

Abstract
Gastrointestinal stromal tumors represent the most common mesenchymal tumor of the digestive tract, driven by gain‐of‐function mutations in KIT. Despite its proven benefits, half of the patients treated with imatinib show disease progression within 2 years due to secondary resistance mutations in KIT. It remains unclear how the genomic and transcriptomic features change during the acquisition of imatinib resistance. Here, we performed exome sequencing and microarray transcription analysis for four imatinib‐resistant cell lines and one cell line briefly exposed to imatinib. We also performed exome sequencing of clinical tumor samples. The cell line briefly exposed to imatinib exhibited few single‐nucleotide variants and copy‐number alterations, but showed marked upregulation of genes related to detoxification and downregulation of genes involved in cell cycle progression. Meanwhile, resistant cell lines harbored numerous genomic changes: amplified genes related to detoxification and deleted genes with cyclin‐dependent kinase activity. Some variants in the resistant samples were traced back to the drug‐sensitive samples, indicating the presence of ancestral subpopulations. The subpopulations carried variants associated with cell death. Pre‐existing cancer cells with genetic alterations promoting apoptosis resistance may serve as a basis whereby cancer cells with critical mutations, such as secondary KIT mutations, can establish full imatinib resistance. © 2017 The Authors Genes, Chromosomes and Cancer Published by Wiley Periodicals, Inc.
INTRODUCTION
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the digestive tract and are characterized by the expression of KIT (also known as CD117) and DOG1. Most GISTs harbor gain‐of‐function mutations in the KIT or PDFGRA genes, which contribute to the development of sporadic GISTs (Hirota et al., 1998; Nishida et al., 1998; Hirota et al., 2003). This knowledge has facilitated the development of targeted therapies with tyrosine kinase inhibitors and the revolutionary chemotherapeutic drug imatinib mesylate (Gleevec®; Novartis Pharmaceuticals). Several clinical studies have demonstrated the efficacy and tolerability of imatinib at the recommended dose of 400 mg/day for advanced, metastatic, or recurrent GIST. In clinical trials, the disease control rate was nearly 85%, and corresponding 2‐year overall survival rates ranged from 70–80%, indicating markedly improved patient outcomes compared with anecdotal data in the pre‐imatinib era (Demetri et al., 2002; Verweij et al., 2004; Blanke et al. 2008). Despite its effectiveness, half of GISTs treated with imatinib develop resistance within 2 years, largely due to the accumulation of additional kinase domain mutations accompanied by concomitant reactivation of the KIT tyrosine kinase, even in the presence of imatinib.
A common laboratory model of acquired resistance involves the development of drug‐resistant tumor cell lines from drug‐sensitive ones (Oxnard et al., 2011; Dhayat et al., 2015; Ito et al., 2015). To establish resistant cell lines, parental drug‐sensitive cells are cultured in a stepwise fashion with increasing drug concentrations until cells emerge that are 100‐fold less sensitive to growth inhibition. Cells are initially treated with a drug concentration at which 90% of the cells die, and, when the resistant cells resume normal growth, the drug concentration is increased. In this fashion, we established KIT‐mutant GIST cell lines resistant to imatinib, reliably identifying clinically relevant imatinib‐resistance mechanisms, such as the KIT D820V and D820Y mutations. Nevertheless, the full spectrum of genomic and transcriptomic changes associated with such resistance mechanisms remains unknown.
We performed an omics‐based analysis by integrating single‐nucleotide variations (SNVs) and copy‐number alterations (CNAs) from next‐generation sequencing (NGS) data with the transcriptome data from microarrays obtained from imatinib‐resistant GIST cell lines. Several GIST cell lines exist, including GIST T1, GIST882, GIST48, and GIST430 (Tuveson et al., 2001; Taguchi et al., 2002; Bauer et al., 2006; Henze et al., 2012), and in this study, we succeeded in generating imatinib‐resistant cell lines from the parental GIST T1 cell line, which was a relevant cell line for our purpose, because it harbors a 57‐nucleotide (V570–Y578) in‐frame deletion in KIT exon 11 (Taguchi et al., 2002), which is often clinically observed in GISTs (Nishida et al., 2009). We also conducted exome sequencing of four imatinib‐resistant clinical samples. Our aims were to comprehensively analyze genomic and transcriptomic changes occurring during the acquisition of resistance and to investigate how cancer cells evolve from a drug‐sensitive state during such acquisition, from an omics viewpoint.
MATERIALS AND METHODS
Cell Culture
We used the established human GIST cell line GIST‐T1, the identity of which was confirmed by DNA fingerprinting through short tandem repeat profiling (Yokoyama et al., 2013). T1 cells were cultured in DMEM 1640 medium supplemented with 10% FBS (HyClone Laboratories, Logan, UT) and 1% penicillin–streptomycin (Nacalai Tesque, Kyoto, Japan) at 37°C in a humidified atmosphere of 5% CO2. To generate imatinib‐resistant cell lines, parental cells were cultured with increasing concentrations of imatinib starting with 10 nM imatinib. Fresh drug was added every 72–96 h. Resistant cells that grew in 10 μM imatinib were established after 20–60 weeks of culture with the drug. Resistant cells were maintained as polyclonal populations under constant 1 μM imatinib selection. DNA‐identity testing on both the parental and resistant cells confirmed that the cells were derived from the same origin.
