

Abstract
Sepsis‐induced acute kidney injury (SI‐AKI) is common and associated with high mortality. Survivors are at increased risk of chronic kidney disease. The precise mechanism underlying SI‐AKI is unknown, and no curative treatment exists. Toll‐like receptor 4 (TLR4) activates the innate immune system in response to exogenous microbial products. The result is an inflammatory reaction aimed at clearing a potential infection. However, the consequence may also be organ dysfunction as the immune response can cause collateral damage to host tissue. The purpose of this review is to describe the basis for how ligand binding to TLR4 has the potential to cause renal dysfunction and the mechanisms by which this may take place in gram‐negative sepsis. In addition, we highlight areas for future research that can further our knowledge of the pathogenesis of SI‐AKI in relation to TLR4 activation. TLR4 is expressed in the kidney. Activation of TLR4 causes cytokine and chemokine release as well as renal leucocyte infiltration. It also results in endothelial and tubular dysfunction in addition to altered renal metabolism and circulation. From a physiological standpoint, inhibiting TLR4 in large animal experimental SI‐AKI significantly improves renal function. Thus, current evidence indicates that TLR4 has the ability to mediate SI‐AKI by a number of mechanisms. The strong experimental evidence supporting a role of TLR4 in the pathogenesis of SI‐AKI in combination with the availability of pharmacological tools to target TLR4 warrants future human studies.
Keywords: acute kidney injury, AKI, renal, sepsis, TLR4, Toll‐like receptor
Toll‐like receptors (TLRs) are crucial for sensing invading microorganisms like bacteria, virus and fungi. As a first line of defence, they initiate an innate immune response that includes activation of polymorphonuclear leucocytes (PMN), monocytes and macrophages (Medzhitov 2001, Hayashi et al. 2003, Farina et al. 2004). They also mediate the release of pro‐inflammatory cytokines and interferons with the ultimate goal to identify and destroy the pathogen (Medzhitov 2001). However, activation of the innate immune system sometimes also causes collateral damage to host tissue and cells, resulting in organ dysfunction and death. In this review, we explore how one specific TLR, Toll‐like receptor 4 (TLR4), may cause acute kidney injury (AKI) in sepsis. Although many TLRs have been described, TLR4 is so far the most extensively studied. A variety of experimental studies have shown beneficial survival effects of blocking this receptor in sepsis. But, as will be briefly outlined in this review, the data from the clinical setting are thus far not convincing.
Toll‐like receptors have previously been implicated as important mediators of disease in various forms of chronic and acute renal failure (Gluba et al. 2010, Valles et al. 2014). Here we will review the experimental evidence supporting a role for TLR4 in sepsis‐induced AKI (SI‐AKI) with emphasis on described mechanisms of action. As will become evident, TLR4 activation may affect both glomerular and tubular processes and experimental data describes a significant improvement in renal function if TLR4 is inhibited in sepsis.
The Toll‐like receptor family
Ten different types of TLRs have been described in humans (TLR1‐10) (Matsushima et al. 2007). They are listed in Table 1 together with known ligands and their presumed role in SI‐AKI. Although TLRs are expressed in most human tissue, they predominate in organs and tissues typically involved in the immune defence such as spleen and blood, as well as those exposed to the external environment (i.e. skin, lung and intestines). Importantly, transcription of TLRs change as a result of an infection and the magnitude and type of TLRs expressed is based on type of invading organism (Zarember & Godowski 2002).
Table 1.
Known human Toll‐like receptors, described agonists and potential involvement in SI‐AKI
| TLR | Example of ligands | Involvement in SI‐AKI |
|---|---|---|
| TLR1 | Triacylated lipopeptides | Pam3cys, an agonist of TLR1 complexed with TLR2 stimulates complement factor B (cfB) production in human proximal tubular cells. Experimental data suggest that this can contribute to SI‐AKI (Li et al. 2016) |
| TLR2 | Bacterial: Glycolipids, phenol‐soluble modulin, lipoprotein, lipoteichoic acid peptidoglycan. Viral: proteins from measles, CMV, HSV‐1. Fungal: lipoarabinomannan, zymosan. Potential endogenous: biglycan, histones, HMBG‐1, hyaluronan, heat‐shock proteins | TLR2‐deficient mice with CLP have less renal hypoxia (Castoldi et al. 2012). Neutralizing histones acting via TLR2 and TLR4 reduces plasma creatinine In murine endotoxemia (Allam et al. 2012). Activation of TLR2 may upregulate cfB that contributes to increased mRNA level of NGAL and KIM‐1 in polymicrobial murine sepsis (Zou et al. 2013). TLR2 inhibits HCO3− reabsorption in the medullary thick ascending limb in response to LPS (Good et al. 2010, 2012). Bacterial lipopeptide acts through TLR2 to increase protein permeability in cultured glomerular endothelial cells and podocytes (Pawar et al. 2009) |
| TLR3 | Viral double‐stranded RNA | Activation of TLR3 may upregulate cfB that contributes to increased mRNA level of NGAL and KIM‐1 in polymicrobial murine sepsis (Zou et al. 2013) |
| TLR4 | Bacterial: LPS. Fungal: Mannan. Viral: Protein from RSV. Potential endogenous: heparin sulphate, fibrinogen, biglycan, histones, HMBG‐1, hyaluronan, heat‐shock proteins | TLR4 mediates reduction in urine output and GFR in a sheep model of E. coli sepsis (Fenhammar et al. 2014). Sepsis causes TLR4 upregulation in the kidney and stimulation results in renal PMN infiltration, release of pro‐inflammatory cytokines and chemokines, glomerular endothelial swelling, tubular ion transport dysfunction and apoptosis. See text for further details and references |
| TLR5 | Bacterial flagellin | Flagellin causes a systemic inflammatory response and liver injury but no renal injury, estimated by change in plasma urea, in mice (Liaudet et al. 2002). TLR5 activation may protect against urinary infection (Andersen‐Nissen et al. 2007) |
| TLR6 | Interacts with TLR2 to recognize bacterial lipopeptides and fungal zymosan | Unknown |
| TLR7 | Single‐stranded RNA from, that is HIV and Influenza virus | TLR7 has been shown to activate B‐lymphocytes and contribute to glomerulonephritis in response to viral agonists (Pawar et al. 2007). Although not verified to be mediated completely by TLR7, AKI often develops in patients with severe influenza infection and this is associated with increased risk of dying (Pettila et al. 2011). |
| TLR8 | ssRNA, synthetic imidazoquinoline derivatives | Unknown. |
| TLR9 | Viral and bacterial CpG DNA motifs | Knockdown of TLR9 by siRNA reduced the increase in plasma creatinine and urea in response to polymicrobial sepsis in mice (Liu et al. 2012). TLR9 knockout reduced AKI in CLP‐induced septic mice (Dear et al. 2006, Yasuda et al. 2008). Possibly via stimulation by mitochondrial DNA (Tsuji et al. 2015) |
| TLR10 | Unknown | Unknown |
TLR4 is a trans‐membrane protein with extracellular leucine‐rich repeats forming a horseshoe‐like shape (Kim et al. 2007). Lipopolysaccharide (LPS) is a major component of the gram‐negative bacterial cell wall and the main agonist of TLR4. On a particular cell, TLR4 is found both on the cell surface and in intracellular phagosomes where TLR4 forms a receptor complex with myeloid differentiation factor 2 (MD2) (Shimazu et al. 1999, Kawai & Akira 2011). Cluster of differentiation 14 (CD14), a co‐receptor, transfers LPS to the TLR4/MD2 complex as well as facilitates endocytosis of TLR4 (Zanoni et al. 2011, Rajaiah et al. 2015). LPS upregulates the production of pro‐inflammatory mediators via MyD88‐ and TRIF‐dependent pathways, which signal from the cell surface and endosomes respectively (Kagan et al. 2008). Both pathways cause translocation of NF‐κB to the cell nucleus resulting in production and release of cytokines and chemokines. TRIF signalling also results in activation of IRF‐3 transcribing interferons (Palsson‐McDermott & O'Neill 2004).
Human cells with TLR4 polymorphism display a reduced inflammatory response to LPS, verifying the LPS‐TLR4 connection also in humans (Tulic et al. 2007).
Sepsis and AKI
More than 5% of hospitalized patients suffer from AKI and thus have an increased risk of dying or developing chronic kidney disease (Wonnacott et al. 2014). In intensive care, AKI is more common and affects more than 50% of the patients admitted, with the severity of renal dysfunction being strongly associated with increased hospital mortality (Hoste et al. 2015).
Sepsis is the most common cause of AKI in intensive care units, and in combination, sepsis and severe renal dysfunction constitute a major risk of dying (50–65% 90‐day mortality) (Bagshaw et al. 2007, Zang & Yan 2013, Cruz et al. 2014). Renal failure in sepsis is characterized by decreased GFR, often estimated by increased plasma creatinine, and oliguria/anuria (Angus & van der Poll 2013). However, the underlying mechanisms are unknown. Compared to other aetiologies of AKI, sepsis is associated with significantly higher mortality rates. This can be attributed to the fact that septic patients often suffer from various comorbidities and generally are severely ill. However, in epidemiological studies, AKI severe enough to require renal replacement therapy has been found to be an independent predictor of mortality in patients with sepsis (Sakhuja et al. 2015). Thus, understanding the causes of renal dysfunction in sepsis and identifying new targets for treatment is pivotal if survival is to be improved in this large group of critically ill patients (Figs 1, 2, 3).
Figure 1.
