

Abstract
Delafloxacin is a novel anionic fluoroquinolone with robust activity against Gram‐positive, Gram‐negative, atypical, and anaerobic bacteria, including methicillin‐resistant S aureus. Delafloxacin is currently being studied for the treatment of acute bacterial skin and skin structure infections and community‐acquired pneumonia. This was a phase 1, open‐label pharmacokinetic and safety study of a single intravenous dose of 300 mg delafloxacin in subjects with mild, moderate, and severe hepatic impairment (Child‐Pugh class A, B, and C, respectively) compared with matched healthy controls. The effects of hepatic impairment were assessed by ANOVA of log‐transformed values for AUC0‐∞, Cmax, and systemic clearance, with hepatic group as a fixed effect. Mean AUC0‐∞ and Cmax in each impairment group were not significantly different from those of the pooled healthy subjects (P > 0.05). The 90% confidence interval (CI) of the percentage ratios of least‐squares means of AUC0‐∞ did not indicate significant differences between the impairment groups and pooled healthy controls: Child‐Pugh class A (mild) 114.4 (CI: 95.6, 137.0), Child‐Pugh class B (moderate) 114.8 (CI: 95.9, 137.4), and Child‐Pugh class C (severe) 115.1 (CI: 96.1, 137.8). A single IV infusion of delafloxacin was generally well tolerated in all treatment groups. The exposure and clearance of delafloxacin in subjects with mild, moderate, or severe hepatic impairment did not significantly differ from those of pooled, matched healthy subjects. Based on these pharmacokinetic data, dose adjustment of delafloxacin in the presence of hepatic impairment is not needed.
Keywords: delafloxacin, fluoroquinolones, pharmacokinetics, hepatic impairment
Delafloxacin is a structurally unique anionic fluoroquinolone with enhanced in vitro activity against Gram‐positive bacteria, including methicillin‐resistant Staphylococcus aureus (MRSA), Gram‐negative bacteria, atypical bacteria, and anaerobic bacteria.1, 2 Delafloxacin is currently being studied for the treatment of acute bacterial skin and skin structure infections (ABSSSI) and hospitalized community‐acquired pneumonia. Recently, delafloxacin was found to be comparable to vancomycin in a phase 3 study to treat patients with ABSSSI. The majority of patients were infected with S aureus, of which half (169/324; 52%) were MRSA. A second phase 3 trial in ABSSSI is ongoing.
Delafloxacin disposition, whether as the intravenous or oral formulation, is defined by a terminal half‐life of ∼12 hours, plasma protein binding of 84%, steady‐state volume of distribution of ∼40 L, and a total clearance of 12.9 L/h.3, 4, 5 Delafloxacin Cmax values increased proportionally, and area under the plasma concentration‐time curve (AUC0‐∞) showed a more than proportional (6‐fold) increase following single intravenous doses ranging from 300 to 1200 mg in healthy human subjects.3 The major route of excretion is renal, with 64% of the dose appearing in the urine as parent compound or glucuronide metabolites over a 24‐hour period following a single intravenous dose.6 The liver is also involved in elimination both through the production of water‐soluble glucuronide conjugates and in the fecal elimination of 29% of an intravenous dose as the unchanged parent compound.
To date, phase 2 and 3 studies have not revealed any deleterious effects on the liver associated with delafloxacin at rates higher than comparators such as vancomycin, tigecycline, and linezolid.7, 8, 9 Given that about 40% of a delafloxacin dose is either apparently cleared unchanged by the liver or conjugated as glucuronide metabolites by the liver, it is important to understand the impact of underlying hepatic impairment on the safety and pharmacokinetics of delafloxacin.
Methods
Study Design
This phase 1, open‐label, 2‐center, single‐IV‐dose study was approved by the Institutional Review Boards of the Orlando Clinical Research Center and the University of Miami prior to receiving signed informed consent from all participants. The study was conducted according to the principles of the International Conference on Harmonisation of Good Clinical Practices. All subjects voluntarily signed informed consent prior to admission into the study.
