

Abstract
Background:
The calcium clearance and reactive oxygen species (ROS) generations in the coronary artery smooth muscle cells in chronic heart failure (HF) have not been fully investigated. Therefore, we attempted to understand the gene expressions underlying the mishandling of calcium clearance and the accumulations of ROS.
Methods:
We initially established an animal model of chronic HF by making the left anterior descending coronary artery ligation (CAL) in rats, and then isolated the coronary artery vascular smooth muscle cells from the ischemic and the nonischemic parts of the coronary artery vessels in 12 weeks after CAL operation. The intracellular calcium concentration and ROS level were measured using flow cytometry, and the gene expressions of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2a), encoding sarcoplasmic reticulum Ca2+-ATPase 2a, encoding sodium-calcium exchanger (NCX), and p47phox encoding a subunit of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase were examined using real-time quantitative reverse transcription polymerase chain reaction and Western blotting, respectively.
Results:
We found that the calcium accumulation and ROS generation in the coronary artery smooth muscle cells isolated from either the ischemic or the nonischemic part of the CAL coronary artery vessel were significantly increased irrespective of blood supply (all P < 0.01). Moreover, these were accompanied by the increased expressions of NCX and p47phox, the decreased expression of SERCA2a, and the increased amount of phosphorylated forms of p47phox in NADPH oxidase (all P < 0.05).
Conclusions:
Our results demonstrated that the disordered calcium clearance and the increased ROS generation occurred in the coronary artery smooth muscle cells in rats with chronic HF produced by ligation of the left anterior descending coronary artery (CAL), and which was found to be disassociated from blood supply, and the increased generation of ROS in the cells was found to make concomitancy to the increased activity of NADPH oxidase in cytoplasm.
Keywords: Calcium Clearance, Chronic Heart Failure, Nicotinamide Adenine Dinucleotide Phosphate Oxidase, Sarco/Endoplasmic Reticulum Ca2+-ATPase, Sodium-calcium Exchanger
Introduction
Chronic heart failure (HF) is a progressive, multifactorial, and debilitating disease that is often characterized by the impairment of systolic function and the loss of calcium homeostasis due to insufficient reuptake of calcium ions back into sarcoplasmic reticulum (SR) using Ca2+-ATPase and/or insufficient inflow of calcium ions into myocytes through using Na+/Ca2+ exchanger.[1,2] Ca2+ is a central player in the excitation-contraction coupling of cardiac myocytes that keeps heart to contract and relax.[3,4,5,6,7,8] Disturbances in Ca2+ homeostasis in myocytes are associated with a diverse range of human pathological conditions, such as myocardial infarction (MI) and HF.
In HF, the impaired cardiac function can cause hemodynamic instability with neurohumoral, cytokine imbalance, and cardiovascular remodeling as a result of multiple phenotypical alterations. For instance, the myocytes isolated often show impaired force development, prolonged action potentials, and slow relaxation. This is due to the altered expression, function, and/or the phosphorylation of Ca2+-regulatory proteins as mentioned above.
Of numerous calcium regulatory proteins, sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2a), a Ca2+-ATPase encoded by SERCA2a gene is characterized to be localized in the SR of myocytes, is responsible for the transportation of Ca2+ back into the SR in the diastole stage of myocytes, while the IP3-activated inositol-1,4,5-trisphosphate receptors, or the ryanodine receptors (RyRs), are responsible for releasing Ca2+ from the SR into cytosol, which normally occurs in the systolic stage of the myocytes. Further, vascular agonists, SR Ca2+ load, as well as the Ca2+ gradient between the SR and the cytosol also affect the Ca2+ release. In addition to SERCA2a, another Na+/Ca2+ exchanger (NCX) distributed in the t-tubular sarcoplasmic-sarcolemmal junctions also play an essential role in maintaining the calcium homeostasis of myocytes. In a physiological condition, this NCX operates in a Ca2+ efflux mode by extruding one Ca2+ in exchange for three Na+ ions. However, under certain pathological conditions, e.g., ischemia hypoxia, there is a substantial rise in cytosolic Na+ level, and the NCX transportation can be reversed to be a Ca2+ influx mode as seen in the constrictions of myofilaments.[9,10]
In cardiac myocytes, the contractile cycle of myofilaments is initiated in the systolic stage by Ca2+ entering the cytosol via voltage-activated L-type Ca2+ channels, such as dihydropyridine receptor, such Ca2+ ions will then bind the RyRs in the SR, and trigger the release of Ca2+ into cytosol from the SR, allowing binding to calmodulin (CAM) and activating the contractions of cardiac myocytes. In the diastolic stage, the cytosolic Ca2+ will be recycled back into the SR via SERCA2a uptaking and/or sarcolemmal NCX efflux, which reduces Ca2+ concentration and causes dissociation of Ca2+ from CAM and a subsequent relaxation of cardiac myocytes.
It is known that abnormal calcium clearance using SERCA2a reuptake and/or NCX efflux is responsible for the diastolic dysfunction of the coronary artery, leading to insufficient myocardial blood supply. Under these circumstances, SERCA2a determines both rate and amplitude of the contraction and relaxation of the cardiac myocytes.
