

Abstract
Omadacycline is a first‐in‐class aminomethylcycline antibiotic being evaluated in phase 3 studies as oral and intravenous monotherapy for bacterial infections. This was a phase 1, randomized, open‐label, 4‐period, crossover study that evaluated the effect of food consumption on the bioavailability of omadacycline. Healthy participant were randomized to 1 of 4 sequences, which included the following predose conditions in different orders (A) ≥6‐hour fast, (B) high‐fat, nondairy meal 4 hours before dosing, (C) high‐fat, nondairy meal 2 hours before dosing, and (D) high‐fat meal containing dairy 2 hours before dosing. Participants received a single 300‐mg oral dose of omadacycline during each treatment period; periods were separated by ≥5 days. Blood samples for pharmacokinetic (PK) analysis were collected over 24 hours after each dose, and safety assessments were performed during each treatment period. Least‐squares mean and 90% confidence intervals were compared for fed state vs fasted state. Thirty‐one participants were included in the PK analysis. Fasted AUC0‐∞, AUC0‐t, and AUC0‐24 were 10.2, 7.2, and 7.2 μg·h/mL, respectively, and Cmax was 0.6 μg/mL. Compared with a fasted dose, bioavailability was reduced by 15% to 17% by a nondairy meal 4 hours before dosing, 40% to 42% by a nondairy meal 2 hours before dosing, and 59% to 63% for a dairy meal 2 hours before dosing. Two participants experienced adverse events (mild nausea, mild somnolence). A 300‐mg oral dose of omadacycline administered within 2 to 4 hours after food had reduced bioavailability compared with the fasted state. Oral omadacycline should be administered in a fasted state.
Keywords: bioavailability, fasted, food effect, omadacycline, pharmacokinetics
Omadacycline is a first‐in‐class aminomethylcycline antibiotic that is characterized by an aminomethyl group at the C9 position on the tetracycline structure, which distinguishes omadacycline from other tetracyclines and results in improved in vitro antimicrobial activity.1 Omadacycline is undergoing clinical development as oral and intravenous (IV) monotherapy for serious community‐acquired bacterial infections. In contrast to earlier‐generation tetracyclines, omadacycline retains activity against bacterial strains characterized by the 2 primary mechanisms of tetracycline resistance: ribosomal protection and efflux.2
Omadacycline demonstrates in vitro activity against Gram‐positive aerobes (including methicillin‐resistant Staphylococcus aureus and vancomycin‐resistant enterococci), some Gram‐negative aerobes, many anaerobes, and atypical pathogens including Legionella spp. and Mycoplasma spp.3, 4, 5, 6, 7, 8 In previous clinical studies in patients with complicated skin and skin structure infection, omadacycline treatment was started with IV administration, after which participants could transition to oral therapy (200 or 300 mg orally once daily, depending on the study) for a total of up to 14 days of treatment.9, 10 In these studies omadacycline was comparable to linezolid for clinical response, and the overall incidence of adverse events also was similar (58% omadacycline vs 63% linezolid).9, 11 The most common adverse event for both drugs was nausea (17% omadacycline vs 15% linezolid).11 Phase 3 studies are ongoing in patients with acute bacterial skin and skin structure infection (ABSSSI) and community‐acquired bacterial pneumonia (CABP), in which omadacycline is started with IV administration, followed by an option to transition to 300 mg orally once daily using a phase 3 tablet formulation. In addition, a phase 3 study in ABSSSI using an oral‐only regimen is planned, and development is being explored for additional uses including treatment of urinary tract infections.