A total of 36 subjects were stratified into 4 groups based on baseline hepatic function as defined by the Child‐Pugh classification system (Table 1):10, 11
Group A: 6 subjects with mild hepatic impairment (Child‐Pugh Class A)
Group B: 6 subjects with moderate hepatic impairment (Child‐Pugh Class B)
Group C: 6 subjects with severe hepatic impairment (Child‐Pugh Class C)
Group D: 6 healthy comparator subjects matched to the subjects in each hepatic impairment group (18 total) with respect to age (±10 years), weight (±20%), and sex. Subjects with hepatic impairment were identified by medical records scored by the Child‐Pugh classification at screening. Healthy subjects were also matched to smoking status and alcohol use where possible.
Table 1.
Child‐Pugh Classification System
| Points Scored for Observed Findings | |||
|---|---|---|---|
| Clinical and Biochemical Measurementsa | 1 | 2 | 3 |
| Hepatic encephalopathy (grade)b | None (Grade 0) | 1 or 2 | 3 or 4c |
| Ascites | Absent | Slight | Moderated |
| Serum bilirubin (mg/dL) | <2.0 | 2.0–3.0 | >3.0 |
| Serum albumin (g/dL) | >3.5 | 2.8–3.5 | <2.8 |
| Prothrombin timee | |||
| Seconds prolonged | <4 | 4–6 | >6 |
| International Normalized Ratio (INR) | <1.7 | 1.7–2.3 | >2.3 |
Child‐Pugh class based upon sum of points: class A, 5–6 points; class B, 7‐9 points; class C, 10‐15 points.
Grades 0 through 4 based on signs, symptoms, and tests.
Stage 3 and 4 encephalopathy were excluded. However, those subjects who received medication to prevent recurrent encephalopathy were scored based on the degree of encephalopathy off treatment.
Moderate or controlled by diuretics.
If a discrepancy between seconds prolonged and INR, points scored for INR were used.
To be included in the study all subjects had to meet the following additional inclusion criteria: age between 18 and 80 years, inclusive; body mass index (BMI) between 18 and 40 kg/m2, inclusive; not child‐bearing or remaining abstinent throughout study; and a baseline blood pressure ≤ 180/100 mm Hg. Subjects were excluded if they had any of the following conditions: a clinically significant abnormality (excluding liver function) in past medical history, including a baseline ECG with clinically significant abnormalities; any surgical or medical condition that would interfere with absorption, distribution, metabolism, or excretion of the study drug; a functional liver transplant; hemoglobin <10 g/dL; a history of recent (6 months) drug and/or alcohol abuse; mental compromise; a history of HIV/AIDS; had received an investigational agent within 30 days of enrolment; a clinically significant drug allergy or hypersensitivity; had consumed any food or beverages that could affect the metabolism and disposition of the study drug; used medications that affect elimination of serum creatinine (eg, trimethoprim, cimetidine) or competitors of tubular secretion (eg, probenecid) within 30 days before dosing; or had clinically significant abnormal findings in serum chemistry, coagulation, hematology, or urinalysis. Subjects with hepatic impairment were also excluded if they were clinically unstable (eg, ascites, nausea) within 14 days of study drug administration, acute viral hepatitis within 1 month of enrollment, active stage 3 or 4 encephalopathy, or severe or acute renal failure. Healthy subjects were excluded with a positive test for hepatitis B or C or if they used over‐the‐counter or prescription drugs within 7 days or antibiotics within 30 days prior to dosing.
Safety assessments, including clinical laboratory results, vital sign measurements, and physical examination findings were recorded through day 4. Adverse events were collected throughout the study period until the follow‐up contact (day 11±1 after study drug administration).
Study Medication
Delafloxacin for injection, 300 mg/vial, is formulated as a sterile, nonpyrogenic, lyophilized cake. Each vial contains the following ingredients: 433 mg delafloxacin meglumine equivalent of 300 mg free acid, 58.56 mg meglumine, 2.4 g sulfobutylether‐β‐cyclodextrin (SBECD), 3.4 mg ethylene‐diamine‐tetra‐acetate disodium (equivalent to 2.6 mg EDTA), and water for injection. Delafloxacin vials were stored at controlled room temperatures (68˚F to 77˚F) and protected from light.
Delafloxacin Pharmacokinetic Plasma Samples
Following the screening period and compliance with the inclusion and exclusion criteria, subjects received a single 1‐hour intravenous infusion of 300 mg delafloxacin, and blood samples were obtained for 72 hours after dosing at 0, 20, 40, and 60 minutes during the infusion and 5 minutes, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 60, and 72 hours after the end of the infusion from the arm opposite that of the infusion site. Pharmacokinetic blood samples were collected in tubes containing dipotassium EDTA and processed for plasma by centrifugation. Samples were stored at –20°C or colder prior to analysis by PPD Bioanalytical Laboratory, Richmond, Virginia (data on file).