Reagents and Antibodies
Imatinib, a selective tyrosine kinase inhibitor of KIT, was synthesized and provided by Novartis Pharmaceuticals (Basel, Switzerland). A primary anti‐KIT antibody (1:1,000) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and an antiphosphotyrosine antibody (clone 4G10) was obtained from Upstate Biotechnology (Lake Placid, NY). The immunoprecipitation and western blotting methods used were described elsewhere (Takahashi et al., 2013).
Clinical Samples
Patients with imatinib‐resistant metastatic GIST were enrolled, and four pairs of primary and imatinib‐resistant tumors from the same patients were selected. The study protocols were approved by the institutional review boards of Osaka University Hospital (14154‐3) and the methods were carried out in accordance with the approved guidelines and with written informed consent from all the patients.
Whole‐Exome Sequencing
Sequence data for the cell lines and clinical samples used in these analyses are available in the DDBJ Sequenced Read Archive (Kodama et al., 2015) under the Accession Nos. DRX033325–DRX033330 and JGAS00000000039. DNA quality was determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA) and the Quant‐iT PicoGreen dsDNA Reagent and Kit (Life Technologies, Carlsbad, CA). Double‐stranded DNA was used for exome‐sequencing library preparation. Exomes were captured using the SureSelect Human All Exon V5 + UTRs Kit (Agilent Technologies, Santa Clara, CA), according to the manufacturer's instructions. The exome capture libraries were sequenced using a HiSeq 1500 system (Illumina, San Diego, CA) to generate 100‐bp paired‐end data.
Microarray Experiments
Microarray data were deposited in the Gene Expression Omnibus database under the GEO Series Accession No. GSE69465. We performed a microarray experiment for each sample without taking replicates. Total RNA was extracted from each cell line and tumor sample using an RNeasy Kit (Qiagen, Valencia, CA). RNA quality was assessed using the Agilent Model 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). Two micrograms of total RNA was processed for microarray experiments using the Agilent Expression Array for One‐Color (Agilent) according to manufacturer's protocols. The resultant biotinylated cRNA was fragmented and then hybridized to the SurePrint G3 Human GE 8x60K v2 Microarray (Agilent). The arrays were washed, stained, and scanned using the Gene Expression Hybridization Kit, the Gene Expression Wash Buffers Pack, and the Agilent DNA Microarray Scanner (G2565CA), respectively. Expression values were generated using Feature Extraction software (Agilent). Each sample and hybridization underwent quality‐control evaluation.
Ultradeep Sequencing
Sequence data for the cell lines used in these analyses are available in the DDBJ Sequenced Read Archive under Accession Nos. DRX033331–DRX033336. In total, 299 candidate SNVs and the whole exome sequences of KIT, KRAS, NRAS, BRAF, NF1, PTEN, PDGFRA, and PTK2 were targeted for capture and deep sequencing. The probes were designed for a final capture size of 94.2 kb using the SureDesign system (Agilent). Target‐sequence enrichment was performed using the Agilent SureSelect Target Enrichment Kit (Agilent), according to manufacturer instructions. The capture libraries were sequenced using a HiSeq 1500 system (Illumina, San Diego, CA) to generate 100‐bp paired‐end data.
SNV/Indel Calling
We used an in‐house program to call SNVs/indels. We removed adaptor sequences from NGS reads and used BWA (Li and Durbin, 2009) to map reads to the human genome. We filtered out PCR‐duplicated reads as well as reads and bases with low mapping and base qualities. The remaining variants were further filtered statistically using Fisher's exact test to compare foreground and background samples, remove germline SNPs/indels, and minimize position‐dependent errors. For cell line samples, we used a daughter cell line as the foreground sample and the parental cell line (T1) as the background sample. For clinical samples, we used samples obtained before and after the acquisition of imatinib resistance as the background and foreground samples, respectively. This statistical filtering process was used to screen for variants with significantly higher variant allele frequencies (VAFs) than present in the background sample. A series of filters was used to remove suspicious variant calls, such as those related to misalignments. Variants for which the allele frequencies were significantly greater than 1% in the binomial test were retained. Regarding indels, we only identified short indels due to the limitation of the BWA alignment. For cell line analysis, variants called in at least one daughter cell line were tabulated and used for downstream analyses.
CNA Calling
We used segments called by both ExomeCNV (Sathirapongsasuti et al., 2011) and Varscan2 (Koboldt et al., 2012) as CNA segments. More specifically, after removing the adaptor sequences from the reads, we used BWA (Li and Durbin, 2009) to map the reads to the human genome. We also filtered out PCR‐duplicated reads. BWA randomly maps reads only once when multiple mapping is possible; hence, we used all mapped reads for CNA calling. From BWA‐mapped BAM files, we ran ExomeCNV following the pipeline recommended on the authors' website. We used a daughter cell line and the parental cell line (T1) as the foreground and background, respectively, for the cell line samples. For the clinical samples, we used samples obtained before and after the development of drug resistance as the foreground and background sets, respectively. Similarly, we ran Varscan2, following the pipeline recommended on the authors' website. We used overlapping segments called by both tools to minimize false positives. CNA “fragments” were defined from CNA segments, as described previously (Kato et al., 2010).
Differentially Expressed Transcript (DET) and Differentially Expressed Gene (DEG) Calling
We drew ratio‐intensity (R–I) plots (Quackenbush 2002) for each cell line, where the background and foreground samples are the parental and daughter lines, respectively (Supporting Information Fig. S1) in the R–I plots. DETs were defined as those for which the fold changes were greater than 2, or greater than 4 when higher stringency was used. DEGs were defined as genes for which transcripts were compressed into a single gene by taking a representative transcript with the maximum expression change.