Results from a number of phase 1 studies in healthy participants were undertaken to characterize the pharmacokinetic (PK) profile of omadacycline.12, 13, 14, 15, 16 After oral administration of a 300‐mg dose in a fasted state, the peak plasma concentration was 0.6 μg/mL, area under the concentration‐time curve was 10.3 h·μg/mL, and terminal elimination half‐life was approximately 17 hours. In a study of the absorption, distribution, metabolism, and elimination of omadacycline, 81% of an oral dose of omadacycline was excreted in the feces, with 14% of dose excreted in the urine; no metabolites of omadacycline were detected in either the urine or the feces. In addition, in vitro studies with human CYP isozymes found omadacycline to be neither a substrate nor an inhibitor of these enzymes.13
During the development process, oral omadacycline formulations have evolved from free base in a capsule through a series of tablet and salt formulations in order to optimize oral bioavailability while improving tolerability. In a study of an early tablet formulation, it was shown that when a light meal was given an hour after oral omadacycline administration to fasted participants, there was a 25% decrease in exposure compared to those who remained fasted and that this decrease was the result of a decrease in extent of omadacycline absorption, because there was no change in its elimination half‐life (data on file, Paratek Pharmaceuticals). The current phase 3 tablet formulation is the tosylate salt of omadacycline, which has been shown to have an absolute bioavailability of 34.5%.12 The primary objective of this study was to evaluate the relative bioavailability of a single oral 300‐mg dose of omadacycline (administered as the phase 3 tablet formulation) at various times after the consumption of food in healthy adult participants.
Methods
The study was conducted according to Good Clinical Practices and the ethical principles of the Declaration of Helsinki. The study was reviewed and approved by the independent Institutional Review Board of the PPD Phase I Clinic (Austin, Texas), where the study was conducted. Each eligible participant provided written informed consent prior to enrollment.
Study Design
This was a phase 1, randomized, open‐label, 4‐period, crossover study conducted in healthy adult participants. The study consisted of a screening period (21 days to 2 days prior baseline), 4 baseline periods (day –1 of each period), 4 treatment periods (days 1 through 2 of each period), and a study completion visit (within 6 to 10 days of the last dose of omadacycline). A washout period of at least 5 days was included between dosing periods.
Participants underwent screening evaluations to determine eligibility within 21 days before dosing in period 1. Participants were admitted to the clinical site on the day before dosing (day −1) in each period for baseline evaluations. Before dosing on day 1 of period 1, participants were randomized to 1 of 4 treatment sequences (Table 1). A 4‐period by 4‐sequence crossover Williams design was used. On day 1 of each period, participants received a single oral dose of 300 mg omadacycline at various times after the consumption of food.
Table 1.
Treatment Sequences
| Sequence | Period 1 | Period 2 | Period 3 | Period 4 |
|---|---|---|---|---|
| ADBC (N = 8) | A | D | B | C |
| BACD (N = 8) | B | A | C | D |
| CBDA (N = 8) | C | B | D | A |
| DCAB (N = 8) | D | C | A | B |
Participants received a single oral 300‐mg dose, administered as 2 150‐mg tablets, in each of 4 treatment periods. Blood samples for PK profiles were collected over 24 hours after each dose, and treatment periods were separated by at least 5 days.
Treatment A: participants fasted overnight (no food or drink except for water for at least 6 hours before dosing); a standard high‐fat (nondairy) meal was served 3 hours after dosing.
Treatment B: a standard high‐fat (nondairy) meal was completed 4 hours before dosing.
Treatment C: a standard high‐fat (nondairy) meal was completed 2 hours before dosing.
Treatment D: a standard high‐fat meal including dairy was completed 2 hours before dosing.
The high‐fat (approximately 50% of total caloric content of the meal) and high‐calorie (approximately 800 to 1000 calories) meal followed Food and Drug Administration guidance recommendations and provided approximately 150, 250, and 500 to 600 calories from protein, carbohydrate, and fat, respectively.17 These meals were to be consumed within 20 minutes. Dose administration for treatments B, C, and D was determined from the end time of the meal. During all 4 treatment periods, participants received no food or drink except water for at least 3 hours after dosing and no dairy products, antacids, or multivitamins for 4 hours after dosing.
The standard high‐fat meal (nondairy) consisted of 2 eggs fried in 2 teaspoonful of olive oil, 2 turkey sausage patties, 2 hash browns fried in 2 teaspoonful of olive oil, and 8 fluid ounces of apple juice (855 calories [kilocalories]). The dietary composition was 57.28 g fat (59.0% of calories), 59.12 g carbohydrate (27.0% of calories), and 30.58 g protein (14.0% of calories).
The standard high‐fat meal including dairy consisted of 2 eggs fried in 2 teaspoonfuls of butter, 2 strips of bacon, 2 slices of white toast with 2 teaspoonfuls of butter, 2 hash browns cooked in 2 teaspoonfuls of butter, and 8 fluid ounces of whole milk (985 calories [kilocalories]). The dietary composition was 64.94 g fat (59.9% of calories), 67.79 g carbohydrate (27.8% of calories), and 29.91 g protein (12.3% of calories).