Delafloxacin was quantitated in human plasma samples using a validated LC‐MS/MS method. The internal standard was deuterated delafloxacin. Briefly, on analysis, a 100‐μL aliquot of each sample was fortified with internal standard working solution. The sample was then extracted by supported liquid‐phase extraction using Biotage Isolute 96‐well SLE+ plates and elution with 30:70 ethyl acetate/hexane, v/v. The eluate was evaporated under a nitrogen stream at approximately 40°C, and the remaining residue was reconstituted with 30:70 acetonitrile/50 mM glycine buffer (pH 9.0), v/v. The final extract was analyzed via HPLC using an XBridge C18 column (2.1 mm × 50 mm, 5 μm) and MS/MS detection using positive‐ion electrospray. A linear, 1/concentration²‐weighted, least‐squares regression algorithm was used to quantitate unknown samples. The nominal concentration range was 5 to 5000 ng/mL.
Acceptable calibration standards exhibited accuracy of ±15% of nominal (±20% at the lower limit of quantitation). The quality control (QC) concentrations were 5, 15, 40, 150, 600, and 3750 ng/mL. During validation, all QC levels passed with inter‐ and intra‐assay accuracy within ±15% of nominal and precision ≤15%. Acceptable stability within ±15% of nominal was demonstrated in stability testing including freeze/thaw, room temperature, and extract stability tests. Frozen storage stability at –20°C and –70°C was demonstrated up to 484 days.
Pharmacokinetic Analyses
The following pharmacokinetic parameters were calculated from plasma concentrations of delafloxacin using noncompartmental analysis: area under the plasma concentration‐time curve (AUC0‐∞) by the trapezoidal rule, maximum observed plasma concentration (Cmax) and time to reach Cmax (Tmax), apparent terminal elimination rate constant and terminal half‐life (λz and t½, respectively), total body clearance (CL) from the ratio of dose to AUC0‐∞, and volume of distribution at steady state (Vss). AUC0‐∞ values were used to estimate exposure since the pharmacokinetic sampling period covered more than 80% of the AUC.
Statistical Analyses
All analyses were performed using SAS® software version 9.2 (SAS Institute, Cary, North Carolina), and Phoenix® WinNonlin® Version 6.2.1 (Pharsight, St. Louis, Missouri) was used for pharmacokinetic analyses. In general, continuous data were summarized by presenting the number of subjects, mean, standard deviation (SD), median, minimum, and maximum, unless otherwise specified. Categorical data were summarized by presenting the number (frequency) and percentage of subjects at each level of response. Geometric mean and geometric coefficients of variation (CV) were also presented for pharmacokinetic data. Baseline data were defined as the last nonmissing measurement (including repeated and unscheduled measurements) before the first dose of study drug unless otherwise specified. Descriptive statistics were calculated for baseline demographics.
To assess the effect of hepatic impairment (group A, subjects with mild hepatic impairment [Child‐Pugh class A]; group B, subjects with moderate hepatic impairment [Child‐Pugh class B]; and group C, subjects with severe hepatic impairment [Child‐Pugh class C]), compared with the healthy subjects (group D), a linear mixed‐effect model was performed on the natural log‐transformed values of AUC0‐∞, Cmax, and CL with hepatic impairment group as a fixed effect. A comparison was performed between each hepatic impairment group and the corresponding healthy matching group. A comparison was also performed between the hepatic impairment groups and the pooled healthy subject groups. The geometric least‐squares (LS) mean ratios of each hepatic impairment group to the corresponding healthy matching group (A:D [mild:match], B:D [moderate:match], and C:D [severe:match]) for AUC0‐∞, Cmax, and CL were calculated by using the antilog of the LS mean difference of the natural log‐transformed values. A 90% confidence interval (CI) for each ratio was constructed as the antilog of the 90% CI of the LS mean difference. No adjustment was made for multiplicity. In addition, the geometric LS means and corresponding 90% CI were computed for AUC0‐∞, Cmax, and CL by taking the antilog of the LS means and corresponding 90% CI from the linear mixed‐effect model on the natural logarithm of the corresponding pharmacokinetic parameters. A similar analysis was performed for the comparison of the hepatic impairment groups to the pooled healthy subjects group. Trend analyses were also performed by 1‐way analysis of variance to detect any trends in pharmacokinetic parameters associated with severity of hepatic impairment.
