

Abstract
Background
Metabolic engineering and synthetic biology of cyanobacteria offer a promising sustainable alternative approach for fossil-based ethylene production, by using sunlight via oxygenic photosynthesis, to convert carbon dioxide directly into ethylene. Towards this, both well-studied cyanobacteria, i.e., Synechocystis sp PCC 6803 and Synechococcus elongatus PCC 7942, have been engineered to produce ethylene by introducing the ethylene-forming enzyme (Efe) from Pseudomonas syringae pv. phaseolicola PK2 (the Kudzu strain), which catalyzes the conversion of the ubiquitous tricarboxylic acid cycle intermediate 2-oxoglutarate into ethylene.
Results
This study focuses on Synechocystis sp PCC 6803 and shows stable ethylene production through the integration of a codon-optimized version of the efe gene under control of the Ptrc promoter and the core Shine–Dalgarno sequence (5′-AGGAGG-3′) as the ribosome-binding site (RBS), at the slr0168 neutral site. We have increased ethylene production twofold by RBS screening and further investigated improving ethylene production from a single gene copy of efe, using multiple tandem promoters and by putting our best construct on an RSF1010-based broad-host-self-replicating plasmid, which has a higher copy number than the genome. Moreover, to raise the intracellular amounts of the key Efe substrate, 2-oxoglutarate, from which ethylene is formed, we constructed a glycogen-synthesis knockout mutant (ΔglgC) and introduced the ethylene biosynthetic pathway in it. Under nitrogen limiting conditions, the glycogen knockout strain has increased intracellular 2-oxoglutarate levels; however, surprisingly, ethylene production was lower in this strain than in the wild-type background.
Conclusion
Making use of different RBS sequences, production of ethylene ranging over a 20-fold difference has been achieved. However, a further increase of production through multiple tandem promoters and a broad-host plasmid was not achieved speculating that the transcription strength and the gene copy number are not the limiting factors in our system.
Keywords: Ethylene, Cyanobacteria, Sustainable, Synechocystis, Glycogen, Oxoglutarate, Arginine
Background
An increasing world population coupled with the need to reduce greenhouse gas emissions is driving the need to explore sustainable and renewable alternatives to the production of bulk fuels and chemicals from fossil sources. Ethylene is one of the most widely produced organic compounds and the raw material for polyethylene for consumables, ethylene oxide/glycol for polyethylene terephthalate (PET)’, resins for PET fiber, bottles and other packaging materials, and ethylene dichloride for PVC plastic uses in construction and piping. The global demand for the above products is leading to increased ethylene consumption which is expected to further grow at about 4% per year over the couple of years [17]. The traditional production methods based on steam cracking of naphtha from petroleum sources, and ethane from natural gas, are under pressure from society, due to the finite oil/gas supplies and large quantities of greenhouse gases produced (1.5–3.0 ton of carbon dioxide for every ton of ethylene produced). Hence there is an urgent need to explore fossil fuel free, sustainable, carbon–neutral ways to produce this important chemical and in this context, microbial biotechnology is leading the way.
Ethylene is biologically synthesized in plants and is an important plant hormone involved in regulation of biological processes like germination and fruit ripening [21, 37]. In higher plants, ethylene is produced from methionine in a three-step reaction via amino-propane carboxylic acid (ACC) in a pathway better known as the ACC pathway or the Yang cycle [1, 10]. Some microbes also produce ethylene, using two distinct pathways, neither of which is found in plants [10]. Most prokaryotes use methionine as the starting substrate and convert it into ethylene in a two-step process via 2-keto-4-methylthiobutyric acid (KMBA), catalyzed by an NADH:Fe-EDTA oxidoreductase (the KMBA pathway) [27]. Many plant pathogens, however, like Pseudomonas syringae and Pencillium digitatum, produce ethylene to weaken their hosts’ defense and they synthesize ethylene directly from 2-oxoglutarate, an important tricarboxylic acid (TCA) cycle intermediate, in an efficient single-step reaction, catalyzed by the ‘ethylene forming enzyme’ (Efe) [20, 26, 40]. This 2-oxoglutarate-dependent pathway has been expressed in a range of bacteria [9, 18, 19], fungi [6, 34] and yeast [28] and this has been covered in detail in a recent review [8].
Out of the various microbial model systems so far studied for heterologous expression of the Efe enzyme, metabolic engineering and synthetic biology of cyanobacteria [16, 31, 38], offers the most promising approach, as these photoautotrophic bacteria can use sunlight to perform oxygenic photosynthesis and thereby sequester and recycle carbon dioxide directly into ethylene. A techno-economic analysis of such a conceptual production process was also recently published [25]. The first studies on production of ethylene in cyanobacteria were carried out in Synechococcus elongatus PCC 7942 (hereafter referred to as Synechococcus), by introducing the efe gene from Pseudomonas syringae pv. phaseolicola PK2 (the Kudzu strain) with the promoter and terminator of the efe gene also from P. syringae [11] into this cyanobacterium. An ethylene production rate of 53 µl/l/h/OD730 was obtained. Replacing the native promoter of the efe gene with the promoter of the psbAI gene (from Synechococcus) and removing the terminator further increased the specific rate of ethylene formation sixfold to 323 µl/l/h/OD730 [30]. However, this strain was unstable due to the occurrence of homologous recombination events between the psbAI gene on the host’s chromosome and the psbAI and efe gene sequences on the plasmid, resulting in a rapid decrease in ethylene production in recombinant cyanobacterial cultures. To circumvent the above problem, the efe gene was then inserted into the chromosomal psbAI locus. This increased the rate of ethylene–formation to 451 µl/l/h/OD730, but the recombinant strain showed a lowered specific growth rate and was genetically unstable due to the presence of mutational hotspots [33]. Recently, stable and continuous ethylene production was achieved in Synechocystis sp. PCC 6803 (hereafter referred to as Synechocystis) in a study in which a codon-optimized efe gene (with mutational hot-spots eliminated) was expressed from a constitutive, pea plant chloroplast psbA promoter, and integrated at the slr0168 neutral-site. A specific ethylene production rate of about 65 µL/L/h/OD730 was achieved in this way, in a strain with one copy of the efe gene, and grown with white light of an intensity of 50 μmol photons/m2/s1, and this production rate was more than doubled (i.e. to about 130 µL/L/h/OD730) in a strain with two copies of the efe gene. By further optimizing growth conditions, a peak volumetric production rate of 5600 µl/l/h and a continuous production rate of 3100 µl/l/h were achieved [35]. Stable ethylene production was also reported in Synechocystis by expressing the Kudzu Efe constitutively from a derivative of the broad-host range low-copy RSF1010 plasmid, under control of the Ptrc promoter, which resulted in production rates of about 170 µl/l/h/OD730 [14]. Very recently, high ethylene production rates were reported in a Synechocystis strain carrying multiple copies of the codon-optimized efe gene from Pseudomonas syringae pv. sesame, under control of the PcpcB promoter (selected after considerable promoter screening efforts) [41]. The authors further increased production rates by optimizing growth conditions and feeding the cells with 2-oxoglutarate, in combination with heterologous expression of a 2-oxoglutarate permease, to achieve an ethylene production rate of 858 µl/l/h/OD730.