Validation
We used ultradeep sequencing data for validating SNVs/indels. We counted the number of candidate variants using mpileup, and calculated the VAFs. Similar to a prior study (Totoki et al., 2014), we generated plots in which the variant positions were sorted according to VAFs or differences in VAFs (Supporting Information Fig. S2), after which thresholds for validation calling were determined.
Heatmap Analysis
All heatmaps were drawn using the R software, where the Euclidean distance was used as the distance metric and the Ward method was used for agglomeration.
Gene Set–Enrichment Analysis
MSigDB (Subramanian et al., 2005) and DAVID (Huang da et al., 2009) were used to identify pathways or gene ontologies in which the genes of an identified group were enriched (P < 0.1). With MSigDB, we used the C2 (canonical pathways) and C5 (biological processes) databases. With DAVID, we used functional annotations to search against the KEGG and REACTOME pathways and gene ontologies (biological processes).
Phylogenetic Tree Analysis
A previously described method was used to reconstruct a phylogenetic tree (Yachida et al., 2016). Specifically, we calculated Nei's genetic distance (Nei, 1978) for synonymous SNVs as follows:
| (1) |
where X and Y represent the two cell lines, p represents the VAF, and i and S indicate the variant site and the number of all variant sites, respectively. We used variant sites (1) at which the depth was 50 or more for reliable P values and (2) at which the variants were determined as called in either of the two cell lines. Then, we used the neighbor‐joining method (Saitou and Nei, 1987) to construct a phylogenetic tree.
RESULTS
Cell Lines and Genomic Experiments
The GIST T1 parental cell line was exposed to imatinib, using doses that were increased in a stepwise manner, and surviving cells were selected until normal cell growth resumed. We thus independently established four imatinib‐resistant GIST cell lines (R2, RA2, R8, and R9), which exhibited imatinib IC50 values (>10 μM) that were >500‐fold higher than the parental cell line (Table 1). The culture time was 20 weeks for R8, 45 weeks for RA2, 52 weeks for R9, and 96 weeks for R2. Western blot analysis demonstrated that R8 and R9 exhibited phosphorylated KIT, which was indicative of KIT reactivation (Supporting Information Fig. S3). Sanger sequencing revealed that R8 harbored KIT D820Y and R9 had KIT D820V as secondary KIT mutations (Table 1). Secondary mutations at D820 (including those just mentioned) are frequently (∼10%) observed in clinical resistance (Antonescu et al., 2005; Nishida et al., 2008, 2009). R2 and RA2 showed no elevated KIT phosphorylation and had no secondary mutations. It took at least 20 weeks for these cell lines to acquire full imatinib resistance, defined as normal cell proliferation in the presence of >10 μM imatinib. Meanwhile, we developed another resistant cell line (T117), which was derived from GIST T1 but was exposed to imatinib for only 7 weeks. T117 showed only slight proliferation when increasing the imatinib concentration from 0.1 to 0.2 μM. We refer to this cell line here as the “briefly exposed” cell line.
Table 1.
Cell Lines
| Cell linea | IM duration (weeks) | IM IC50 (µM) | KIT phosphorylation under IM (1 mM) | Secondary KIT mutation |
|---|---|---|---|---|
| T1 | N/A | 0.02 | − | None |
| T117 | 7 | N/A | − | None |
| R8 | 20 | >10 | + | Exon17 D820Y |
| RA2 | 45 | >10 | − | None |
| R9 | 52 | >10 | + | Exon17 D820V |
| R2 | 60 | >10 | − | None |
Four resistant cell lines (R8, RA2, R9, and R2) were established from the T1 parental cell line harboring the KIT exon 11 deletion. T117 was exposed to imatinib (IM) for 7 weeks and had not yet acquired full resistance. R8 and R9 had secondary mutations, which were confirmed by Sanger sequencing.
We performed exome sequencing and microarray expression profiling in all cell lines. The average depth of exome sequencing was 83 (Supporting Information Table S1). We identified SNVs/indels and CNAs specifically in the daughter cell lines, using the parental T1 line as a reference. During microarray analysis, we obtained log2 expression ratios (exRs) for each daughter cell line by comparison to the parental line. We validated 92% (262/285) of the SNV/indel calls by ultradeep sequencing with an average sequencing depth of 60,323 (Supporting Information Table S2).
Genomic Features
The briefly exposed cell line T117 had far fewer genetic changes (SNVs and CNAs) than did the resistant cell lines (Fig. 1). That is, the number of SNVs and CNAs and the total size of the CNAs were significantly lower (chi‐squared test, P < 0.01) in T117 than in the other cell lines. The VAFs of the SNVs were significantly lower in T117 for both nonsilent and synonymous SNVs (Fig. 1, Kolmogorov–Smirnov test, P < 0.01). Log values of copy‐number ratios (cnRs), which represent the degrees of amplification and deletion, were significantly lower for amplification in T117 (Fig. 1, Kolmogorov–Smirnov test, P < 0.01). In contrast, transcriptome analysis showed that the most drastic expression changes occurred in T117, as shown by the number of DETs (Fig. 1 and Supporting Information Fig. S4; binomial test, P < 0.01). Except for the number of twofold upregulated DETs, there was no obvious distinction between the cell lines with or without secondary KIT mutations (Fig. 1).