Participant Selection
Male and female participants ages 18 to 55 years (inclusive) with a body mass index ≥18 to ≤30 kg/m2 and in good health based on medical history, physical examination, vital signs, clinical laboratory measurements, and 12‐lead electrocardiogram (ECG) were eligible. Participants were required to be afebrile and have blood pressure and pulse rate within normal ranges (systolic 90–140 mm Hg; diastolic 50–90 mm Hg; heart rate 40–90 beats/min). Women were required to have a negative serum pregnancy test and to use a highly effective form of contraception during the study.
Participants were excluded for use of other investigational drugs within 30 days of screening or any prescription drugs, herbal supplements, or over‐the‐counter drugs within 2 to 4 weeks of day –1; recent smoking history or history of bronchospastic disease; history of autonomic dysfunction; history of clinically significant cardiac rhythm abnormality; history or evidence of immunodeficiency or a positive test for human immunodeficiency virus; hypersensitivity or allergic reaction to any tetracycline; or history of any clinically significant medical condition (eg, gastrointestinal disease or major gastrointestinal surgery; hepatitis B or hepatitis C infection or other liver disease; renal impairment or urinary obstruction; pancreatitis) that could interfere with the conduct or interpretation of the study.
Study Assessments
Blood samples for PK assessments of omadacycline were collected from all participants before dosing (predose) and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 16, and 24 hours after dosing in each period. The following PK parameters were calculated for omadacycline from plasma concentration vs actual time data for each participant using noncompartmental analysis: area under the plasma concentration‐time curve (AUC) from time 0 to 24 hours after dosing (AUC0‐24), AUC from time 0 to the last quantifiable concentration (AUC0‐t), AUC time 0 extrapolated to infinity (AUC0‐∞), maximum (peak) observed plasma concentration (Cmax), time to reach Cmax (Tmax), and terminal elimination half‐life (T1/2).
Safety and tolerability were assessed by monitoring and recording of adverse events (AEs), clinical laboratory test results (hematology, serum chemistry, and urinalysis), vital sign measurements, physical examination findings, and 12‐lead ECG results.
Statistical Analysis
The individual PK parameters for omadacycline were summarized by treatment using descriptive statistics: number, mean, standard deviation (SD), coefficient of variation (CV), median, minimum, and maximum. Geometric means were included for AUC and Cmax. The individual plasma concentration‐vs‐time data for omadacycline were used to derive the PK parameters using noncompartmental analysis, as deemed appropriate, using Phoenix® WinNonlin® (Pharsight Corp, St. Louis, Missouri), Version 6.2.1. Actual sampling times, rather than scheduled sampling times, were used in the computation of PK parameters.
A linear mixed‐effect model (SAS PROC MIXED) with treatment condition, sequence, and period as fixed effects and participant nested within sequence as a random effect was fitted to the natural‐log‐transformed PK parameters AUC0‐24, AUC0‐t, AUC0‐∞, and Cmax for estimation of effects and construction of confidence intervals (CI) for the test treatments (fed states: treatments B, C, and D) compared with the reference treatment (fasted state: treatment A).
Absence of the effect of food was concluded if the 90%CI for the test‐to‐reference ratios (B/A, C/A, or D/A) of geometric means were entirely contained within the criterion interval of 80% to 125% for AUC0‐24, AUC0‐t, AUC0‐∞, and Cmax. For Tmax, the Wilcoxon signed‐rank test was performed. A P‐value ≤.05 was considered a statistically significant difference between the reference and test treatments.
The sample size determination was based on consideration of the precision of the estimate of the geometric mean ratio (GMR) of PK parameters in the fed state compared with those in the fasted state. Given the study design, 24 evaluable participants would provide a 90% probability that the 90% confidence interval (CI) of the GMR would be within 87% and 115% of the point estimate. These precision estimates were based on an assumption that the PK parameters are log‐normally distributed, with intersubject CVs no greater than 24% (as had been observed in prior PK studies). Allowing for up to 2 participants per sequence to discontinue before completing all study periods, the total planned enrollment was 32 participants.