Results
Subject Demographics
A total of 39 subjects were enrolled, and 36 (92.3%) completed the study. The 3 discontinuations were due to possibly or probably (n = 2) related treatment‐emergent adverse events (TEAEs), which were either mild or moderate in severity. One subject with mild hepatic impairment had a mild TEAE of drug hypersensitivity (limited urticarial rash not at site of injection) and was discontinued 31 minutes after start of infusion. The other 2 discontinuations were in the healthy control group: 1 with presyncope and the other with drug hypersensitivity (itching and hives). All 3 cases resolved before or by the end of study. Thus, 39 subjects were included in the safety analysis, and 36 subjects were included in the pharmacokinetic population.
Demographics and baseline characteristics are listed in Table 2. Overall, demographic and baseline characteristics were similar across groups with the exception of severity of liver disease. As expected with subjects with hepatic impairment, more conditions/diagnoses were documented at screening for medical history compared with the healthy group (group D). However, none of the medical history findings at screening precluded any subject from entering the study. Of the causes for hepatic impairment, viral disease (hepatitis B or C) was responsible for disease in 4, 3, and 4 subjects, and alcohol abuse in 2, 3, and 2 subjects in groups A, B, and C, respectively; 1 subject had an unknown etiology for hepatic impairment in group A. All subjects with hepatic impairment who reported prior medications were on a stable drug regimen for at least a week before study drug dosing, and the medications were allowed per protocol.
Table 2.
Summary of Mean (SD) Subject Demographics and Baseline Characteristicsa
| Group A Mild (n = 7) | Group D Match (n = 7) | Group B Moderate (n = 6) | Group D Match (n = 7) | Group C Severe (n = 6) | Group D Match (n = 6) | Group D Overall (n = 20) | Overall(n = 39) | |
|---|---|---|---|---|---|---|---|---|
| Age (years) | 54.4 (6.8) | 51.7 (6.5) | 56.8 (7.3) | 56.4 (7.0) | 54.3 (5.4) | 53.7 (7.7) | 54.0 (7.0) | 54.5 (6.6) |
| Male, no. (%) | 6 (85.7) | 6 (85.7) | 5 (83.3) | 6 (85.7) | 6 (100.0) | 6 (100.0) | 18 (90.0) | 35 (89.7) |
| Race (white), No. (%) | 7 (100.0) | 5 (71.4) | 5 (83.3) | 4 (57.1) | 5 (83.3) | 5 (83.3) | 14 (70.0) | 31 (79.5) |
| Height (cm) | 169 (9.5) | 172 (8.6) | 176 (12.0) | 171 (8.0) | 172 (6.7) | 175 (7.1) | 173 (7.7) | 172 (8.5) |
| Weight (kg) | 80 (7.1) | 82 (11.6) | 93 (17.5) | 90 (7.1) | 97 (25.8) | 91 (20.8) | 87 (13.7) | 88 (16.1) |
| BMI (kg/m2) | 28.1 (5.2) | 27.7 (3.5) | 30.4 (6.0) | 30.7 (3.0) | 32.1 (6.5) | 29.5 (4.7) | 29.3 (3.7) | 30.0 (4.8) |
| Child‐Pugh Class Score | 5.7 (0.5) | NA | 8.5 (0.6) | NAb | 10.8 (1.0) | NA | NA | NA |
| Median (range) | 6 (5‐6) | 8.5 (8‐9) | 10.5 (10‐12) | |||||
| Serum creatinine (mg/dL) | 0.69 (0.15) | 0.73 (0.14) | 0.68 (0.23) | 0.73 (0.25) | 0.57 (0.08) | 0.78 (0.16) | 0.75 (0.18) | NA |
aAll subjects including 3 subjects who received delafloxacin but discontinued.
bNA, not applicable.