Figure 1.
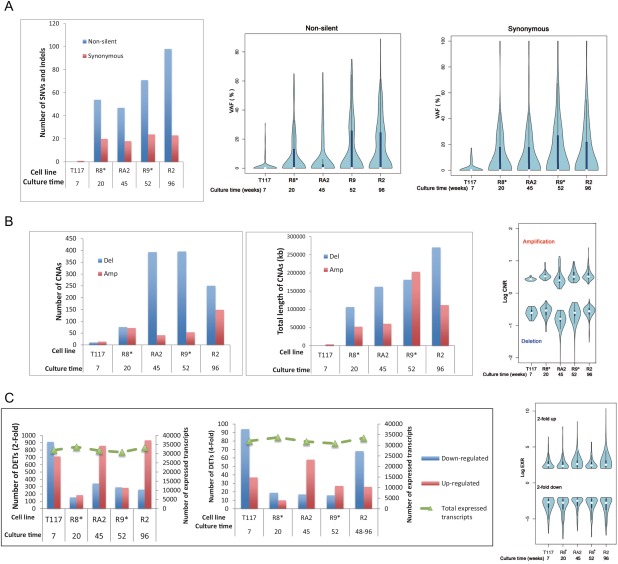
Statistics of SNVs/indels, CNAs, and transcripts. The parental cell line (T1) was used as the background set to identify all types of alterations. The culture time shown is expressed in weeks. Asterisks (*) indicate cell lines with secondary KIT mutations known to cause drug resistance. (A) Graphs regarding SNVs/indels, which show the number of SNVs/indels and the distributions of their VAFs. Nonsilent mutations were composed of nonsynonymous, stop gain/loss, frame‐shift, and splicing mutations. (B) Graphs depicting CNAs with the number and total length of CNA segments and the distribution of their log cnRs. (C) Graphs illustrating gene expression, which show the numbers of DETs with >2‐ and 4‐fold changes and the distribution of log exRs for genes with >2‐fold changes. For reference, we drew a line indicating the number of all expressed transcripts in microarrays on the right axis. [Color figure can be viewed at wileyonlinelibrary.com]
We then generated heat maps to identify the associations of these alterations between multiple cell lines (Fig. 2 and Supporting Information Fig. S5). As shown in the SNV heatmap, most SNVs found in T117 were also found in the other cell lines. Ultradeep sequencing results showed that almost all SNVs found in T117 (average VAF: 8%) were also present in T1 at smaller VAFs (5%) and in all other cell lines at much higher VAFs (30%) (Supporting Information Table S3), though the resistant cell lines carried a unique set of variants (Fig. 2). Gene set‐enrichment analysis (GSEA) showed that most genes shared by all cell lines were associated with the functional terms “calcium‐dependent cell–cell adhesion” and “responses to cell death” (Fig. 2). The cell death‐related genes included KLK8, NEFH, OBSCN, and UNC5D.
Figure 2.
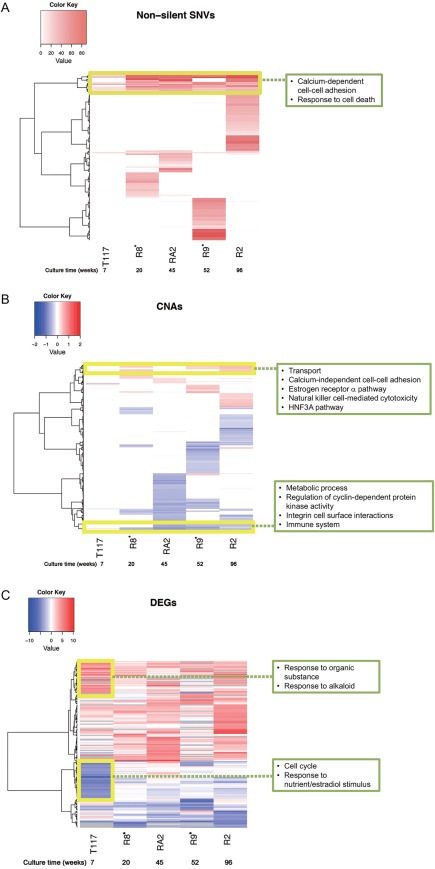
Heatmaps across cell lines. Boxes represent the results of GSEA. (A) SNVs: The SNV heatmap depicts VAFs across cell lines for nonsilent SNVs observed in at least one cell line. The vertical axis represents genes. The heatmap for synonymous SNVs is shown in Supporting Information Figure S5. (B) CNAs: the CNA heatmap depicts log cnRs for the CNA‐fragment regions. The vertical axis represents CNA‐fragment regions called in at least one cell line. (C) Expression levels: the DEG heatmap depicts log exRs for genes with >4‐fold expression changes. The vertical axis represents genes with expression changes in at least one cell line. The gray color in the heatmap indicates values filtered by data‐quality check. [Color figure can be viewed at wileyonlinelibrary.com]
DEGs showed drastic changes in T117 and lesser changes in other cell lines (Fig. 2). No gene clusters maintained the same degree of expression changes across all cell lines. Strongly upregulated genes (shown in dark red) in T117 were mostly associated with the functional terms “response to organic substance,” “negative regulation of defense responses,” and “responses to alkaloids.” The set of strongly downregulated genes (dark blue) was enriched in “cell cycle” and “response to nutrient/estradiol stimulus.” Expression changes for these genes were less drastic in the resistant cell lines. In T117, which showed much slower proliferation than did the other resistant cell lines, many downregulated genes (e.g., PLK1, AURKB, and CCNA2) were related to cell cycle regulation.