In HF condition, the generations of reactive oxygen species (ROS) by either cardiac myocytes or endothelium cells can be enhanced upon ischemia hypoxia.[11,12,13] The exact roles of the ROS produced have not been fully understood yet. Several lines of evidence suggested that elevated ROS might be responsible for inducing the necrosis of myocardial tissue in the ischemia hypoxia situations, especially in combination with an overexpressed sarcolemmal NCX. However, some other investigations suggested that the increased ROS in HF could be protective in a sense of stimulating NCX activity by overcoming Na+-dependent NCX inactivation. Nevertheless, ROS are critical in regulating the redox condition in cardiovascular system both in physiological and in pathological conditions, especially when heart is in ischemic condition. For example, a recent study revealed that expressions of 630 genes in early hypoxia were regulated by ROS, including p65, NF-κB, Ca2+-handling proteins, such as calsequestrin, CAM, and calreticulin, and ion channels, including NCX, Na+-K+-ATPase, SERCA2a, and PLB; as well as the stress markers, such as RyR2, ANP, and BNP.[14,15,16]
So far, studies on the alterations of the expressions/functions of SERCA2a gene and NCX gene in HF have been controversy.[17,18,19] For example, it has been shown that reduction in the expression of SERCA2a gene may be associated with the overexpression of NCX gene, which may compensate the loss of the reuptake of Ca2+ and be beneficial to the removal of Ca2+ in the impaired diastolic myocytes.[20] However, studies have also shown that a reduction in SERCA2a protein disrupted the balance of Ca2+ transportation between SERCA2a and NCX, resulting in increased reliance on NCX gene in the failing human myocytes. Hasenfuss et al. suggested that in some HF patients with impaired systolic function and preserved diastolic function, large increase in NCX gene expression and modest decrease in the SERCAactivity coexisted.[21] However, other researchers have also observed marked SERCA downregulation without any significant increase in NCX gene expression.[22] Finally, a recent study has demonstrated that improved contractility after left ventricular (LV) assist device implantation reflects a decrease in NCX protein levels and Ca2+ transportation without changes in SERCA gene expression.[23] Herein, we have established a rat chronic HF model through left anterior descending coronary artery ligation (CAL). We found that both Ca2+ concentration and ROS level were enhanced in the coronary artery smooth muscle cells in an ischemia independent manner. We also detected a decrease in expression of SERCA2a gene and increases in NCX and p47phox gene expressions, as well as an increase in phosphorylated p47phox protein in the chronic HF rats.
Methods
Animals
Male Sprague Dawley rats (aged 3-month-old and weighing 250–300 g) were purchased from Vital River Laboratory Animal Co., Ltd., Beijing, China. All experiments involving the rats were performed in conformity with the institutional guidelines for care and use of laboratory animals, the surgery was done using isoflurane anesthesia, and every effort was made to minimize the suffering, and all experimental protocols used in this study were approved by the Committee for Laboratory Animal Welfare and Ethics of Hebei University.
Animal grouping and induction of chronic heart failure
Thirty rats were randomly divided into control (n = 10) and HF groups (n = 20). Chronic HF symptoms were induced by carrying out ligation of the left anterior descending coronary artery of the rats (CAL) as previously described.[24,25,26,27,28] In 12 weeks after operation, 10 control and 10 CAL rats were selected for the subsequent experimentations, separately. Three rats in either sham group or CAL group were used to measure the intracellular concentrations of Ca2+ and ROS, the amounts of the mRNA molecules using real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) and the proteins using Western blotting, respectively.
Evaluation of cardiac function
In 12th week after operation, rats from both control and CAL groups were anesthetized by intraperitoneal injection of 10% chloral hydrate (300 mg/kg), and then the right common carotid arteries of the heart were exposed by separating from the neck tissues. To determine blood flow dynamics, including left ventricular systolic pressure (LVSP), left ventricular end diastolic pressure (LVEDP), heart rate (HR), LV pressure and maximum rate of rise (dp/dtmax), LV pressure rate of decline (dp/dtmin), a pressure sensor and a signal acquisition and analysis system were used with connecting to a polyethylene catheter inserted into the left ventricle through the right common carotid artery.[29,30]
Isolation of coronary artery smooth muscle cells
The coronary arteries were taken from the rats and soaked in HEPES-buffered saline (HBS) containing 130 mmol/L NaCl, 5.0 mmol/L KCl, 1.2 mmol/L MgCl2, 10 mmol/L glucose, and 10 mmol/L HEPES (pH 7.3, adjusted by NaOH) at 4°C. The coronary arteries were then digested in a solution containing Type I collagenase (1.6 mg/ml) for 10 min. To obtain the tunica media vasorums, adventitias and endothelium were carefully stripped off using ophthalmological tweezers under a stereomicroscope.[30] To isolate the smooth muscle cells, the tunica media vasorums obtained were digested using a HBS solution containing 1.8 mg/ml papain, 1.5 mg/ml dithiothreitol, and 1.4 mg/ml albumin for 25 min (at 37°C), and then washed three times using HBS buffer. The tunica media vasorums were further incubated in a HBS solution containing 3 mg/ml Type II collagenase, 1.4 mg/ml albumin at 37°C for 45 min.[31,32,33,34] After the treatments, cells were cultured in a Petri dish containing Dulbecco's Modified Eagle Medium and incubated at 37°C with 5% CO2.
Calponin immunofluorescence staining
The smooth muscle cells were fixed using polyformaldehyde for 20 min, perforated using 1% Triton for 5 min, and blocked using 10% goat serum for 20 min, before being incubated at 4°C with anti-calponin antibody (GeneTex Inc., USA) overnight.[35] Then, the cells were washed and incubated with fluorescence-conjugated secondary antibody by 1:50 dilution for 30 min under dark condition. The 4',6-diamidino-2-phenylindole was used to counter stain the nuclei. The cells were examined using a laser confocal microscopy (Zeiss 710, Zeiss Inc., Germany).
Measurement of intracellular Ca2+ concentration by flow cytometry
The coronary artery smooth muscle cells were cultured for 48 h and trypsinized and collected by centrifugation at 400 ×g for 5 min. The cells were stained with Fluo-4 AM (4 μmol/L) for 30 min in dark at 37°C. Fluorescent signals were detected and recorded using the 10,000 cells FL1 channel of BD small flow cytometry (BD Accuri C6, BD Inc., USA).[36]
Measurements of reactive oxygen species using flow cytometry
Coronary artery vascular smooth muscle cells (CAVSMCs) obtained from the sham and the CAL rats were inoculated in 96-well plates and cultured for 48h, and the adherent cells were trypsinized and collected by centrifugation. The cell pellets were resuspended using 1.5 ml of 4 µmol/L 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA) working fluid and incubated at 37°C for 30 min in dark. The DCFH-DA was removed through centrifugation at 400 ×g for 10 min, and the cell pellets were washed twice with HBS. Finally, cell pellets were resuspended in 200 µl HBS, and their fluorescence signals were detected using FL1 channel in small flow cytometry BD.[37]
Measurements of mRNA expression of sarco/endoplasmic reticulum Ca2+-ATPase, Na+/Ca2+ exchanger, and p47phox using real-time quantitative reverse transcription polymerase chain reaction
The attached cells in each group were selected and digested with pancreatic enzyme (0.25%). Then, 1 ml trizol lysis buffer was added. RNA extraction was carried out by following the manufacture instruction. The A260/A280 ratio range was from 1.8 to 2.0. The cDNA was synthesized by reverse transcription according to the instruction of PrimeScript RT kits (Takara, Japan).[38] Amplification and detection of cDNA were performed using a real-time fluorescent quantitative PCR instrument by following the instruction of SYBR qPCR mix (Takara, Japan). Primer sequences of target gene were as follows: SERCA2a Forward primer: 5'-CTAGGCCTCCGGTCCTAACT-3'; and reverse primer: 5'-TGTGAGGAACTGAACCGACG-3'; NCX forward primer: 5'-GGTGAGTGGATTCGGGATCG-3'; and Reverse primer: 5'-CCGTCTCAGCTCTCATGCTT-3'; p47 phox, forward primer: 5'-TCCCAACTACGCAGGTGA AC-3'; and reverse primer: 5'-CCTGGGTTATCTCCTCCC CA-3'; GAPDH forward primer: 5'-ACCACAGTCCATGC CATCAC-3'; and reverse primer: 5'-TCCACCACCCTGTT GCTGTA-3'. Data arrangement and statistical analysis were performed using 2−ΔΔct analysis method and combined with SPSS16.0 statistical software.