One healthy matched subject was erroneously enrolled twice into the study. The subject was first enrolled in the mild impairment match group and, approximately 3 months later, in the severe impairment match group. This subject received the delafloxacin 300 mg IV dose twice but was excluded from the safety and pharmacokinetic analysis populations after the second dose in the severe impairment match group. No adverse events were reported by this subject after the second dose. Subsequently, another matched healthy subject was enrolled to replace the duplicate subject in the severe impairment match group for safety and pharmacokinetic analyses.
Delafloxacin Pharmacokinetics
The mean plasma delafloxacin concentration‐time profiles for the 3 hepatic impairment groups and the matching control subjects are presented in Figure 1. During a single 1‐hour infusion of delafloxacin 300 mg, mean plasma concentrations of delafloxacin rapidly increased. After the end of infusion, mean plasma concentrations of delafloxacin exponentially decreased. As visually inspected, the overall shape of the mean plasma concentration‐time profile of delafloxacin was similar for each hepatic function group and matched the data of the healthy subject control group.
Figure 1.
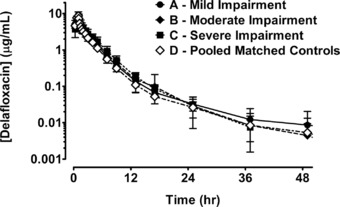
Mean (± SD) plasma concentrations of delafloxacin (μg/mL) vs time profiles by hepatic group: group A mild (●), group B moderate (♦), group C severe (■), and pooled matched controls (◊).
Mean plasma pharmacokinetic parameters of delafloxacin for cases and controls are presented in Table 3. Not surprisingly, Tmax values were at 1 hour in all study groups, coinciding with the end of the infusion of study drug. Exposures, as measured by AUC0‐∞, were slightly increased in those subjects with mild hepatic impairment (group A) compared with healthy controls (group D) as the geometric LS mean ratios were approximately 110% but not significantly different. Mean clearance was slightly lower (ratio 92%; CI 74‐114) in this population. Similarly, there were no significant differences in AUC0‐∞, Cmax, CL, or Vss in patients with moderate hepatic impairment (group B). Total exposures for plasma delafloxacin were increased in subjects with severe hepatic impairment (group C) compared with matched healthy subjects, as the geometric LS mean ratios for AUC0‐∞ were 138 (CI 112‐169), indicating a lack of significant change with severe hepatic impairment. Clearance for delafloxacin decreased in subjects with severe hepatic impairment compared with matched controls, as the geometric LS mean ratio for CL was approximately 73% (CI 59‐89). Mean delafloxacin AUC0‐∞, Cmax, CL, and Vss values were similar between those subjects with hepatic impairment, regardless of severity, and matched healthy control subjects (Figure 2).
Table 3.
Mean (CV) Plasma Pharmacokinetic Parameters of Delafloxacin by Hepatic Function Group
| Parameter | Group A Mild (n = 6) | Group D Match (n = 6) | Group B Moderate (n = 6) | Group D Match (n = 6) | Group C Severe (n = 6) | Group D Match (n = 6) | Group D Overall (n = 18) |
|---|---|---|---|---|---|---|---|
| AUC0‐∞ (μg · h/mL) | 23.5 (16.3) | 21.7 (17.3)a | 23.4 (9.2) | 23.6 (19.9) | 24.4 (28.7) | 17.2 (15.2) | 20.8 (21.9)b |
| Cmax (μg/mL) | 9.2 (15.5) | 9.3 (17.3) | 7.9 (9.8) | 9.0 (19.9) | 8.4 (30.6) | 8.3 (21.3) | 8.9 (18.9) |
| t½ (hours) | 13.5 (58.7) | 8.2 (62.5)a | 9.1 (92.5) | 9.3 (50.9) | 5.3 (24.4) | 7.1 (80.1) | 8.2 (60.4)b |
| CL (L/h) | 13.1 (17.9) | 14.1 (14.3)a | 12.9 (9.1) | 13.2 (22.7) | 13.6 (41.3) | 17.9 (17.4) | 15.1 (22.3)b |
| Vss (L) | 50.9 (20.2) | 44.8 (18.6)a | 48.7 (20.2) | 51.5 (25.3) | 49.7 (42.5) | 47.9 (23.5) | 48.3 (22.5)b |
n = 5.
n = 17.
Figure 2.