In this study, we achieved stable ethylene production by integrating the codon-optimized efe gene into Synechocystis under control of the Ptrc promoter and the core Shine-Dalgarno sequence (5′-AGGAGG-3′) as the ribosome binding site (RBS), at the slr0168 neutral site. We subsequently increased the ethylene production rate twofold through RBS screening. To better understand the bottlenecks towards further improving the rate of ethylene production, we studied gene expression using multiple consecutive promoters, and we used a broad host self-replicating plasmid, which has been reported to have about one to three times the copy number of the genome [4]. Moreover, instead of expressing a transporter and feeding the cells with 2-oxoglutarate, from which ethylene is formed by the Efe enzyme directly, we have engineered the cells to raise the intracellular generation of this key metabolite by knocking out the pathway towards glycogen synthesis and starving the cells for nitrogen.
Methods
Strains and growth conditions
Escherichia coli strains XL-1 Blue (Stratagene) or EPI400 (Epicentre Biotechnologies) were grown in lysogeny broth (LB) medium at 37 °C in a shaking incubator at 200 rpm or on plates augmented with 1.5% (w/v) agar. Antibiotics, kanamycin (50 µg/ml), chloramphenicol (35 µg/ml) and ampicillin (100 µg/ml) were added alone or in combination when needed.
Synechocystis sp. PCC 6803, obtained from D. Bhaya, University of Stanford, Stanford CA, was routinely cultivated in liquid BG-11 medium (Sigma-Aldrich), at 30º C, in a shaking incubator, under constant white light illumination (30 μmol photons/m2/s1). Cultures were supplemented with 20 mM TES-KOH, pH 8.0. Kanamycin resistant strains were grown in medium containing 50 µg/ml kanamycin. BG-11 plates were made with 1.5% (w/v) agar and additionally supplemented with 10 mM TES-KOH, pH 8.0, 0.3% (w/v) sodium thiosulphate and 50 µg/ml kanamycin for resistant strains. For nitrogen starvation experiments, nitrogen free BG-11 medium (BG-11No) was used and supplemented with 1 mM NaNO3. Cells were routinely stored at −80 °C in BG-11 medium supplemented with 5% (v/v) DMSO. Growth was monitored by following the optical density at 730 nm (OD730).
Natural transformation, in wild-type Synechocystis, as well as in the glycogen-synthesis knock-out strain, was performed as described previously [2, 3, 7]. Transformants were subjected to increasing concentrations of antibiotics to drive chromosome segregation. Conjugation of the pVZ derived plasmids from E. coli XL-1 to Synechocystis (both wild-type and the glycogen-synthesis knock-out strain) was carried out by tri-parental mating using E. coli J53 (pRP4) as helper strain as reported previously [32]. Kanamycin (50 µg/ml) was used to select for positive clones. Correct insertion of genes or plasmids was verified by colony PCR’s using Taq DNA polymerase (Thermo-Scientific).
Codon optimization and gene synthesis
The gene sequence of the ethylene-forming enzyme from Pseudomonas syringae pv. phaseolicola (Genebank: AAD16440.1) was taken from the NCBI protein database. Codon optimization and removal of restriction sites in the gene to be used for further cloning procedures was done using the OPTIMIZER application [29] and the codon usage table described in [32]. The gene was synthesized by GenScript (NJ, USA) and delivered as pSEQ_efe.
Plasmid construction
Integration plasmids: The efe gene was amplified from pSEQ-efe using efe-bb-fwd (contains the RBS-aggagg) and efe-bb-rev (Table 1), cut with EcoRI/SpeI (insert) and ligated into pSB1AC3_TT cut with EcoR1/Xba1 (vector) to obtain pSB1AC3_TT_efe. The rbs_efe-tt construct from above plasmid was cloned in line with the Ptrc promoter in plasmid pSEQ_trc [2] to get plasmid pSEQ-Ptrc_efe-tt. The whole efe expression cassette Ptrc_efe_tt was then subcloned into the pHKH001 integration plasmid [2]. For integration of plasmids to express efe under different RBS sequences, the efe gene was amplified with primers containing the RBS sequences (Table 1) and cloned back into the pHKH001 integration vectors. The 3xPtrc and 5xPtrc constructs were synthesized and directly inserted into pHKH001 by GenScript. The efe gene amplified with the Pbs-30-containing primer was then cloned behind the multiple promoter sequences to obtain the plasmids pHKH-3xtrc_rbs30_efe-tt and pHKH-5xtrc_rbs30_efe-tt.
Table 1.
Cloning parts, plasmids, strains and primers used in this study
| Cloning parts, plasmids, strains and primers | Description | Source/remark |
|---|---|---|
| RBS a | ||
| Rbs-O | AGGAGGACTAGCATG | [2] |
| Rbs-34 | ATTAAAGAGGAGAAAACTAGCATG | [15] |
| Rbs-30 | AAAGAGGAGAAAACTAGCATG | [15] |
| Rbs-H | TAGTGGAGGTACTAGCATG | RBS* from [15] |
| Rbs-C9 | AAAGGAGGTGATAGCATG | This study |
| Rbs-C10 | AAAGGAGGTGATTAGCATG | This study |
| Rbs-C11 | AAAGGAGGTGATCTAGCATG | This study |
| Promoter | ||
| Ptrc | TTGACAATTAATCATCCGGCTCGTATAATGTGTGGAATTGTGAGCGGATAACAATTTCACAC | [2] |
| 3 × Ptrc | TTGACAATTAATCATCCGGCTCGTATAATGTGTGGAATTGTGTTGACAATTAATCATCCGGCTCGTATAATGTGTGGAATTGTGTTGACAATTAATCATCCGGCTCGTATAATGTGTGGAATTGTGAGCGGATAACAATTTCACACA | This study |
| 5 × Ptrc | TTGACAATTAATCATCCGGCTCGTATAATGTGTGGAATTGTGTTGACAATTAATCATCCGGCTCGTATAATGTGTGGAATTGTGTTGACAATTAATCATCCGGCTCGTATAATGTGTGGAATTGTGTTGACAATTAATCATCCGGCTCGTATAATGTGTGGAATTGTGTTGACAATTAATCATCCGGCTCGTATAATGTGTGGAATTGTGAGCGGATAACAATTTCACACA | This study |