Measurements of sarco/endoplasmic reticulum Ca2+-ATPase, Na+/Ca2+ exchanger, p47phox, and p-p47phox using Western blotting
Cultured coronary artery smooth muscle cells were used for carrying out Western blotting analysis.[39] In specific, cultured cells were rinsed three times using HBS buffer and incubated on ice, lysis solution was then added and the cells were incubated further on ice for 10 min. 25 µl of each cell lysates were resolved by running a polyacrylamide gel electrophoresis, and then transferred onto a polyvinylidene fluoride (PVDF) membrane. After being blocked using skim milk, PVDF membrane was incubated overnight at 4°C in a blocking buffer containing primary antibodies of anti-SERCA2a, anti-NCX, anti-p47phox, anti-p-p47phox, and anti-actin, respectively. The PVDF membranes were then rinsed and incubated in a blocking buffer containing horseradish peroxidase-conjugated secondary antibody for 2 h. The chemiluminescence on the membrane was analyzed using a JS-860B gel analyzer (Tianneng, Shanghai, China).
Statistical analysis
SPSS version 16.0 statistical software (SPSS Inc., USA) was used for statistical analysis. Data were expressed as mean ± standard error (SE). Single factor analysis of variance was used for the mean comparison between different experimental groups. A value of P < 0.05 was considered statistically significant.
Results
Establishments of the rat chronic heart failure model and evaluation of the cardiac functions
Chronic HF models of rats were established through making a ligation of the left anterior descending coronary artery (CAL) in rats [Figure 1]. The cardiac functions of the CAL rats were characterized after 12 weeks of operation. As shown in Table 1, compared with the rats in the sham group, both HRs and LVSP of the CAL rats decreased significantly (P < 0.05 for both cases). The LVEDP of the CAL rats were found to be increased significantly (P < 0.05). Moreover, the LV pressure, maximum rate of rise (dp/dtmax), and pressure decline rate (dp/dtmin) were all significantly reduced (P < 0.05 for all cases). The above data indicated that both systolic and diastolic functions in the CAL rats were impaired, and that the chronic HF rat models were successfully established.
Figure 1.
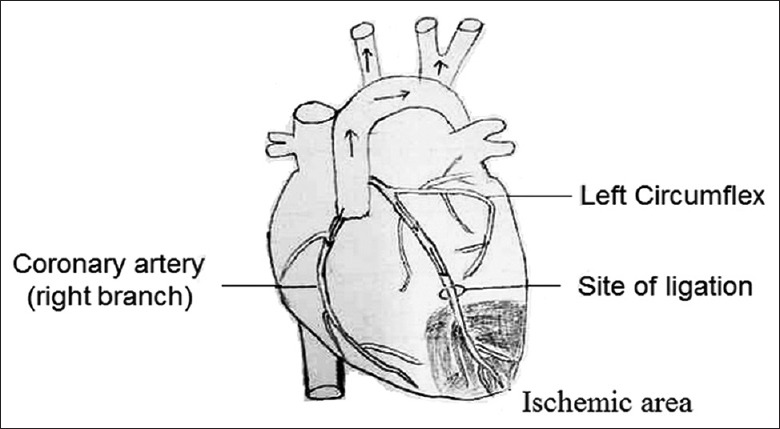
Illustration of coronary artery ligation operation.
Table 1.
Cardiac functions of coronary artery ligation and sham rats
| Group | HR | LVSP | LVEDP | dp/dtmax | dp/dtmin |
|---|---|---|---|---|---|
| Sham (n = 10) | 384.0 ± 9.0 | 138.0 ± 7.0 | 6.0 ± 1.0 | 4320 ± 122 | 2748 ± 134 |
| CAL (n = 10) | 343.0 ± 10.0 | 86.0 ± 7.0 | 10.7 ± 1.3 | 2267 ± 104 | 1913 ± 100 |
| t | 9.60 | 15.51 | 9.00 | 40.45 | 15.79 |
| P | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
Data are shown as mean ± SE. CAL: Coronary artery ligation; HR: Heart rate; LVSP: Left ventricular systolic pressure; LVEDP: Left ventricular end diastolic pressure; SE: Standard error.
Preparation and characterization of the isolated coronary artery smooth muscle cells
CAVSMCs were isolated and cultivated as illustrated in the material and methods. They were verified by immunofluorescent staining of calponin, a smooth muscle-specific protein. As shown in Figure 2, the cell nuclei were stained blue and the silhouette of the CAVSMCs was stained green, showing the typical characteristics of smooth muscle cells. These cells were then used for the subsequent experiments.
Figure 2.