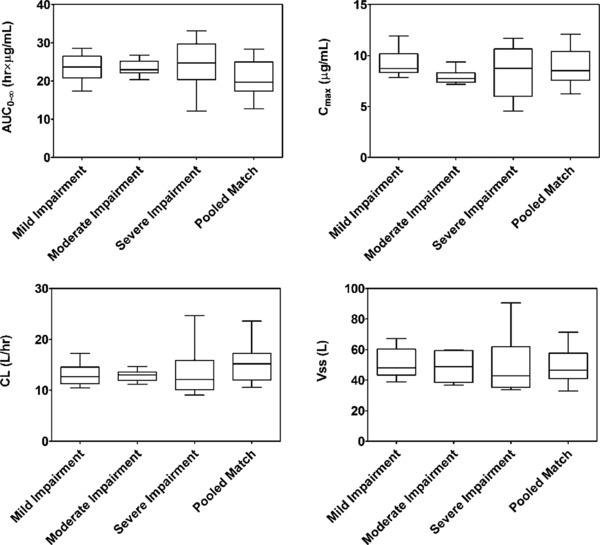
Box‐and‐whisker plots of mean delafloxacin AUC0‐∞, Cmax, CL, and Vss values by hepatic function. The whiskers represent the 5th and 95th percentiles.
Safety Assessment
Overall, delafloxacin was well tolerated. A total of 11 TEAEs were reported, and 10 of 39 subjects (26%) reported at least 1 TEAE after receiving 300 mg IV delafloxacin. TEAEs were reported by 1 of 7 subjects (14%) in the mild hepatic impairment group, no subjects in the moderate hepatic impairment group, 2 of 6 subjects (33%) in the severe hepatic impairment group, and 7 of 20 subjects (35%) overall in the healthy subjects group. Of these 11 events, the majority were considered probably or possibly related to study drug by the investigator; no subject had a TEAE that was considered definitely related to study drug. Adverse events included infections (n = 3: hordeolum, tinea, nasopharyngitis), drug hypersensitivity (n = 2), headache and presyncope (n = 2), eye pain, abdominal pain, musculoskeletal pain, and infusion site pain (n = 1 each). All TEAEs were mild to moderate in severity and resolved by the end of study. There were no deaths or serious adverse events reported in this study. Further, there were no apparent treatment‐related trends or clinically significant findings observed in the clinical laboratory assessments, vital sign measurements, electrocardiogram results, or physical examination findings.
Discussion
Hepatic impairment did not significantly affect systemic exposures of delafloxacin. AUC0‐∞ exposures for plasma delafloxacin were slightly increased by approximately 1.1‐fold in subjects with mild hepatic impairment (Child‐Pugh class A) compared with healthy subjects, but the change was not significant. In the population with moderate hepatic insufficiency (Child‐Pugh class B), there were no appreciable differences in mean AUC0‐∞, Cmax, t½, CL, or Vss. The pharmacokinetic parameters derived in patients with severe hepatic impairment (Child‐Pugh class C) resulted in an increase of 1.1‐ to 1.4‐fold in AUC0‐∞ exposures compared with healthy controls as a result of decreased total body clearance. It is important to note that the subjects in the severe impairment matched control group (Table 3) had markedly and unusually low systemic exposures of delafloxacin that likely contributed to apparent, yet statistically insignificant, differences in systemic exposures with those subjects with severe hepatic impairment. When compared with the overall healthy subject group and previous pharmacokinetic data in healthy subjects,3, 4 there were no appreciable differences in exposure. Cmax, t½, and Vss values were relatively unchanged between the 2 study groups.
An important pharmacokinetic/pharmacodynamic (PK/PD) measure that relates delafloxacin exposure to its antibiotic effect is the ratio of the delafloxacin 24‐hour plasma AUC to the minimum inhibitory concentration (MIC) of the strain of bacteria responsible for infection.12 This PK/PD measure (AUC24/MIC) is typical of the fluoroquinolone class of antibiotics. The current study was conducted to investigate whether hepatic impairment would adversely affect delafloxacin AUC, in particular whether AUC would be decreased and, if so, impact efficacy. It was tempting to speculate that if there were impairment of hepatic metabolism, any impact would be in the direction of decreasing the clearance of delafloxacin and therefore increasing delafloxacin AUC, which would not be problematic for delafloxacin efficacy. The results of the current study showed no impact of hepatic impairment, positive or negative, on delafloxacin AUC.