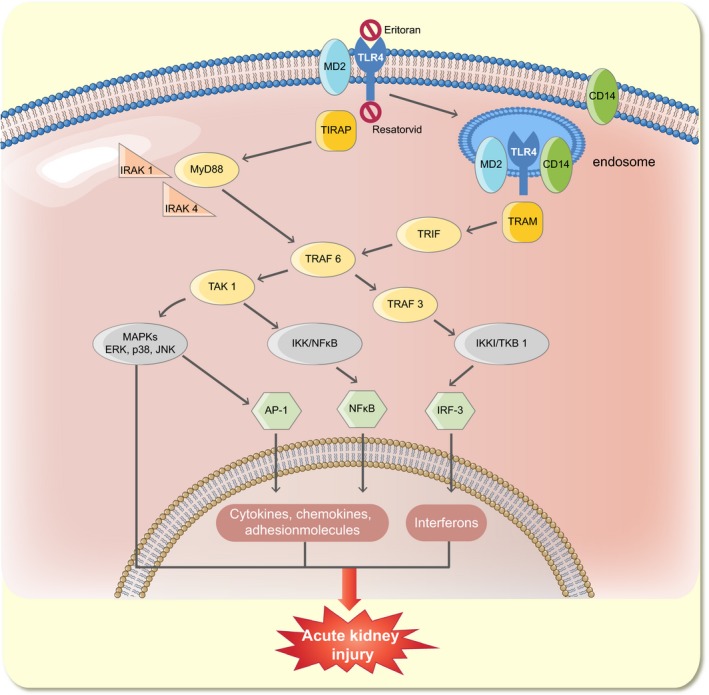
TLR4 signalling in SI‐AKI. LPS binding to the extracellular domain of TLR4 and MD2 is facilitated by CD14 and results in activation of both the MyD88‐ and TRIF‐dependent pathways. In the proximal tubule, it has been described that upon activation the entire TLR4/MD2/CD14 complex is subject to endocytosis. The downstream effects include upregulation of transcription factors NFkB, AP‐1 and IRF‐3 resulting in transcription of pro‐inflammatory genes including cytokines, chemokines, adhesion molecules and interferons. Furthermore, activation of MAPKs such as p38, ERK and JNK takes place. The ensuing overall renal effects include endothelial and tubular dysfunction in addition to altered renal metabolism and circulation (not shown in figure), giving rise to acute kidney injury. The two methods of inhibiting TLR4 signalling are shown above. Eritoran (E5564), a synthetic lipopolysaccharide, binds to cell‐surface TLR4‐MD2 without activating the receptor and thus inhibits signalling by bacterial LPS. Resatorvid (TAK‐242) binds to TLR4's intracellular domain and blocks further signalling downstream. TRIF (Toll/IL‐1 receptor domain containing adaptor inducing IFN‐β); IRF‐3 (interferon regulatory factor 3); NFkB (nuclear factor kappa‐light‐chain enhancer of activated B cells); AP‐1 (activator protein 1); TRAF (TNF receptor associated factor); IRAK (impairment of IL‐1R associated kinase 1 activity); TAK1 (transforming growth factor beta‐activated kinase 1); TBK1 (tank binding kinase 1); IKK (I‐kappa B kinase complex); MAPKs (mitogen activated protein kinases).
Figure 2.
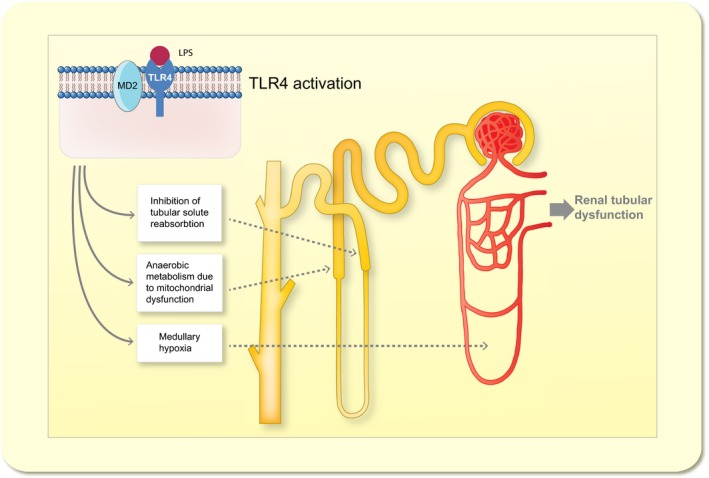
Toll‐like receptor 4 (TLR4)‐mediated renal tubular dysfunction. Selection of proposed mechanisms by which TLR4 activation contributes to tubular dysfunction in S‐AKI (for details see text). LPS (Lipopolysaccharide); TLR4 (Toll‐like receptor 4).
Figure 3.
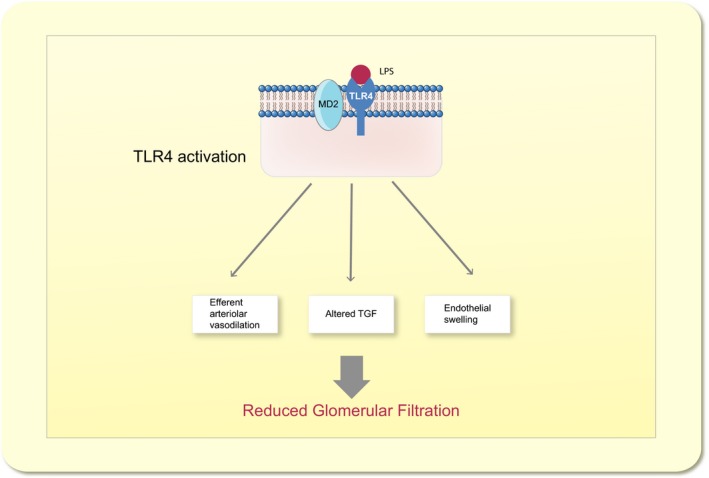
Toll‐like receptor 4 (TLR4)‐mediated reduction in glomerular filtration rate. Selection of proposed mechanisms by which TLR4 activation contributes to reduced glomerular filtration in S‐AKI (for details see text). LPS (Lipopolysaccharide); TLR4 (Toll‐like receptor 4); TGF (Tubuloglomerular feedback).
Acute kidney injury is a functional classification and the underlying causes likely differ. Many relevant hypotheses for the mechanisms of AKI have been described (Persson 2013). The most common cause of AKI is assumed to be changes in renal perfusion causing ischaemia and acute tubular necrosis (ATN) (Hultstrom 2013). For long, this concept also included SI‐AKI (Schrier & Wang 2004). However, a significant major reduction in renal blood flow has been difficult to verify in humans with sepsis (Brenner et al. 1990, Prowle et al. 2012) and post‐mortem analysis of kidneys from patients with severe sepsis and AKI have failed to show widespread ATN (Lerolle et al. 2010). On the contrary, in large animal models of septic shock, it has been shown that AKI develops even though renal blood flow is increased or unchanged (Langenberg et al. 2006, Frithiof et al. 2011). This has led to the hypothesis that local renal and/or systemic inflammation contributes to SI‐AKI and that the kidney failure often seen in association with sepsis not necessarily has to involve inadequate global renal perfusion (Lipcsey & Bellomo 2011).
Targeting TLR4 in experimental models of SI‐AKI
The recent shift in focus from SI‐AKI being a cardiovascular problem to becoming an inflammatory consequence has made TLR4 a potential mediator of SI‐AKI. Indeed, recent experimental evidence point strongly towards that TLR4 is of significant importance in the development of AKI in gram‐negative sepsis. Pre‐treatment with TAK‐242, which inhibits the effect of TLR4, in conscious sheep subjected to an intravenous LPS infusion completely abolished the AKI that developed in the vehicle treated animals (Fenhammar et al. 2011). In a follow‐up study Fenhammar et al. (2014) demonstrated that TAK‐242 reversed a pronounced decline in urine output and GFR, when used as intravenous treatment twelve hours into ovine hyperdynamic E. coli sepsis. This was associated with reduced endothelial swelling in the glomeruli, but compared to sham‐treated animals, there were no differences with regard to blood pressure, RBF or renal microcirculation, suggesting that hemodynamic factors were not pivotal for the SI‐AKI to develop.
Although renal function cannot be monitored with the same temporal resolution, similar detrimental effects of TLR4 as in sheep have been described in murine SI‐AKI. TLR4‐deficient mice were protected from the increase in BUN and serum creatinine caused by LPS and cisplatin in wild‐type mice (Ramesh et al. 2007). These results were recently confirmed in endotoxemic TLR4‐knockout mice (Smith et al. 2015). Furthermore, TLR4‐deficient mice subjected to polymicrobial sepsis had reduced increase in plasma creatinine and urea compared to wild‐type mice (Castoldi et al. 2012).
Thus, either pharmacological inhibition or genetic deletion of TLR4 significantly reduces SI‐AKI in various species and experimental models. In the following sections, we will review detailed mechanisms for how TLR4 may mediate renal dysfunction in sepsis.
Renal expression of TLR4
Several studies have verified renal TLR4 expression in both cortex and medulla (Tsuboi et al. 2002, Wolfs et al. 2002, Zarember & Godowski 2002, Laestadius et al. 2003, Samuelsson et al. 2004, El‐Achkar et al. 2006, 2007, Pulskens et al. 2008, Zhang et al. 2008, Chen et al. 2011, Kalakeche et al. 2011, Liu et al. 2014a) On a cellular level, TLR4 is mainly located in tubular epithelium but is also found in the glomeruli, including podocytes and mesangial cells, as well as vascular endothelium (Wolfs et al. 2002, Brown et al. 2007, El‐Achkar et al. 2007, Chen et al. 2011). Renal expression appears dynamic with a low constitutive level that increases in response to LPS. El‐Achkar et al. could show a fluctuating expression in rodent kidneys when subjected to polymicrobial sepsis. An initially low expression was increased throughout the tubuli, glomeruli and renal vasculature followed by a slower retreat to baseline within 3 days (El‐Achkar et al. 2006). In general, TLR4 is often situated on the apical cell membrane and would therefore present on the luminal side suggesting optimal ligand exposure in blood, filtrate and urine respectively (Wolfs et al. 2002). CD14 expression has also been noted to increase in response to sepsis. Polymicrobial sepsis‐induced CD14 predominantly in proximal tubuli where it co‐localized with TLR4 at the apical cell membrane and in endosomes in rat kidneys (El‐Achkar et al. 2006). Internalization of TLR4 into endosomes has been shown to be mediated by both CD14‐dependent and CD14‐independent processes (Zanoni et al. 2011, Rajaiah et al. 2015).
TLR4, cytokines and chemokines in SI‐AKI
Cytokines and chemokines are released during sepsis and implicated in the pathogenesis of SI‐AKI. Renal cells and circulating immune cells release several cytokines in response to TLR4‐LPS signalling, including pro‐inflammatory TNF‐α, IL‐6 and IL‐1β (Cunningham et al. 2004, Castoldi et al. 2012, Han et al. 2012). During polymicrobial sepsis, TLR4‐, TLR2‐ and MyD88‐deficient mice displayed lower levels of cytokines, in turn correlated with less renal inflammation and renal dysfunction (Castoldi et al. 2012). However, as mice without tubular TLR4 but with significantly increased systemic cytokine levels are protected against LPS‐induced renal injury, the local inflammatory response appears most important for SI‐AKI to occur (Hato et al. 2015).