| Terminator | ||
| tt | Same as B0014 from http://www.partsregistry.org | [2] |
| Plasmids | ||
| pHKH001 | Integration vector disrupting slr0168 in Synechocystis genome with kanamycin resistance cassette a selection marker | [2] |
| pHKH_BB | Bio-brick compatible derivative of pHKH001 carrying the Ptrc promoter and tt-transcription terminator | This study |
| pHKH_rbsO_efe | Integration vector carrying efe under Ptrc promoter and Rbs-O | This study |
| pHKH_rbs34_efe | Integration vector carrying efe under Ptrc promoter and Rbs-34 | This study |
| pHKH_rbs30_efe | Integration vector carrying efe under Ptrc promoter and Rbs-30 | This study |
| pHKH_rbsH_efe | Integration vector carrying efe under Ptrc promoter and Rbs-H | This study |
| pHKH_rbsc9_efe | Integration vector carrying efe under Ptrc promoter and Rbs-C9 | This study |
| pHKH_rbsc10_efe | Integration vector carrying efe under Ptrc promoter and Rbs-C10 | This study |
| pHKH_rbsc11_efe | Integration vector carrying efe under Ptrc promoter and Rbs-C11 | This study |
| pHKH_3xPtrc_efe | Integration vector carrying efe under 3xPtrc promoter and Rbs-30 | This study |
| pHKH_5xPtrc_efe | Integration vector carrying efe under 5x Ptrc promoter and Rbs-30 | This study |
| pVZ_LS | A bio-brick compatible version of mobilizable broad host self-replication plasmid pVZ321 containing the RSF1010 replicon, and carrying the Ptrc promoter, Rbs30 and tt-transcription terminator | Plasmid pVZ321 from [42] |
| pVZ-efe | pVZ plasmid carrying efe gene under Ptrc promoter and rbs 30 | This study |
| pVZ_Nhis-efe | pVZ plasmid carrying N-terminal hexa-histidine tagged efe under promoter Ptrc and Rbs-30 | This study |
| Strains | ||
| Wild-type (WT) | Synechocystis sp. PCC6803, glucose tolerant, naturally transformable | Bhaya (Stanford) |
| VPV1 | WT, Slr0168::Ptrc-rbsO-efe-Kmr | This study |
| VPV2 | WT, Slr0168::Ptrc-rbs34-efe-Kmr | This study |
| VPV3 | WT, Slr0168::Ptrc-rbs30-efe-Kmr | This study |
| VPV4 | WT, Slr0168::Ptrc-rbsH-efe-Kmr | This study |
| VPV5 | WT, Slr0168::Ptrc-rbsc9-efe-Kmr | This study |
| VPV6 | WT, Slr0168::Ptrc-rbsc10-efe-Kmr | This study |
| VPV7 | WT, Slr0168::Ptrc-rbsc11-efe-Kmr | This study |
| VPV40 | WT, Slr0168::3xPtrc-rbsO-efe-Kmr | This study |
| VPV43 | WT, Slr0168::5xPtrc-rbsO-efe-Kmr | This study |
| SAW11 | Glycogen knock out strain | [36] |
| VPV55 | WT carrying plasmid pVZ _efe | This study |
| VPV56 | SAW11 carrying plasmid pVZ _efe | This study |
| VPV58 | SAW11 carrying plasmid pVZ_Nhis_efe | This study |
| VPV62 | WT carrying plasmid pVZ_Nhis_efe | This study |
| VPV65 | SAW11, Slr0168::Ptrc-rbs30-efe-Kmr | This study |
| Primers | ||
| efe-bb-fwd | GCGGAATTCGCGGCCGCTTCTAGAGGAGGACTAGCATGACCAACTTGCAAACCTTTGAAT | |
| efe-bb-r | GTACTGCAGCGGCCGCTACTAGTATTAGGAGCCGGTGGCGCG | |
| vpv-1-st-Hind-R | CTAGTAAAGCTTATCAATACTTTCCACCCC | |
| VPV-efe-Bamh1-f-noatg | CCATCACGGATCCACCAACTTGCAAACCTTTGAATTGC | |
| VPV-efe-st-Hind3-rev | TAATTAAGCTTATCAGGAGCCGGTGGCGCGGGTATCGG | |
| V-rbs30-efe-f | GACAGCTAGCATTAAAGAGGAGAAAACTAGCATGACCAACTTGCAAACC | |
| V-rbs34-efe-f | GACAGCTAGCAAAGAGGAGAAAACTAGCATGACCAACTTGCAAACC | |
| V-rbsC10-efe-f | GACAGCTAGCAAAGGAGGTGATTAGCATGACCAACTTGCAAACC | |
| V-rbsC11-efe-f | GACAGCTAGCAAAGGAGGTGATCTAGCATGACCAACTTGCAAACC | |
| V-rbsC9-efe-f | GACAGCTAGCAAAGGAGGTGATAGCATGACCAACTTGCAAACC | |
| V-rbsH-efe-f | GACAGCTAGCTAGTGGAGGTACTAGCATGACCAACTTGCAAACC | |
| v-bbs-pUC-r | CGGGATCCGATCCAATCTGCAGCGGCCGCTACTAGTA | |
| H1-SEG-VF | TGTCGCCGCTAAGTTAGACCGC | |
| H2-SEG-VR | CTGTGGGTAGTAAACTGGCAATGCC | |
| H1-SEQ-VF | CGGCAATGGTCCCAAAAT | |
| KanF-SEQ-VR | AGACGTTTCCCGTTGAAT | |
| pVZ321-SEQ-F | CGCAGGGCTTTATTGATT | |
| pVZ321-SEQ-R | CCCCCCCCACTCTATTGTA | |
| efe-n–H-f | CAGATATGACATATGCATCATCATCATCATCATACCAACTTGCAAACCTTTGAATTGC | |
| efe-ba-r | ACCTAGGTCAGTAGGATCCTTATTAGGAGCCGGTGGCGCGGGTATCG |
a RBS sequence underlined and start codon ATG is shown in bold
Self-replicating plasmids: a bio-brick compatible pVZ321 derivative, already carrying the Ptrc promoter and Rbs-30, was used as the starting point. The efe gene was amplified, and inserted into the NdeI/BamHI site of the pVZ321 derivative to obtain plasmids pVZ-efe.
Measurement of the biomass-specific rate of ethylene production
Ethylene production experiments were carried out in triplicate cultures that were in the late exponential growth phase prior to harvesting. Cultures were harvested around OD730 = 1 and were assayed. A 4 ml culture volume was transferred to a 20 ml crimp cap glass vial and 40 µl of 1 M sodium bicarbonate solution (resulting in 10 mM final concentration) was added. The vial was sealed with a butyl rubber stopper, and incubated at room temperature on a flat orbital shaker placed under a panel of fluorescent white lights, with a light intensity of about 250–300 μmol photons/m2/s1, for 2 h, after which the culture was heated to 80 °C for 10 min to stop ethylene production and all further metabolism. The quantitative determination of the amount of ethylene produced was carried out on an Agilent trace-ultra gas analyzer, by injecting 1 ml of headspace and using a standard program for ethylene analysis. An ethylene standard curve was made by injecting known volumes of pure ethylene into blank 20 ml crimp cap glass vials, carrying 4 ml of BG-11 medium and 40 µl of 1 M sodium bicarbonate solution.