Results
Thirty‐two participants were enrolled in the study and were included in the PK and safety analyses. The overall mean age of participants was 32.3 years, with a range of 21 to 50 years (Table 2). The majority of participants were white (24 participants, 75%), and 15 participants (47%) were male. One participant was discontinued from the study because of a positive alcohol screen and did not receive treatments D and A. Another participant requested to be withdrawn and did not receive treatments B and C. Therefore, PK data were available for 31 participants for all treatments.
Table 2.
Baseline Demographics
| Participants (N = 32) | |
|---|---|
| Age, yearsa | 32.3 (8.0) |
| Age range, years | 21‐50 |
| Male, n (%) | 15 (46.9) |
| Race, n (%) | |
| White | 24 (75.0) |
| Black/African American | 8 (25.0) |
| Hispanic/Latino | 12 (37.5) |
| Height, cma | 168.0 (9.5) |
| Weight, kga | 71.5 (13.4) |
| BMI, kg/m2 a | 25.2 (3.2) |
BMI, body mass index.
Mean (standard deviation).
Pharmacokinetic Analysis
Fasted AUC0‐∞, AUC0‐t, and AUC0‐24 were 10.2, 7.2, and 7.2 μg·h/mL, respectively, and Cmax was 0.6 μg/mL (Table 3). A notable reduction in systemic exposure to omadacycline was observed for the 3 test treatments (treatments B, C, and D) versus treatment A (Figure 1). The effect of food was more pronounced when a high‐fat meal was consumed closer to dosing and when dairy was included in the meal. Across all treatment sequences, mean T1/2 ranged from 13.5 to 13.8 hours.
Table 3.
Plasma Pharmacokinetic Parameters for Omadacycline After a Single 300‐mg Oral Dose
| Mean (Coefficient of Variation) | ||||
|---|---|---|---|---|
| Parameter | Treatment A (N = 31) | Treatment B (N = 31) | Treatment C (N = 31) | Treatment D (N = 31) |
| AUC0‐24, μg·h/mL | 7.2 (28.1) | 6.1 (26.3) | 4.2 (23.4) | 2.8 (44.3) |
| AUC0‐t, μg·h/mL | 7.2 (28.1) | 6.1 (26.3) | 4.2 (23.4) | 2.8 (44.4) |
| AUC0‐∞, μg·h/mL | 10.2 (27.0)b | 8.8 (29.2) | 6.0 (25.4) | 4.0 (44.1) |
| Cmax, μg/mL | 0.6 (25.3) | 0.6 (25.0) | 0.4 (22.4) | 0.3 (42.6) |
| Tmax, hoursa | 2.5 (1.5, 4.1) | 2.9 (1.0, 6.0) | 2.9 (1.0, 6.0) | 2.9 (1.0, 6.0) |
| T1/2, hours | 13.8 (10.3)b | 13.6 (12.7) | 13.6 (12.2) | 13.5 (14.7) |
Treatment A: participants fasted overnight (no food or drink except for water for at least 6 hours before dosing); a standard high‐fat (nondairy) meal was served 3 hours after dosing.
Treatment B: a standard high‐fat (nondairy) meal was completed 4 hours before dosing.
Treatment C: a standard high‐fat (nondairy) meal was completed 2 hours before dosing.
Treatment D: a standard high‐fat meal including dairy was completed 2 hours before dosing.
Median (minimum, maximum) values.
N = 30.
Figure 1.
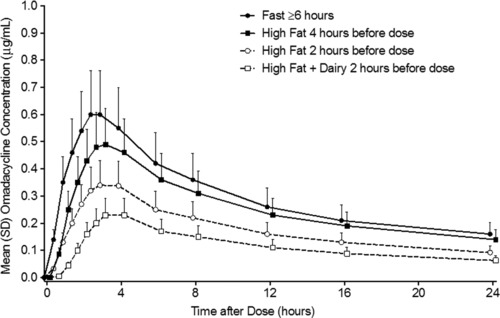
Plasma concentration‐time curve for omadacycline.