Studies have shown renal chemokine release induced by TLR4 activation. Tubular epithelial cells expressed monocyte chemoattractant protein‐1 (MCP‐1) and RANTES when exposed to synthetic lipid A and released CXCL1 and CXCL2 in response to LPS (Tsuboi et al. 2002, Brown et al. 2007). Furthermore, urinary tract epithelial cells also released CXCL2 via TLR4 signalling and thus recruited leucocytes to the infected urinary tract (Patole et al. 2005). In addition to these inflammatory mediators, adhesion molecules are upregulated as a result of TLR4 activation. ICAM‐1 expression was increased on tubular epithelial cells and glomerular endothelial cells in murine endotoxemia (Cunningham et al. 2004). Hence, current data suggest that cytokines, chemokines and adhesion molecules are released due to TLR4 signalling. This contributes to renal leucocyte infiltration and renal injury in sepsis.
Leucocyte infiltration, TLR4 and SI‐AKI
In the experimental setting, renal leucocyte infiltration is associated with renal dysfunction, and in post‐mortem biopsies, from septic patients, leucocytes were observed in the glomeruli, capillaries and tubular lumens (Lerolle et al. 2010). TLR4 activation has repeatedly been shown to be involved in renal inflammation, as in murine polymicrobial sepsis, murine endotoxemia and ovine E. coli sepsis where the phenomenon is associated with renal dysfunction (Cunningham et al. 2004, Castoldi et al. 2012, Fenhammar et al. 2014). Stimulated leucocytes may both participate in eradicating invading pathogens as well as causing collateral damage to renal tissues. In the previously mentioned studies, inhibiting TLR4 signalling or depleting leucocytes improved renal function. Other data support the opposite scenario where PMN depleted mice subjected to LPS displayed greater kidney injury compared to mice with normal PMN count (Wulfert et al. 2012).
Several groups have studied the significance of local renal or systemic TLR4 in leucocyte infiltration and renal dysfunction. In an experimental set‐up of wild‐type and TLR4‐deficient mice, single kidney transplants were made between the two strains before treatment with LPS. Wild‐type animals with TLR4‐deficient kidneys developed renal inflammation and AKI, emphasizing the need for systemic TLR4 in order for SI‐AKI to develop (Cunningham et al. 2004). In contrast, renal TLR4 was necessary for AKI to develop in a different murine LPS models (Hato et al. 2015).
In summary, activation of TLR4 promotes renal leucocyte infiltration. Data supporting both beneficial and detrimental effects on the kidney as a result of this have been described. It is reasonable to assume that an inflammatory process would be able to cause endogenous tissue damage but at the same time aid in clearing an infection that has reached the kidney.
Renal endothelial effects of TLR4 activation
Endothelial dysfunction in sepsis is a complex event causing abnormal vascular tone, hyperpermeability, hypercoagulability and leucocyte migration (Aird 2003). Activation of TLR4 may be a part of the mechanism mediating endothelial dysfunction in the kidney. This is supported by reports demonstrating that TLR4‐deficient mice subjected to caecal ligation and puncture have an attenuated increase in renal vascular permeability (Castoldi et al. 2012). Furthermore, LPS‐TLR4 signalling in podocytes has been noted to cause diminished foot processes and hence proteinuria (Reiser et al. 2004).
Glomerular endothelial swelling and diminished glomerular endothelial fenestrae were noted in mice subjected to LPS or TNF‐α as well as in sheep subjected to live E. coli infusion (Fenhammar et al. 2014, Xu et al. 2015). Normal GFR is dependent on an intact and normally functioning glomerular filtration barrier, thus reduced filtration area due to these effects reduce GFR. The endothelial changes and reduction in GFR were attenuated in mice by inhibiting the effect of TNF‐α and in sheep with the TLR4 inhibitor TAK‐242.
A possible mechanism for TLR4‐induced endothelial swelling is through increased reactive oxygen species as Becerra et al. (2011) noted a ROS‐dependent influx of sodium causing cellular oedema in gram‐negative sepsis.
TLR4‐induced tubular dysfunction
Tubular dysfunction is frequently described in experimental models of sepsis and is often due to tubular cell injury (Di Sole & Girardi 2011). TLR4 and LPS interactions in tubular cells cause release of inflammatory mediators (as described above), cellular uptake of LPS oxidative stress, reduction of ion reabsorption and reduced tubular flow.
Systemically administered LPS in rats reaches the kidneys within minutes and although homogenously filtered, LPS distributes heterogeneously amongst the nephrons (El‐Achkar et al. 2006). Furthermore, LPS interacts differently with distinct tubular sections. In S1 tubular segments, systemically administered LPS is selectively taken up and neutralized after filtration into the lumen via TLR4‐CD14 influenced endocytosis, yet injures tubular segments downstream through TLR4‐dependent oxidative stress (Kalakeche et al. 2011, Zanoni et al. 2011).
It has also been suggested that the tubular cells adapt to cellular stress in sepsis by conserving energy and thus prioritizing cell survival above maintaining organ function (Gomez et al. 2014). Sodium transport is the major energy‐consuming process in tubular epithelial cells; thus, reducing sodium transport activity decreases energy expenditure and promotes cell survival (Gomez et al. 2015). Downregulation of renal sodium, chloride and glucose transporters in response to LPS and inflammatory cytokines has been described (Schmidt et al. 2007a,b,c). Later reports have demonstrated TLR4 as directly involved in inhibition of bicarbonate absorption in the medullary thick ascending limb (mTAL) (Watts et al. 2011, 2013b, Good et al. 2012). A reduction in chloride and sodium reabsorption during sepsis would in turn reduce GFR through tubuloglomerular feedback (Morrell et al. 2014). In a recent study in endotoxemic mice, reduced tubular flow was demonstrated to be TLR4‐dependent. LPS accumulation in proximal tubular lumens was seen within hours of administration, and within 1 day, luminal obstruction by LPS was observed. This mechanism may contribute to reduced urine output in gram‐negative sepsis (Nakano et al. 2015). To summarize, LPS‐induced TLR4 activation in tubular cells seems to entail inflammatory, adaptive as well as directly injurious effects.
TLR4, renal apoptosis and acute tubular necrosis
TLR4 activation may cause tubular cell injury, as mentioned above, yet the available human material to date displays renal histopathological changes in SI‐AKI discordant with the often pronounced loss of renal function (Takasu et al. 2013). Acute tubular necrosis (ATN) is commonly seen in ischaemic kidney injury but not in SI‐AKI. In a systemic review, ATN was only noted in 25 of 184 patient samples with SI‐AKI (Langenberg et al. 2008). Two later studies with post‐mortem biopsies from patients with SI‐AKI have both observed moderate tubular injury in a majority of patients. Lerolle et al. (2010) noted loss of brush border, flattening of cytoplasm, tubular vacuolization and mitochondrial injury. Takasu et al. noted merely focal tubular injury with two distinct histological profiles correlated with the expression of kidney injury molecule‐1 (KIM‐1) and mammalian target of rapamycin (mTOR) respectively (Lerolle et al. 2010, Takasu et al. 2013). Expression of both mTOR and KIM‐1 has been described as a downstream effect of TLR4 activation (Castoldi et al. 2012, Watts et al. 2013a). Thus, tubular necrosis is rare and tubular injury is observed although not extensive enough to explain the renal dysfunction seen in SI‐AKI.
The role of apoptosis in SI‐AKI is debated. Lerolle et al. found increased apoptosis in the tubuli but Takasu et al. could not (Lerolle et al. 2010, Takasu et al. 2013). Data from animal models are also inconsistent. Several experimental settings have found increased apoptosis in tubular epithelial cells, including murine endotoxemia and murine polymicrobial sepsis (Cunningham et al. 2004, Castoldi et al. 2012, Lee et al. 2012). Further, pro‐apoptotic signalling by TLR4 has been proposed including caspase induction (Cunningham et al. 2004, Castoldi et al. 2012, Liu et al. 2014b, Hsu et al. 2016), However, other studies involving ovine E. coli sepsis and murine polymicrobial sepsis could not show increased apoptosis (Dear et al. 2006, Langenberg et al. 2014). In human SI‐AKI, neither necrosis nor apoptosis are a dominant feature making the significance of the experimental observations of those events uncertain. The limited histological damage seen in human SI‐AKI may perhaps be explained by a TLR4‐mediated adaptive mechanism as discussed previously. SI‐AKI could also be mainly a physiological and not a morphological problem.
Renal macrocirculatory alterations as cause of SI‐AKI
Sepsis‐induced acute kidney injury (SI‐AKI) has traditionally been viewed as a result of hypoperfusion due to distributional hypovolemia and shock (Schrier & Wang 2004). Experimental and clinical observations have challenged this view, and a more complex perspective including cellular dysfunction and microcirculatory alterations has recently been postulated (Gomez et al. 2014). Mediators such as nitric oxide (NO), cytokines and reactive oxygen species (ROS) induce alterations in macro‐ and microcirculation and are released due to TLR4 activation (Patzak et al. 2004, Holmqvist et al. 2005).
Clinical observational studies have detected a correlation between lower mean arterial pressure (MAP) and the risk of progression of AKI (Poukkanen et al. 2013). There is also a correlation between the aggregated time of hypotension defined as MAP < 65 and the risk of progression of AKI (Janssen van Doorn et al. 2014). In experimental studies, shorter periods of hypoperfusion alone seldom result in AKI (Saotome et al. 2010). In the clinical setting, the observation of a low frequency of AKI after cardiac arrests supports this view (Chua et al. 2012). The susceptibility to low arterial blood pressure might, however, differ in an inflamed kidney where hypotension might be considered a second insult. As hypotension is a common feature of septic shock, it might be a clinically relevant mechanism of SI‐AKI development. In a large randomized controlled study of different target blood pressures (MAP 65–70 vs. 80–85), a decreased risk of AKI with higher blood pressures was observed in patients with a previous history of hypertension but not otherwise in healthy adults (Asfar et al. 2014). Fenhammar et al. (2011) reported that TLR4 inhibition both prevented LPS‐induced hypotension and AKI but merely treating the low blood pressure with vasopressors had no effect on the development of renal dysfunction. This indicates that hypotension is not the sole cause of TLR4‐mediated SI‐AKI. Data further supporting this view was presented in a recent porcine experimental sepsis study were renal failure developed in some animals but not others, even though perfusion pressure was identical in both groups (Benes et al. 2011).