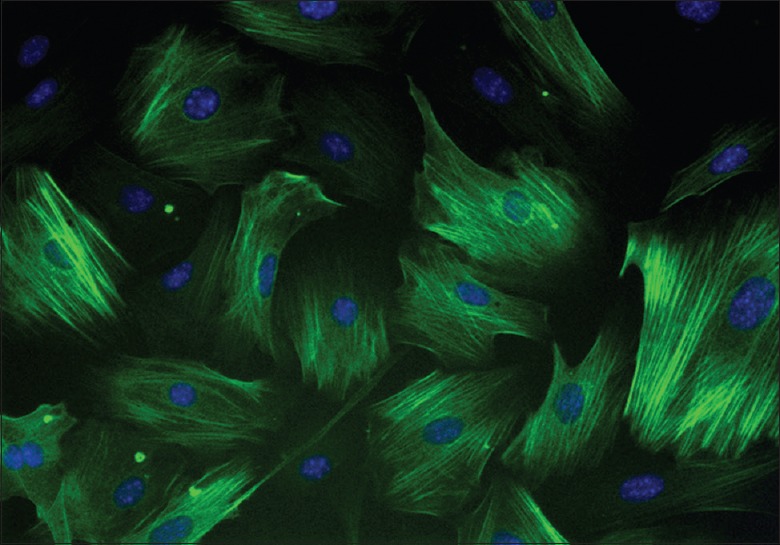
Immunofluorescent staining of calponin in the coronary artery smooth muscle cells of rats (original magnification ×400).
Elevated intracellular Ca2+ concentration in the coronary artery vascular smooth muscle cells
The calcium concentrations in the CAVSMCs isolated either from nonischemic or ischemic areas (IAs) of the CAL rats were significantly higher than those of the sham rats (all P < 0.01). No significant difference in the calcium level was noted between the myocytes isolated from nonischemic areas (NIAs) and IAs in the CAL rats (P > 0.05) [Figure 3]. This suggested with a disassociation of calcium overload from the ischemic status of the coronary artery smooth muscles cells.
Figure 3.

The relative fluorescence intensity of calcium ions in the coronary artery smooth muscle cells. (a) The relative fluorescence intensity of Fluo4-AM in the sham and the CAL rats; (b) the relative fluorescence intensity of Fluo4-AM in the sham and the CAL rats. CAL: Coronary artery ligation; NIA: Nonischemic area; IA: Ischemic area. *P < 0.01 vs. sham group; n.s: Nonsignificant.
Increased levels of reactive oxygen species in the coronary artery vascular smooth muscle cells
ROS are often generated as a result of cellular respiration in mitochondria and are maintained normally at low level using antioxidant molecules and enzymes. Multiple lines of evidence indicated that ROS were increased in cardiovascular diseases (CVDs) such as MI, stroke, and HF. ROS were recognized not only as cytotoxic molecules but also as cell signaling mediators. A study on the effects of the early hypoxia on the gene expressions using primary neonatal rat ventricular cardiomyocytes revealed that 630 genes including those encoding ion channels, such as NCX, SERCA2a, and PLB, were differentially regulated by ROS.[16]
However, there is little known about the effects of ROS relative to the signaling in CVD. Animal studies show that ROS contribute to the improvement of CVD, including angiogenesis following ischemia. Compared to rats in the sham group, the ROS' levels in the CAVSMCs in both NIA and IA in the CAL rats were significantly increased (all P < 0.01) [Figure 4]; however, the elevations in ROS were not significantly different between the two types of CAVSMCs (P > 0.05) [Figure 4].
Figure 4.

The relative fluorescence intensity of DCFH-DA in the smooth muscle cells of the coronary artery (n = 3). (a) Intensity of DCFH-DA in the sham and the CAL rats; (b) Averaged relative fluorescence intensity of DCFH-DA. *P < 0.01 vs. sham group; n.s: Nonsignificant. CAL: Coronary artery ligation; NIA: Nonischemic area; IA: Ischemic area; DCFH-DA: 2',7'-dichlorodihydrofluorescein diacetate.
Decreased mRNA transcription of sarco/endoplasmic reticulum Ca2+-ATPase and increased mRNA transcription of Na+/Ca2+ exchanger in the coronary artery vascular smooth muscle cells
The elevation of intracellular Ca2+ is a multiphase process that involves Ca2+ release from additional intracellular stores, such as endoplasmic reticulum (ER) and Ca2+ influx across the plasma membrane. Key components in regulating such calcium include NCX and plasma membrane Ca2+ ATPase (PMCA) channels, which are responsible for the active removal of Ca2+ out of cells, and SERCA2a that sequesters Ca2+ in the ER.[40,41,42] Since we detected an increase in Ca2+ in the coronary artery smooth muscle cells of the CAL rats, we then measured the gene transcriptions of SERCA2a and NCX, respectively. We found that the mRNA expressions of SERCA2a in the CAVSMCs, obtained from both the NIAs and the IAs, were downregulated significantly when compared to those isolated from the rats in the sham group (P < 0.01). However, the mRNA transcriptions of NCX gene in the CAVSMCs, isolated from either the NIAs or from the IAs, were increased significantly (P < 0.01). Once again, there is no significant difference in mRNA transcriptions of the SERCA2a gene and the NCX gene in the CAVSMCs isolated from the ischemic and the nonischemic regions of the CAL rats [Figure 5].
Figure 5.

The relative mRNA expressions of SERCA2a and NCX in the cells isolated from nonischemic and ischemic area of the coronary artery of the CAL rats. (a) The expressions of NCX in the sham and the CAL rats; (b) The level of SERCA2a in the sham and the CAL rats. *P < 0.05 vs. sham group; n.s: Nonsignificant; CAL: Coronary artery ligation; NIA: Nonischemic area; IA: Ischemic area; SERCA2a: Sarco/endoplasmic reticulum Ca2+-ATPase; NCX: Na+/Ca2+ exchanger.
The relative levels of sarco/endoplasmic reticulum Ca2+-ATPase and Na+/Ca2+ exchanger in the coronary artery smooth muscle cells
Compared with the rats in the sham group, the protein levels of SERCA2a in CAL rats both decreased significantly (P < 0.01) in the NIA and the IA [Figure 6a]. By contrast, the protein levels of NCX in the NIA and in the IAs in CAL rats were both significantly upregulated when compared with those of the sham rats (P < 0.01) [Figure 6b]. Once again, there were no significant differences in the NCX or SERCA2a in the cells isolated from NIA and IAs of the CAL rats (P > 0.05) [Figure 6a and 6b].
Figure 6.