The between‐participant variability in systemic exposure to omadacycline was similar between treatment A (no meal before dosing) and treatments B and C (nondairy meal 4 and 2 hours before dosing, respectively), with CV ranging from 22.4% to 29.2% for Cmax, AUC0‐24, AUC0‐t, and AUC0‐∞. By contrast, for treatment D (meal containing dairy 2 hours before dosing), the corresponding CVs were higher, ranging from 42.6% to 44.4%.
For the tested PK parameters (Cmax, AUC0‐t, AUC0‐∞, AUC0‐24, and Cmax), an absence of food effect was not confirmed because the 90%CI of the geometric least‐squares mean ratios (test/reference) was not contained within the criterion interval of 80% to 125% (Table 4). Compared with a fasted dose, omadacycline exposure (Cmax and AUC) was reduced by 15% to 17% for a nondairy meal 4 hours before dosing, by 40% to 42% for a nondairy meal 2 hours before dosing, and by 59% to 63% for a dairy meal 2 hours before dosing. A slight difference in median Tmax was observed between treatment A (2.5 hours) and treatments B, C, and D (2.9 hours); however, the difference was not statistically significant.
Table 4.
Statistical Analysis of the Effect of Food on Plasma Pharmacokinetic Parameters of Omadacycline (N = 31)
| Parameter | Treatment | Geometric LS Mean | Treatment Comparison | Ratio of Geometric LS Mean (%) | 90%CI of Ratio (%) |
|---|---|---|---|---|---|
| AUC0‐24, μg·h/mL | A | 7.4 | |||
| B | 6.2 | B/A | 83.4 | 74.9, 92.7 | |
| C | 4.3 | C/A | 57.7 | 51.9, 64.2 | |
| D | 2.8 | D/A | 37.3 | 33.6, 41.5 | |
| AUC0‐t, μg·h/mL | A | 7.4 | |||
| B | 6.2 | B/A | 83.3 | 74.9, 92.7 | |
| C | 4.3 | C/A | 57.7 | 51.9, 64.1 | |
| D | 2.8 | D/A | 37.3 | 33.5, 41.4 | |
| AUC0‐∞, μg·h/mL | Aa | 10.6 | |||
| B | 9.0 | B/A | 84.7 | 75.8, 94.6 | |
| C | 6.2 | C/A | 58.4 | 52.3, 65.3 | |
| D | 4.0 | D/A | 37.9 | 34.0, 42.3 | |
| Cmax, μg/L | A | 0.66 | |||
| B | 0.56 | B/A | 84.5 | 75.9, 94.1 | |
| C | 0.39 | C/A | 60.1 | 54.0, 66.9 | |
| D | 0.27 | D/A | 40.7 | 36.5, 45.2 |
CI, confidence interval; LS, least squares.
N = 30, a terminal monoexponential phase could not be identified for 1 participant.
Safety and Tolerability
Two participants experienced treatment emergent adverse events (AEs) (1 reported nausea, 1 reported somnolence). Both events were of mild intensity, and both participants continued in the study. No participant discontinued the study for an AE, and no participant experienced a serious AE. No abnormal physical examination or findings were reported, and there were no clinically significant changes in clinical laboratory tests or ECG findings.
A slight increase from baseline in heart rate (median 8 to 10 beats/min at 4 to 6 hours postdose) was observed for treatment A (ie, the group with highest omadacycline exposure). In all other treatment groups the median change from baseline in heart rate was ≤3 beats/min at all measured time points. No notable changes in blood pressure were observed.
Discussion
The pharmacokinetics of tetracyclines vary widely,18 possibly related to chemical structure and structure‐activity relationships of older‐ vs newer‐generation products. When administered with food, absorption, and thus systemic exposure with older tetracyclines is reduced by 50%, whereas systemic exposure to the newer‐generation oral doxycycline is reduced by only 20%.18 In contrast, administration with food has almost no effect on the systemic exposure to oral minocycline.18 Oral absorption of the most recent additions to the tetracycline class of antibiotics also is widely variable. Tigecycline is poorly absorbed orally, and therefore, it is only available for IV administration.19, 20 In contrast, a study of the effect of food on the PK profile of eravacycline after oral administration reported a 62% reduction in AUC.21
Previous clinical studies using earlier oral formulations of omadacycline showed that dosing with food notably decreased exposure to omadacycline but had no effect on its elimination half‐life, indicating that food consumption had an impact on the amount of the omadacycline dose absorbed but not on the elimination processes (data on file, Paratek Pharmaceuticals). This study was designed to evaluate the effect of food on the most current phase 3 tablet formulation and to quantify any differences in exposure based on varying the time between a meal and a subsequent oral dose. The PK results obtained with a 300‐mg oral dose of omadacycline in the fasted state were consistent with results from an earlier study of this phase 3 tablet formulation,12 thereby supporting these fasted data as the “reference” values. Single doses of omadacycline were well tolerated in all dosing periods in this study, also consistent with the overall safety profile observed in prior clinical studies.