For ethylene production under nitrogen limitation conditions 35 ml cultures of OD730 = 1.0 were collected, washed once with BG11No supplemented with 1 mM NaNO3 and 10 mM sodium bicarbonate, and re-suspended in the same medium, and incubated at low light intensity (30 μmol photons/m2/s1) at 30 °C. After 18 h, 4 mL of each culture was added to gas chromatography (GC) vials in duplicate, with 10 mM bicarbonate each, and incubated at low light (30 μmol photons/m2/s1) for 3 h and growth was stopped by heating at 80 °C. Low light was used in this case because of the sensitivity of the glycogen-synthesis knockout strain (i.e. ∆glgC strain and its derivatives) to high light. The same was repeated at 21, 24, 40, 46 and 63 h to study the dynamics elicited by nitrogen limitation. All figures were made using Grafit [22].
Method for measuring 2-oxoglutarate
2-oxoglutarate levels were measured as reported earlier [36]. Briefly, after measuring the ethylene in the headspace, the glass vials were de-capped and culture centrifuged for 10 min at 12,000 rpm and the supernatant subsequently filtered (Sartorius Stedim Biotech, Minisart SRP4, 0.45 μm). Separation of organic acids was achieved by application of a 20-μL aliquot on a Rezex ROA-Organic Acid H+ (8%) column (Phenomenex), coupled to a refractive index detector (Jasco, RI-1530), using a flow of 0.5 mL/min and a column temperature of 45 °C.
Ethylene production in aerated batch culture
Ethylene production under aeration was tested with the best strain, i.e. VPV3. This was carried out in a 1 l flat bottle fitted with a gas dispersion tube with a bottom frit at room temperature. A late exponential seed culture was diluted into 1 l of BG-11 medium and an initial OD730 of around 0.5 and cultivated for the next 7 days. The culture was continuously bubbled with CO2:AIR = 1:100 and the flow was maintained around 100 ml/min, Light was gradually increased from 30 to 220 μmol photons/m2/s1. From the third day onwards, 4 ml culture was withdrawn (in triplicate) and transferred to a 20 ml glass vial and sealed and incubated under the same conditions as the flat bottle reactor for 1 h, after which the culture was heated to 80 °C for 10 min to stop ethylene production (and all further metabolism). The amount of ethylene produced was determined as reported above.
Results
Introduction of the efe gene under control of the Ptrc promoter in Synechocystis
The amino-acid sequence encoding the ethylene-forming enzyme from Pseudomonas syringae pv. phaseolicola (Genebank: AAD16440.1) was codon optimized for the preferred codon usage of Synechocystis and synthesized. The gene was introduced under the control of the Ptrc promoter and the ribosome binding site (RBS) AGGAGG (Rbs-O, which is the six-base consensus sequence found in case of 57% of Escherichia coli genes and 26% of Synechocystis genes contain the same [24], Table 1), in combination with the transcription terminator BB0014 (parts.igem.org) in the pHKH integration [2] plasmid using standard bio-brick assembly techniques. The final plasmid construct was confirmed by sequencing and transformed into Synechocystis by natural transformation for integration of the efe expression cassette and a kanamycin resistance cassette at the neutral docking site slr0168. Transformants were selected on kanamycin plates and segregated. Complete segregation was confirmed by colony PCR. Ethylene accumulation in the headspace of a closed culture vessel was quantified by gas chromatography with a flame ionization detector (GC-FID). The specific rate of rate of ethylene production observed for this strain (i.e. VPV1; see Table 1) was 103 ± 1 µl/l/h/OD730 (Fig. 1B).
Fig. 1.
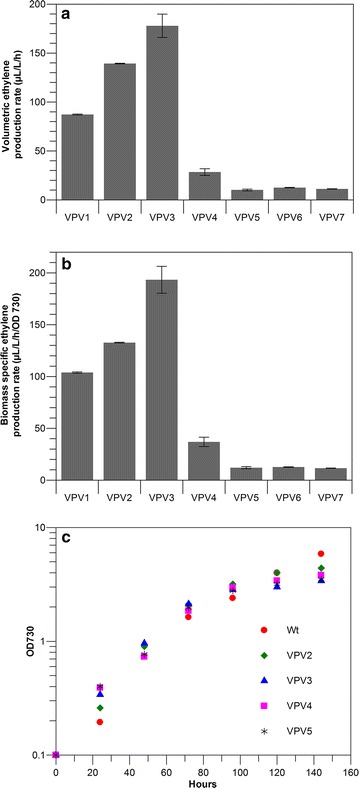
Rate of ethylene production in Synechocystis strains expressing the efe gene under control of the Ptrc promoter and various RBSs. a Comparison of volumetric ethylene production rates; b comparison of biomass specific ethylene production rates; c typical growth curves of the wild-type and ethylene producing Synechocystis strains. The genetic makeup of the VPV strains is detailed in Table 1. Error bars indicate standard deviation for triplicate measurements
Optimization of the ribosome binding site
Using PCR amplification with primers with overhangs, we generated and subsequently cloned 6 RBS sequences: Rbs-34, Rbs-30, Rbs-H, Rbs-C9, Rbs-C10 and Rbs-C11 in front of the efe gene, replacing the initially used Rbs-O sequence. Rbs-34, Rbs-30, Rbs-H were reported in literature (BBa_B0034, BBa_B0030 and Rbs* respectively from [15]. The Rbs-C sequence is the exact complement to the 3′-end of the 16S RNA sequence of Synechocystis. Rbs-C9, Rbs-C10 and Rbs-C11 differed in the optimal aligned spacing (OAS) between the RBS and the start codon, as described in [24], to determine whether the OAS has an impact on the level of efe expression and hence ethylene production. Using the above constructs, integration plasmids were made and transformed into Synechocystis. Transformants were obtained and they were also subjected to the procedure for segregation between the wild type- and mutant genotype. Successful segregation was confirmed by colony PCR. The strains carrying Rbs-34 (VPV2), Rbs-30 (VPV3), Rbs-H (VPV4), and Rbs-C9 (VPV5), Rbs-C10 (VPV6) and Rbs-C11 (VPV7) were grown in batch culture, to compare their growth (rate) with that of the wild type (Fig. 1). No significant growth defects were observed for any of the constructs, indicating that overexpression of the Efe enzyme has no severe effect on growth (rate) under the measured conditions. GC analysis for ethylene detection was carried out as described in the Methods section. All strains carrying the efe gene produced ethylene, and the biomass specific rate of ethylene production was quantified (Fig. 1). The strains based on Rbs-C, which is exactly the complement of the consensus 3′-end of the 16S ribosome sequence of Synechocystis, produced the lowest amount of ethylene (Fig. 1. Moreover those strains (VPV5, VPV6 and VPV7) produced ethylene at similar rates indicating that the OAS does not have a strong impact on the expression of Efe. Strain VPV3 showed the maximum specific ethylene production rate, followed by VPV2 and VPV4. This is contrary to current literature, where Rbs-H (VPV4) is reported to be the RBS resulting in strongest expression amongst this set of sequences [15]. This may be explained by the notion that translation efficiency from the RBS is also dependent on its local sequence context, because of the secondary structure formed by the mRNA at the 3′ region, and hence is also dependent on the gene sequence around the RBS. The maximum rate of ethylene formation observed for strain VPV3 (i.e. with Rbs-30), was 195 ± 12 µl/l/h/OD730, which is double that of strain VPV1 (i.e. with Rbs-O). To validate if the differences in ethylene production rate can be indeed linked to the level of the Efe enzyme in the cells, cell free extracts were prepared and loaded on SDS-PAGE gel subsequently stained with Coomassie brilliant blue. However, no clear bands for the Efe protein could be detected in any of the strains tested (data not shown).