The relative levels of SERCA2a and NCX in the cells isolated from nonischemic and ischemic area of the coronary artery of the CAL rats. (a) The levels of SERCA2a in the Sham and the CAL rats; (b) the levels of NCX in the Sham and the CAL rats. *P < 0.05 vs. sham group. n.s: Nonsignificant; CAL: Coronary artery ligation; NIA: Nonischemic area; IA: Ischemic area; SERCA2a: Sarco/endoplasmic reticulum Ca2+-ATPase; NCX: Na+/Ca2+ exchanger.
The relative levels of p47phox and p-p47phox in the coronary artery smooth muscle cells
Elevated ROS generation was associated with almost all CVD diseases. ROS can be generated through the Complex I and III of the respiratory chain in mitochondria[43,44,45,46] and through nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system,[47,48,49,50,51,52,53] cytochrome P450, peroxidase system, NO synthase,[54] and xanthine oxidases in cell membrane and cytoplasm.[55] In HF, several studies have indicated that elevated ROS may be produced mainly through NADPH oxidase in cell membrane and cytoplasm. NADPH oxidase is composed of multiple subunits, including gp91phox, phoxP22 in cell membrane and p47phox, p67phox, p40phox, small molecule GTP binding proteins (Rac1 and Rac-2) in the cytoplasm.[56,57] Since we have detected a significant increase in ROS production in the cytoplasm of the artery vascular smooth muscle cells in CAL rats, we then attempted to measure the mRNA transcription and translation of p47phox gene using qRT-PCR and Western blotting, respectively. These results are presented in Figure 7. As shown in Figure 7a and 7b, the mRNA transcription of p47phox gene was significantly upregulated, and the protein ofp47phox was also significantly elevated irrespective of bloody supply when compared to those in sham rats [Figure 7a and 7b]. We then measured the relative level of phosphorylated p47phox (p-p47phox) using Western blotting. Once again, we found that the p-p47phox was also significantly enhanced compared to that of sham rats, suggesting that enhanced NADPH oxidase activity was directly associated with the elevated ROS generation in the CAVSMCs in the CAL rats.
Figure 7.

The relative levels of p47phox and p-p47phox in the cells isolated from the nonischemic and the ischemic areas of the coronary artery of the CAL rats. (a) The relative mRNA transcription of p47phox in the ischemic cells and the nonischemic cells. (b) The levels of p47phox in the sham rats and the CAL rats, †P < 0.05 vs. sham group. (c) The levels of p-p47phox in the sham and CAL rats. *P < 0.05 vs. sham group; †P < 0.05 vs. sham group; n.s: Nonsignificant; CAL: Coronary artery ligation; NIA: Nonischemic area; IA: Ischemic area.
Discussion
HF has been characterized by contractile dysfunction and a high incidence of sudden death from nonreentrant ventricular arrhythmias, both of which involve altered intracellular calcium handling. In this work, we have investigated the calcium mishandling in the coronary artery smooth muscle cells of rats with chronic HF symptoms. The gene expressions of two ion channel genes, SERCA2a and NCX, as well as the gene, p47phox encoding a subunit of NADPH oxidase, were characterized using real-time qRT-PCR and their protein levels by Western blotting, respectively. We found that increments in intracellular calcium concentrations and ROS generation in the coronary artery smooth muscle cells of the CAL rats (P < 0.01) were irrespective of blood supply when compared with those of the sham rats (P > 0.05). More specifically, we found that NCX was upregulated in the failing and hypertrophic heart in both activity and expression. The increased NCX has been found to be involved in the regulation of several parameters of cardiac excitation-contraction coupling, such as cytosolic Ca2+ concentration, repolarization, and contractility.[5,6,8,10,15] Therefore, increased NCX activity has been identified as a mechanism promoting HF, cardiac ischemia, and arrhythmia. Increased amount or activity of NCX may transfer more extracellular calcium ions into the cytoplasm through the reverse mode of transport, increasing calcium level in coronary artery smooth muscle cell and impair the decline of Ca2+ level in diastole stage of the myocytes. However, it is unknown whether the increased expression of NCX is a cause or a consequence of the chronic HF. Increased NCX activity promoted arrhythmia and exacerbated ischemic necrosis in human HF, while some investigations suggested that the increased NCX could also provide an adaptive mechanism that can be used to against the diastolic Ca2+ overload.[17,18,19,20,21] Yet, it may also be possible that the increased NCX in the failing heart competes with the reduced SERCA2a of failing myocardium for cytosolic Ca2+, showing a maladaptive mechanism contributing to sarcoplasmic reticular Ca2+ loss and reduced contractility. In this latter case, we have indeed detected a reduced expressions of SERCA2a in both transcription level and protein level, which could be relevant to the high intracytoplasmic Ca2+ concentration in the diastolic coronary artery smooth muscle cells, leading to the observed phenomena such as coronary artery vascular smooth muscle diastolic dysfunction, slowly reduced intracytoplasmic Ca2+ concentration, prolonged coronary artery vasoconstriction time, abnormal coronary artery perfusion, and insufficient myocardial blood supply.
Nevertheless, maintenance of an appropriate level of calcium in smooth muscle cells is essential to their contraction and expansion in a physiological condition. However, in the disease condition, the abnormal increase in free Ca2+ concentration in the cytoplasm either by decreased SERCA2a or increased NCX or both made Ca2+-CaM binding much easier in the activated state of the myocytes that activates the myosin light-chain kinase. Under such a circumstance, myosin light-chain was then phosphorylated by Mg2+ ATPase, causing cross-bridge sliding and the myofilaments contracting in the smooth muscle cells. However, the decline of free Ca2+ concentration in the cytoplasm was retarded because of the malfunctions of SERCA2a, NCX, or their combination, for which the myosin light-chain kinase was unable to be inactivated and dephosphorylated in time in subsequent diastole stage, causing a delay in the relaxations in the smooth muscle cells.