CNAs exhibited the least change in the briefly exposed cell line, while the resistant cell lines exhibited large‐scale changes (Fig. 2). The CNAs were mostly cell‐line specific, with few regions shared across the resistant cell lines. GSEA showed that the amplified regions were associated with “transport,” “calcium‐independent cell–cell adhesion,” “the estrogen receptor α pathway,” “natural killer cell‐mediated cytotoxicity,” and “the HNF3A pathway.” Deletions were related to “metabolic process,” “regulation of cyclin‐dependent protein kinase activity,” “integrin–cell surface interactions,” and “the immune system.” In the resistant cell lines, many genes related to cell–cell contact were amplified, including CLDN8, CLDN14, and CLDN17. In contrast, CDKN2C and CDKN2D, which regulate cyclin‐dependent protein kinase activity, showed copy‐number loss.
Evolutionary Viewpoint
We reconstructed a phylogenetic tree (Fig. 3A), in which branch lengths represent evolutionary time measured by VAF increments. The resistant cell lines appear to have branched from nearly the same point, which stretches back to the T1 and T117 cell lines, and branch lengths roughly correlated with culture time. The briefly exposed cell line (T117) was very close to T1. Interestingly, some SNVs were found in all cell lines studied (Fig. 3A). A notable gene with a traceable SNV is KLK8, a cell death‐related gene expressed in several types of malignancies, including ovarian, cervical, head and neck, and salivary gland cancers (Kishi et al., 2003; Cane et al., 2004; Darling et al., 2008). We checked T1 and T117 for the secondary KIT mutations found in R8 and R9, but did not observe them at the SNV detection limit of NGS, that is, a VAF of ∼0.1% (Supporting Information Table S3).
Figure 3.
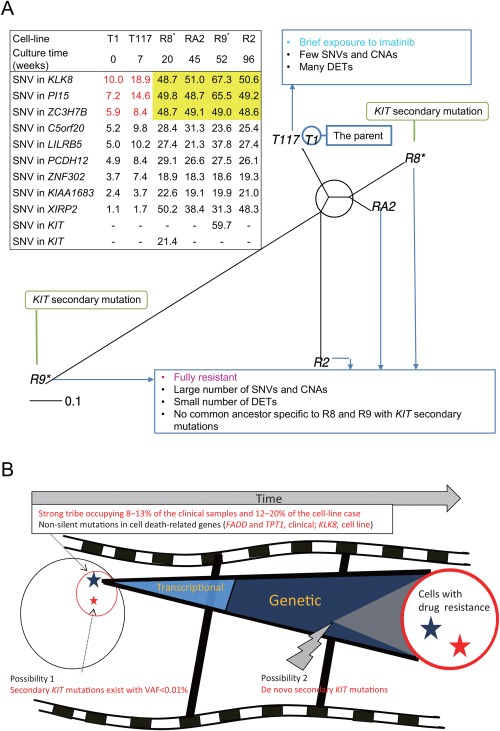
Evolutionary analysis and model of GIST imatinib resistance. (A) Phylogenetic tree and SNVs traceable to T1. The root of the tree can be assumed as T1 or located near T1. SNVs that can be tracked back to T1 are shown in the table, where the percentages of VAFs from ultra‐deep sequencing are filled in the cells. VAFs shown in yellow are nearly 50%, which indicates that all cells in the cell lines should have a variant. VAFs shown in red correspond to the VAFs of the same SNVs in T117 and T1. For reference, we listed the secondary KIT mutations, where the symbol “–” indicates VAFs below the detection limit of NGS. (B) A possible evolutionary model. A strong tribe pre‐exists at the indicated percentages, resisting cell death under imatinib exposure. A small number of cells in the tribe with secondary KIT mutations (possibility 1 shown in the figure) or cells that have acquired the mutations (possibility 2) ultimately dominate the population. During the evolutionary process, they may change their early transcriptional responses and be later selected based on genetic alterations. [Color figure can be viewed at wileyonlinelibrary.com]
Sequencing in Clinical Samples
We also performed exome sequencing of GIST samples taken from four patients before and after the acquisition of imatinib resistance. The average depths were 148, 131, 63, and 21 (Supporting Information Table S4). We called SNVs/indels that were present specifically in the resistant samples, finding 35, 19, 10, and 35 nonsilent SNVs in the four samples. Three samples had V654A in exon 13 of KIT and one sample had Y823D in exon 17 of KIT, suggesting it as a clear source of resistance. From the exome data, we also called CNAs that were present specifically in the resistant samples. When we selected amplifications overlapped in at least two out of the four samples, we found totally 134 Mb amplification regions. GSEA showed an intensely significant involvement of drug metabolism and detoxification (KEGG “metabolism of xenobiotics by cytochrome p450” with an FDR q‐value of 4 × 10−8; REACTOME “glucuronidation” with an FDR q value of 6 × 10−7). Deletions were only 0.5 Mb, and GSEA did not show any significant pathways or gene functions.
We looked at nonsilent SNVs in the resistant samples that were also present in the initial pre‐resistance samples. Excluding SNVs detected by only one sequence read, we found only six SNVs in two samples. Among the functions of genes with the SNVs, apoptosis was the only gene function that was shared between the two samples through FADD and TPT1 (Table 2). This finding is interesting because our cell‐line results showed that some SNVs in cell death‐related genes (such as KLK8) were already present in the parental cell line T1.