Large animal experiments have described an early phase of septic AKI with normal or increased renal blood flow (RBF) during sepsis where renal hypoperfusion in a global sense reasonably cannot explain the reduction in GFR (Langenberg et al. 2006). Haemodynamically, this hyperdynamic state is characterized by an increased cardiac output (CO) and a low systemic vascular resistance, similar to what is commonly observed in early clinical sepsis (Langenberg et al. 2005). Human data of RBF during sepsis are scarce as the unreliability of PAH clearance in sepsis until recently mandated for more invasive measuring (Brenner et al. 1990, Prowle et al. 2012). In the studies by Brenner et al. and Prowle et al., including a total of 18 septic patients, absolute values of RBF exhibit large variations, although some common patterns were observed. The renal fraction of the cardiac output was decreased and was by Brenner et al. suggested to correlate to GFR, although the small number of participating patients makes this conclusion uncertain. Whereas high CO and AKI in early human sepsis seem to be common, animal models of septic AKI can be categorized as either hyper‐ or hypodynamic depending on the effects of CO (Langenberg et al. 2005). Hypodynamic circulation is most often accompanied by reduced RBF where hyperdynamic models often exhibit less affected or increased RBF. In the previous mentioned ovine study, where sepsis was induced by E. coli infusion, TLR4 inhibition significantly improved renal function without affecting renal blood flow (Fenhammar et al. 2014).
Renocirculatory dysregulation in S‐AKI
There are several proposed mechanisms that might further explain the macrocirculatory alterations and their relevance to renal function.
Glomerular filtration is dependent of the perfusion pressure regulated by the tone of the afferent and efferent arteriole. A reduced renal blood flow, either caused by low systemic pressure or increased intrarenal resistance due to pathological afferent arteriolar constriction, may reduce GFR due to lowered filtration pressure. In the hyperdynamic models with increased RBF, filtration pressure has not been directly assessed to our knowledge, but may theoretically be reduced as well. The efferent arterial tone is crucial for maintaining an adequate filtration pressure, and several mediators linked to TLR4 activation such as NO, prostaglandins and endothelin are known to influence vascular tone (El‐Achkar et al. 2007, Fenhammar et al. 2008). During septic shock, there is a massive release of NO. NO counteracts the constricting effects of angiotensin II on the efferent arteriole (Patzak et al. 2004). This may cause a reduction in the efferent arteriolar tone thus reducing filtration pressure resulting in the typical decrease in urine output and increase in plasma creatinine observed in SI‐AKI. Improvement of renal function after angiotensin II administration, although RBF is decreased, further strengthens this hypothesis (Wan et al. 2009). Increased expression of iNOS and eNOS in these septic models is found in the renal cortex with a paradoxically decreased expression in the renal medulla (Langenberg et al. 2014). This contrasting difference in expression pattern may explain why blockade of iNOS do not restore renal function (Ishikawa et al. 2012).
Increased intrarenal vascular resistance is, however, also linked to septic AKI in man (Dewitte et al. 2012). Some endotoxemic large animal models (Fenhammar et al. 2008) and small animal models (Nitescu et al. 2008) improve renal function by mainly vasodilatory interventions, indicating renal vasoconstriction by TLR4. This may be due to a hypodynamic experimental phenotype, but an increased intrarenal resistance is also demonstrated in hyperdynamic septic pigs with AKI, compared with those lacking AKI (Benes et al. 2011). An increase in intrarenal resistance has been suggested to reflect a different mechanism in septic AKI which may be more dominant later in the clinical course because manifest AKI in man usually is accompanied by a reduced RBF (Prowle et al. 2012).
One of the key features of the renal circulation is autoregulation of blood flow within a wide range of systemic blood pressures. Clinical data suggest that this intrinsic control may be disrupted during sepsis where renal blood flow seems to correlate to cardiac output (Prowle et al. 2012). It has been reported that endotoxemia attenuates the dynamic response of the tubuloglomerular feedback mechanism (TGF) in rats (Nitescu et al. 2010). TLR4 might be a key component as CLP‐treated mice upregulates cTAL COX‐2 and while developing AKI, but not if TLR4 deficient (El‐Achkar et al. 2007). COX‐2 upregulation in cTAL and adjacent macula densa affects the TGF in a proconstrictive manner (Araujo & Welch 2009).
Altered microcirculation during endotoxemia and TLR4 activation
As mentioned earlier, renal tubular cells express TLR4 and can be directly activated by LPS. Also, an impaired microcirculation in the peri‐tubular capillary network has been described during endotoxemia in mice (Tiwari et al. 2005, Wu et al. 2007). Others report a peri‐tubular hypervelocity of red blood cells (Burban et al. 2013) during CLP‐induced sepsis in rat with a reduction in GFR, which in turn was reversed with noradrenaline infusion. Peri‐tubular capillary dysfunction is suggested to be an early feature of SI‐AKI in mice after CLP (Wang et al. 2012). Reduced tubular flow and impaired peri‐tubular microcirculation resulting in AKI have recently been demonstrated in LPS‐treated mice and are likely TLR4 dependent, but apparently by a pathway independent of TNF‐α (Nakano et al. 2015).
Several authors have investigated changes in renal microcirculation in general and if this is linked to the development of SI‐AKI. There are a number of methods to assess microcirculation, which may explain the variability in the findings. Authors using laser Doppler probes (Di Giantomasso et al. 2003, Porta et al. 2006, Chvojka et al. 2008, Fenhammar et al. 2014, Calzavacca et al. 2015) have described both variable, regionally reduced and regionally increased tissue perfusion in their experiments. Interestingly, TLR4 antagonism did not increase neither cortical nor medullar perfusion while still attenuating AKI (Fenhammar et al. 2014). Kidneys of endotoxemic rats evaluated with laser speckle imaging after fluid resuscitation present a pattern of increased heterogeneity regarding microcirculation (Legrand et al. 2011). There are both hypoperfused and normally perfused areas in conjunction with leucocyte infiltration and increased iNOS expression. These findings have generated a hypothesis of a heterogenous microcirculatory defect in gram‐negative SI‐AKI, where hypoperfused areas generate micro‐ischaemia despite normal total renal blood flow (Gomez et al. 2014). This may in turn explain differences and variability measured with laser Doppler probes besides different experimental design.
Renal oxygenation after TLR4 activation
In human postoperative AKI, there is a characteristic change in oxygen demand in comparison with sodium reabsorption (Redfors et al. 2010). Sodium resorption is the result of the most oxygen demanding process in the kidney and oxygen demand per reabsorbed molecule of sodium increases during AKI in a typical manner. Similar findings are observed in fluid resuscitated endotoxemic rats (Johannes et al. 2006) and may be commonplace in AKI in general. During the initial hours after LPS administration, oxygen consumption, QO2 or VO2, seems to remain fairly unchanged in both small and large animals (Johannes et al. 2006, Porta et al. 2006, Dyson et al. 2011). An exception is non‐resuscitated animal models of sepsis, both large and small, where significant reduction in RBF and oxygen delivery (DO2) occurs (Gullichsen et al. 1989, Johannes et al. 2009a). Increasing arterio‐venous oxygen difference has been described in the initial phase of endotoxemia (Gullichsen et al. 1989) and may to a certain extent compensate a reduced oxygen delivery by increasing oxygen extraction (Porta et al. 2006). Whereas renal QO2 may be measured quite reliably, the actual demand from a cellular or regional perspective is not always identical. Microscopic hypoxic areas have been described (Johannes et al. 2009a,b, Dyson et al. 2011), at least in conjuncture with reduced RBF, and this has been proposed to contribute to the development of AKI. Intrarenal oxygen shunting (Leong et al. 2007) is suggested to maintain tissue oxygenation constant during normal physiologic conditions and preventing hyperoxia despite altered renal blood flows necessary for GFR regulation. Besides overall renal DO2 and QO2, this would be a third determinant of tissue oxygenation. BOLD‐MRI is used to determine oxygenation in tissue, but intrarenal oxygen shunting may add uncertainty to this measurement in the kidney (Evans et al. 2007, Niendorf et al. 2015). BOLD‐MRI was used to demonstrate intact tissue oxygenation after 18 h LPS and TLR4 stimulation in mice (Tran et al. 2011), but in that study the lack of hypoxia was also indicated by an absence of elevated HIF‐1‐α expression, which is a significant marker for tubular hypoxia. However, regionally lower tissue oxygenation in the medulla paired with lower medullary perfusion is found in hyperdynamic gram‐negative sepsis in sheep (Calzavacca et al. 2015). This despite elevated RBF and maintained cortical oxygenation and perfusion, which may reflect intrarenal redistribution of blood flow during sepsis.
TLR4 activation and renal oxygen utilization
Lactate/pyruvate ratios reflect anaerobic metabolism and when increased imply a difference between actual demand and consumption of oxygen. Increased intrarenal ratios have been observed both cortically and medullary after TLR4 activation by either E. coli or LPS (Levy et al. 2003, Fenhammar et al. 2014). TLR4 blocking during E. coli infusion attenuates the increased lactate/pyruvate ratio and restores renal function. In LPS‐treated rats (Levy et al. 2003), the increased lactate/pyruvate ratio reflects an actual decrease in intrarenal ATP levels when accompanied by a profound reduction in RBF.
Increased anaerobic metabolism could theoretically occur despite normal oxygen delivery if mitochondrial function is impaired. As the kidney is second only to the heart in mitochondrial density, a role for mitochondrial dysfunction in septic AKI seems plausible. However, when Porta et al. (2006) investigated mitochondrial dysfunction in pigs after 24 h of TLR4 stimulation by LPS infusion, no mitochondrial dysfunction was found in the kidney. Others have described morphological changes of the proximal tubular mitochondria after LPS exposure in mice, such as swelling and rarefied cristae, and these have been linked to functional impairment assessed by cytochrome c oxidase activity, the last enzyme in the respiratory electron transport chain (Tran et al. 2011). The same authors described that mice lacking PGC‐1‐α, an important regulator of, mitochondrial biogenesis, exhibited a lack of recovery in renal function after TLR4 activation by LPS administration. Interestingly, mice exposed to LPS developed AKI where LPS seemed to suppress PGC‐1‐α and contribute to mitochondrial dysfunction (Smith et al. 2015). This was not observed in TLR4‐deficient mice who also maintained normal renal function. Autophagy is also induced in renal tubular epithelial cells by LPS through a TLR4‐dependent mechanism and seems to contribute to renoprotection (Leventhal et al. 2016). Renoprotective effects by autophagy have also been supported in other models of SI‐AKI (Hsiao et al. 2012). Species and experimental phenotype may explain different findings regarding mitochondrial dysfunction. In the experiments by Tran et al., RBF decreased early and significantly, in contrast to the study by Porta et al., which may explain the different findings. Also, in the later, GFR is not assessed and may therefore reflect a lack of AKI development.