This study demonstrated a significant reduction in exposure when oral omadacycline 300 mg was administered in various fed states compared with the fasted state. Based on AUC, omadacycline exposure varied by timing with a 17% decrease in exposure when participants were fed a nondiary meal 4 hours prior to dosing and a 42% decrease when fed a nondiary meal 2 hours before dosing. When dairy was included in the meal 2 hours before dosing, exposure was decreased by 63% compared to fasting administration. The elimination half‐life of omadacycline was unaffected by food intake, with values ranging from 13.5 to 13.8 hours across all the study arms. Thus, the food effect was more pronounced when a meal was consumed closer to oral dosing, with an even greater effect when dairy was included in the meal. The latter findings are consistent with the known tetracycline characteristic of binding to calcium as well as other multivalent cations. Older tetracycline compounds including tetracycline, oxytetracycline, and chlortetracycline, which exhibit oral absorption ranging from 25% to >80%, form an insoluble complex with calcium, magnesium, aluminum, and iron that markedly reduces absorption after oral administration.18 Although food does not impact the oral absorption of minocycline, absorption is reduced by administration with iron‐containing products and antacids containing calcium and magnesium.18 Various antacid preparations and multivitamin products may contain 1 or more of the multivalent cations (calcium, magnesium, aluminum, iron, bismuth, or zinc), and therefore, coadministration of these products with oral tetracyclines should be avoided. In conclusion, based on the food effect observed in this study, omadacycline tablets should be taken in a fasted state, and coadministration with dairy products, cation‐containing antacids, or multivitamins should be avoided.
Disclosures
E.T., A.M., S.V., S.K.T., S.B., and E.L. are employees of Paratek Pharmaceuticals. The authors acknowledge the editorial assistance of Richard S. Perry, PharmD, in the preparation of this manuscript, which was supported by Paratek Pharmaceuticals, King of Prussia, Pennsylvania. Rebecca N. Wood‐Horrall, MD, PPD Inc, was the principal investigator at the study site.
References
- 1. Honeyman L, Ismail M, Nelson M, et al. Structure‐activity relationship of the aminomethylcyclines and the discovery of omadacycline. Antimicrob Agents Chemother. 2015;59:7044–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Draper MP, Weir S, Macone A, et al. Mechanism of action of the novel aminomethylcycline antibiotic omadacycline. Antimicrob Agents Chemother. 2014;58:1279‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dubois J, Dubois M, Martel J‐F, Tanaka SK. In vitro activity of omadacycline against Legionella pneumophila. Presented at 55th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; September 17‐21, 2015; San Diego, CA. Poster P‐1992.
- 4. Flamm RK, Rhomberg PR, Huband MD, Farrell DJ. Activity of omadacycline tested against Enterobacteriaceae causing urinary tract infections from a global surveillance program (2014). Presented at 55th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; September 17‐21, 2015; San Diego, CA. Poster C‐614.
- 5. Flamm RK, Rhomberg PR, Huband MD, Farrell DJ. Activity of omadacycline tested against Streptococcus pneumoniae from a global surveillance program (2014). Presented at 55th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; September 17‐21, 2015; San Diego, CA. Poster C‐554.
- 6. Flamm RK, Rhomberg PR, Huband MD, Farrell DJ. Activity of omadacycline tested against Staphylococcus aureus from a global surveillance program (2014). Presented at 55th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; September 17‐21, 2015; San Diego, CA. Poster F‐289.
- 7. Kim O, Leahy RG, Traczewski M, Macone A, Steenbergen J, Tanaka SK. Activity and efficacy of omadacycline against Clostridium difficile . Presented at 26th European Congress on Clinical Microbiology and Infectious Diseases (ECCMID); April 9‐12, 2016; Amsterdam, the Netherlands. Poster P‐1325.