The levels of ROS are increased in almost every CVD, such as in mitochondria under ischemia reperfusion as seen in human MI or stroke, the increased generation of ROS by NADPH oxidase in angiotensin II, in HF when cytokines became up-regulated, and in atherosclerotic plaques by macrophage-derived foam cells.[44,46,47,49,50,58] In this work, we have indeed detected an upregulated expression of p47phox gene in both transcription and translation, and also an increased phosphorylation of p47phox expressed, consistent with the increments in ROS production as seen in CAL rats. Generally speaking, there are two roles of ROS in CVDs, one is to directly promote myocardial tissue necrosis due to ischemia and hypoxia, exacerbating cell damage and death; the other is to act as a regulator in redox modification of protein and changing protein function. For example, ROS could increase active VEGFa through stabilizing HIF-1α and inhibiting anti-angiogenic factor, sFlt1 using S-glutathionylation modification on these transcription factors, which will then boost VEGFa signaling in endothelial cells through inhibition of PTPs and activation of SERCA2a.[59] As second messenger, ROS could regulate the expressions of 630 CVD-related genes.[16] In our case, the increments in ROS in the CAL rats with chronic HF may tightly be linked to the altered expressions of SERCA2a, NCX and p47phox rather than the shortage in blood supply. Indeed, according to a recent study, both expressions of SERCA2a and NCX are regulated by ROS in HF rats.[60] Therefore, we argued a closed interplay between the reduced expression of SERCA2a, the increased expression of NCX, and the loss of calcium homeostasis as well as the overgeneration of ROS in the cardiac artery smooth muscle cells of the CAL rats. However, the exact mechanisms underlined remained to be further elucidated.
In conclusion, our results argued a dissociation of the calcium accumulation and ROS generation from the blood supply and revealed associations of the mishandled calcium with the increased expression of NCX, and the reduced expression of SERCA2a, and the associations of the ROS increments with the increased transcription, translation, and phosphorylation of p47phox in the coronary artery smooth muscle cells of the rats with chronic HF.
Financial support and sponsorship
This work was supported by grants from the Hebei Provincial Key Research Programs for Medical Science (No. 20160051) and the Beijing Natural Science Foundation (No. 5132014).
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
We are particularly grateful to Dr. Zhi-Lian Yue, Intelligent Polymer Research Institute, AIIM Facility, University of Wollongong, Australia, for her thorough reading of the manuscript.
Footnotes
Edited by: Qiang Shi
References
- 1.Mitrovic S, Nogueira C, Cantero-Recasens G, Kiefer K, Fernández-Fernández JM, Popoff JF, et al. TRPM5-mediated calcium uptake regulates mucin secretion from human colon goblet cells. Elife. 2013;2:e00658. doi: 10.7554/eLife.00658. doi: 10.7554/eLife.00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnáiz-Cot JJ, Damon BJ, Zhang XH, Cleemann L, Yamaguchi N, Meissner G, et al. Cardiac calcium signalling pathologies associated with defective calmodulin regulation of type 2 ryanodine receptor. J Physiol. 2013;591:4287–99. doi: 10.1113/jphysiol.2013.256123. doi: 10.1113/jphysiol.2013.256123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zile MR, Baicu CF, Gaasch WH. Diastolic heart failure – Abnormalities in active relaxation and passive stiffness of the left ventricle. ACC Curr J Rev. 2004;13:1953–9. doi: 10.1056/NEJMoa032566. doi: 10.1056/NEJMoa032566. [DOI] [PubMed] [Google Scholar]
- 4.Clapham DE. Calcium signaling. Cell. 2007;131:1047–58. doi: 10.1016/j.cell.2007.11.028. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 5.Marchand A, Abi-Gerges A, Saliba Y, Merlet E, Lompré AM. Calcium signaling in vascular smooth muscle cells: From physiology to pathology. Adv Exp Med Biol. 2012;740:795–810. doi: 10.1007/978-94-007-2888-2_35. doi: 10.1007/978-94-007-2888-2_35. [DOI] [PubMed] [Google Scholar]
- 6.Dodd AN, Kudla J, Sanders D. The language of calcium signaling. Annu Rev Plant Biol. 2010;61:593–620. doi: 10.1146/annurev-arplant-070109-104628. doi: 10.1146/annurev-arplant-070109-104628. [DOI] [PubMed] [Google Scholar]
- 7.Naziroglu M. Role of selenium on calcium signaling and oxidative stress-induced molecular pathways in epilepsy. Neurochem Res. 2009;34:2181–91. doi: 10.1007/s11064-009-0015-8. doi: 10.1007/s11064-009-0015-8. [DOI] [PubMed] [Google Scholar]
- 8.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: Dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–29. doi: 10.1038/nrm1155. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 9.Winslow RL, Walker MA, Greenstein JL. Modeling calcium regulation of contraction, energetics, signaling, and transcription in the cardiac myocyte. Wiley Interdiscip Rev Syst Biol Med. 2016;8:37–67. doi: 10.1002/wsbm.1322. doi: 10.1002/wsbm.1322. [DOI] [PubMed] [Google Scholar]
- 10.Zhu J, Dong X, Liu Q, Wu C, Wang Q, Long Z, et al. Hydrophobic bile acids relax rat detrusor contraction via inhibiting the opening of the Na+/Ca²+ exchanger. Sci Rep. 2016;6:21358. doi: 10.1038/srep21358. doi: 10.1038/srep21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. Adynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–74. doi: 10.1016/j.cell.2008.02.048. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, et al. Aredox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell. 2008;133:978–93. doi: 10.1016/j.cell.2008.04.041. doi: 10.1016/j.cell.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 13.Burgoyne JR, Mongue-Din H, Eaton P, Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res. 2012;111:1091–106. doi: 10.1161/CIRCRESAHA.111.255216. doi: 10.1161/CIRCRESAHA.111.255216. [DOI] [PubMed] [Google Scholar]