Clinical trials of substances targeting TLR4 in sepsis
Two compounds interfering with LPS‐TLR4 signalling have been used in clinical trials: Eritoran (also known as E5564) and Resatorvid (also known as TAK‐242); however, none have focused specifically on SI‐AKI. Eritoran is a synthetically produced lipopolysaccharide that binds to cell‐surface TLR4‐MD2 receptor without activating it and thereby blocks the effects of bacterial LPS. Resatorvid attaches to the intracellular domain of TLR4 and inhibits the signal transduction leading to NFκB activation. A randomized, double‐blind, placebo‐controlled trial of TAK‐242 showed a trend towards a reduced 28‐day mortality in patients with both septic shock and respiratory failure in the treatment group, but the result was not significant (Rice et al. 2010). No data on renal function were presented separately, but the Sequential Organ Failure Assessment score (SOFA, in which plasma creatinine is included) did not differ between groups. Eritoran was first investigated in a prospective, randomized, double‐blind, placebo‐controlled, multi‐centre, ascending‐dose trial in which a high dose (105 mg) showed a tendency to reduce mortality in patients with severe sepsis (Tidswell et al. 2010). In a follow‐up phase III study, Eritoran did not increase survival in septic patients compared to placebo (Opal et al. 2013). Furthermore, patients with SI‐AKI did not have reduced mortality if treated with Eritoran. This may be due to the facts that also gram‐positive infections were treated with Eritoran, a relatively low mortality rate in both the treatment and placebo group, treatment was initiated too late or that low levels of circulating LPS was present. It may also indicate that TLR4 activation does not cause AKI in these patients or that separate inflammatory mediators, acting via pathways different from TLR4, also contributes to SI‐AKI. Different study designs may, however, truly test the hypothesis that inhibition of TLR4 attenuates SI‐AKI in human gram‐negative sepsis.
Conclusion
TLR4 is central in the inflammatory signalling cascade triggered by infection, as it constitutes the main sensor of gram‐negative bacteria, which initiates an immune reaction causing host damage. In the experimental setting, TLR4 stimulation of the innate immune system can cause renal injury and dysfunction. What is more, TLR4‐signalling blockade in various sepsis models blunts or even abolishes AKI.
Experimentally, TLR4 activation entails both glomerular and tubular effects reducing GFR and impairing tubular function. Glomerular endothelial swelling in combination with decreased filtration pressure (due to either pre‐glomerular vasoconstriction or post‐glomerular vasodilation) plays a role in diminishing GFR. TLR4‐mediated mitochondrial dysfunction and an adaptive reduction in bicarbonate reabsorption further compromises tubular function. In human sepsis, the mechanisms underlying renal dysfunction remain unknown, and so is the exact role of LPS mediated TLR4‐activation. For completely blocking the detrimental effects of the immune system in SI‐AKI, it appears likely that further signalling pathways other than those elicited via TLR4 require targeting.
Today, plenty pre‐clinical data support targeting TLR4 for preventing or treating AKI in human gram‐negative sepsis. Strict selection of patients with gram‐negative infections and assessment of circulating LPS is recommended at this stage.
Conflict of interest
The authors report no conflicting interests.
We would like to thank the Stiftelsen Nordisk Fysiologi (SNF) and the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) for their generous support for the AP Symposium on ‘RENOPROTECTION’. RF was supported by funds from the Swedish Research Council (grant 523‐2014‐2569).
References
- Aird, W.C. 2003. The hematologic system as a marker of organ dysfunction in sepsis. Mayo Clin Proc, 78, 869–881. [DOI] [PubMed] [Google Scholar]
- Allam, R. , Scherbaum, C.R. , Darisipudi, M.N. , Mulay, S.R. , Hagele, H. , Lichtnekert, J. , Hagemann, J.H. , Rupanagudi, K.V. , Ryu, M. , Schwarzenberger, C. et al 2012. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol 23, 1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen‐Nissen, E. , Hawn, T.R. , Smith, K.D. , Nachman, A. , Lampano, A.E. , Uematsu, S. , Akira, S. & Aderem, A. 2007. Cutting edge: Tlr5−/− mice are more susceptible to Escherichia coli urinary tract infection. J Immunol 178, 4717–4720. [DOI] [PubMed] [Google Scholar]
- Angus, D.C. & van der Poll, T. 2013. Severe sepsis and septic shock. N Engl J Med 369, 840–851. [DOI] [PubMed] [Google Scholar]
- Araujo, M. & Welch, W.J. 2009. Cyclooxygenase 2 inhibition suppresses tubuloglomerular feedback: roles of thromboxane receptors and nitric oxide. Am J Physiol Renal Physiol 296, F790–F794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asfar, P. , Meziani, F. , Hamel, J.F. , Grelon, F. , Megarbane, B. , Anguel, N. , Mira, J.P. , Dequin, P.F. , Gergaud, S. , Weiss, N. et al 2014. High versus low blood‐pressure target in patients with septic shock. N Engl J Med 370, 1583–1593. [DOI] [PubMed] [Google Scholar]
- Bagshaw, S.M. , Uchino, S. , Bellomo, R. , Morimatsu, H. , Morgera, S. , Schetz, M. , Tan, I. , Bouman, C. , Macedo, E. , Gibney, N. , Tolwani, A. , Oudemans‐van Straaten, H.M. , Ronco, C. , Kellum, J.A. & Beginning & Ending Supportive Therapy for the Kidney, I . 2007. Septic acute kidney injury in critically ill patients: clinical characteristics and outcomes. Clin J Am Soc Nephrol, 2, 431–439. [DOI] [PubMed] [Google Scholar]
- Becerra, A. , Echeverría, C. , Varela, D. , Sarmiento, D. , Armisén, R. , Nuñez‐Villena, F. , Montecinos, M. & Simon, F. 2011. Transient receptor potential melastatin 4 inhibition prevents lipopolysaccharide‐induced endothelial cell death. Cardiovasc Res 91, 677–684. [DOI] [PubMed] [Google Scholar]
- Benes, J. , Chvojka, J. , Sykora, R. , Radej, J. , Krouzecky, A. , Novak, I. & Matejovic, M. 2011. Searching for mechanisms that matter in early septic acute kidney injury: an experimental study. Crit Care 15, R256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner, M. , Schaer, G.L. , Mallory, D.L. , Suffredini, A.F. & Parrillo, J.E. 1990. Detection of renal blood flow abnormalities in septic and critically ill patients using a newly designed indwelling thermodilution renal vein catheter. Chest 98, 170–179. [DOI] [PubMed] [Google Scholar]
- Brown, H.J. , Lock, H.R. , Wolfs, T.G. , Buurman, W.A. , Sacks, S.H. & Robson, M.G. 2007. Toll‐like receptor 4 ligation on intrinsic renal cells contributes to the induction of antibody‐mediated glomerulonephritis via CXCL1 and CXCL2. J Am Soc Nephrol 18, 1732–1739. [DOI] [PubMed] [Google Scholar]
- Burban, M. , Hamel, J.F. , Tabka, M. , de La Bourdonnaye, M.R. , Duveau, A. , Mercat, A. , Cales, P. , Asfar, P. & Lerolle, N. 2013. Renal macro‐ and microcirculation autoregulatory capacity during early sepsis and norepinephrine infusion in rats. Crit Care 17, R139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzavacca, P. , Evans, R.G. , Bailey, M. , Bellomo, R. & May, C.N. 2015. Cortical and Medullary Tissue Perfusion and Oxygenation in Experimental Septic Acute Kidney Injury. Crit Care Med 43, e431–e439. [DOI] [PubMed] [Google Scholar]
- Castoldi, A. , Braga, T.T. , Correa‐Costa, M. , Aguiar, C.F. , Bassi, E.J. , Correa‐Silva, R. , Elias, R.M. , Salvador, F. , Moraes‐Vieira, P.M. , Cenedeze, M.A. , Reis, M.A. , Hiyane, M.I. , Pacheco‐Silva, A. , Goncalves, G.M. & Saraiva Camara, N.O. 2012. TLR2, TLR4 and the MYD88 signaling pathway are crucial for neutrophil migration in acute kidney injury induced by sepsis. PLoS One 7, e37584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Hartono, J.R. , John, R. , Bennett, M. , Zhou, X.J. , Wang, Y. , Wu, Q. , Winterberg, P.D. , Nagami, G.T. & Lu, C.Y. 2011. Early interleukin 6 production by leukocytes during ischemic acute kidney injury is regulated by TLR4. Kidney Int 80, 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua, H.R. , Glassford, N. & Bellomo, R. 2012. Acute kidney injury after cardiac arrest. Resuscitation 83, 721–727. [DOI] [PubMed] [Google Scholar]
- Chvojka, J. , Sykora, R. , Krouzecky, A. , Radej, J. , Varnerova, V. , Karvunidis, T. , Hes, O. , Novak, I. , Radermacher, P. & Matejovic, M. 2008. Renal haemodynamic, microcirculatory, metabolic and histopathological responses to peritonitis‐induced septic shock in pigs. Crit Care 12, R164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz, M.G. , Dantas, J.G. , Levi, T.M. , Rocha Mde, S. , de Souza, S.P. , Boa‐Sorte, N. , de Moura, C.G. & Cruz, C.M. 2014. Septic versus non‐septic acute kidney injury in critically ill patients: characteristics and clinical outcomes. Rev Bras Ter Intensiva 26, 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, P.N. , Wang, Y. , Guo, R. , He, G. & Quigg, R.J. 2004. Role of Toll‐like receptor 4 in endotoxin‐induced acute renal failure. J Immunol 172, 2629–2635. [DOI] [PubMed] [Google Scholar]