The findings of this study are consistent with our understanding of the disposition of delafloxacin in man. Following an intravenous dose of [14C]delafloxacin in healthy subjects in a mass balance study, delafloxacin and its glucuronide metabolites were primarily excreted by the renal route, and only 29% excreted unchanged in the feces.6 A fraction of a delafloxacin dose is conjugated by the liver into glucuronidated metabolites for excretion by the kidneys, and this process is facilitated through the liver.13 However, unlike hepatic metabolism through the cytochrome P450 system, phase 2 conjugation by the liver is rarely impacted by hepatic disease until there is severe damage.14, 15 Therefore, one might predict that hepatic disease would not be a major factor in the disposition of delafloxacin and its glucuronide metabolites. However, because a large percentage of delafloxacin is cleared by the kidneys (∼65%), whether as intact molecule or the conjugated metabolites, we also could have a confounding variable in subjects with varying degrees of renal function. Nonetheless, as noted in Table 2, baseline serum creatinine values were similar among all groups including the matched controls. The mean serum creatinine values in the severe hepatic impairment group (0.56 mg/dL) were about 25% lower than those of the matched controls (0.78 mg/dL), but the difference is of little clinical significance.
When studying the impact of organ dysfunction on the disposition of any drug, one needs to consider the many capabilities of the liver. As discussed, the liver is responsible for intrinsic metabolism of drugs through the cytochrome P450 system. We completed a multiple oral dose study of delafloxacin on the pharmacokinetics of a single oral dose of midazolam, a sensitive substrate for CYP3A.16 No interaction was found that is consistent with the aforementioned mass balance study6 and in vitro studies conducted with human liver microsomes (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4/5) (data on file). In contrast, other antibiotics, such as ciprofloxacin, erythromycin, and clarithromycin, are potent inhibitors of CYP3A4 that result in drug‐drug interactions with drugs metabolized by this isoenzyme (eg, hydrocodone).17
Another function of the liver is the production of albumin, an important element in plasma protein binding of many drugs. For example, warfarin is greater than 95% protein bound, and doses need to be reduced in those patients with end‐stage liver disease due to the increased availability of free (not bound to albumin) warfarin. Delafloxacin is 84% bound to albumin, and as a result, hepatic impairment could have a role in the amount of available free drug. Although the extent of protein binding was not evaluated in this study, a surrogate measure of protein binding, volume of distribution,18 was relatively similar across all hepatic impairment groups and healthy matched controls. Finally, the liver serves a role in presystemic metabolism or first‐pass effect on oral medications. Given that the bioavailability of oral delafloxacin is approximately 58% and a small fraction of administered dose undergoes metabolism,5, 6 it is unlikely that the drug undergoes significant first‐pass effect by the liver.
This study is noteworthy in that the pharmacokinetics of delafloxacin are described in subjects with severe hepatic impairment (Child‐Pugh class C). The majority of pharmacokinetic studies in hepatic impairment are completed in subjects with mild to moderate hepatic impairment (Child‐Pugh classes A and B, respectively), and investigators only speculate on drug disposition in severe hepatic impairment. In our study there was a small increase in plasma delafloxacin exposure relative to the matched controls in the severe hepatic impairment group; however, exposure was similar to that in those subjects in the mild and moderate hepatic impairment groups and not different from the overall matched healthy subjects and historical data.3, 4 This study involved a single dose, which is appropriate because delafloxacin does not show time‐dependent pharmacokinetics.
In conclusion, the pharmacokinetics of a single intravenous dose of delafloxacin was unchanged by hepatic impairment. Delafloxacin was generally well tolerated by subjects with hepatic impairment and by matched healthy subjects. When delafloxacin is administered to patients with mild, moderate, or severe hepatic impairment, no initial dose adjustment appears warranted.
Four authors (M.Q., L.E.L., D.R.L., S.K.C.) are employed by Melinta Therapeutics, Inc; all research was funded by Melinta Therapeutics, Inc. The other authors were compensated for their work on this study.
These data were presented in part during ID Week 2015 in San Diego CA (poster #911).
This trial has been registered at ClinicalTrials.gov under registration number NCT02245243.