Table 2.
Nonsilent SNVs in Apoptosis‐Related Genes Found in Both Drug‐Resistant and Initial Pretreatment Clinical Samples
| Sample ID | Chra | Position | Variant | Gene | Amino acid change | VAF (%) in sensitive cells | VAF (%) in resistant cells | KIT mutationb VAF (%) in sensitive cells | KIT mutationb VAF (%) in resistant cells |
|---|---|---|---|---|---|---|---|---|---|
| S001 | 11 | 70052259 | A | FADD | V103I | 4.0 | 30.0 | ∼0c | 27.8 |
| S003 | 13 | 45914267 | A | TPT1 | A52V | 2.4 | 31.3 | ∼0c | 31.8 |
The two samples had only six, including these SNVs, nonsilent SNVs identified in both drug‐sensitive and ‐resistant states. No gene functions other than apoptosis for genes harboring the six SNVs were shared between the two samples.
Chr, chromosome.
For reference, we have also listed the secondary KIT V654A mutation, which was the only nonsilent SNV independently found in the two (and another) resistant samples, thereby indicating a source of the resistance.
Values below the NGS detection limit.
DISCUSSION
Resistance from a Genomic Viewpoint
We were surprised to find drastic gene‐expression changes, but only a few SNV/CNA changes in the briefly exposed cell line T117. In contrast, the resistant cell lines showed numerous genomic changes but less pronounced differences in their gene‐expression profiles. There were no clear common—or heritable—Transcriptional changes between the cell lines, unlike the common patterns observed in the SNV heatmap (Fig. 2). These observations suggested that most GIST cells first respond to imatinib by altering transcription; subsequently, those cells harboring genetic variants conducive to drug resistance (e.g., changing a protein conformation to interfere with drug binding) survive and the transcriptomic response, now unnecessary, subsides. It is also possible that cells carrying these genomic variants have simply survived without transcription alterations.
A previous investigation of resistant cell lines focused on the importance of CNAs in generating drug resistance (erlotinib and afatinib in EGFR‐mutated lung cancers) (Jia et al., 2013). Using a briefly exposed cell line, we identified the importance of transcriptional responses prior to the acquisition of full drug resistance. Interestingly, the altered transcriptional programming in the briefly exposed cell line appears to have slowed cell cycle progression and strengthened the resistance to stress (based on responses to alkaloids and biological oxidation; Fig. 2). The cell line also showed upregulated expression of γ‐amino butyric acid (GABA)‐related genes (ABAT and GABRR1). Because GABA regulates the proliferation of pluripotent and neural stem cells in embryonic and adult tissues (Young and Bordey, 2009), these observations suggested that the briefly exposed cell line has stem cell characteristics.
Critical mutations such as secondary KIT mutations have been identified as factors in drug resistance (Corless, 2014; Nishida et al., 2014); however, our findings lead us to suggest that other subsidiary SNVs/CNAs may assist in generating drug‐resistant cancer cells. GSEA indicated an importance of pathways that could enable cancer cells to escape cell death (“response to cell death” and “natural killer cell mediated cytotoxicity”), to repress cell cycle activities (“regulation of cyclin‐dependent protein kinase activity”), and to activate detoxification (“transport”, “HNF3A pathway”). We did not identify any decisive pathways that were specific to cell lines without secondary KIT mutations, for which the acquisition of additional samples with imatinib resistance would be necessary.
Resistance from a Cancer Evolutionary Viewpoint
Our analysis of VAFs (Fig. 3A) showed that SNVs saturated (approximately 50% VAFs) in all resistant cell lines had lower VAFs in T1 cells than in T117, supporting an evolutionary dynamic in which a small subpopulation of T1 cells gradually grew in the briefly exposed cell line, after which the population was overtaken by the resistant cells (Fig. 3B). Considering that the common SNVs across all the cell lines were enriched in genes that mediate responses to cell death, there might be a cell death‐resistant “tribe” among T1 cells (Fig. 3B). Most importantly, we also found evidence of such a “strong tribe” in clinical samples: the nonsilent SNVs in FADD and TPT1, apoptosis‐related genes, was present at a low percentage in the sample prior to the emergence of imatinib resistance.
The tribe seemed to substantially occupy the parental drug‐sensitive cell populations. We estimated the tribe fraction to be 12–20% in the parental cell line, as VAFs in T1 for the saturated SNVs (Fig. 3A) were 6–10% (assuming they are heterozygous and not in CNAs). Similarly, the tribe fraction in the clinical initial sample was estimated to be 8–13% (2.4/31.3–4.0/30.0), because the SNV VAFs in the initial and resistant samples were 2.4 and 31.3% for TPT1 and 4.0% and 30.0% for FADD (these resistant samples' VAFs were similar to the VAFs of the KIT V654A mutation), respectively (assuming the same tumor purities in the clinical samples before and after treatment).
Compared with the tribe fraction, the presence of the secondary KIT mutations seems negligible. Our resistant cell lines, which were independently cultured, did not always carry the same secondary KIT mutation. This indicates that the secondary mutations were newly acquired or present at a sufficiently low fraction in T1 or T117 to be easily influenced by chance. The prevalence of this fraction was <0.1% (the detection limit of NGS), or potentially below 0.01%, because T117, in which the secondary mutations were not detected, were derived from <10% of the T1 cells. For reference, a previous study of colorectal cancer with KRAS mutations related to panitumumab resistance theoretically predicted the prevalence of the drug‐resistant fraction to be on the order of 0.0001% (2,000–3,000 cells out of one billion cells) (Diaz et al., 2012).