Multiple promoters
Use of repeating identical promoters has been shown to increase expression of genes of interest in E. coli [23]. We have used a similar approach in Synechocystis and constructed two integration plasmids, one containing a triple Ptrc promoter (3×-Ptrc) and another one containing a quintuple Ptrc promoter (5×-Ptrc) in front of the Rbs-30-efe sequence, were made and confirmed by sequencing (Sequences see Table 1). The plasmids were transformed and the transformants were segregated as above, to obtain the strains VPV40 (3×-Ptrc) and VPV43 (5×-Ptrc). These strains were tested for their specific rate of ethylene production. Contrary to expectation, use of the in-tandem promoters did not lead to an increase in ethylene production (Fig. 2). Both strains VPV40 (3×-Ptrc) and VPV43 (5×-Ptrc) showed no significant difference in ethylene production compared to VPV3 (1×-Ptrc). This may suggest that these in-tandem promoter sequences do not increase expression significantly in our ethylene producing Synechocystis strains. Increased expression is expected to increase production as comparable ethylene producing systems [14, 35] show a corresponding increase in productivity. In those examples gene dosage has been increased through the introduction of two copies of the efe gene into the hosts’ genome.
Fig. 2.
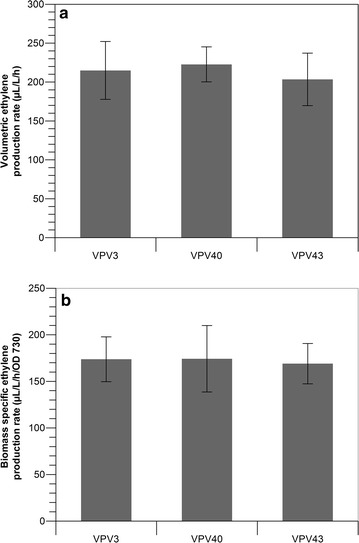
Rates of ethylene production form Synechocystis strains expressing the efe gene under control of in-tandem promoters. a Comparison of the volumetric ethylene production rate; b comparison of the biomass-specific rate of ethylene production. Error bars indicate standard deviation for triplicate measurements
Comparison of an integrated efe construct with expression of efe from a plasmid
The broad host-range self-replicating plasmid pVZ321 (based on the RSF1010 replicon, [42] has been reported to be present in Synechocystis in numbers three times as high as the copy number of the genome, and has also been shown to express higher levels of protein as compared to the expression from an equivalent construct with chromosomal integration [3, 4]. So, inserting the efe expression cassette Ptrc-Rbs30-efe in this plasmid could lead to a higher level of Efe expression than in the equivalent strain with chromosomal insertion. The codon-optimized efe gene was inserted behind a Ptrc promoter and Rbs30 in a bio-brick compatible pVZ321 derivative, to obtain the plasmid pVZ-efe. The vector was transformed into Synechocystis by conjugation. Transformants were selected on kanamycin plates and uptake of the plasmid was confirmed by colony PCR, to obtain the strain VPV55 (containing pVZ-efe). The strain was tested for ethylene production and compared with the chromosomal integration strain VPV3 (Fig. 3a). Interestingly, the strain carrying the plasmid produced less ethylene than the strain with the chromosomally integrated efe gene. Comparison of growth curves of these strains does not show a significant difference in the growth rate nor in the growth yield between the strain with the integrated gene (i.e. VPV3) and the plasmid carrying strains VPV55 and VPV62 (Fig. 3b). We therefore interpret this to be a consequence of the modest control that the amount of Efe enzyme will have on the in vivo rate of ethylene formation when the efe gene is expressed from the strong trc promotor in combination with a strong RBS.
Fig. 3.
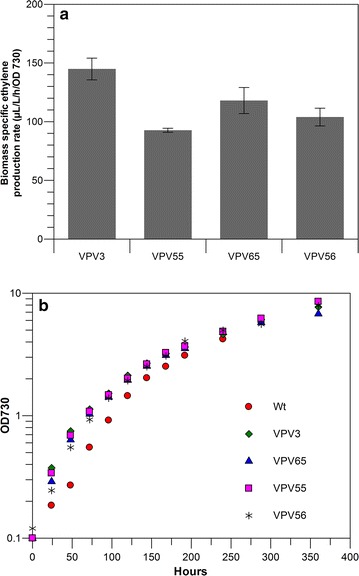
Comparison of rate of ethylene production between wild-type Synechocystis, expressing efe from a plasmid (VPV55) and as chromosomal integration (VPV3), and comparison with the corresponding ∆glgC strains (VPV56 and VPV65). a Biomass specific production rates; b typical growth curves of ethylene producing Synechocystis strains mentioned above in comparison to their wild-type. Error bars in a indicate standard deviation for triplicate measurements
Ethylene production in a ∆glgC strain under standard growth conditions
It is reported in literature that the deletion of the glgC gene (slr1176, which encodes glucose-1-phosphate adenyl transferase) of the glycogen-synthesis pathway, leads to accumulation of 2-oxoglutarate under nitrogen-limiting growth conditions [5, 13]. As 2-oxoglutarate is the substrate for the Efe enzyme for the production of ethylene, improving the intracellular concentration of 2-oxoglutarate may improve the rate of ethylene production. A glycogen synthesis knock-out strain was made by replacing the glgC gene with a chloramphenicol resistance cassette (SAW011), as has been reported elsewhere [36]. This strain was transformed with the pHKH_Ptrc_Rbs30_efe, to insert the efe gene into the slr0168 locus, as was earlier done for wild type Synechocystis. Transformants were selected on kanamycin and segregated. The segregated strain was confirmed by colony PCR to obtain strain VPV65 (∆glgC-efe). The plasmid pVZ-efe was also conjugated into SAW011 and selected on kanamycin to obtain the corresponding strain VPV56 (∆glgC-pVZ-efe). The strains were then tested under standard growth conditions for the rate of ethylene production (Fig. 3a). Here again, it is observed that the strains carrying efe on a plasmid produce less ethylene than the strain in which the efe gene is integrated into the genome. However, the difference between VPV65 and VPV56 is much less pronounced than between the respective wild-type background strains (VPV3 and VPV55). Interestingly, the glycogen synthesis knockout strains, carrying the efe gene on a plasmid, produce more than the corresponding wild-type background strain carrying the same plasmid, while their growth curves do not show any pronounced negative effect on growth rate nor growth yield (Fig. 3b).