References
- 1. Bassetti M, Della Siega P, Pecori D, Scarparo C, Righi E. Delafloxacin for the treatment of respiratory and skin infections. Expert Opin Investig Drugs. 2015;24:433–442. [DOI] [PubMed] [Google Scholar]
- 2. Van Bambeke F. Delafloxacin, a non‐zwitterionic fluoroquinolone in phase III of clinical development: evaluation of its pharmacology, pharmacokinetics, pharmacodynamics, and clinical efficacy. Future Microbiol. 2015;10:1111–1123. [DOI] [PubMed] [Google Scholar]
- 3. Hoover R, Hunt T, Benedict M, et al. Safety, tolerability, and pharmacokinetic properties of intravenous delafloxacin after single and multiple doses in healthy volunteers. Clin Ther. 2016;38:53–65. [DOI] [PubMed] [Google Scholar]
- 4. Hoover R, Hunt T, Benedict M, et al. Single and multiple ascending‐dose studies of oral delafloxacin: effects of food, sex, and age. Clin Ther. 2016;38:39–52. [DOI] [PubMed] [Google Scholar]
- 5. Hoover R, Lawrence L, Benedict M, et al. Pharmacokinetics and relative bioavailability of intravenous and oral formulations of delafloxacin (DLX) in healthy subjects [poster]. Presented at Interscience Conference of Antimicrobial Agents and Chemotherapy; September 5, 2014; Washington, DC.
- 6. McEwen A, Lawrence L, Hoover R, et al. Disposition, metabolism and mass balance of delafloxacin in healthy human volunteers following intravenous administration. Xenobiotica. 2015;45:1054–1062. [DOI] [PubMed] [Google Scholar]
- 7. O'Riordan W, Mehra P, Manos P, Kingsley J, Lawrence L, Cammarata S. A randomized phase 2 study comparing two doses of delafloxacin with tigecycline in adults with complicated skin and skin‐structure infections. Int J Infect Dis. 2015;30:67–73. [DOI] [PubMed] [Google Scholar]
- 8. Kingsley J, Mehra P, Lawrence LE, et al. A randomized, double‐blind, phase 2 study to evaluate subjective and objective outcomes in patients with acute bacterial skin and skin structure infections treated with delafloxacin, linezolid or vancomycin. J Antimicrob Chemother. 2016;71:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cammarata S, Gardovskis J, Farley B, et al. Results of a global phase 3 study of delafloxacin (DLX) compared to vancomycin with aztreonam (VAN) in acute bacterial skin and skin structure infections (ABSSSI). Presented at IDWeek; October 7, 2015; San Diego, CA.
- 10. Child CG, Turcotte JG. Surgery and portal hypertension. Major Probl Clin Surg. 1964;1:1–85. [PubMed] [Google Scholar]
- 11. Pugh RN, Murray‐Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–649. [DOI] [PubMed] [Google Scholar]
- 12. Ambrose PG, Bhavnani SM, Rubino CM, et al. Pharmacokinetics‐pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin Infect Dis. 2007;44:79–86. [DOI] [PubMed] [Google Scholar]
- 13. King CD, Rios GR, Green MD, Tephly TR. UDP‐glucuronosyltransferases. Curr Drug Metab. 2000;1:143–161. [DOI] [PubMed] [Google Scholar]
- 14. Pacifici GM, Viani A, Franchi M, et al. Conjugation pathways in liver disease. Br J Clin Pharmacol. 1990;30:427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yeung CK, Yoshida K, Kusama M, et al. Organ impairment—drug‐drug interaction database: A tool for evaluating the impact of renal or hepatic impairment and pharmacologic inhibition on the systemic exposure of drugs. CPT Pharmacometr Syst Pharmacol. 2015;4:489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lawrence L, Quintas M, Hoover R, et al. A phase 1, open‐label study of the effect of delafloxacin (DLX) on the pharmacokinetics (PK) of a single oral dose of midazolam (MID). Presented at the American Society for Microbiology, Microbe 2016; June 16, 2016; Boston, MA.
- 17. http://www.drugs.com/drug-interactions/ciprofloxacin-index.html?filter=3&generic_only. Accessed May 18, 2016.
- 18. Keller F, Maiga M, Neumayer HH, Lode H, Distler A. Pharmacokinetic effects of altered plasma protein binding of drugs in renal disease. Eur J Drug Metab Pharmacokinet. 1984;9:275–282. [DOI] [PubMed] [Google Scholar]