Considering these fractions and their functional importance, a combination of cell death resistance and secondary KIT mutations would have provided cells with a better growth advantage as compared with secondary KIT mutations alone. The tribe may have endured exposure to imatinib initially, changing its transcriptional responses (as in T117), and then having acquired secondary mutations that conferred full resistance to the cancer cells (Fig. 3B). Another alternative is that the critical mutations initially existed as well, and the combination of these mutations with alterations promoting cell death resistance provided cells with full resistance to imatinib (Fig. 3B). We proposed these models in imatinib‐resistant GISTs from genome‐scale observation analysis. The next step will be to test them by molecular‐intervention experiments on multiple cell lines and to verify them using a critical number of clinical samples.
Disclosure
T. Nishida received a grant from Novartis Pharma KK and honoraria from Pfizer, Bayer, and Novartis. T. Naka received a grant from Novartis Pharma KK.
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
Supporting Information Table 3.
Supporting Information
ACKNOWLEDGMENTS
We thank Maiko Ako at NIBIO and Isao Kurosaka, Momoko Nagai, and Hanako Ono at NCC for providing technical assistance.
Contributor Information
Toshirou Nishida, Email: tnishida@ncc.go.jp.
Mamoru Kato, Email: mamkato@ncc.go.jp.
REFERENCES
- Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, Leversha MA, Jeffrey PD, Desantis D, Singer S, Brennan MF, Maki RG, DeMatteo RP. 2005. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 11:4182–4190. [DOI] [PubMed] [Google Scholar]
- Bauer S, Yu LK, Demetri GD, Fletcher JA. 2006. Heat shock protein 90 inhibition in imatinib‐resistant gastrointestinal stromal tumor. Cancer Res 66:9153–9161. [DOI] [PubMed] [Google Scholar]
- Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki RG, Tanaka M, Hecht JR, Heinrich MC, Fletcher CD, Crowley JJ, Borden EC. 2008. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol 26:626–632. [DOI] [PubMed] [Google Scholar]
- Cane S, Bignotti E, Bellone S, Palmieri M, De las Casas L, Roman JJ, Pecorelli S, Cannon MJ, O'Brien T, Santin AD. 2004. The novel serine protease tumor‐associated differentially expressed gene‐14 (KLK8/Neuropsin/Ovasin) is highly overexpressed in cervical cancer. Am J Obstet Gynecol 190:60–66. [DOI] [PubMed] [Google Scholar]
- Corless CL. 2014. Gastrointestinal stromal tumors: what do we know now? Mod Pathol 27:S1–S16. [DOI] [PubMed] [Google Scholar]
- Darling MR, Tsai S, Jackson‐Boeters L, Daley TD, Diamandis EP. 2008. Human kallikrein 8 expression in salivary gland tumors. Head Neck Pathol 2:169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. 2002. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 347:472–480. [DOI] [PubMed] [Google Scholar]
- Dhayat SA, Mardin WA, Seggewiss J, Strose AJ, Matuszcak C, Hummel R, Senninger N, Mees ST, Haier J. 2015. MicroRNA profiling implies new markers of gemcitabine chemoresistance in mutant p53 pancreatic ductal adenocarcinoma. PLoS One 10:e0143755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA, Jr ., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. 2012. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486:537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze J, Muhlenberg T, Simon S, Grabellus F, Rubin B, Taeger G, Schuler M, Treckmann J, Debiec‐Rychter M, Taguchi T, Fletcher JA, Bauer S. 2012. p53 modulation as a therapeutic strategy in gastrointestinal stromal tumors. PLoS One 7:e37776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. 1998. Gain‐of‐function mutations of c‐kit in human gastrointestinal stromal tumors. Science 279:577–580. [DOI] [PubMed] [Google Scholar]
- Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, Shinomura Y, Kitamura Y. 2003. Gain‐of‐function mutations of platelet‐derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 125:660–667. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Kikuchi E, Tanaka N, Kosaka T, Suzuki E, Mizuno R, Shinojima T, Miyajima A, Umezawa K, Oya M. 2015. Down‐regulation of NF kappa B activation is an effective therapeutic modality in acquired platinum‐resistant bladder cancer. BMC Cancer 15:324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia P, Jin H, Meador CB, Xia J, Ohashi K, Liu L, Pirazzoli V, Dahlman KB, Politi K, Michor F, Zhao Z, Pao W. 2013. Next‐generation sequencing of paired tyrosine kinase inhibitor‐sensitive and ‐resistant EGFR mutant lung cancer cell lines identifies spectrum of DNA changes associated with drug resistance. Genome Res 23:1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Kawaguchi T, Ishikawa S, Umeda T, Nakamichi R, Shapero MH, Jones KW, Nakamura Y, Aburatani H, Tsunoda T. 2010. Population‐genetic nature of copy number variations in the human genome. Hum Mol Genet 19:761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi T, Grass L, Soosaipillai A, Scorilas A, Harbeck N, Schmalfeldt B, Dorn J, Mysliwiec M, Schmitt M, Diamandis EP. 2003. Human kallikrein 8, a novel biomarker for ovarian carcinoma. Cancer Res 63:2771–2774. [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. 2012. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22:568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama Y, Mashima J, Kosuge T, Katayama T, Fujisawa T, Kaminuma E, Ogasawara O, Okubo K, Takagi T, Nakamura Y. 2015. The DDBJ Japanese Genotype–phenotype archive for genetic and phenotypic human data. Nucleic Acids Res 43:(Database issue):D18–D22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. 1978. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida T, Doi T, Naito Y. 2014. Tyrosine kinase inhibitors in the treatment of unresectable or metastatic gastrointestinal stromal tumors. Expert Opin Pharmacother 15:1979–1989. [DOI] [PubMed] [Google Scholar]
- Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, Kanakura Y, Tanaka T, Takabayashi A, Matsuda H, Kitamura Y. 1998. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet 19:323–324. [DOI] [PubMed] [Google Scholar]
- Nishida T, Kanda T, Nishitani A, Takahashi T, Nakajima K, Ishikawa T, Hirota S. 2008. Secondary mutations in the kinase domain of the KIT gene are predominant in imatinib‐resistant gastrointestinal stromal tumor. Cancer Sci 99:799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida T, Takahashi T, Miyazaki Y. 2009. Gastrointestinal stromal tumor: A bridge between bench and bedside. Gastric Cancer 12:175–188. [DOI] [PubMed] [Google Scholar]
- Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Miller VA, Pao W. 2011. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res 17:5530–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush J. 2002. Microarray data normalization and transformation. Nat Genet 32:496–501. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. 1987. The neighbor‐joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. [DOI] [PubMed] [Google Scholar]
- Sathirapongsasuti JF, Lee H, Horst BA, Brunner G, Cochran AJ, Binder S, Quackenbush J, Nelson SF. 2011. Exome sequencing‐based copy‐number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics 27:2648–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: A knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi T, Sonobe H, Toyonaga S, Yamasaki I, Shuin T, Takano A, Araki K, Akimaru K, Yuri K. 2002. Conventional and molecular cytogenetic characterization of a new human cell line, GIST‐T1, established from gastrointestinal stromal tumor. Lab Invest 82:663–665. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Serada S, Ako M, Fujimoto M, Miyazaki Y, Nakatsuka R, Ikezoe T, Yokoyama A, Taguchi T, Shimada K, Kurokawa Y, Yamasaki M, Miyata H, Nakajima K, Takiguchi S, Mori M, Doki Y, Naka T, Nishida T. 2013. New findings of kinase switching in gastrointestinal stromal tumor under imatinib using phosphoproteomic analysis. Int J Cancer 133:2737–2743. [DOI] [PubMed] [Google Scholar]
- Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H, Yamamoto S, Shinbrot E, Hama N, Lehmkuhl M, Hosoda F, Arai Y, Walker K, Dahdouli M, Gotoh K, Nagae G, Gingras MC, Muzny DM, Ojima H, Shimada K, Midorikawa Y, Goss JA, Cotton R, Hayashi A, Shibahara J, Ishikawa S, Guiteau J, Tanaka M, Urushidate T, Ohashi S, Okada N, Doddapaneni H, Wang M, Zhu Y, Dinh H, Okusaka T, Kokudo N, Kosuge T, Takayama T, Fukayama M, Gibbs RA, Wheeler DA, Aburatani H, Shibata T. 2014. Trans‐ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet 46:1267–1273. [DOI] [PubMed] [Google Scholar]
- Tuveson DA, Willis NA, Jacks T, Griffin JD, Singer S, Fletcher CD, Fletcher JA, Demetri GD. 2001. STI571 inactivation of the gastrointestinal stromal tumor c‐KIT oncoprotein: biological and clinical implications. Oncogene 20:5054–5058. [DOI] [PubMed] [Google Scholar]
- Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, Issels R, van Oosterom A, Hogendoorn PC, Van Glabbeke M, Bertulli R, Judson I. 2004. Progression‐free survival in gastrointestinal stromal tumours with high‐dose imatinib: Randomised trial. Lancet 364:1127–1134. [DOI] [PubMed] [Google Scholar]
- Yachida S, Wood LD, Suzuki M, Takai E, Totoki Y, Kato M, Luchini C, Arai Y, Nakamura H, Hama N, Elzawahry A, Hosoda F, Shirota T, Morimoto N, Hori K, Funazaki J, Tanaka H, Morizane C, Okusaka T, Nara S, Shimada K, Hiraoka N, Taniguchi H, Higuchi R, Oshima M, Okano K, Hirono S, Mizuma M, Arihiro K, Yamamoto M, Unno M, Yamaue H, Weiss MJ, Wolfgang CL, Furukawa T, Nakagama H, Vogelstein B, Kiyono T, Hruban RH, Shibata T. 2016. Genomic Sequencing identifies ELF3 as a driver of ampullary carcinoma. Cancer Cell 29:229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama T, Enomoto T, Serada S, Morimoto A, Matsuzaki S, Ueda Y, Yoshino K, Fujita M, Kyo S, Iwahori K, Fujimoto M, Kimura T, Naka T. 2013. Plasma membrane proteomics identifies bone marrow stromal antigen 2 as a potential therapeutic target in endometrial cancer. Int J Cancer 132:472–484. [DOI] [PubMed] [Google Scholar]
- Young SZ, Bordey A. 2009. GABA's control of stem and cancer cell proliferation in adult neural and peripheral niches. Physiology (Bethesda) 24:171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
Supporting Information Table 3.
Supporting Information