Ethylene production in a glycogen knock-out strain under nitrogen limitation
The ethylene producing wild-type background strains VPV3 (with an efe gene integrated on the genome) and VPV55 (with the efe gene on a plasmid) and the glycogen synthesis knock-out strains VPV65 (with an efe gene integrated on the genome) and VPV56 (with the efe gene on a plasmid) were selected for tests of ethylene production in nitrogen-limitation experiments. Exponential phase cultures were washed with nitrate free BG-11 medium (BG-11No), supplemented with 1 mM sodium nitrate, and then resuspended in the same medium. Samples were withdrawn after 18, 21, 24, 40, 46 and 63 h incubation at low incident light intensity (50 μmol photon/m2/s1) and ethylene production rates were determined in the same low-light conditions (Fig. 4). A clear drop in ethylene production is observed between 24 and 40 h even though the cultures show a positive change in OD730 indicating that (non-exponential) growth still continues. As expected, the wild type strain grows to an OD of about twice that of the glycogen synthesis deficient strains at 63 h, confirming observations reported in literature [13], on the ability of the wild-type strain to undergo a final division cycle under N-limitation. The glycogen synthesis knock-out strains VPV65 and VPV56, under nitrogen limitation, show higher 2-oxoglutarate levels than the wild-type background strain at the 46 and 63 h time points (Fig. 4b), and hence were expected to have produced more ethylene. However, at all time points, the wild-type background strains produce more than the glycogen synthesis knock-out strains, indicating that other factors such as a likely drop in the intracellular arginine concentration, or overall protein levels upon nitrogen starvation, now may be affecting the production rate of ethylene.
Fig. 4.
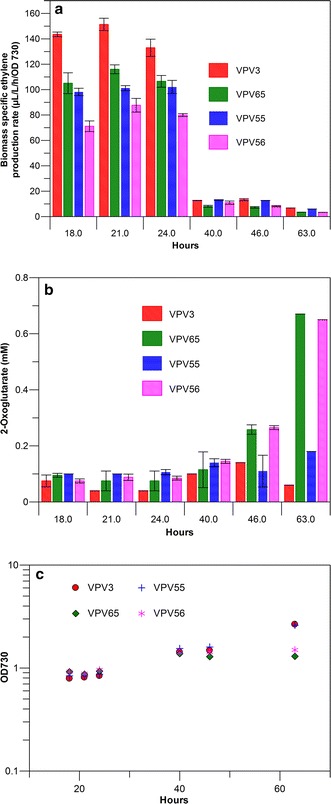
Comparison of ethylene production in wild type and in a ∆glgC strain, with a plasmid- and a chromosome-incorporated efe gene. a Comparison of Biomass-specific ethylene production rates between wild-type Synechocystis strains expressing the efe gene from a plasmid (VPV55) or as a chromosomal integration construct (VPV3), in comparison to the glycogen-synthesis deficient Synechocystis strains expressing the efe gene on a plasmid (VPV56) or as a chromosomal integration construct (VPV65) under nitrogen limitation conditions, induced by growing the cells in nitrogen-restricted medium. b 2-Oxoglutarate measured in the supernatants of the ethylene-producing strains after measurement of the rate of ethylene-production. c Typical growth data of the mutant strains during the period in which the ethylene production experiments were conducted. Error bars indicate standard deviation for triplicate measurements for (a) and standard deviation for duplicate measurements for (b)
Ethylene production in an aerated batch culture
The best ethylene-producing strain (VPV3, with an integrated efe gene and Rbs-30) was chosen for further production experiments. This strain was grown in a specially-designed 1 L flat glass bottle, fitted with a fine sparger, and grown for 7 days while the light intensity was gradually increased from 30 to 220 μmol photon/m2/s1. The volumetric ethylene production was then determined. We were able to achieve a maximum volumetric ethylene production rate of 443 µl/l/h at the higher light intensity of 220 μmol photon/m2/s1 (Fig. 5). This value is similar to the values reported by others [35] using a single copy of the efe gene but almost double the value reported for efe gene under the PcpcB promoter [41] indicating that the Ptrc promoter may be a stronger promoter than the PcpcB promoter. A similar observation is reported recently for isoprene production in Synechococcus elongatus [12].
Fig. 5.
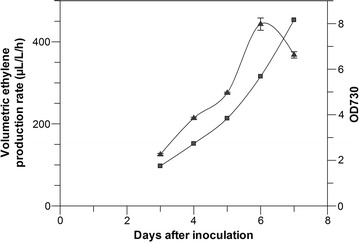
Ethylene production and growth with aerated batch cultivation. Volumetric ethylene production (filled triangles, levels indicated on the left y-axis) and OD730 (filled triangles, levels indicated on the right y-axis) plotted against time after inoculation. Error bars indicate standard deviation for triplicate measurements. Lines in the figure serve as a guide to the eye
Discussion
Here we test several classical approaches from metabolic engineering with the aim to increase ethylene production in a genetically modified cyanobacterium. We systematically compare 7 different ribosomal binding sites with respect to ethylene production levels. We find a different ranking of the ribosomal binding sites compared to a previous expression study in Synechocystis which quantified heterologous expression level by GFP fluorescence using partially the same (overall 4) ribosomal binding sites [15]. Accordingly, we show that a broad range of productivities (20-fold difference) can be achieved through different RBS sequences, However, this variation is not a function of the RBS sequence alone; i.e. it is also dependent on the gene sequence following the RBS, and hence the way forward will be to further engineer ribosome binding sites to achieve higher productivities. Indeed, using an engineered ribosome binding site, [39] have recently shown 3.9-fold higher ethylene productivity in a Synechocystis strain carrying a single copy of efe gene per genome. Next we adopted the strategy of adding identical promoters repeatedly upstream the gene of interest, an approach which had resulted in successful subsequent GFP-overexpression in E. coli [23]. However, in our case a single copy of Ptrc, three and even five copies, all result in similar ethylene production rates. It has been suggested that a RSF1010-based plasmid, which is widely used in cyanobacterial research, reaches higher a copy-number per cell than the genome [4] and results in higher expression of a heterologous gene with subsequent higher rates of product formation [3]. Here, however, we do not find higher ethylene production. Thus the best-performing RBS in combination with subsequent transcription-increasing strategies (in-tandem promoters, plasmid-based expression) does not increase the productivity. These experiments are therefore most straightforwardly interpreted by assuming that the gene copy number is not the limiting factor in our system. Rather, the activity of the ethylene forming enzyme may have reached such a (high) level already so that the control by the amount of Efe activity over the flux towards the product is low or even absent. Therefore, we then asked if increased substrate availability would increase the rate of ethylene production: N-limitation in a glycogen-knockout strain results in intra- and extracellular accumulation of 2-oxoglutarate; however, also this did not result in higher ethylene production levels suggesting that the bottleneck is elsewhere in metabolism. A key factor could be the levels of arginine in the cells. The ethylene-forming enzyme uses arginine both as a cofactor as well as a substrate and this has a direct influence on the rate of ethylene production. Finally we use the best performing strain for ethylene production in 1 l batch cultures and achieved stable ethylene production for 1 week with maximum volumetric ethylene production rate of 443 µl/l/h at a light intensity of 220 μmol photon/m2/s1.