- 8. Macone AB, Caruso BK, Leahy RG, et al. In vitro and in vivo antibacterial activities of omadacycline, a novel aminomethylcycline. Antimicrob Agents Chemother. 2014;58:1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Noel GJ, Draper MP, Hait H, Tanaka SK, Arbeit RD. A randomized, evaluator‐blind, phase 2 study comparing the safety and efficacy of omadacycline to those of linezolid for treatment of complicated skin and skin structure infections. Antimicrob Agents Chemother. 2012;56:5650–5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Noel GJ, Draper MP, Hait H, Tanaka SK. Safety and efficacy of PTK 0796 (omadacycline) as treatment of complicated skin and soft tissue infection (cSSTI). Presented at 23rd European Congress on Clinical Microbiology and Infectious Diseases, (ECCMID); March 31‐April 3, 2012, London, UK. Poster P‐694.
- 11. Tanaka SK, Chitra S, Garrity‐Ryan L, Tzanis E, Loh E. A pooled analysis of two randomized multicenter, evaluator‐blind studies comparing the safety and efficacy of omadacycline and linezolid for the treatment of complicated skin and skin structure infections. Presented at 26th European Congress on Clinical Microbiology and Infectious Diseases (ECCMID); April 9‐12, 2016, Amsterdam, the Netherlands. Poster P‐1326.
- 12. Sun H, Maietta R, Machineni S, et al. A single‐dose study to evaluate the pharmacokinetics, safety, and tolerability of multiple formulations of PTK 0796 in healthy subjects. Presented at 21st European Congress on Clinical Microbiology and Infectious Diseases (ECCMID); May 7‐11, 2011, Milan, Italy. Poster P‐1423.
- 13. Flarakos J, Du Y, Gu H, et al. Clinical disposition, metabolism and in vitro drug‐drug interaction properties of omadacycline [published online ahead of print August 8, 2016]. Xenobiotica. [DOI] [PubMed] [Google Scholar]
- 14. Tanaka SK, Tzanis E, Villano S. Effect of age and gender on the pharmacokinetics of the oral and IV omadacycline, A new class of aminomethylcyclines. Presented at 26th European Congress on Clinical Microbiology and Infectious Diseases (ECCMID); April 9‐12, 2016, Amsterdam, the Netherlands. Poster P‐1318.
- 15. Ting L, Kovacs SJ, Prestgaard J, et al. An open‐label study of the pharmacokinetics and safety of single IV and oral doses of omadacycline in subjects with mild, moderate, and severe hepatic impairment. Presented at 52nd Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; September 9‐12, 2012; Washington, DC. Poster A‐1282.
- 16. Ting L, Sun H, Kovacs SJ, Klausner K, Tanaka SK. Pharmacokinetics of intravenous and oral PTK796, a new aminomethylcycline antibiotic. Presented at 50th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; September 12‐15, 2010; Boston, MA. Abstract K‐124.
- 17. Department of Health and Human Services , Food and Drug Administration (DHHS) , Center for Drug Evaluation and Research (US). Guidance for industry: food‐effect bioavailability and fed bioequivalence studies. December 2002.
- 18. Agwuh KN, MacGowan A. Pharmacokinetics and pharmacodynamics of the tetracyclines including glycylcyclines. J Antimicrob Chemother. 2006;58:256–265. [DOI] [PubMed] [Google Scholar]
- 19. MacGowan AP. Tigecycline pharmacokinetic/pharmacodynamic update. J Antimicrob Chemother. 2008;62(Suppl 1):i11–i16. [DOI] [PubMed] [Google Scholar]
- 20. Meagher AK, Ambrose PG, Grasela TH, Ellis‐Grosse EJ. The pharmacokinetic and pharmacodynamic profile of tigecycline. Clin Infect Dis. 2005;41(Suppl 5):S333–S340. [DOI] [PubMed] [Google Scholar]
- 21. Horn PT, Sutcliffe JA, Walpole SM, Leighton A. Pharmacokinetics, safety and tolerability of a novel fluorocycline, TP‐434, following multiple dose administration. Presented at 49th Infectious Diseases Society of America Annual Meeting; October 20‐23, 2011; Boston, MA.