- Dear, J.W. , Yasuda, H. , Hu, X. , Hieny, S. , Yuen, P.S. , Hewitt, S.M. , Sher, A. & Star, R.A. 2006. Sepsis‐induced organ failure is mediated by different pathways in the kidney and liver: acute renal failure is dependent on MyD88 but not renal cell apoptosis. Kidney Int 69, 832–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewitte, A. , Coquin, J. , Meyssignac, B. , Joannes‐Boyau, O. , Fleureau, C. , Roze, H. , Ripoche, J. , Janvier, G. , Combe, C. & Ouattara, A. 2012. Doppler resistive index to reflect regulation of renal vascular tone during sepsis and acute kidney injury. Crit Care 16, R165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giantomasso, D. , Morimatsu, H. , May, C.N. & Bellomo, R. 2003. Intrarenal blood flow distribution in hyperdynamic septic shock: effect of norepinephrine. Crit Care Med 31, 2509–2513. [DOI] [PubMed] [Google Scholar]
- Di Sole, F. & Girardi, A.C. 2011. Uncovering the pathway of sepsis‐induced renal tubular dysfunction. Focus on “Basolateral LPS inhibits NHE3 and HCOFormula absorption through TLR4/MyD88‐dependent ERK activation in medullary thick ascending limb”. Am J Physiol Cell Physiol 301, C1290–C1292. [DOI] [PubMed] [Google Scholar]
- Dyson, A. , Bezemer, R. , Legrand, M. , Balestra, G. , Singer, M. & Ince, C. 2011. Microvascular and interstitial oxygen tension in the renal cortex and medulla studied in a 4‐h rat model of LPS‐induced endotoxemia. Shock 36, 83–89. [DOI] [PubMed] [Google Scholar]
- El‐Achkar, T.M. , Huang, X. , Plotkin, Z. , Sandoval, R.M. , Rhodes, G.J. & Dagher, P.C. 2006. Sepsis induces changes in the expression and distribution of Toll‐like receptor 4 in the rat kidney. Am J Physiol Renal Physiol 290, F1034–F1043. [DOI] [PubMed] [Google Scholar]
- El‐Achkar, T.M. , Plotkin, Z. , Marcic, B. & Dagher, P.C. 2007. Sepsis induces an increase in thick ascending limb Cox‐2 that is TLR4 dependent. Am J Physiol Renal Physiol 293, F1187–F1196. [DOI] [PubMed] [Google Scholar]
- Evans, R.G. , Leong, C.L. , Anderson, W.P. & O'Connor, P.M. 2007. Don't be so BOLD: potential limitations in the use of BOLD MRI for studies of renal oxygenation. Kidney Int, 71, 1327–1328; author reply 1328. [DOI] [PubMed] [Google Scholar]
- Farina, C. , Theil, D. , Semlinger, B. , Hohlfeld, R. & Meinl, E. 2004. Distinct responses of monocytes to Toll‐like receptor ligands and inflammatory cytokines. Int Immunol 16, 799–809. [DOI] [PubMed] [Google Scholar]
- Fenhammar, J. , Andersson, A. , Frithiof, R. , Forestier, J. , Weitzberg, E. , Sollevi, A. & Hjelmqvist, H. 2008. The endothelin receptor antagonist tezosentan improves renal microcirculation in a porcine model of endotoxemic shock. Acta Anaesthesiol Scand 52, 1385–1393. [DOI] [PubMed] [Google Scholar]
- Fenhammar, J. , Rundgren, M. , Forestier, J. , Kalman, S. , Eriksson, S. & Frithiof, R. 2011. Toll‐like receptor 4 inhibitor TAK‐242 attenuates acute kidney injury in endotoxemic sheep. Anesthesiology 114, 1130–1137. [DOI] [PubMed] [Google Scholar]
- Fenhammar, J. , Rundgren, M. , Hultenby, K. , Forestier, J. , Taavo, M. , Kenne, E. , Weitzberg, E. , Eriksson, S. , Ozenci, V. , Wernerson, A. & Frithiof, R. 2014. Renal effects of treatment with a TLR4 inhibitor in conscious septic sheep. Crit Care 18, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frithiof, R. , Soehnlein, O. , Eriksson, S. , Fenhammar, J. , Hjelmqvist, H. , Lindbom, L. & Rundgren, M. 2011. The effects of isoflurane anesthesia and mechanical ventilation on renal function during endotoxemia. Acta Anaesthesiol Scand 55, 401–410. [DOI] [PubMed] [Google Scholar]
- Gluba, A. , Banach, M. , Hannam, S. , Mikhailidis, D.P. , Sakowicz, A. & Rysz, J. 2010. The role of Toll‐like receptors in renal diseases. Nat Rev Nephrol 6, 224–235. [DOI] [PubMed] [Google Scholar]
- Gomez, H. , Ince, C. , De Backer, D. , Pickkers, P. , Payen, D. , Hotchkiss, J. & Kellum, J.A. 2014. A unified theory of sepsis‐induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock 41, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez, H. , Jin, K. & Kellum, J.A. 2015. The Role of Energy Regulation in the Tubular Epithelial Cell Response to Sepsis. Nephron 131, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good, D.W. , George, T. & Watts, B.A. III 2010. Toll‐like receptor 2 mediates inhibition of HCO(3)(‐) absorption by bacterial lipoprotein in medullary thick ascending limb. Am J Physiol Renal Physiol 299, F536–F544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good, D.W. , George, T. & Watts, B.A. III 2012. Toll‐like receptor 2 is required for LPS‐induced Toll‐like receptor 4 signaling and inhibition of ion transport in renal thick ascending limb. J Biol Chem 287, 20208–20220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullichsen, E. , Nelimarkka, O. , Halkola, L. & Niinikoski, J. 1989. Renal oxygenation in endotoxin shock in dogs. Crit Care Med 17, 547–550. [DOI] [PubMed] [Google Scholar]
- Han, M. , Li, Y. , Liu, M. , Li, Y. & Cong, B. 2012. Renal neutrophil gelatinase associated lipocalin expression in lipopolysaccharide‐induced acute kidney injury in the rat. BMC Nephrol 13, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hato, T. , Winfree, S. , Kalakeche, R. , Dube, S. , Kumar, R. , Yoshimoto, M. , Plotkin, Z. & Dagher, P.C. 2015. The macrophage mediates the renoprotective effects of endotoxin preconditioning. J Am Soc Nephrol 26, 1347–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, F. , Means, T.K. & Luster, A.D. 2003. Toll‐like receptors stimulate human neutrophil function. Blood 102, 2660–2669. [DOI] [PubMed] [Google Scholar]
- Holmqvist, B. , Olsson, C.F. , Svensson, M.L. , Svanborg, C. , Forsell, J. & Alm, P. 2005. Expression of nitric oxide synthase isoforms in the mouse kidney: cellular localization and influence by lipopolysaccharide and Toll‐like receptor 4. J Mol Histol 36, 499–516. [DOI] [PubMed] [Google Scholar]
- Hoste, E.A. , Bagshaw, S.M. , Bellomo, R. , Cely, C.M. , Colman, R. , Cruz, D.N. , Edipidis, K. , Forni, L.G. , Gomersall, C.D. , Govil, D. et al 2015. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI‐EPI study. Intensive Care Med 41, 1411–1423. [DOI] [PubMed] [Google Scholar]
- Hsiao, H.W. , Tsai, K.L. , Wang, L.F. , Chen, Y.H. , Chiang, P.C. , Chuang, S.M. & Hsu, C. 2012. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock 37, 289–296. [DOI] [PubMed] [Google Scholar]
- Hsu, S.P. , Chen, C.C. & Chien, C.T. 2016. Pretreatment of sialic acid efficiently prevents lipopolysaccharide‐induced acute renal failure and suppresses TLR4/gp91‐mediated apoptotic signaling. Kidney Blood Press Res 41, 267–277. [DOI] [PubMed] [Google Scholar]
- Hultstrom, M. 2013. Neurohormonal interactions on the renal oxygen delivery and consumption in haemorrhagic shock‐induced acute kidney injury. Acta Physiol (Oxf) 209, 11–25. [DOI] [PubMed] [Google Scholar]
- Ishikawa, K. , Calzavacca, P. , Bellomo, R. , Bailey, M. & May, C.N. 2012. Effect of selective inhibition of renal inducible nitric oxide synthase on renal blood flow and function in experimental hyperdynamic sepsis. Crit Care Med 40, 2368–2375. [DOI] [PubMed] [Google Scholar]
- Janssen van Doorn, K. , Verbrugghe, W. , Wouters, K. , Jansens, H. & Jorens, P.G. 2014. The duration of hypotension determines the evolution of bacteremia‐induced acute kidney injury in the intensive care unit. PLoS One 9, e114312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes, T. , Mik, E.G. , Nohe, B. , Raat, N.J. , Unertl, K.E. & Ince, C. 2006. Influence of fluid resuscitation on renal microvascular PO2 in a normotensive rat model of endotoxemia. Crit Care 10, R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes, T. , Mik, E.G. & Ince, C. 2009a. Nonresuscitated endotoxemia induces microcirculatory hypoxic areas in the renal cortex in the rat. Shock 31, 97–103. [DOI] [PubMed] [Google Scholar]
- Johannes, T. , Mik, E.G. , Klingel, K. , Dieterich, H.J. , Unertl, K.E. & Ince, C. 2009b. Low‐dose dexamethasone‐supplemented fluid resuscitation reverses endotoxin‐induced acute renal failure and prevents cortical microvascular hypoxia. Shock 31, 521–528. [DOI] [PubMed] [Google Scholar]
- Kagan, J.C. , Su, T. , Horng, T. , Chow, A. , Akira, S. & Medzhitov, R. 2008. TRAM couples endocytosis of Toll‐like receptor 4 to the induction of interferon‐beta. Nat Immunol 9, 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalakeche, R. , Hato, T. , Rhodes, G. , Dunn, K.W. , El‐Achkar, T.M. , Plotkin, Z. , Sandoval, R.M. & Dagher, P.C. 2011. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol 22, 1505–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai, T. & Akira, S. 2011. Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650. [DOI] [PubMed] [Google Scholar]
- Kim, H.M. , Park, B.S. , Kim, J.I. , Kim, S.E. , Lee, J. , Oh, S.C. , Enkhbayar, P. , Matsushima, N. , Lee, H. , Yoo, O.J. & Lee, J.O. 2007. Crystal structure of the TLR4‐MD‐2 complex with bound endotoxin antagonist Eritoran. Cell 130, 906–917. [DOI] [PubMed] [Google Scholar]
- Laestadius, A. , Soderblom, T. , Aperia, A. & Richter‐Dahlfors, A. 2003. Developmental aspects of Escherichia coli‐induced innate responses in rat renal epithelial cells. Pediatr Res 54, 536–541. [DOI] [PubMed] [Google Scholar]
- Langenberg, C. , Bellomo, R. , May, C. , Wan, L. , Egi, M. & Morgera, S. 2005. Renal blood flow in sepsis. Crit Care 9, R363–R374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenberg, C. , Wan, L. , Egi, M. , May, C.N. & Bellomo, R. 2006. Renal blood flow in experimental septic acute renal failure. Kidney Int 69, 1996–2002. [DOI] [PubMed] [Google Scholar]
