. 2012;102:128–34. doi: 10.1094/PHYTO-06-11-0162. [DOI] [PubMed] [Google Scholar]
- 72.Talas F, Wurschum T, Reif JC, Parzies HK, Miedaner T. Association of single nucleotide polymorphic sites in candidate genes with aggressiveness and deoxynivalenol production in Fusarium graminearum causing wheat head blight. BMC Genet. 2012;13:14. doi: 10.1186/1471-2156-13-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Menke J, Weber J, Broz K, Kistler HC. Cellular development associated with induced mycotoxin synthesis in the filamentous fungus Fusarium graminearum. PLoS One. 2013;8 doi: 10.1371/journal.pone.0063077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kistler HC, Broz K. Cellular compartmentalization of secondary metabolism. Front Microbiol. 2015;6:68. doi: 10.3389/fmicb.2015.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pinson-Gadais L, Foulongne-Oriol M, Ponts N, Barreau C, Richard-Forget F. The French Fusarium Collection: a living resource for mycotoxin research. Fungal Genet Rep. 2013;60(Suppl):295. [Google Scholar]
- 76.Trail F, Common R. Perithecial development by Gibberella zeae: a light microscopy study. Mycologia. 2000;92:130. doi: 10.2307/3761457. [DOI] [Google Scholar]
- 77.O’Donnell K, Kistler HC, Tacke BK, Casper HH. Gene genealogies reveal global phylogeographic structure and reproductive isolation among lineages of Fusarium graminearum, the fungus causing wheat scab. Proc Natl Acad Sci U S A. 2000;97:7905–10. doi: 10.1073/pnas.130193297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. [Google Scholar]
- 79.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and Indels. PLoS One. 2012;7 doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stajich JE, Harris T, Brunk BP, Brestelli J, Fischer S, Harb OS, et al. FungiDB: an integrated functional genomics database for fungi. Nucleic Acids Res. 2012;40:D675–81. doi: 10.1093/nar/gkr918. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Strains are available from the International Center for Microbial Resources (CIRM, (http://www6.inra.fr/cirm)), with codes 1974, 1977, 1981, 1988, 1999 and 2013 for strains INRA-156, INRA-159, INRA-164, INRA-171, INRA-181 and INRA-195 respectively. The entire set of raw reads that was generated in this project has been deposited in fastq format at the GenBank Sequence Read Archive under the accession number SRP064374. The description and annotation of variant supporting the conclusions of this article is accessible in Additional file 3: Table S3.