- 14.Del Toro R, Levitsky KL, López-Barneo J, Chiara MD. Induction of T-type calcium channel gene expression by chronic hypoxia. J Biol Chem. 2003;278:22316–24. doi: 10.1074/jbc.M212576200. doi: 10.1074/jbc.M212576200. [DOI] [PubMed] [Google Scholar]
- 15.Xie H, Zhang YQ, Pan XL, Wu SH, Chen X, Wang J, et al. Decreased calcium-activated potassium channels by hypoxia causes abnormal firing in the spontaneous firing medial vestibular nuclei neurons. Eur Arch Otorhinolaryngol. 2015;272:2703–11. doi: 10.1007/s00405-014-3158-4. doi: 10.1007/s00405-014-3158-4. [DOI] [PubMed] [Google Scholar]
- 16.Kim DK, Choi E, Song BW, Cha MJ, Ham O, Lee SY, et al. Differentially regulated functional gene clusters identified in early hypoxic cardiomyocytes. Mol Biol Rep. 2012;39:9549–56. doi: 10.1007/s11033-012-1819-1. doi: 10.1007/s11033-012-1819-1. [DOI] [PubMed] [Google Scholar]
- 17.Leszek P, Szperl M, Klisiewicz A, Janas J, Biederman A, Rywik T, et al. Alteration of myocardial sarcoplasmic reticulum Ca2+-ATPase and Na+-Ca2+ exchanger expression in human left ventricular volume overload. Eur J Heart Fail. 2007;9:579–86. doi: 10.1016/j.ejheart.2007.01.011. doi: 10.1016/j.ejheart.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 18.Ottolia M, Torres N, Bridge JH, Philipson KD, Goldhaber JI. Na+/Ca2+ exchange and contraction of the heart. J Mol Cell Cardiol. 2013;61:28–33. doi: 10.1016/j.yjmcc.2013.06.001. doi: 10.1016/j.yjmcc.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shattock MJ, Ottolia M, Bers DM, Blaustein MP, Boguslavskyi A, Bossuyt J, et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J Physiol. 2015;593:1361–82. doi: 10.1113/jphysiol.2014.282319. doi: 10.1113/jphysiol.2014.282319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang WB, Kwan CY. Pharmacological evidence that potentiation of plasmalemmal Ca(2+)-extrusion is functionally coupled to inhibition of SR Ca(2+)-ATPases in vascular smooth muscle cells. Naunyn Schmiedebergs Arch Pharmacol. 2016;389:447–55. doi: 10.1007/s00210-016-1209-7. doi: 10.1007/s00210-016-1209-7. [DOI] [PubMed] [Google Scholar]
- 21.Hasenfuss G, Meyer M, Schillinger W, Preuss M, Pieske B, Just H. Calcium handling proteins in the failing human heart. Basic Res Cardiol. 1997;92(Suppl 1):87–93. doi: 10.1007/BF00794072. doi: 10.1007/BF00794072. [DOI] [PubMed] [Google Scholar]
- 22.Lu YM, Huang J, Shioda N, Fukunaga K, Shirasaki Y, Li XM, et al. CaMKIIdB mediates aberrant NCX1 expression and the imbalance of NCX1/SERCA in transverse aortic constriction-induced failing heart. PLoS One. 2011;6:e24724. doi: 10.1371/journal.pone.0024724. doi: 10.1371/journal.pone.0024724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Logantha SJ, Stokke MK, Atkinson AJ, Kharche SR, Parveen S, Saeed Y, et al. Ca(2+)-Clock-dependent pacemaking in the sinus node is impaired in mice with a cardiac specific reduction in SERCA2 abundance. Front Physiol. 2016;7:197. doi: 10.3389/fphys.2016.00197. doi: 10.3389/fphys.2016.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, Cao C, Hui L, Liu T, Wang Y, Gao S, et al. A study of myocardial ischemia model induced by left coronary artery ligation in rats. World J Cardiovasc Dis. 2016;6:133–42. doi: 10.4236/wjcd.2016.65014. [Google Scholar]
- 25.Klocke R, Tian W, Kuhlmann MT, Nikol S. Surgical animal models of heart failure related to coronary heart disease. Cardiovasc Res. 2007;74:29–38. doi: 10.1016/j.cardiores.2006.11.026. doi: 10.1016/j.cardiores.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 26.Muthuramu I, Lox M, Jacobs F, De Geest B. Permanent ligation of the left anterior descending coronary artery in mice: A model of post-myocardial infarction remodelling and heart failure. J Vis Exp. 2014;94:e52206. doi: 10.3791/52206. doi: 10.3791/52206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Wang Q, Guo W, Zhu YZ. Hydrogen sulfide attenuates cardiac dysfunction in a rat model of heart failure: A mechanism through cardiac mitochondrial protection. Biosci Rep. 2011;31:87–98. doi: 10.1042/BSR20100003. doi: 10.1042/BSR20100003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang X, Tang W, Sun S, Huang L, Chang YT, Huang Z, et al. Cardiopulmonary resuscitation in a rat model of chronic myocardial ischemia. J Appl Physiol. 2006;101:1091–6. doi: 10.1152/japplphysiol.01487.2005. doi: 10.1152/japplphysiol.01487.2005. [DOI] [PubMed] [Google Scholar]
- 29.Rennison JH, McElfresh TA, Okere IC, Vazquez EJ, Patel HV, Foster AB, et al. High-fat diet postinfarction enhances mitochondrial function and does not exacerbate left ventricular dysfunction. Am J Physiol Heart Circ Physiol. 2007;292:H1498–506. doi: 10.1152/ajpheart.01021.2006. doi: 10.1152/ajpheart.01021.2006. [DOI] [PubMed] [Google Scholar]
- 30.Baker M, Robinson SD, Lechertier T, Barber PR, Tavora B, D’Amico G, et al. Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc. 2011;7:89–104. doi: 10.1038/nprot.2011.435. doi: 10.1038/nprot.2011.435. [DOI] [PubMed] [Google Scholar]
- 31.Essin K, Welling A, Hofmann F, Luft FC, Gollasch M, Moosmang S. Indirect coupling between Cav1.2 channels and ryanodine receptors to generate Ca2+ sparks in murine arterial smooth muscle cells. J Physiol. 2007;584(Pt 1):205–19. doi: 10.1113/jphysiol.2007.138982. doi: 10.1113/jphysiol.2007.138982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santana LF, Navedo MF. Molecular and biophysical mechanisms of Ca2+ sparklets in smooth muscle. J Mol Cell Cardiol. 2009;47:436–44. doi: 10.1016/j.yjmcc.2009.07.008. doi: 10.1016/j.yjmcc.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill-Eubanks DC, Werner ME, Heppner TJ, Nelson MT. Calcium signaling in smooth muscle. Cold Spring Harb Perspect Biol. 2011;3:a004549. doi: 10.1101/cshperspect.a004549. doi: 10.1101/cshperspect.a004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hopson KP, Truelove J, Chun J, Wang Y, Waeber C. S1P activates store-operated calcium entry via receptor- and non-receptor-mediated pathways in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2011;300:C919–26. doi: 10.1152/ajpcell.00350.2010. doi: 10.1152/ajpcell.00350.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang JW, Hung SP, Wu HH, Wu WM, Yang AH, Tsai HL, et al. Therapeutic effects of umbilical cord blood-derived mesenchymal stem cell transplantation in experimental lupus nephritis. Cell Transplant. 2011;20:245–57. doi: 10.3727/096368910X520056. doi: 10.3727/096368910X520056. [DOI] [PubMed] [Google Scholar]