Authors' contribution
VPV carried out the molecular genetics work, constructed the strains, cultured the strains, and performed the ethylene and oxoglutarate measurements, participated in the design of the study and drafted the manuscript. SAA contributed to the strain construction and oxoglutarate measurements, participated in the design of the study, and drafted the manuscript. KJH oversaw the study, participated in the design of the study, and drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Alejandra de Almeida for help with the design of the multiple promoter constructs and the oxoglutarate detection assay and Kornel Golebski and the Molecular Microbial Physiology Group for helpful discussions. SAA and KJH are supported by the research program of BioSolar Cells, co-financed by the Dutch Ministry of Economic Affairs, Agriculture and Innovation.
Competing interests
VPV and SAA declare that they have no competing interests. KJH is scientific advisor at Photanol B.V., a University of Amsterdam spin-off company which aims at commercializing cyanobacterial biotechnology.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Contributor Information
Vinod Puthan Veetil, Email: vinodpv17@gmail.com.
S. Andreas Angermayr, Email: andreas.angermayr@ist.ac.at.
Klaas J. Hellingwerf, Email: K.J.Hellingwerf@uva.nl
References
- 1.Adams DO, Yang SF. Ethylene biosynthesis: identification of 1-aminocyclopropane-1-carboxylic acid as an intermediate in the conversion of methionine to ethylene. Proc Natl Acad Sci USA. 1979;76:170–174. doi: 10.1073/pnas.76.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angermayr SA, Paszota M, Hellingwerf KJ. Engineering a cyanobacterial cell factory for production of lactic acid. Appl Environ Microbiol. 2012;78(19):7098–7106. doi: 10.1128/AEM.01587-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angermayr SA, van der Woude AD, Correddu D, Vreugdenhil A, Verrone V, Hellingwerf KJ. Exploring metabolic engineering design principles for the photosynthetic production of lactic acid by Synechocystis sp. PCC6803. Biotechnol Biofuels. 2014;7:99. doi: 10.1186/1754-6834-7-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berla BM, Saha R, Immethun CM, Maranas CD, Moon TS, Pakrasi HB. Synthetic biology of cyanobacteria: unique challenges and opportunities. Front Microbiol. 2013;4:246. doi: 10.3389/fmicb.2013.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carrieri D, Paddock T, Maness PC, Seibert M, Yu J. Photo-catalytic conversion of carbon dioxide to organic acids by a recombinant cyanobacterium incapable of glycogen storage. Energy Environ Sci. 2012;5:9457–9461. doi: 10.1039/c2ee23181f. [DOI] [Google Scholar]
- 6.Chen X, Liang Y, Hua J, Tao L, Qin W, Chen S. Overexpression of bacterial ethylene-forming enzyme gene in Trichoderma reesei enhanced the production of ethylene. Int J Biol Sci. 2010;6:96–106. doi: 10.7150/ijbs.6.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eaton-Rye JJ. Construction of gene interruptions and gene deletions in the cyanobacterium Synechocystis sp. strain PCC 6803. Methods Mol Biol. 2011;684:295–312. doi: 10.1007/978-1-60761-925-3_22. [DOI] [PubMed] [Google Scholar]
- 8.Eckert C, Xu W, Xiong W, Lynch S, Ungerer J, Tao L, Gill RT, Maness P, Yu J. Ethylene-forming enzyme and bioethylene production. Biotechnol Biofuels. 2014;7(1):33. doi: 10.1186/1754-6834-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukuda H, Ogawa T, Ishihara K, Fujii T, Nagahama K, Omata T, Inoue Y, Tanase S, Morino Y. Molecular cloning in Escherichia coli, expression, and nucleotide sequence of the gene for the ethylene-forming enzyme of Pseudomonas syringae Pv phaseolicola Pk2. Biochem Bioph Res Co. 1992;188:826–832. doi: 10.1016/0006-291X(92)91131-9. [DOI] [PubMed] [Google Scholar]
- 10.Fukuda H, Ogawa T, Tanase S. Ethylene production by micro-organisms. Adv Microb Physiol. 1993;35:275–306. doi: 10.1016/S0065-2911(08)60101-0. [DOI] [PubMed] [Google Scholar]
- 11.Fukuda H, Sakai M, Nagahama K, Fujii T, Matsuoka M, Inoue Y, Ogawa T. Heterologous expression of the gene for the ethylene-forming enzyme from Pseudomonas syringae in the cyanobacterium Synechococcus. Biotechnol Lett. 1994;16:1–6. doi: 10.1007/BF01022614. [DOI] [Google Scholar]
- 12.Gao X, Gao F, Liu D, Zhang H, Nie X, Yang C. Engineering the methylerythritol phosphate pathway in cyanobacteria for photosynthetic isoprene production from CO2. Energy Environ Sci. 2016;9:1400–1411. doi: 10.1039/C5EE03102H. [DOI] [Google Scholar]
- 13.Gründel M, Scheunemann R, Lockau W, Zilliges Y. Impaired glycogen synthesis causes metabolic overflow reactions and affects stress responses in the cyanobacterium Synechocystis sp. PCC 6803. Microbiol Read Engl. 2012;158:3032–3043. doi: 10.1099/mic.0.062950-0. [DOI] [PubMed] [Google Scholar]
- 14.Guerrero F, Carbonell V, Cossu M, Correddu D, Jones PR. Ethylene synthesis and regulated expression of recombinant protein in Synechocystis sp PCC 6803. PLoS ONE. 2012;7(11):e50470. doi: 10.1371/journal.pone.0050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heidorn T, Camsund D, Huang HH, Lindberg P, Oliveira P, Stensjö K, Lindblad P. Synthetic biology in cyanobacteria engineering and analyzing novel functions. Methods Enzymol. 2011;497:539–579. doi: 10.1016/B978-0-12-385075-1.00024-X. [DOI] [PubMed] [Google Scholar]
- 16.Hellingwerf KJ, Teixeira de Mattos MJ. Alternative routes to biofuels: light-driven biofuel formation from CO2 and water based on the ‘photanol’ approach. J Biotechnol. 2009;142(1):87–90. doi: 10.1016/j.jbiotec.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 17.IHS, Ethylene-Chemical Economics Handbook (2016) https://www.ihs.com/products/ethylene-chemical-economics-handbook.html.