- Langenberg, C. , Bagshaw, S.M. , May, C.N. & Bellomo, R. 2008. The histopathology of septic acute kidney injury: a systematic review. Crit Care 12, R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenberg, C. , Gobe, G. , Hood, S. , May, C.N. & Bellomo, R. 2014. Renal histopathology during experimental septic acute kidney injury and recovery. Crit Care Med 42, e58–e67. [DOI] [PubMed] [Google Scholar]
- Lee, S.Y. , Lee, Y.S. , Choi, H.M. , Ko, Y.S. , Lee, H.Y. , Jo, S.K. , Cho, W.Y. & Kim, H.K. 2012. Distinct pathophysiologic mechanisms of septic acute kidney injury: role of immune suppression and renal tubular cell apoptosis in murine model of septic acute kidney injury. Crit Care Med 40, 2997–3006. [DOI] [PubMed] [Google Scholar]
- Legrand, M. , Bezemer, R. , Kandil, A. , Demirci, C. , Payen, D. & Ince, C. 2011. The role of renal hypoperfusion in development of renal microcirculatory dysfunction in endotoxemic rats. Intensive Care Med 37, 1534–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong, C.L. , Anderson, W.P. , O'Connor, P.M. & Evans, R.G. 2007. Evidence that renal arterial‐venous oxygen shunting contributes to dynamic regulation of renal oxygenation. Am J Physiol Renal Physiol 292, F1726–F1733. [DOI] [PubMed] [Google Scholar]
- Lerolle, N. , Nochy, D. , Guerot, E. , Bruneval, P. , Fagon, J.Y. , Diehl, J.L. & Hill, G. 2010. Histopathology of septic shock induced acute kidney injury: apoptosis and leukocytic infiltration. Intensive Care Med 36, 471–478. [DOI] [PubMed] [Google Scholar]
- Leventhal, J.S. , Ni, J. , Osmond, M. , Lee, K. , Gusella, G.L. , Salem, F. & Ross, M.J. 2016. Autophagy limits endotoxemic acute kidney injury and alters renal tubular epithelial cell cytokine expression. PLoS One 11, e0150001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, B. , Mansart, A. , Bollaert, P.E. , Franck, P. & Mallie, J.P. 2003. Effects of epinephrine and norepinephrine on hemodynamics, oxidative metabolism, and organ energetics in endotoxemic rats. Intensive Care Med 29, 292–300. [DOI] [PubMed] [Google Scholar]
- Li, D. , Zou, L. , Feng, Y. , Xu, G. , Gong, Y. , Zhao, G. , Ouyang, W. , Thurman, J.M. & Chao, W. 2016. Complement factor B production in renal tubular cells and its role in sodium transporter expression during polymicrobial sepsis. Crit Care Med 44, e289–e299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaudet, L. , Murthy, K.G. , Mabley, J.G. , Pacher, P. , Soriano, F.G. , Salzman, A.L. & Szabo, C. 2002. Comparison of inflammation, organ damage, and oxidant stress induced by Salmonella enterica serovar Muenchen flagellin and serovar Enteritidis lipopolysaccharide. Infect Immun 70, 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipcsey, M. & Bellomo, R. 2011. Septic acute kidney injury: hemodynamic syndrome, inflammatory disorder, or both? Crit Care 15, 1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. , Li, Y. , Hu, Z. , Su, J. , Huo, Y. , Tan, B. , Wang, X. & Liu, Y. 2012. Small interfering RNA targeting Toll‐like receptor 9 protects mice against polymicrobial septic acute kidney injury. Nephron Exp Nephrol 122, 51–61. [DOI] [PubMed] [Google Scholar]
- Liu, P. , Li, F. , Qiu, M. & He, L. 2014a. Expression and cellular distribution of TLR4, MyD88, and NF‐kappaB in diabetic renal tubulointerstitial fibrosis, in vitro and in vivo . Diabetes Res Clin Pract 105, 206–216. [DOI] [PubMed] [Google Scholar]
- Liu, X.J. , Tan, Y. , Geng, Y.Q. , Wang, Z. , Ye, J.H. , Yin, X.Y. & Fu, B. 2014b. Proximal tubule toll‐like receptor 4 expression linked to inflammation and apoptosis following hypoxia/reoxygenation injury. Am J Nephrol 39, 337–347. [DOI] [PubMed] [Google Scholar]
- Matsushima, N. , Tanaka, T. , Enkhbayar, P. , Mikami, T. , Taga, M. , Yamada, K. & Kuroki, Y. 2007. Comparative sequence analysis of leucine‐rich repeats (LRRs) within vertebrate toll‐like receptors. BMC Genom 8, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov, R. 2001. Toll‐like receptors and innate immunity. Nat Rev Immunol 1, 135–145. [DOI] [PubMed] [Google Scholar]
- Morrell, E.D. , Kellum, J.A. , Hallows, K.R. & Pastor‐Soler, N.M. 2014. Epithelial transport during septic acute kidney injury. Nephrol Dial Transplant 29, 1312–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, D. , Doi, K. , Kitamura, H. , Kuwabara, T. , Mori, K. , Mukoyama, M. & Nishiyama, A. 2015. Reduction of tubular flow rate as a mechanism of oliguria in the early phase of endotoxemia revealed by intravital imaging. J Am Soc Nephrol 26, 3035–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niendorf, T. , Pohlmann, A. , Arakelyan, K. , Flemming, B. , Cantow, K. , Hentschel, J. , Grosenick, D. , Ladwig, M. , Reimann, H. , Klix, S. , Waiczies, S. & Seeliger, E. 2015. How bold is blood oxygenation level‐dependent (BOLD) magnetic resonance imaging of the kidney? Opportunities, challenges and future directions. Acta Physiol (Oxf) 213, 19–38. [DOI] [PubMed] [Google Scholar]
- Nitescu, N. , Grimberg, E. & Guron, G. 2008. Low‐dose candesartan improves renal blood flow and kidney oxygen tension in rats with endotoxin‐induced acute kidney dysfunction. Shock 30, 166–172. [DOI] [PubMed] [Google Scholar]
- Nitescu, N. , DiBona, G.F. , Grimberg, E. & Guron, G. 2010. Angiotensin II type 1 receptor antagonism attenuates abnormalities in dynamic renal blood flow autoregulation in rats with endotoxin‐induced acute kidney injury. Kidney Blood Press Res 33, 200–208. [DOI] [PubMed] [Google Scholar]
- Opal, S.M. , Laterre, P.F. , Francois, B. , LaRosa, S.P. , Angus, D.C. , Mira, J.P. , Wittebole, X. , Dugernier, T. , Perrotin, D. , Tidswell, M. et al 2013. Effect of eritoran, an antagonist of MD2‐TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA 309, 1154–1162. [DOI] [PubMed] [Google Scholar]
- Palsson‐McDermott, E.M. & O'Neill, L.A. 2004. Signal transduction by the lipopolysaccharide receptor, Toll‐like receptor‐4. Immunology 113, 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patole, P.S. , Schubert, S. , Hildinger, K. , Khandoga, S. , Khandoga, A. , Segerer, S. , Henger, A. , Kretzler, M. , Werner, M. , Krombach, F. , Schlondorff, D. & Anders, H.J. 2005. Toll‐like receptor‐4: renal cells and bone marrow cells signal for neutrophil recruitment during pyelonephritis. Kidney Int 68, 2582–2587. [DOI] [PubMed] [Google Scholar]
- Patzak, A. , Kleinmann, F. , Lai, E.Y. , Kupsch, E. , Skelweit, A. & Mrowka, R. 2004. Nitric oxide counteracts angiotensin II induced contraction in efferent arterioles in mice. Acta Physiol Scand 181, 439–444. [DOI] [PubMed] [Google Scholar]
- Pawar, R.D. , Ramanjaneyulu, A. , Kulkarni, O.P. , Lech, M. , Segerer, S. & Anders, H.J. 2007. Inhibition of Toll‐like receptor‐7 (TLR‐7) or TLR‐7 plus TLR‐9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol 18, 1721–1731. [DOI] [PubMed] [Google Scholar]
- Pawar, R.D. , Castrezana‐Lopez, L. , Allam, R. , Kulkarni, O.P. , Segerer, S. , Radomska, E. , Meyer, T.N. , Schwesinger, C.M. , Akis, N. , Grone, H.J. & Anders, H.J. 2009. Bacterial lipopeptide triggers massive albuminuria in murine lupus nephritis by activating Toll‐like receptor 2 at the glomerular filtration barrier. Immunology 128, e206–e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson, P.B. 2013. Mechanisms of acute kidney injury. Acta Physiol (Oxf) 207, 430–431. [DOI] [PubMed] [Google Scholar]
- Pettila, V. , Webb, S.A. , Bailey, M. , Howe, B. , Seppelt, I.M. & Bellomo, R. 2011. Acute kidney injury in patients with influenza A (H1N1) 2009. Intensive Care Med 37, 763–767. [DOI] [PubMed] [Google Scholar]
- Porta, F. , Takala, J. , Weikert, C. , Bracht, H. , Kolarova, A. , Lauterburg, B.H. , Borotto, E. & Jakob, S.M. 2006. Effects of prolonged endotoxemia on liver, skeletal muscle and kidney mitochondrial function. Crit Care 10, R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poukkanen, M. , Wilkman, E. , Vaara, S.T. , Pettila, V. , Kaukonen, K.M. , Korhonen, A.M. , Uusaro, A. , Hovilehto, S. , Inkinen, O. , Laru‐Sompa, R. , Hautamaki, R. , Kuitunen, A. & Karlsson, S. 2013. Hemodynamic variables and progression of acute kidney injury in critically ill patients with severe sepsis: data from the prospective observational FINNAKI study. Crit Care 17, R295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prowle, J.R. , Molan, M.P. , Hornsey, E. & Bellomo, R. 2012. Measurement of renal blood flow by phase‐contrast magnetic resonance imaging during septic acute kidney injury: a pilot investigation. Crit Care Med 40, 1768–1776. [DOI] [PubMed] [Google Scholar]
- Pulskens, W.P. , Teske, G.J. , Butter, L.M. , Roelofs, J.J. , van der Poll, T. , Florquin, S. & Leemans, J.C. 2008. Toll‐like receptor‐4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS One 3, e3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaiah, R. , Perkins, D.J. , Ireland, D.D. & Vogel, S.N. 2015. CD14 dependence of TLR4 endocytosis and TRIF signaling displays ligand specificity and is dissociable in endotoxin tolerance. Proc Natl Acad Sci USA 112, 8391–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh, G. , Zhang, B. , Uematsu, S. , Akira, S. & Reeves, W.B. 2007. Endotoxin and cisplatin synergistically induce renal dysfunction and cytokine production in mice. Am J Physiol Renal Physiol 293, F325–F332. [DOI] [PubMed] [Google Scholar]
- Redfors, B. , Bragadottir, G. , Sellgren, J. , Swärd, K. & Ricksten, S.‐E. 2010. Acute renal failure is NOT an “acute renal success”—a clinical study on the renal oxygen supply/demand relationship in acute kidney injury. Crit Care Med 38, 1695–1701. [DOI] [PubMed] [Google Scholar]