- 36.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–27. doi: 10.1161/01.HYP.0000258594.87211.6b. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Posey AD, Kawalekar OU, June CH. Measurement of intracellular ions by flow cytometry. Curr Protoc Immunol. 2015;72:9.8.1–9.8.21. doi: 10.1002/0471142956.cy0908s72. doi: 10.1002/0471142956.cy0908s72. [DOI] [PubMed] [Google Scholar]
- 38.Melo SF, Barauna VG, Neves VJ, Fernandes T, Lara Lda S, Mazzotti DR, et al. Exercise training restores the cardiac microRNA-1 and – 214 levels regulating Ca2+ handling after myocardial infarction. BMC Cardiovasc Disord. 2015;15:166. doi: 10.1186/s12872-015-0156-4. doi: 10.1186/s12872-015-0156-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choy JS, Lu X, Yang J, Zhang ZD, Kassab GS. Endothelial actin depolymerization mediates NADPH oxidase-superoxide production during flow reversal. Am J Physiol Heart Circ Physiol. 2014;306:H69–77. doi: 10.1152/ajpheart.00402.2013. doi: 10.1152/ajpheart.00402.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:379–83. doi: 10.1038/415198a. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 41.Rhodes JD, Sanderson J. The mechanisms of calcium homeostasis and signalling in the lens. Exp Eye Res. 2009;88:226–34. doi: 10.1016/j.exer.2008.10.025. doi: 10.1016/j.exer.2008.10.025. [DOI] [PubMed] [Google Scholar]
- 42.Roe AT, Frisk M, Louch WE. Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr Pharm Des. 2015;21:431–48. doi: 10.2174/138161282104141204124129. doi: 10.2174/138161282104141204124129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nickel A, Kohlhaas M, Maack C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol. 2014;73:26–33. doi: 10.1016/j.yjmcc.2014.03.011. doi: 10.1016/j.yjmcc.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 44.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–35. doi: 10.1161/01.res.88.5.529. doi: 10.1161/01.RES.88.5.529. [DOI] [PubMed] [Google Scholar]
- 45.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 46.Münzel T, Gori T, Keaney JF, Jr, Maack C, Daiber A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur Heart J. 2015;36:2555–64. doi: 10.1093/eurheartj/ehv305. doi: 10.1093/eurheartj/ehv305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565–70. doi: 10.1073/pnas.1002178107. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lassègue B, Griendling KK. NADPH oxidases: Functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–61. doi: 10.1161/ATVBAHA.108.181610. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vendrov AE, Vendrov KC, Smith A, Yuan J, Sumida A, Robidoux J, et al. NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid Redox Signal. 2015;23:1389–409. doi: 10.1089/ars.2014.6221. doi: 10.1089/ars.2014.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun QA, Runge MS, Madamanchi NR. Oxidative stress, NADPH oxidases, and arteries. Hamostaseologie. 2016;36:77–88. doi: 10.5482/HAMO-14-11-0076. doi: 10.5482/HAMO-14-11-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee MY, San Martin A, Mehta PK, Dikalova AE, Garrido AM, Datla SR, et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. 2009;29:480–7. doi: 10.1161/ATVBAHA.108.181925. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gupte SA, Kaminski PM, George S, Kouznestova L, Olson SC, Mathew R, et al. Peroxide generation by p47phox-Src activation of Nox2 has a key role in protein kinase C-induced arterial smooth muscle contraction. Am J Physiol Heart Circ Physiol. 2009;296:H1048–57. doi: 10.1152/ajpheart.00491.2008. doi: 10.1152/ajpheart.00491.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–90. doi: 10.1161/CIRCRESAHA.111.243972. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mollnau H, Oelze M, August M, Wendt M, Daiber A, Schulz E, et al. Mechanisms of increased vascular superoxide production in an experimental model of idiopathic dilated cardiomyopathy. Arterioscler Thromb Vasc Biol. 2005;25:2554–9. doi: 10.1161/01.ATV.0000190673.41925.9B. doi: 10.1161/01.ATV.0000190673.41925.9B. [DOI] [PubMed] [Google Scholar]
- 55.Baldus S, Müllerleile K, Chumley P, Steven D, Rudolph V, Lund GK, et al. Inhibition of xanthine oxidase improves myocardial contractility in patients with ischemic cardiomyopathy. Free Radic Biol Med. 2006;41:1282–8. doi: 10.1016/j.freeradbiomed.2006.07.010. doi: 10.1016/j.freeradbiomed.2006.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 57.Brandes RP, Weissmann N, Schröder K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic Biol Med. 2014;76:208–26. doi: 10.1016/j.freeradbiomed.2014.07.046. doi: 10.1016/j.freeradbiomed.2014.07.046. [DOI] [PubMed] [Google Scholar]
- 58.Cao YY, Chen ZW, Gao YH, Wang XX, Ma JY, Chang SF, et al. Exenatide Reduces Tumor Necrosis Factor-α-induced Apoptosis in Cardiomyocytes by Alleviating Mitochondrial Dysfunction. Chin Med J. 2015;128:3211–8. doi: 10.4103/0366-6999.170259. doi: 10.4103/0366-6999.170259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Watanabe Y, Cohen RA, Matsui R. Redox regulation of ischemic angiogenesis-Another aspect of reactive oxygen species. Circulation J. 2016;80:1278–84. doi: 10.1253/circj.CJ-16-0317. doi: 10.1253/circj.CJ-16-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eigel BN, Gursahani H, Hadley RW. ROS are required for rapid reactivation of Na+/Ca2+ exchanger in hypoxic reoxygenated guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol. 2004;286:H955–63. doi: 10.1152/ajpheart.00721.2003. doi: 10.1152/ajpheart.00721.2003.286. [DOI] [PubMed] [Google Scholar]