- 18.Ishihara K, Matsuoka M, Inoue Y, Tanase S, Ogawa T, Fukuda H. Overexpression and in vitro reconstitution of the ethylene-forming enzyme from Pseudomonas syringae. J Ferment Bioeng. 1995;79:205–211. doi: 10.1016/0922-338X(95)90604-X. [DOI] [Google Scholar]
- 19.Ishihara K, Matsuoka M, Ogawa T, Fukuda H. Ethylene production using a broad-host-range plasmid in Pseudomonas syringae and Pseudomonas putida. J Ferment Bioeng. 1996;82:509–511. doi: 10.1016/S0922-338X(97)86994-2. [DOI] [Google Scholar]
- 20.Jacobsen DW, Wang CH. The biogenesis of ethylene in Penicillium digitatum. Plant Physiol. 1968;43(12):1959–1966. doi: 10.1104/pp.43.12.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson PR, Ecker JR. The ethylene gas signal transduction pathway: a molecular perspective. Annu Rev Genet. 1998;32:227–254. doi: 10.1146/annurev.genet.32.1.227. [DOI] [PubMed] [Google Scholar]
- 22.Leatherbarrow RJ. GraFit Version 7. Horley: Erithacus Software Ltd.; 2009. [Google Scholar]
- 23.Li M, Wang J, Geng Y, Li Y, Wang Q, Liang Q, Qi Q. A strategy of gene overexpression based on tandem repetitive promoters in Escherichia coli. Microb Cell Fact. 2012;11:19. doi: 10.1186/1475-2859-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma J, Campbell A, Karlin S. Correlations between Shine-Dalgarno sequences and gene features such as predicted expression levels and operon structures. J Bacteriol. 2002;184(20):5733. doi: 10.1128/JB.184.20.5733-5745.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markham JN, Tao L, Davis R, Voulis N, Angenent TL, Ungerer J, Yu J. Techno-economic analysis of a conceptual biofuel production process from bioethylene produced by photosynthetic recombinant cyanobacteria. Green Chem. 2016 [Google Scholar]
- 26.Nagahama K, Ogawa T, Fujii T, Tazaki M, Tanase S, Morino Y, Fukuda H. Purification and properties of an ethylene-forming enzyme from Pseudomonas syringae pv. phaseolicola PK2. J Gen Microbiol. 1991;137:2281–2286. doi: 10.1099/00221287-137-10-2281. [DOI] [PubMed] [Google Scholar]
- 27.Ogawa T, Takahashi M, Fujii T, Tazaki M, Fukuda H. The role of NADH:Fe(III)EDTA oxidoreductase in ethylene formation from 2-keto-4-methylthiobutyrate. J Ferment Bioeng. 1990;69:287–291. doi: 10.1016/0922-338X(90)90107-8. [DOI] [Google Scholar]
- 28.Pirkov I, Albers E, Norbeck J, Larsson C. Ethylene production by metabolic engineering of the yeast Saccharomyces cerevisiae. Metab Eng. 2008;10:276–280. doi: 10.1016/j.ymben.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Puigbò P, Guzmán E, Romeu A, Garcia-Vallvé S. OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. 2007;35:W126–W131. doi: 10.1093/nar/gkm219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakai M, Ogawa T, Matsuoka M, Fukuda H. Photosynthetic conversion of carbon dioxide to ethylene by the recombinant cyanobacterium, Synechococcus sp. PCC 7942, which harbors a gene for the ethylene-forming enzyme of Pseudomonas syringae. J Ferment Bioeng. 1997;84:434–443. doi: 10.1016/S0922-338X(97)82004-1. [DOI] [Google Scholar]
- 31.Savakis P, Hellingwerf KJ. Engineering cyanobacteria for direct biofuel production from CO2. Curr Opin Biotechnol. 2014;33C:8–14. doi: 10.1016/j.copbio.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 32.Savakis PE, Angermayr SA, Hellingwerf KJ. Synthesis of 2,3-butanediol by Synechocystis sp. PCC6803 via heterologous expression of a catabolic pathway from lactic acid- and enterobacteria. Metab Eng. 2013;20:121–130. doi: 10.1016/j.ymben.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 33.Takahama K, Matsuoka M, Nagahama K, Ogawa T. Construction and analysis of a recombinant cyanobacterium expressing a chromosomally inserted gene for an ethylene-forming enzyme at the psbAI locus. J Biosci Bioeng. 2003;95:302–305. doi: 10.1016/S1389-1723(03)80034-8. [DOI] [PubMed] [Google Scholar]
- 34.Tao L, Dong HJ, Chen X, Chen SF, Wang TH. Expression of ethylene-forming enzyme (EFE) of Pseudomonas syringae pv. glycinea in Trichoderma viride. Appl Microbiol Biotechnol. 2008;80:573–578. doi: 10.1007/s00253-008-1562-7. [DOI] [PubMed] [Google Scholar]
- 35.Ungerer J, Tao L, Davis M, Ghirardi M, Maness PC, Yu JP. Sustained photosynthetic conversion of CO2 to ethylene in recombinant cyanobacterium Synechocystis 6803. Energ Environ Sci. 2012;5:8998–9006. doi: 10.1039/c2ee22555g. [DOI] [Google Scholar]
- 36.van der Woude AD, Angermayr SA, Puthan Veetil V, Osnato A, Hellingwerf KJ. Carbon sink removal: increased photosynthetic production of lactic acid by Synechocystis sp. PCC6803 in a glycogen storage mutant. J Biotechnol. 2014;184:100–102. doi: 10.1016/j.jbiotec.2014.04.029. [DOI] [PubMed] [Google Scholar]
- 37.Wang KL, Li H, Ecker JR. Ethylene biosynthesis and signaling networks. Plant Cell. 2002;14(Suppl):S131–S151. doi: 10.1105/tpc.001768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wijffels RH, Kruse O, Hellingwerf KJ. Potential of industrial biotechnology with cyanobacteria and eukaryotic microalgae. Curr Opin Biotechnol. 2013;24:405–413. doi: 10.1016/j.copbio.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Xiong W, Morgan JA, Ungerer J, Wang B, Maness PC, Yu J. The plasticity of cyanobacterial metabolism supports direct CO2 conversion to ethylene. Nat Plants. 2015;1:5053. [Google Scholar]
- 40.Young RE, Pratt HK, Biale JB. Identification of ethylene as a volatile product of the fungus Penicillium digitatum. Plant Physiol. 1951;26:304–310. doi: 10.1104/pp.26.2.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu T, Xie X, Li Z, Tan X, Lu X. Enhancing photosynthetic production of ethylene in genetically engineered Synechocystis sp. PCC 6803. Green Chem. 2015;17:421–434. doi: 10.1039/C4GC01730G. [DOI] [Google Scholar]
- 42.Zinchenko VV, Piven IV, Melnik VA, Shestakov SV. Vectors for the complementation analysis of cyanobacterial mutants. Russ J Genet. 1999;35:228–232. [Google Scholar]