- Reiser, J. , von Gersdorff, G. , Loos, M. , Oh, J. , Asanuma, K. , Giardino, L. , Rastaldi, M.P. , Calvaresi, N. , Watanabe, H. , Schwarz, K. et al 2004. Induction of B7‐1 in podocytes is associated with nephrotic syndrome. J Clin Invest 113, 1390–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, T.W. , Wheeler, A.P. , Bernard, G.R. , Vincent, J.L. , Angus, D.C. , Aikawa, N. , Demeyer, I. , Sainati, S. , Amlot, N. , Cao, C. , Ii, M. , Matsuda, H. , Mouri, K. & Cohen, J. 2010. A randomized, double‐blind, placebo‐controlled trial of TAK‐242 for the treatment of severe sepsis. Crit Care Med 38, 1685–1694. [DOI] [PubMed] [Google Scholar]
- Sakhuja, A. , Kumar, G. , Gupta, S. , Mittal, T. , Taneja, A. & Nanchal, R.S. 2015. Acute kidney injury requiring dialysis in severe sepsis. Am J Respir Crit Care Med 192, 951–957. [DOI] [PubMed] [Google Scholar]
- Samuelsson, P. , Hang, L. , Wullt, B. , Irjala, H. & Svanborg, C. 2004. Toll‐like receptor 4 expression and cytokine responses in the human urinary tract mucosa. Infect Immun 72, 3179–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome, T. , Ishikawa, K. , May, C.N. , Birchall, I.E. & Bellomo, R. 2010. The impact of experimental hypoperfusion on subsequent kidney function. Intensive Care Med 36, 533–540. [DOI] [PubMed] [Google Scholar]
- Schmidt, C. , Hocherl, K. & Bucher, M. 2007a. Regulation of renal glucose transporters during severe inflammation. Am J Physiol Renal Physiol 292, F804–F811. [DOI] [PubMed] [Google Scholar]
- Schmidt, C. , Hocherl, K. , Schweda, F. & Bucher, M. 2007b. Proinflammatory cytokines cause down‐regulation of renal chloride entry pathways during sepsis. Crit Care Med 35, 2110–2119. [DOI] [PubMed] [Google Scholar]
- Schmidt, C. , Hocherl, K. , Schweda, F. , Kurtz, A. & Bucher, M. 2007c. Regulation of renal sodium transporters during severe inflammation. J Am Soc Nephrol 18, 1072–1083. [DOI] [PubMed] [Google Scholar]
- Schrier, R.W. & Wang, W. 2004. Acute renal failure and sepsis. N Engl J Med 351, 159–169. [DOI] [PubMed] [Google Scholar]
- Shimazu, R. , Akashi, S. , Ogata, H. , Nagai, Y. , Fukudome, K. , Miyake, K. & Kimoto, M. 1999. MD‐2, a molecule that confers lipopolysaccharide responsiveness on Toll‐like receptor 4. J Exp Med 189, 1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J.A. , Stallons, L.J. , Collier, J.B. , Chavin, K.D. & Schnellmann, R.G. 2015. Suppression of mitochondrial biogenesis through toll‐like receptor 4‐dependent mitogen‐activated protein kinase kinase/extracellular signal‐regulated kinase signaling in endotoxin‐induced acute kidney injury. J Pharmacol Exp Ther 352, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasu, O. , Gaut, J.P. , Watanabe, E. , To, K. , Fagley, R.E. , Sato, B. , Jarman, S. , Efimov, I.R. , Janks, D.L. , Srivastava, A. , Bhayani, S.B. , Drewry, A. , Swanson, P.E. & Hotchkiss, R.S. 2013. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med 187, 509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidswell, M. , Tillis, W. , Larosa, S.P. , Lynn, M. , Wittek, A.E. , Kao, R. , Wheeler, J. , Gogate, J. , Opal, S.M. & Eritoran Sepsis Study, G . 2010. Phase 2 trial of eritoran tetrasodium (E5564), a toll‐like receptor 4 antagonist, in patients with severe sepsis. Crit Care Med, 38, 72–83. [DOI] [PubMed] [Google Scholar]
- Tiwari, M.M. , Brock, R.W. , Megyesi, J.K. , Kaushal, G.P. & Mayeux, P.R. 2005. Disruption of renal peritubular blood flow in lipopolysaccharide‐induced renal failure: role of nitric oxide and caspases. Am J Physiol Renal Physiol 289, F1324–F1332. [DOI] [PubMed] [Google Scholar]
- Tran, M. , Tam, D. , Bardia, A. , Bhasin, M. , Rowe, G.C. , Kher, A. , Zsengeller, Z.K. , Akhavan‐Sharif, M.R. , Khankin, E.V. , Saintgeniez, M. et al 2011. PGC‐1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest 121, 4003–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi, N. , Yoshikai, Y. , Matsuo, S. , Kikuchi, T. , Iwami, K. , Nagai, Y. , Takeuchi, O. , Akira, S. & Matsuguchi, T. 2002. Roles of toll‐like receptors in C‐C chemokine production by renal tubular epithelial cells. J Immunol 169, 2026–2033. [DOI] [PubMed] [Google Scholar]
- Tsuji, N. , Tsuji, T. , Ohashi, N. , Kato, A. , Fujigaki, Y. & Yasuda, H. 2015. Role of mitochondrial DNA in septic AKI via toll‐like receptor 9. J Am Soc Nephrol 2009–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulic, M.K. , Hurrelbrink, R.J. , Prele, C.M. , Laing, I.A. , Upham, J.W. , Le Souef, P. , Sly, P.D. & Holt, P.G. 2007. TLR4 polymorphisms mediate impaired responses to respiratory syncytial virus and lipopolysaccharide. J Immunol 179, 132–140. [DOI] [PubMed] [Google Scholar]
- Valles, P.G. , Lorenzo, A.G. , Bocanegra, V. & Valles, R. 2014. Acute kidney injury: what part do toll‐like receptors play? Int J Nephrol Renovasc Dis 7, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, L. , Langenberg, C. , Bellomo, R. & May, C.N. 2009. Angiotensin II in experimental hyperdynamic sepsis. Crit Care 13, R190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Holthoff, J.H. , Seely, K.A. , Pathak, E. , Spencer, H.J. III , Gokden, N. & Mayeux, P.R. 2012. Development of oxidative stress in the peritubular capillary microenvironment mediates sepsis‐induced renal microcirculatory failure and acute kidney injury. Am J Pathol 180, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts, B.A. 3rd , George, T. , Sherwood, E.R. & Good, D.W. 2011. Basolateral LPS inhibits NHE3 and HCOFormula absorption through TLR4/MyD88‐dependent ERK activation in medullary thick ascending limb. Am J Physiol Cell Physiol 301, C1296–C1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts, B.A. III , George, T. & Good, D.W. 2013a. Lumen LPS inhibits HCO3(‐) absorption in the medullary thick ascending limb through TLR4‐PI3K‐Akt‐mTOR‐dependent inhibition of basolateral Na+/H+ exchange. Am J Physiol Renal Physiol 305, F451–F462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts, B.A. III , George, T. , Sherwood, E.R. & Good, D.W. 2013b. A two‐hit mechanism for sepsis‐induced impairment of renal tubule function. Am J Physiol Renal Physiol 304, F863–F874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfs, T.G. , Buurman, W.A. , van Schadewijk, A. , de Vries, B. , Daemen, M.A. , Hiemstra, P.S. & van ‘t Veer, C. 2002. In vivo expression of Toll‐like receptor 2 and 4 by renal epithelial cells: IFN‐gamma and TNF‐alpha mediated up‐regulation during inflammation. J Immunol, 168, 1286–1293. [DOI] [PubMed] [Google Scholar]
- Wonnacott, A. , Meran, S. , Amphlett, B. , Talabani, B. & Phillips, A. 2014. Epidemiology and outcomes in community‐acquired versus hospital‐acquired AKI. Clin J Am Soc Nephrol 9, 1007–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, L. , Tiwari, M.M. , Messer, K.J. , Holthoff, J.H. , Gokden, N. , Brock, R.W. & Mayeux, P.R. 2007. Peritubular capillary dysfunction and renal tubular epithelial cell stress following lipopolysaccharide administration in mice. Am J Physiol Renal Physiol 292, F261–F268. [DOI] [PubMed] [Google Scholar]
- Wulfert, F.M. , van Meurs, M. , Kurniati, N.F. , Jongman, R.M. , Houwertjes, M.C. , Heeringa, P. , Struys, M.M. , Zijlstra, J.G. & Molema, G. 2012. Age‐dependent role of microvascular endothelial and polymorphonuclear cells in lipopolysaccharide‐induced acute kidney injury. Anesthesiology 117, 126–136. [DOI] [PubMed] [Google Scholar]
- Xu, S. , Chen, Y.H. , Tan, Z.X. , Xie, D.D. , Zhang, C. , Xia, M.Z. , Wang, H. , Zhao, H. , Xu, D.X. & Yu, D.X. 2015. Vitamin D3 pretreatment alleviates renal oxidative stress in lipopolysaccharide‐induced acute kidney injury. J Steroid Biochem Mol Biol 152, 133–141. [DOI] [PubMed] [Google Scholar]
- Yasuda, H. , Leelahavanichkul, A. , Tsunoda, S. , Dear, J.W. , Takahashi, Y. , Ito, S. , Hu, X. , Zhou, H. , Doi, K. , Childs, R. , Klinman, D.M. , Yuen, P.S. & Star, R.A. 2008. Chloroquine and inhibition of Toll‐like receptor 9 protect from sepsis‐induced acute kidney injury. Am J Physiol Renal Physiol 294, F1050–F1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang, Z.D. & Yan, J. 2013. An analysis of clinical characteristics of septic acute kidney injury by using criteria of Kidney Disease: improving Global Outcomes. Zhonghua Nei Ke Za Zhi 52, 299–304. [PubMed] [Google Scholar]
- Zanoni, I. , Ostuni, R. , Marek, L.R. , Barresi, S. , Barbalat, R. , Barton, G.M. , Granucci, F. & Kagan, J.C. 2011. CD14 controls the LPS‐induced endocytosis of Toll‐like receptor 4. Cell 147, 868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarember, K.A. & Godowski, P.J. 2002. Tissue expression of human Toll‐like receptors and differential regulation of Toll‐like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol 168, 554–561. [DOI] [PubMed] [Google Scholar]
- Zhang, B. , Ramesh, G. , Uematsu, S. , Akira, S. & Reeves, W.B. 2008. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J Am Soc Nephrol 19, 923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, L. , Feng, Y. , Li, Y. , Zhang, M. , Chen, C. , Cai, J. , Gong, Y. , Wang, L. , Thurman, J.M. , Wu, X. , Atkinson, J.P. & Chao, W. 2013. Complement factor B is the downstream effector of TLRs and plays an important role in a mouse model of severe sepsis. J Immunol 191, 5625–5635. [DOI] [PMC free article] [PubMed] [Google Scholar]